

Abstract
Huntington's disease (HD) is characterized by the selective loss of striatal projection neurons. In early stages of HD, neurodegeneration preferentially occurs in the lateral globus pallidus (LGP) and substantia nigra (SN), two regions in which the axons of striatal neurons terminate. Here we report that in mice expressing full-length mutant huntingtin and modeling early stages of HD, neuropil aggregates form preferentially in the LGP and SN. The progressive formation of these neuropil aggregates follows intranuclear accumulation of mutant huntingtin and becomes prominent from 11 to 27 months after birth. Neuropil aggregates, but no intranuclear inclusions, were observed in the LGP and SN, suggesting that huntingtin aggregates are formed in the axons of striatal projection neurons. In the LGP and SN, we observed degenerated axons in which huntingtin aggregates were associated with dark, swollen organelles that resemble degenerated mitochondria. Neuritic aggregates also form in cultured striatal neurons expressing mutant huntingtin, block protein transport in neurites, and cause neuritic degeneration before nuclear DNA fragmentation occurs. These findings suggest that the early neuropathology of HD originates from axonal dysfunction and degeneration associated with huntingtin aggregates.
Keywords: Huntingtin, polyglutamine, axon, degeneration, transport, mitochondria
Studies of postmortem brains from patients with Huntington's disease (HD) have indicated that medium spiny neurons are selectively degenerated (Ferrante et al., 1985;Graveland et al., 1985; Vonsattel et al., 1985). The majority of these neurons extend their axons to the lateral globus pallidus (LGP), the medial globus pallidus (MGP) (also known as the internal globus pallidus), and the substantia nigra (SN). Striatal neurons projecting to the LGP are enriched in enkephalins, whereas striatal neurons projecting to the MGP and SN are enriched in substance P (Graybiel, 1990). The decreased density of enkephalin-immunoreactive fibers in the LGP and substance P-immunoreactive neuropils in the SN is found in early HD patients. In addition, the LGP and SN are more significantly degenerated than the MGP during early stages of HD (Reiner et al., 1988; Richfield et al., 1995). More importantly, such early neuropil degeneration has also been observed in presymptomatic HD patients (Albin et al., 1990, 1992), suggesting that degeneration of striatal projection neurons may be initiated from their axons.
The widespread expression of the HD protein, huntingtin, provides no clues to the selective HD pathology. In HD, mutant huntingtin accumulates in the nucleus, in contrast to the predominantly cytoplasmic distribution of normal huntingtin. Dystrophic neurites, which are reported to be extracellular structures containing axonal processes, also contain mutant huntingtin in HD patient brains (DiFiglia et al., 1997). Although intranuclear accumulation of mutant huntingtin affects gene expression in HD animal and cell models (Cha et al., 1998; S. H. Li et al., 1999; Luthi-Carter et al., 2000;Nucifora et al., 2001), nuclear polyglutamine inclusions (NIs) do not correlate with neurodegeneration (Klement et al., 1998; Saudou et al., 1998; Kuemmerle et al., 1999; Warrick et al., 1999; Kazemi-Esfarjani and Benzer, 2000). Recently, we have identified small huntingtin aggregates in dendrites and axons and named them neuropil aggregates (Gutekunst et al., 1999; H. Li et al., 1999, 2000). Similarly, in the brains of HD patients, N-terminal huntingtin accumulates and forms puncta in corticostriatal fibers (Sapp et al., 1999). In the R6/2 HD mouse model that expresses N-terminal mutant huntingtin, the formation of neuropil aggregates is highly correlated with disease progression in the absence of obvious neurodegeneration (H. Li et al., 1999).
Although our previous study showed that huntingtin aggregates are enriched in striatal medium spiny neurons in HD mice (H. Li, et al., 2000), it is unclear whether neuropil aggregates are selectively localized in those neurons that are preferentially affected by HD and whether these aggregates are associated with early neuropathological changes. HD-repeat knock-in mice that express full-length mutant huntingtin under the endogenous mouse Hdhpromoter do not show obvious neurological symptoms (Levine et al., 1999; Shelbourne et al., 1999; Wheeler et al., 2000), providing a good animal model to uncover neuropathological changes in presymptomatic or early HD. Using these HD mice, we demonstrate that huntingtin aggregates are preferentially formed in the striatal projection neurons that degenerate during the early stages of HD. In cultured neurons, huntingtin aggregates cause neuritic degeneration before nuclear DNA fragmentation.
MATERIALS AND METHODS
Light microscopic examination. R6/2 mice (B6CBA-TgN (HDexon1)62) that express exon1 of the human mutant HD gene with 115–150 CAGs under the human HD gene promoter (Mangiarini et al., 1996). These mice were obtained from The Jackson Laboratory (Bar Harbor, ME) at 4–12 weeks of age. HD-repeat knock-in mice that express 72–80 CAGs in the endogenous mouse HD gene were generated as described previously (Shelbourne et al., 1999). Mice were anesthetized and then perfused intracardially with PBS, pH 7.2, for 30 sec followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB) at a pH of 7.2. Brains were removed, cryoprotected in 30% sucrose at 4°C, and sectioned at 40 μm using a freezing microtome. Free-floating sections were preblocked in 4% normal goat serum (NGS) in PBS, 0.1% Triton X-100, and avidin (10 μg/ml) and then incubated with EM48, a rabbit polyclonal antibody against the first 256 amino acids of human huntingtin (Gutekunst et al., 1999; H.Li et al., 2000), at 4°C for 48 hr. The EM48 immunoreactive product was visualized with an avidin–biotin complex kit (Vector Laboratories, Burlingame, CA).
Electron microscopic immunocytochemistry. Immunogold labeling was performed as described previously (H. Li et al., 1999,2000). Briefly, mice were fixed by perfusion with PBS containing 4% paraformaldehyde and 0.2% glutaraldehyde. After perfusion, the brain was removed, post-fixed with 4% paraformaldehyde in PB for 6–8 hr, and sectioned using a vibratome. Brain sections were incubated with EM48 in PBS containing 4% NGS for 24–60 hr at 4°C and then with Fab fragments of goat anti-rabbit secondary antibodies (1:50) conjugated to 1.4 nm gold particles (Nanoprobes Inc., Stony Brook, NY) in PBS with 4% NGS overnight at 4°C. After rinsing in PBS, sections were fixed again in 2% glutaraldehyde in PB for 1 hr, silver intensified using the IntenSEM kit (Amersham International, Buckinghamshire, UK), osmicated in 1% OsO4 in PB, and stained overnight in 2% aqueous uranyl acetate.
