

Abstract
Inflammation in Alzheimer's disease (AD) patients is characterized by increased cytokines and activated microglia. Epidemiological studies suggest reduced AD risk associates with long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs). Whereas chronic ibuprofen suppressed inflammation and plaque-related pathology in an Alzheimer transgenic APPSw mouse model (Tg2576), excessive use of NSAIDs targeting cyclooxygenase I can cause gastrointestinal, liver, and renal toxicity. One alternative NSAID is curcumin, derived from the curry spice turmeric. Curcumin has an extensive history as a food additive and herbal medicine in India and is also a potent polyphenolic antioxidant. To evaluate whether it could affect Alzheimer-like pathology in the APPSw mice, we tested a low (160 ppm) and a high dose of dietary curcumin (5000 ppm) on inflammation, oxidative damage, and plaque pathology. Low and high doses of curcumin significantly lowered oxidized proteins and interleukin-1β, a proinflammatory cytokine elevated in the brains of these mice. With low-dose but not high-dose curcumin treatment, the astrocytic marker GFAP was reduced, and insoluble β-amyloid (Aβ), soluble Aβ, and plaque burden were significantly decreased by 43–50%. However, levels of amyloid precursor (APP) in the membrane fraction were not reduced. Microgliosis was also suppressed in neuronal layers but not adjacent to plaques. In view of its efficacy and apparent low toxicity, this Indian spice component shows promise for the prevention of Alzheimer's disease.
Keywords: Alzheimer's disease, inflammation, oxidative damage, anti-oxidant, microglia, plaque, interleukin-1β, Tg2576, APPswedish
Alzheimer's disease (AD) involves a chronic CNS inflammatory response that is associated with both head injury and β-amyloid (Aβ) pathology (Rogers et al., 1996). In populations with prolonged use of nonsteroidal anti-inflammatory drugs (NSAIDs), including the common over-the-counter medication ibuprofen, the risk for AD is significantly reduced (Breitner et al., 1995; Stewart et al., 1997). Consistent with this epidemiological association, chronic ibuprofen treatment significantly suppressed inflammation and the development of β-amyloid pathology in an animal model for Alzheimer's disease, the Tg2576 APPSw transgenic mouse (Lim et al., 2000). However, a principal limitation precluding widespread NSAID use for prevention of AD is gastrointestinal and occasional liver and kidney toxicity caused by inhibiting cyclooxygenase I (Bjorkman, 1998; Tomoda et al., 1998; Cappell and Schein, 2000; McGettigan and Henry, 2000; Sung et al., 2000). Side effect issues could be overcome using alternative anti-inflammatory drugs directed against different inflammatory targets.
Significant oxidative damage occurs in AD (Smith et al., 1991;Friedlich and Butcher, 1994; Smith et al., 1997; Montine et al., 1999). Because antioxidants can protect neurons from β-amyloid toxicity in vitro (Behl et al., 1994; Mattson and Goodman, 1995), a clinical trial was performed to test the ability of vitamin E to slow down the progression of AD (Sano et al., 1997). However, the limited success of this high-dose α-tocopherol trial has generated interest in other antioxidants because α-tocopherol (unlike γ-tocopherol) is a poor scavenger of the nitric oxide (NO)-based free radicals produced during inflammation (Christen et al., 1997) and elevated in AD (Adams et al., 1991; Smith et al., 1997). One phenolic antioxidant alternative is curcumin, a yellow curry spice derived from turmeric. This spice is used as a food preservative and herbal medicine in India (Kelloff et al., 1991; Kelloff et al., 2000), where the prevalence of AD in patients between 70 and 79 years of age is 4.4-fold less than that of the United States (Ganguli et al., 2000). Curcumin is several times more potent than vitamin E as a free radical scavenger (Zhao et al., 1989), protects the brain from lipid peroxidation (Martin-Aragon et al., 1997), and scavenges NO-based radicals (Sreejayan and Rao, 1997). Based on these considerations, we tested curcumin for its ability to inhibit the combined inflammatory and oxidative damage that occur as a response to amyloid in the transgenic mouse model APPSw. This model, which carries a human familial AD gene (amyloid precursor protein with the “Swedish” double mutation) (Hsiao et al., 1996), displays age-related neuritic plaque pathology, a quantifiable inflammatory response (Frautschy et al., 1998), oxidative damage (Perry and Smith, 1997; Pappolla et al., 1998; Smith et al., 1998), and age-related memory deficits linked to defective long-term potentiation (LTP) (Chapman et al., 1999).