All sections used for electron microscopy (EM) were dehydrated in ascending concentrations of ethanol and propylene oxide/Eponate 12 (1:1) and embedded in Eponate 12 (Ted Pella, Redding, CA). Ultrathin sections (60 nm) were cut using a Leica Ultracut S ultramicrotome (Leica, Nussloch, Germany). Thin sections were counterstained with 5% aqueous uranyl acetate for 5 min followed by Reynolds lead citrate for 5 min and examined using a Hitachi H-7500 electron microscope (Hitachi, Tokyo, Japan).
Quantification of aggregates in the brain. We quantified the immunoreactive aggregates in sections using light microscopy at 63× magnification with a Zeiss microscope (Axioskop 2; Zeiss, Thornwood, NY) and video system (Dage-MTI Inc. Michigan City, IN). All huntingtin aggregates in the captured images (8455 μm2/frame) were counted. The aggregates were categorized as intranuclear or neuropil aggregates. Neuropil aggregates are localized outside the cell body and their size is usually smaller, whereas nuclear aggregates or NIs appeared as single inclusions within the nucleus. Data analysis was performed using SigmaPlot 4.11E and Student's t tests.
Primary neuronal culture and transfection. Neurons were isolated from the striatum of embryonic day 14–16 rats and cultured in neurobasal/B27 medium according to the method used in our previous study (S. H. Li et al., 2000). After culturing for 3–4 d, the striatal neurons were transfected with huntingtin cDNAs using CaPO4 transfection. We obtained a 2–5% transfection rate. Huntingtin exon1 cDNA encoding 20 (GFP-20Q) or 120 (GFP-120Q) glutamines was fused in-frame to the C terminus of green fluorescent protein (GFP) in the GFP expression vector (Clontech, Palo Alto, CA). After 24–48 hr of transfection, cells were used for immunofluorescence analysis. Cotransfection of striatal neurons with GFP-120Q and red fluorescent protein (RFP) (Clontech) was also performed. For immunofluorescent double labeling, we used mouse antibodies to tubulin (Sigma, St. Louis, MO) and rabbit antibody EM48 to huntingtin. To examine fluorescence in living cells, cell-cultured dishes (60 mm) were placed on a temperature-controlled stage (20/20 Technology, Mississauga, Canada) and cells were maintained at 37°C with a constant CO2 flow that maintained the medium pH at ∼7.4. A Zeiss fluorescent microscope (Axiovert S100) and video system (Dage-MTI Inc.) were used to capture images. The captured images were stored and processed using Adobe Photoshop software (Adobe Systems, San Jose, CA). Quantitative analysis of neurons containing aggregates and neuritic degeneration was performed by counting 324–435 GFP-120Q-transfected neurons and 113–215 GFP-20Q- or GFP vector-transfected neurons in two to three independent experiments. Data were expressed as the mean ± SD and the statistical analysis was performed with the Student's t test.
RESULTS
Selective accumulation of huntingtin aggregates in striatal neurons in HD mice expressing full-length mutant huntingtin protein
Previous studies have shown that HD transgenic mice expressing N-terminal fragments of mutant huntingtin have more severe neurological symptoms than HD-repeat knock-in mice, which express full-length mutant huntingtin (Davies et al., 1997; Levine et al., 1999; Schilling et al., 1999; Shelbourne et al., 1999; Wheeler et al., 2000). Because N-terminal mutant huntingtin is toxic and forms aggregates in the brain (Davies et al., 1997; DiFiglia et al., 1997; Gutekunst et al., 1999;Hodgson et al., 1999; H. Li et al., 2000), we compared aggregate formation in various brain regions of these two types of HD mice. For immunocytochemical studies, we used the EM48 antibody, which specifically recognizes mutant huntingtin and its aggregates (Gutekunst et al., 1999; H. Li et al., 1999, 2000). In R6/2 mice, which express the HD exon1 mutant huntingtin with 115–150 glutamines (Davies et al., 1997), nuclear huntingtin aggregates are widely distributed in all of the brain regions examined, including the hippocampus and cerebellum, two regions that are relatively unaffected in HD. In contrast, HD-repeat knock-in mice, which express 72–80 CAGs in the endogenous mouse Hdh gene (Shelbourne et al., 1999), show the highest density of nuclear huntingtin aggregates in the striatum. The cortex, hippocampus, and cerebellum display weak or no nuclear huntingtin staining (Fig. 1). Thus, the context of full-length mutant huntingtin clearly influences the cell-specific formation of aggregates.
Fig. 1.
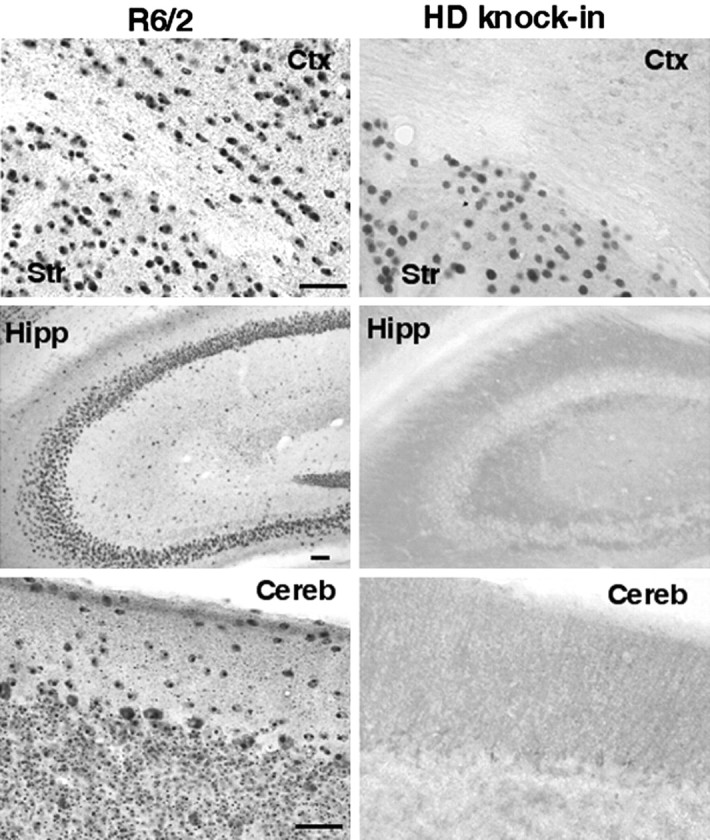
Selective accumulation of nuclear huntingtin aggregates in striatal neurons in HD-repeat knock-in mice. Low-magnification micrographs of brain sections from an R6/2 mouse at 12 weeks of age and a HD-repeat knock-in mouse at 24 months of age are shown. The sections were stained with EM48. Nuclear huntingtin aggregates are widely distributed in R6/2 mice, which express N-terminal mutant huntingtin. In the HD-repeat knock-in mouse brain, which expresses full-length mutant huntingtin, only the striatal neurons (Str) were immunoreactive with EM48.Ctx, Cortex; Hipp, hippocampus;Cereb, cerebellum. Scale bars, 25 μm.