MATERIALS AND METHODS
Animals. Ten-month-old male and female APPSw Tg+ and Tg− mice from 12 litters were randomly split between treatment groups. Tg+ mice were fed either chow (PMI Feeds Inc., St. Louis, MO) containing a low dose of curcumin (160 ppm; n = 9; Sigma, St. Louis, MO), a high dose of curcumin (5000 ppm;n = 6), or no drug (n = 8) for 6 months before being killed. Tg− littermates were fed chow containing no drug (n = 5). At the time of death, neither the weights nor the ages of the mice were significantly different, and there were no indications of diet-related toxicities. Mice were perfused with 0.9% normal saline, followed by HEPES buffer, pH 7.2, containing 5 mg/ml each leupeptin and aprotinin and 2 mg/ml pepstatin A. Brain regions were dissected from one hemisphere using mouse brain atlas coordinates (Franklin and Paxinos, 1997) as reported previously (Lim et al., 2000). Thalamic, cortical, and hippocampal regions, as well as entorhinal cortex and piriform cortex–amygdala sections, were dissected out and snap frozen in liquid nitrogen. Biochemical measurements were performed in the hippocampus, entorhinal cortex, piriform cortex–amygdala, and residual cortex (cortex region without frontal, entorhinal, or piriform areas).
Tissue preparation. Tissue samples were processed as described previously (Lim et al., 2000). Briefly, samples were homogenized in 10 vol of TBS containing a cocktail of protease inhibitors (20 μg/ml each pepstatin A, aprotinin, phosphoramidon, and leupeptin, 0.5 mm PMSF, and 1 mm EGTA). Samples were sonicated briefly (two times for 10 sec) and centrifuged at 100,000 × g for 20 min at 4°C. The soluble fraction (supernatant) was used for interleukin-1β (IL-1β) or soluble Aβ ELISAs, whereas the TBS-insoluble pellet was sonicated in 10 vol of 2% SDS. The resulting homogenate was centrifuged at 100,000 × g for 20 min at 20°C. The SDS-soluble fraction was used for Western analysis of GFAP and APP but not for Aβ ELISA. To analyze insoluble Aβ, the SDS-insoluble pellet was solubilized and sonicated in 70% formic acid. The resulting extract was neutralized with 0.25 mTris containing 30% acetonitrile and 5 mNaOH.
Sandwich ELISA for Aβ. Our sandwich ELISA for total Aβ has been described previously (Lim et al., 2000). Briefly, the assay uses monoclonal 4G8 against Aβ17–24 (Senentek, Napa, CA) as the capture antibody (3 μg/ml), biotinylated 10G4 against Aβ1–15 as the detecting antibody, and a reporter system using streptavidin–alkaline phosphatase and AttoPhos (JBL Scientific Inc., San Luis Obispo, CA) as the substrate (excitation, 450 nm; emission, 580 nm).
Sandwich ELISA for IL-1β. This assay using polyclonal antibody against mouse IL-1β (Endogen, Woburn, MA) for capture and monoclonal anti-mouse IL1-β (Endogen) for detection can measure IL-β down to 0.5 pg under most conditions (Lim et al., 2000).
Measurement of oxidized proteins. Amounts of oxidized proteins containing carbonyl groups were measured using an Oxyblot kit (Intergen, Purchase, NY). Briefly, 10 μg of protein from the SDS extract were reacted with 1× dinitrophenylhydrazine (DNPH) for 15–30 min, followed by neutralization with a solution containing glycerol and β-mercaptoethanol. These samples were electrophoresed on a 10% Tris–glycine gel, transferred, and blocked. The blot was incubated overnight with a rabbit anti-DNPH antibody (1:150) at 4°C, followed by incubation in goat anti-rabbit (1:300) for 1 hr at room temperature. Bands were visualized using chemiluminescent techniques with nonsaturating exposures and quantified.
Immunoblots for GFAP and APP. Levels of GFAP and APP (rabbit polyclonal 681–695; Kang sequence) were determined on immunoblots containing 40 μg of SDS-soluble brain homogenate. Blots were performed as described previously (Lim et al., 2000).
Immunostaining and image analysis. Immunohistochemistry and image analysis of anti-amyloid-stained deposits and microglia was performed on coronal brain sections from Tg+ untreated and Tg+ animals treated with low-dose curcumin as described previously (Lim et al., 2000). Tissue from the high-dose curcumin (HD curcumin) group was taken for electrophysiology and not available. Briefly, 10 μm hemibrain cryostat sections cut from the posterior pole to the anterior margin of the hippocampus were incubated overnight at 4°C (1:100) in “DAE” polyclonal antibody (anti-Aβ1–13) made against synthetic peptides Aβ1–13 or antibody against phosphotyrosine (PT), which serves as a rodent microglia marker (Frautschy et al., 1998). Briefly, antigen retrieval was accomplished by incubating sections in an unmasking solution (Vector Laboratories, Burlingame, CA) for 30 sec in a pressure cooker. Sections were allowed to cool down to room temperature before they were washed with TBS, and endogenous peroxidase was quenched with 0.3% hydrogen peroxide for 15 min. After blocking with normal serum, sections were incubated with anti-PT (1:400) at 37°C for 40 min. Slides were incubated in biotinylated goat anti-rabbit antibodies for DAE staining (1:1000) or biotinylated goat anti-mouse antibodies for PT staining (1:1000), followed by ABC reagent, each for 30 min at 37°C. Sections were developed with metal enhanced DAB (Pierce, Rockford, IL). On adjacent sections, DAE yielded similar results to monoclonal antibodies Aβ17–24 (4G8) or Aβ37–42 (2G9, 7A3). However, the rabbit polyclonal DAE was used for image analysis because it lacked the occasional vascular artifacts present when mouse monoclonals are used on mouse tissue.
Image analysis of DAE was performed on two coronal sections per brain. All images were acquired from an Olympus Optical (Tokyo, Japan) Vanox-T microscope with an Optronix Engineering LX-450A CCD video system. The video signal was routed into a Macintosh computer via a Scion Corp. (Frederick, MD) AG-5 averaging frame grabber, and these digitized images were analyzed with NIH Image public domain software. Custom Pascal macro subroutines were written to calculate various parameters of Aβ immunostaining, which included number of plaques, mean diameter, mean area, mean percentage area, and total area of plaques. Plaque burden was defined as percentage of total hippocampal and cortical area stained for Aβ, excluding identifiable vascular labeling. Individual plaque areas were assessed at 20× optical magnification, an analysis requiring acquisition of several microscopic fields. Total brain region areas were determined with 1× magnification. Then plaque areas per hemibrain section were totaled, and this sum area was divided by total hippocampal and cortical area of the relevant hemibrain section.
Image analysis was also used to obtain percentage of PT (DAB-labeled) area in rings of one and two plaque radii around anti-Aβ–vector blue-labeled plaques and in cortical and hippocampal neuronal layers (layers 1–6 of parietal, occipital, and temporal cortex; layers 1–3 of entorhinal cortex and hippocampal stratum oriens and lacunosum, outer and inner molecular layers, and hilus of the dentate).