Neuropil aggregates are formed in projection neurons
Next, we examined the distribution of neuropil aggregates in the LGP, MGP, and SN of HD-repeat knock-in mice. All three of these regions receive axons from striatal projection neurons. The SN is composed of the substantia nigra pars reticulate (SNr) and the substantia nigra pars compacta. Because the SNr constitutes the major part of the SN, we focused on the SNr and refer to it as the SN. We also included an R6/2 mouse brain for comparison.
As expected, both neuropil and intranuclear aggregates were observed in the striatum (Fig. 2). Because striatal neuronal cell bodies also contain huntingtin aggregates, it is difficult to determine whether neuropil aggregates in the striatum are from cortical projection neurons or from the neurons within the striatum. However, only neuropil aggregates, but not intranuclear inclusions, were seen in the striatal projection regions (LGP, SN, and MGP) in HD-repeat knock-in mice. This is consistent with observations that intranuclear accumulation of N-terminal huntingtin does not occur in the globus pallidus region of the human HD brain (Sapp et al., 1999). The lack of intranuclear staining in the projection regions suggests that neurons within these regions do not accumulate mutant huntingtin fragments. Thus, neuropil aggregates seen in the projection regions are likely to be the aggregates that are formed in the axons from striatal projection neurons. Interestingly, neuropil aggregates are more abundant in the LGP and SN than in the MGP. The LGP and SN are reported to show the earliest degenerative changes in HD patient brains (Reiner et al., 1988; Richfield et al., 1995). The high density of neuropil aggregates in the LGP and SN suggests that striatal neurons projecting to these two regions contain more axonal aggregates, whereas the lower frequency of neuropil aggregates in the MGP suggests that striatal neurons projecting to the internal globus pallidus form fewer aggregates in their axons.
Fig. 2.
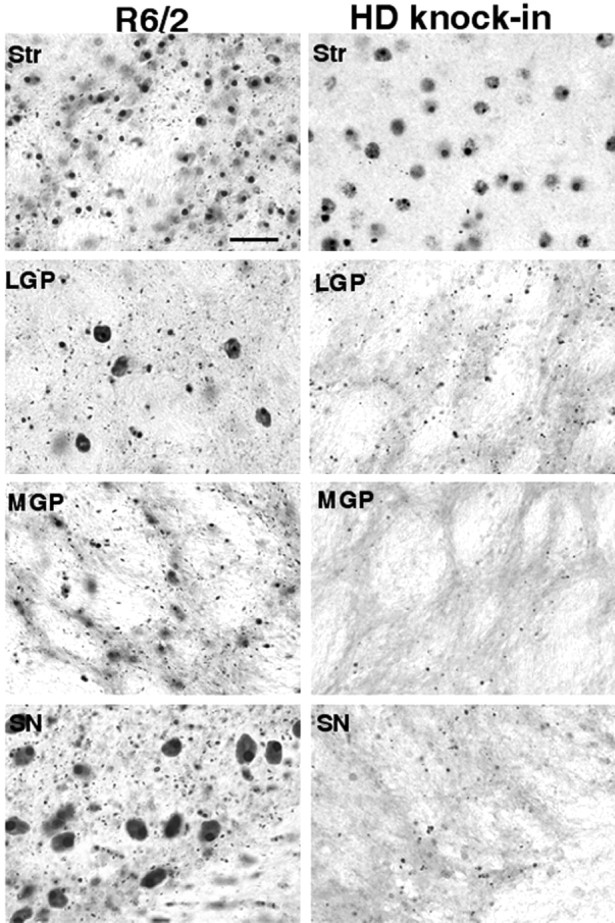
Selective distribution of neuropil aggregates in striatal projection regions in the HD-repeat knock-in mouse brain. Micrographs of the striatum (Str), LGP, MGP, and SN from an R6/2 mouse at 12 weeks of age and a HD-repeat knock-in mouse at 21 months of age are shown. In the R6/2 mouse, intranuclear and neuropil aggregates are widely distributed in all brain regions. In the HD-repeat knock-in mouse, the LGP and SN contain only neuropil aggregates. Note that the neuropil aggregates are more abundant in the LGP and SN than in the MGP. Scale bar, 20 μm.
Progressive formation of neuropil aggregates in striatal projection neurons
If neuropil aggregates are involved in the progressive neuropathology of HD, their formation may also be progressive and precede neurodegeneration. Therefore, we examined the temporal relationship between the formation of neuropil aggregates and the age of HD-repeat knock-in mice. At 4 months of age, we only saw very faint and diffuse nuclear staining in the striatum. No obvious neuropil aggregates were seen at this age. At 7–8 months, a few neuropil aggregates appeared in both the striatum and the LGP. At 11–12 months, significant neuropil aggregates were observed in the striatal projection regions, such as the LGP and SN. At 21–24 months, neuropil aggregates were even more abundant. The formation of neuropil aggregates in the sections of the LGP is illustrated in Figure3A. Quantification of neuropil aggregates (Fig. 3B) confirms that these aggregates are much more abundant in the LGP and SN than in other regions at all ages examined. As in previous studies (H. Li et al., 2000; Wheeler et al., 2000), nuclear aggregates were always more frequent in the striatum compared with other regions. They are also more abundant than neuropil aggregates (data not shown). These findings suggest that mutant huntingtin first accumulates in the nucleus of striatal neurons to form intranuclear inclusions and then forms aggregates in their processes.
Fig. 3.