Statistical analyses. A two-factor ANOVA (treatment × region or transgene × region) was performed to analyze differences in levels of IL-1β, GFAP, soluble and insoluble Aβ, carbonyl proteins (Oxyblot), and image analysis data. For evaluating microglial response to plaques in the ring analysis, a 2 × 2 ANOVA of microglial staining (percentage area) was performed, with treatment and ring being analyzed. Ring 1 labeling measures microglial staining within plaques, whereas ring 2 labeling measures microglial staining in the immediate vicinity but outside of plaques. A “ring effect” would signify that microglial staining is dependent on proximity to plaques. A treatment by ring interaction would imply that treatment effect may be dependent on ring. Post hoccomparisons between regions were performed using Fisher's PLSD. Bartlett's test for homogeneity of variances was also performed to determine whether variances were equal. Some analyses required logarithmic or square root transformations to establish homogeneity.p < 0.05 was considered significant.
RESULTS
Curcumin decreases IL-1β levels in Tg+ mice
Accumulating evidence implicates interleukin-1 in AD pathogenesis (Sheng et al., 1996; Griffin et al., 1998, 2000). We chose to assay interleukin-1β because it is not only involved in AD and elevated in Tg2576 (Lim et al., 2000) but also because age-related elevations of IL-1β in rodents have been implicated in age-related memory loss and defective LTP (Murray and Lynch, 1998). Levels of IL-1β were measured from the soluble fraction of three diet groups: Tg+ untreated, Tg+ low-dose curcumin, and Tg+ HD curcumin. Measurements were made in four brain regions (hippocampus, entorhinal cortex, piriform cortex, and residual cortex) and analyzed by two-factor ANOVA (diet × region). Our previous studies revealed a significant transgene effect in Tg+ mice compared with Tg− mice, in which IL-1β levels were elevated 2.4–6.7-fold in various regions of the brain (Lim et al., 2000). In the current study, two-way ANOVA showed a significant treatment effect with low-dose curcumin, decreasing IL-1β expression by 61.8% (F(1,57) = 19.6;p < 0.0001) (Fig.1A), with a significant treatment–region interaction. In addition, high doses of curcumin also significantly lowered IL-1β levels in Tg+ mice by 57% (F(1,48) = 31.8; p < 0.0001) (Fig. 1B). Therefore, both low and high doses of curcumin can decrease levels of IL-1β in Tg+ mice.
Fig. 1.

Effect of curcumin on IL-1β on APPSw brains.A, Effect of low-dose curcumin on IL-1β. Protein levels were measured in TBS-extracted supernatant from Tg+ mice fed low-dose curcumin diet and untreated Tg+ mice. Levels of IL-1β were significantly decreased by 61.8%. in curcumin-treated animals. *p < 0.05. B, Effect of high-dose curcumin on IL-1β. Protein levels were measured in TBS-extracted supernatant from Tg+ mice fed a high-dose curcumin diet and untreated Tg+ mice. Two-way ANOVA revealed a 57% reduction in IL-1β levels in Tg+ mice receiving a high dose of curcumin compared with untreated animals. ***p < 0.001.
Immunoblot analysis was used to determine whether curcumin could lower levels of GFAP, an astrocyte marker that is often elevated in inflammatory conditions and is increased in amyloid-forming APP transgenic mice (Irizarry et al., 1997). Levels of GFAP were significantly increased by 20% in APPSw mice (Lim et al., 2000). Two-way ANOVA demonstrated a significant treatment effect with low-dose curcumin, in which levels were decreased by 16.5% (F(1,58) = 4.8; p = 0.03). No significant treatment effects were observed with high-dose curcumin (data not shown).
Microglial activation was estimated by measuring the percentage of PT-stained area on cryostat sections in untreated and curcumin-treated (low-dose) animals. Two-way ANOVA (diet × region) demonstrated a diet effect in cortical and hippocampal layers in treated animals, in which percentage of PT-stained area was significantly reduced by 31% (p < 0.0001) (Fig.2A,B). Curcumin had the greatest impact on the outer molecular layer of the hippocampus and layer two of the cortex (data not shown). As shown in Figure 2, C and D, curcumin did not significantly reduce microglial staining within plaques (ring 1) and even significantly increased plaque-associated PT immunolabeling immediately outside of plaques (ring 2).