Age dependence of neuropil aggregates in HD-repeat knock-in mice. A, EM48 immunostaining of the LGP in HD knock-in mice at 4, 8, 12, and 24 months of age. The density of neuropil aggregates is greater in older mice. Scale bar, 10 μm.B, Quantification of neuropil aggregate density in various brain regions from HD-repeat knock-in mice at different ages (7–24 months). Values are expressed as the mean ± SD and were obtained by counting aggregates in three to five images (8455 μm2/per image) for each brain region.Ctx, Cortex; STR, striatum.
Mutant huntingtin aggregates are associated with axonal degeneration
Because the formation of neuropil aggregates correlates with the regions in which early neurodegeneration occurs in HD, we investigated whether any axonal or dendritic degeneration is associated with neuropil aggregates. To do so, we used EM48 immunogold labeling, because this method allowed us to better reveal huntingtin aggregates and the ultrastructure of neuronal processes.
EM examination relies primarily on the preservation of brain tissue and does not allow for a large-scale screen, so we focused on those brain tissues that had a well-preserved ultrastructure. EM examination of younger HD mice (<11 months) did not reveal any obvious neuropil degeneration (data not shown). However, two of three HD mice aged between 17 and 27 months showed neuropil degeneration in the LGP (Fig.4) and SN (data not shown) regions. Several pieces of evidence suggest that these neuropils are degenerated axons of striatal projection neurons (Fig. 4). First, the plasma membrane around the neuropils is clearly visible, making these neuronal processes distinct from other cellular structures and organelles. Second, because nuclear accumulation of mutant huntingtin precedes the formation of neuropil aggregates in these HD mice and because there is no nuclear staining of mutant huntingtin in the LGP, neuropil aggregates seen in the LGP should be in axons of striatal projection neurons. Moreover, no huntingtin aggregates were found in postsynaptic terminals in the striatal projection regions, suggesting that only axons of striatal neurons to the LGP contain huntingtin aggregates. Third, although degenerating axons might lose their identifying synapses and vesicles, some synaptic vesicles were still visible in the degenerated processes (Fig. 4C, arrow). Fourth, many dark, swollen organelles were seen within the degenerated processes, providing evidence for axonal degeneration. The size of these dark structures suggests that they may be mitochondria that have degenerated and lost their double membrane. These structures also resemble autolysosomes, which contain degenerated and dark cytoplasmic organelles including lysosomes and mitochondria (Dunn, 1990; Mizushima et al., 2001) and were also found in huntingtin-transfected cells (Kegel et al., 2000). In an axonal terminal containing clusters of immunogold particles or microaggregates, a degenerated mitochondrion is more clearly demonstrated in Figure5B. A number of immunogold particles are also associated with the surface of synaptic vesicles. We noticed that some degenerated axons had fewer huntingtin immunogold particles (Fig. 4C). It is possible that ultrathin sections of the brain used in electronic microscopy may not always be in the correct plane or of sufficient thickness to reveal aggregates in degenerated axons.
Fig. 4.
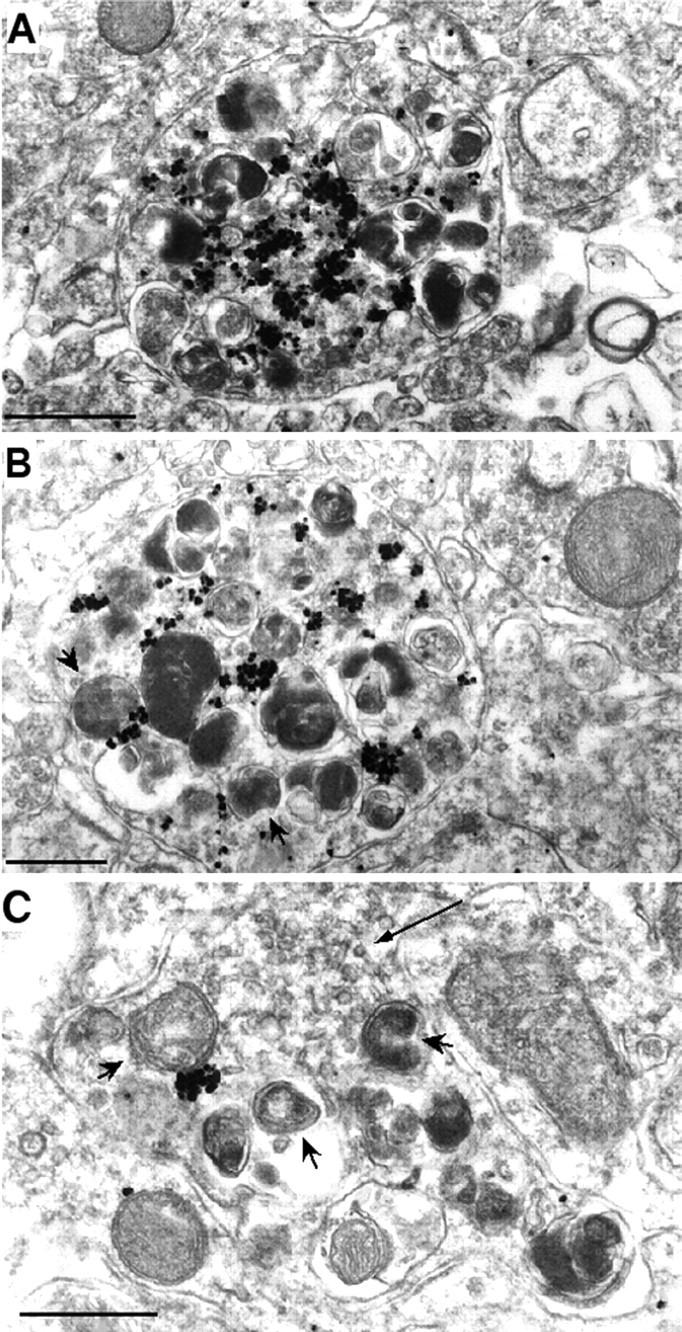
Degeneration of LGP axons containing huntingtin aggregates. EM48 immunogold labeling shows three degenerated axons in the LGP of a HD-repeat knock-in mouse at 17 months of age. The axons contain immunogold-labeled aggregates and many dark and swollen organelles, suggesting an association of huntingtin aggregates with degenerated axons. Some of the degenerated organelles have double membranes and resemble mitochondria (arrowheads). InC, synaptic vesicles (arrow) are evident in the degenerated axon. Scale bars, 0.5 μm.
Fig. 5.