Fig. 2.
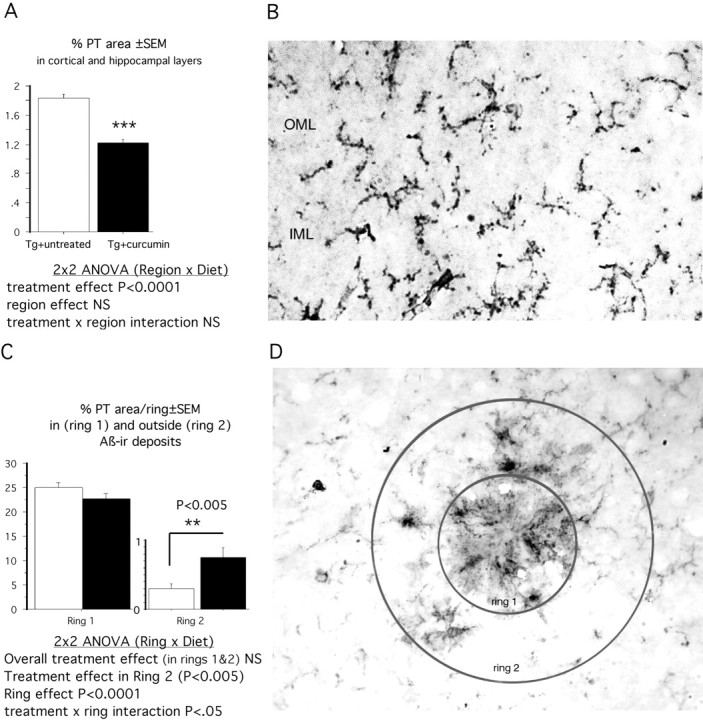
Reductions in percentage of stained microglia in response to dietary curcumin (160 ppm) were observed in neuronal layers in every region examined except the hilus of the hippocampus (A). Layers 1 of the cortex and the stratum oriens of the hippocampus were minimally affected, whereas the most robust reductions were observed in layer 2 of the cortex (40% reduction) and the outer molecular layer (OML) of the dentate gyrus (53% reduction). An example of the PT staining quantified in the OML is shown in B. D, Percentage of PT staining was quantified within plaques (ring 1) and associated within 1 plaque radius (ring 2). Whether curcumin altered the association of microglia with plaques was analyzed by analysis of the staining within these rings, using a 2 × 2 ANOVA (treatment diet × ring) of microglial staining (percentage area). A ring effect signifies that microglial staining is dependent on proximity to the plaque. The treatment × ring interaction signifies that curcumin treatment effects depend on the ring analyzed. As shown in C, microglial PT staining was not reduced within plaques (ring 1) but was even increased in ring 2 around plaques in curcumin-treated animals.IML, Inner molecular layer.
Oxidative damage is reduced in curcumin-treated mice
Oxidative damage was assessed in Tg− untreated, Tg+ untreated, and Tg+ HD curcumin groups using Western blot analysis, in which carbonyl groups on oxidized proteins were derivitized with DNPH and detected using an anti-DNP antibody. A representative example of an Oxyblot is shown in Figure 3. Two-way ANOVA (transgene × region) revealed a significant transgene effect in oxidized protein levels, which were increased 10.7-fold in Tg+ untreated mice (F(1,36) = 47.6;p < 0.0001) (Fig. 3B), consistent with previous reports of oxidative damage in APPSw mice. A combined regional analysis of all four brain regions revealed that animals treated with high doses of curcumin had a significantly lower level of oxidized proteins (46.3%) compared with untreated animals (F(1,47) = 6.3; p = 0.01) (Fig. 3C). Oxidative damage was measured in two brain regions (residual cortex and piriform cortex) of animals treated with the low dose of curcumin. Two-way ANOVA revealed a significant treatment effect with low-dose curcumin, in which oxidized protein levels were reduced 61.5% in treated animals (F(1,31) = 7.31; p = 0.01) (Fig. 3D). Therefore, both low and high doses of curcumin significantly decreased levels of oxidized proteins in the APPSw mice.