Ultrastructure of EM48 immunoreactive aggregates in axon terminals. Micrographs indicate that immunogold particles are clustered in axon terminals in the LGP. A, A HD mouse at 27 months of age. A large huntingtin aggregate is present in an axon in which the postsynaptic density (double arrows) is evident. The aggregate has a fibrous structure and has EM48 immunogold labeling. It occupies most of the area of the axonal terminal, and no synaptic vesicles can be identified. In normal axonal terminals without huntingtin aggregates, however, many synaptic vesicles (arrows) are observed. B, A HD mouse at 17 months of age. Huntingtin aggregates are associated with degenerated mitochondria (arrowhead) in an axonal terminal. Synaptic vesicles in this terminal are indicated by arrows. The postsynaptic terminal (double arrows) contains normal mitochondria and no huntingtin aggregates. Scale bars, 0.5 μm.
We noticed that small huntingtin aggregates often accumulate in axons. In some cases, EM examination revealed large huntingtin aggregates that were present in axonal terminals. More convincing evidence to support the above ideas is that axon terminals, which show clear synapses and vesicles, contain huntingtin aggregates with a filament profile (Fig.5). A striking finding is that the size of a huntingtin aggregate can be so large that it almost occupies the entire presynaptic terminal (Fig. 5A). Often, these synapses with aggregates contained fewer vesicles compared with those synapses without huntingtin aggregates (Fig. 5A). The plasma membrane of the synapses near the aggregates is often not intact or appears to be ruffled (data not shown), suggesting that a morphological change may result from the early degeneration of a synapse. The observation that degenerated mitochondria are also evident in axonal terminals (Fig. 5B) is consistent with the idea that degenerated mitochondria are often associated with huntingtin aggregates (Fig. 4).
Several additional lines of evidence support the specific association of these huntingtin aggregates with degenerated axons in the striatal projection regions. First, similar degenerated axons in association with huntingtin aggregates are also found in the SN (data not shown). Second, we did not observe degenerated axons in the cortex and striatum of the same HD mouse brain using EM examination or terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Third, we never saw enlarged axons with dark and degenerated organelles in the brains of age-matched wild-type mice. More importantly, we identified these degenerated axons by using EM48 that labels mutant huntingtin and its aggregates.
We compared neuropil aggregate distribution in different brain regions (Table 1). The density of neuropil aggregates is well correlated with the brain regions that degenerate in early HD patients or show degenerated axons in HD-repeat knock-in mice. However, in R6/2 mice that normally die at 12–14 weeks, we did not observe degenerated axons associated with huntingtin aggregates. The context of full-length huntingtin and age-dependent changes in cellular function may contribute to the axonal degeneration seen in HD-repeat knock-in mice.
Table 1.
Neuropil aggregates and early neuropathology
| Cortex | Striatum | LGP | MGP | SN | |
|---|---|---|---|---|---|
| Neuropil degeneration in presymptomatic HD patients1-a | ND1-b | ND | Yes | ND | Yes |
| Neuropil aggregate density in HD knock-in mice | Very low | Low | High | Low | High |
| Axonal degeneration in HD knock-in mice | No | No | Yes | No | Yes |
ND, Not detected.
Huntingtin aggregates block neuritic transport in cultured striatal neurons
The large-sized aggregates may affect neuronal transport in axons and neuronal processes. To provide evidence for this idea, we examined cultured striatal neurons that were transfected with GFP fusion proteins containing the HD exon1 protein with a 120 glutamine repeat (GFP-120Q). We observed that neuritic aggregates could be formed within 2 hr in living cells (Fig.6A). In contrast, neurons transfected with huntingtin containing a 20 glutamine repeat (GFP-20Q) did not show such neuritic aggregates. The relatively rapid formation of aggregates allowed us to examine the effect of huntingtin aggregates on protein transport in neurites of cultured cells. Because fluorescence proteins are transported from the cell body to the nerve terminals of cultured neurons, we cotransfected GFP-huntingtin with RFP to examine the influence of huntingtin aggregates on the transport of RFP in living cells. When mutant huntingtin was diffuse and did not form large aggregates in the neurites, RFP was also diffusely distributed through all neurites (Fig. 6B). Once neurite aggregates were formed 6 hr later, RFP existed as puncta that were colocalized with huntingtin aggregates. More importantly, no diffuse RFP localization within neurites was seen. Thus, mutant huntingtin aggregates might retain RFP by blocking its transport in neurites.
Fig. 6.
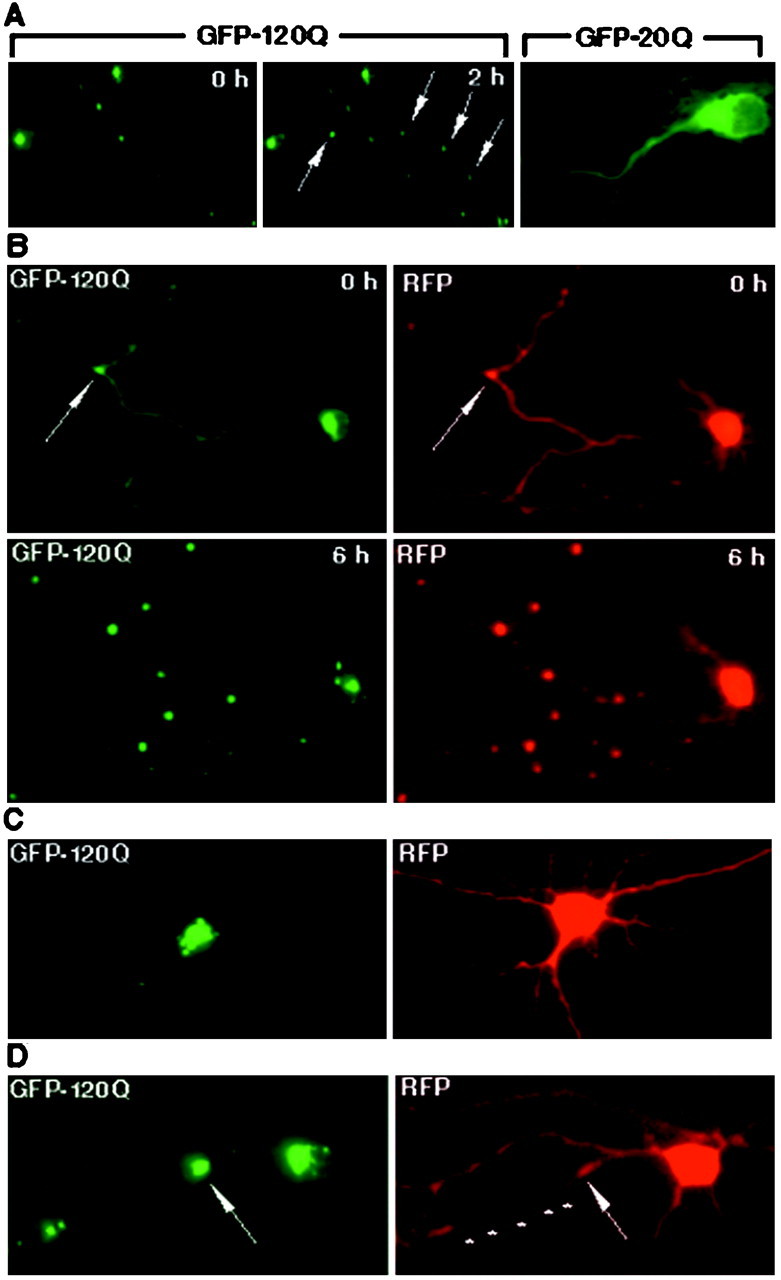
Blockage of RFP transport by huntingtin aggregates in neurites of cultured striatal neurons. A, Time course recording the formation of neuritic aggregates in living striatal neurons transfected with GFP-120Q. Newly synthesized aggregates after 2 hr of observation are indicated by arrows. A control cell expressing GFP-20Q after 2 hr of observation did not form such neuritic aggregates. B, Time-course examination of RFP and GFP-120Q distribution in living striatal neurons. Note that at the beginning (0 hr) of the examination, most of the RFP and GFP-120Q was diffuse in the neurites, with some huntingtin aggregates formed (arrow). After 6 hr, GFP-120Q formed a number of neuritic aggregates. RFP was colocalized with these aggregates, and no obvious diffuse distribution of RFP and GFP-120Q in neurites was seen.C, D, RFP- and GFP-120Q-transfected cells were fixed and then examined. Intranuclear huntingtin aggregates did not affect RFP distribution in neurites (C), but a large neuritic aggregate (arrow) prevented the distribution of RFP in the distal region of the neurite (D).
To confirm the observation on living cells, we also fixed cells and examined the distribution of RFP in those cells that contained huntingtin aggregates (Fig. 6C,D). Huntingtin aggregates in the nucleus or the cell body of striatal neurons did not significantly affect RFP distribution in neurites (Fig. 6C). In the neurites in which large huntingtin aggregates were present, however, there was much less RFP present in the region distal to huntingtin aggregates (Fig. 6D). Degeneration of neurons became obvious after transfection for 2 d, and the number of degenerated neurons in GFP-120Q transfection is significantly higher than that in GFP vector or GFP-20Q transfection. These findings suggest that huntingtin aggregates block the transport of RFP from the cell body to the neurites.
Neuritic degeneration precedes nuclear fragmentation in huntingtin transfected striatal neurons
Next, we wanted to know whether neuritic aggregates caused neuritic degeneration that might occur earlier than degeneration of the cell body (Fig. 7). We used tubulin staining to examine neurite integrity (Fig. 7B) and Hoechst nuclear staining (Fig. 7D) to examine nuclear DNA fragmentation that represents cell-body degeneration. After 17–24 hr of transfection, transfected neurons containing huntingtin aggregates in their cell body (Fig. 7A,E) often had intact tubulin staining in their neurites and intact nuclear DNA staining. However, in neurons that contained large neuritic aggregates, tubulin staining of neurites was often discontinuous, reflecting the degeneration of neurites. Despite such neuritic degeneration, these neurons did not show obvious nuclear DNA fragmentation (Fig. 7D,F). Thus, neuritic degeneration could occur in the absence of cell-body degeneration.
Fig. 7.
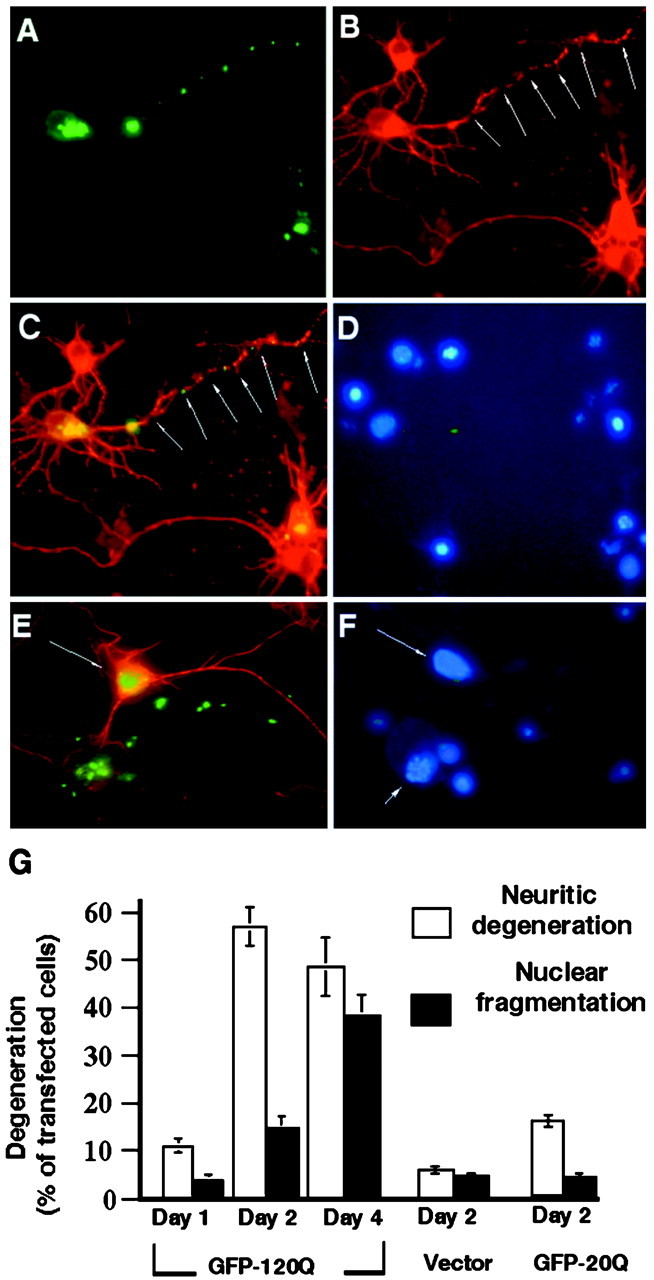
Neuritic degeneration of huntingtin-transfected striatal neurons before nuclear fragmentation. A–D, Cells were transfected with GFP-120Q for 24 hr and then stained with antibodies to huntingtin (A) and tubulin (B). In the merged image(C), neuritic aggregates are associated with the fragmentation of neurites that display discontinuous tubulin labeling.D, Nuclear staining did not show any DNA fragmentation in the neuron that has degenerated neurites. Small nuclei (no arrows) of glial cells are also shown. E, F, Amerged image of striatal neurons transfected with GFP-120Q for 48 hr. The cells were stained with antibodies against huntingtin (green) and tubulin (red). Note that a cell containing intranuclear huntingtin has intact nuclear staining (arrow) and neuritic tubulin labeling. A transfected cell showing DNA fragmentation (arrowhead), however, displays neuritic aggregates and has lost neuritic tubulin staining. G, Quantitative measurement of GFP-120Q-, GFP-20Q-, or GFP vector-transfected cells that displayed neuritic degeneration and nuclear degeneration. The data were obtained by counting transfected cells in two to three independent transfections.
To quantitatively examine the effect of huntingtin aggregates on neurites, we counted huntingtin-transfected neurons that showed neuritic degeneration or nuclear DNA fragmentation. The result revealed that during early transfection time (days 1–2), more neuritic degeneration was observed than nuclear degeneration (Fig.7G). As transfection time was prolonged (day 4), the number of cells with nuclear DNA fragmentation was significantly increased. These findings suggest that N-terminal fragments of mutant huntingtin induce neuritic degeneration that occurs earlier than nuclear degeneration.
DISCUSSION
Selective neurodegeneration has been a puzzling issue in all polyglutamine disorders. The present study demonstrates that the context of full-length huntingtin protein confers a selective accumulation of mutant huntingtin in striatal projection neurons, precisely the neurons that are preferentially affected in HD. More importantly, the distribution of huntingtin aggregates in the axons of these striatal projection neurons is specifically associated with axonal degeneration. In vitro experiments also show that neuritic aggregates affect protein transport in neurites and cause neuritic degeneration before neurons die. Thus, the present study demonstrates for the first time the association between neuropil aggregate formation and the selective neuropathology in early HD and suggests that HD pathology originates in axons.
The above conclusion is supported by the comparison of huntingtin aggregates in R6/2 mice that express N-terminal mutant huntingtin and HD-repeat knock-in mice that express full-length mutant huntingtin. R6/2 mice show a widespread distribution of intranuclear and neuropil aggregates, whereas HD-repeat knock-in mice display a selective distribution of huntingtin aggregates in striatal neurons. It was shown recently that the length of the huntingtin CAG repeat tends to expand in striatal tissue in the HD-repeat knock-in mice that we examined in this study, suggesting that the instability of this very large CAG repeat may contribute to cell-specific neuropathology (Kennedy and Shelbourne, 2000). Another possibility is that the selective processing of full-length mutant huntingtin in striatal neurons may lead to the accumulation and aggregation of N-terminal mutant huntingtin in these neurons. Consistent with the latter idea, our previous study has shown that N-terminal huntingtin fragments form huntingtin aggregates in HD-repeat knock-in mice (H. Li et al., 2000).
Because the presence of nuclear inclusions does not correlate with patterns of HD pathology (Saudou et al., 1998; Kuemmerle et al., 1999), the present study focused on the role of mutant huntingtin in neuronal processes. Neuropil aggregates appear to be different from nuclear inclusions, because most of them are not labeled by anti-ubiquitin antibodies (Gutekunst et al., 1999; H. Li et al., 1999). In addition, we were unable to label these neuropil aggregates with antibodies against heat shock protein 70 (Hsp70), Hsp40, or the transcription factor cAMP response element binding protein binding protein (data not shown). If neuropil aggregates are involved in the selective neuropathology of HD, they should, at least, have the following two features. First, the development and distribution of neuropil aggregates should correlate with the early neurodegeneration pattern. Second, aggregated huntingtin should associate with early pathological changes. The first feature is supported in this study by the temporal and regional studies of neuropil aggregate formation in the regions in which striatal neurons extend their axons. We have confirmed that the LGP and SN, which show degeneration earlier than other regions, contain the highest density of neuropil aggregates. Conversely, the MGP, which degenerates later, contains fewer neuropil aggregates. It is possible that the MGP contains fewer axons from the striatum so it has fewer neuropil aggregates. Nevertheless, the preferential localization of huntingtin aggregates in axons of striatal projection neurons agrees well with the degeneration of the striatal projection neurons in HD patient brains, which was identified based on the decreased enkephalin and substance P immunostaining of terminals within the LGP and SN (Reiner et al., 1988; Albin et al., 1990, 1992;Richfield et al., 1995).
The second and most significant finding is that aggregates are associated with the degeneration of axons. Electron microscopic examination revealed that many huntingtin aggregates were in axonal terminals but not in dendrites or postsynaptic terminals. Degenerated organelles, including mitochondria, are found to be present in the axons containing huntingtin aggregates. Mitochondrial degeneration fits well with the notion that mitochondrial dysfunction and oxidative stress are involved in HD pathogenesis (Browne et al., 1997; Sawa et al., 1999; Schapira, 1999; Beal, 2000). Although it remains to be investigated whether mutant huntingtin or its aggregates directly affect the function of mitochondria and other organelles or whether some of these degenerated organelles are autolysosomes, as reported by others (Dunn, 1990; Kegel et al., 2000; Mizushima et al., 2001), our finding provides the morphological evidence for mitochondrial degeneration in early HD.
The presence of large-sized aggregates in axons and their terminals suggests that huntingtin aggregates may block the transport of organelles or vesicles in axons. This blockage could impair axonal function, either by creating a physical barrier or through biochemical interactions of mutant huntingtin with other proteins. Consistent with this idea, huntingtin aggregates block RFP transport in neurites of transfected striatal neurons. Blocking axonal transport could also contribute to the degeneration of mitochondria and other organelles and ultimately lead to cell-body degeneration. It is also possible that mutant huntingtin may directly bind to synaptic vesicles and affect synaptic transmission before it forms large aggregates. This may explain why in the hippocampal region in HD-repeat-knock-in mice, where no obvious huntingtin aggregates were observed, an impairment of synaptic transmission was also observed (Usdin et al., 1999). Whether aggregated huntingtin produces more adverse effects on synaptic transmission in striatal neurons remains to be investigated. If these pathological changes occur in a subset of neurons, they may not be sufficient to induce obvious degeneration of cell bodies but may be able to cause the behavioral phenotype observed in these HD mice (Shelbourne et al., 1999) or the motor dysfunction observed in presymptomatic HD patients (Smith et al., 2000). In HD exon1 transgenic mice, however, the widely distributed and abundant neuropil aggregates may contribute to their more obvious synaptic dysfunction (Murphy et al., 2000) and mitochondrial abnormalities (Tabrizi et al., 2000).
Previous studies using HD transgenic mice expressing mutant huntingtin under exogenous promoters showed that cell bodies of striatal neurons were degenerating in symptomatic mice (Reddy et al., 1998; Hodgson et al., 1999). Because these mouse models used different promoters and constructs, the expression levels and localization of mutant huntingtin in transgenic mice are not the same as that in HD knock-in mice. Mutant huntingtin, when expressed at a higher level, could also affect neuronal function or induce neuronal degeneration before forming obvious aggregates (Reddy et al., 1998; Hodgson et al., 1999). The present study demonstrates that when expression of full-length mutant huntingtin is driven by endogenous mouse Hdh regulatory elements, axonal degeneration selectively occurs in HD-affected neurons in mice modeling early stages of the disease. Despite the abundance of nuclear aggregates in the striatum, striatal neurons do not show obvious cell-body degeneration. It is worthy pointing out that axonal degeneration was found in corticostriatal pathways in the brains of HD patients (Sapp et al., 1999). We did not observe such degeneration in HD-knock-in mice, suggesting that axonal degeneration of striatal neurons may occur earlier than neuronal degeneration in other brain regions. All of these findings also point out the possibility that HD pathology originates in axons of these striatal neurons. This possibility is also supported by in vitroexperiments showing that neuritic degeneration precedes cell-body degeneration in cultured striatal neurons. Moreover, many other studies have shown that polyglutamine aggregates in the nucleus are not associated with neurodegeneration (Klement et al., 1998; Saudou et al., 1998; Kuemmerle et al., 1999; Warrick et al., 1999; Kazemi-Esfarjani and Benzer, 2000). However, cytoplasmic polyglutamine aggregates can induce apoptosis (Sanchez et al., 1999). The association of neuropil aggregates with degenerated axons implies an important role of axonal aggregates in the selective neurodegeneration of HD. The pathological role of axonal aggregates is also supported by the fact that axonal damage is the cause of a variety of nervous system diseases, such as stroke (Sawlani et al., 1997), spongiform encephalopathies (Liberski and Gajdusek, 1997), Guillain–Barré syndrome (Trojaborg, 1998), insulin-dependent diabetic neuropathy (Said et al., 1992), and multiple sclerosis (Arnold, 1999; Trapp et al., 1999). Because axonal injury usually results in delayed cell death because of the relatively long distance between the site of injury and the cell body, our findings also suggest that prevention of this early neuropathologic change may provide an effective therapeutic approach to the treatment of HD.
Footnotes
This work was supported by grants from the National Institutes of Health (AG19206) and the Hereditary Disease Foundation. H.L. is a recipient of the postdoctoral fellowship of the Hereditary Disease Foundation and is partly supported by the National Natural Science Foundation of China.
Correspondence should be addressed to Dr. Xiao-Jiang Li, Department of Genetics, Emory University School of Medicine, 1462 Clifton Road Northeast, Atlanta, GA 30322. E-mail: xiaoli@genetics.emory.edu.
REFERENCES
- 1.Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-d-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;322:1293–1298. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- 2.Albin RL, Reiner A, Anderson KD, Dure L, IV, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- 3.Arnold DL. Magnetic resonance spectroscopy: imaging axonal damage in MS. J Neuroimmunol. 1999;98:2–6. doi: 10.1016/s0165-5728(99)00074-0. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 5.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 6.Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 9.Dunn WA., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 11.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 12.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 13.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 15.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 16.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy L, Shelbourne PF. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum Mol Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- 18.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 19.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 20.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- 21.Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum Mol Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 22.Li SH, Cheng AL, Li H, Li XJ. Cellular defects and altered gene expression in PC12 cells stably expressing mutant huntingtin. J Neurosci. 1999;19:5159–5172. doi: 10.1523/JNEUROSCI.19-13-05159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 24.Li SH, Li H, Torre ER, Li XJ. Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci. 2000;16:168–183. doi: 10.1006/mcne.2000.0858. [DOI] [PubMed] [Google Scholar]
- 25.Liberski PP, Gajdusek DC. Myelinated axon undergoes complete demyelination in the panencephalopathic–but it is merely subjected to the Wallerian degeneration in the polioencephalopathic type of transmissible spongiform encephalopathies. Pol J Pathol. 1997;48:163–171. [PubMed] [Google Scholar]
- 26.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 27.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 28.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20:5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 31.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 32.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol. 1995;38:852–861. doi: 10.1002/ana.410380605. [DOI] [PubMed] [Google Scholar]
- 34.Said G, Goulon-Goeau C, Slama G, Tchobroutsky G. Severe early-onset polyneuropathy in insulin-dependent diabetes mellitus. A clinical and pathological study. N Engl J Med. 1992;326:1257–1263. doi: 10.1056/NEJM199205073261905. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 36.Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol. 1999;58:165–173. doi: 10.1097/00005072-199902000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 38.Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 39.Sawlani V, Gupta RK, Singh MK, Kohli A. MRI demonstration of Wallerian degeneration in various intracranial lesions and its clinical implications. J Neurol Sci. 1997;146:103–108. doi: 10.1016/s0022-510x(96)00299-7. [DOI] [PubMed] [Google Scholar]
- 40.Schapira AH. Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia, and Friedreich's ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- 41.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 42.Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RM. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8:763–774. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- 43.Smith MA, Brandt J, Shadmehr R. Motor disorder in Huntington's disease begins as a dysfunction in error feedback control. Nature. 2000;403:544–549. doi: 10.1038/35000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
- 45.Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Trojaborg W. Acute and chronic neuropathies: new aspects of Guillain-Barre syndrome and chronic inflammatory demyelinating polyneuropathy, an overview and an update. Electroencephalogr Clin Neurophysiol. 1998;107:303–316. doi: 10.1016/s0013-4694(98)00096-0. [DOI] [PubMed] [Google Scholar]
- 47.Usdin MT, Shelbourne PF, Myers RM, Madison DV. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- 48.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 50.Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]