Fig. 3.

Measurement of oxidized proteins in APPSw mice.A, Representative example of Oxyblot using 10 μg of protein from Tg− untreated and Tg+ untreated entorhinal cortex samples. B, Transgene effect on oxidized proteins, as measured by Oxyblot, of SDS-extracted supernatant from hippocampus and entorhinal cortex of Tg− untreated (n = 9) and Tg+ untreated (n = 6). Two-way ANOVA showed a highly significant transgene effect, in which the levels of oxidized proteins were 12-fold higher in Tg+ animals compared with Tg− animals. ***p < 0.001. C, High-dose curcumin effect. Levels of oxidized proteins in oxyblots of SDS-extracted supernatant from hippocampal, entorhinal, and piriform cortices of Tg+ untreated (n = 6) and Tg+ high-dose curcumin (n = 8). Two-way ANOVA showed that levels of oxidized proteins were 46% lower in mice fed a diet containing a high-dose of curcumin (HD curcumin). *p < 0.05. D, Low-dose curcumin effect. Amounts of oxidized proteins in residual cortex and piriform cortex of Tg+ untreated and Tg+ curcumin-treated animals. Two-way ANOVA revealed a significant treatment effect. *p < 0.05. OD, Optical density.
Low doses of curcumin reduce TBS-soluble Aβ and SDS-insoluble Aβ (amyloid)
Previous studies have shown that a chronic dose of ibuprofen significantly reduces insoluble and soluble Aβ in APPSw mice (Lim et al., 2000). We tested whether this same effect was seen in curcumin-treated animals. Levels of SDS-insoluble Aβ were measured by ELISA in entorhinal cortex, hippocampus, and residual cortex regions. Two-way ANOVA (treatment × region) revealed a significant reduction in insoluble Aβ (F(1,36) = 10.97; p = 0.002), in which amounts were lowered by 39.2% (Fig.4A). There was also a significant region-dependent effect (F(2,36) = 13.7; p < 0.0001), in which insoluble Aβ levels were decreased in all regions except in residual cortex (data not shown). High doses of curcumin, on the other hand, did not alter the level of insoluble Aβ in the brains of treated mice (data not shown).
Fig. 4.

Effect of low-dose curcumin on SDS-insoluble Aβ and plaque pathology. A, Formic acid extracted (SDS-insoluble) Aβ as measured by sandwich ELISA. Aβ was measured in the three regions of the brain: hippocampus, entorhinal cortices, and residual cortex. A two-way ANOVA (treatment × region) showed significant treatment effects in insoluble Aβ levels (***p < 0.001). Homogeneity of variance was obtained using a natural log transformation of square root transformed values. B, Plaque burden (percentage of hippocampal and cortical area stained with amyloid) in Tg+ untreated and Tg+ low-dose curcumin mice. Image analysis was performed on amyloid-positive structures (DAE-positive) in hemibrain cryostat sections. Two-way ANOVA revealed a significant 43% reduction in plaque burden in curcumin-treated animals (*p = 0.03).C, Soluble Aβ in Tg+ untreated and Tg+ low-dose curcumin mice as measured by sandwich ELISA. Aβ levels were measured in hippocampus, entorhinal cortex, piriform cortex, and residual cortex regions. Two-way ANOVA (treatment - region) showed significant treatment effects in decreasing the levels of soluble Aβ (*p < 0.05).
Two-way ANOVA (treatment × region) revealed a significant reduction in the level of soluble Aβ in animals treated with low-dose curcumin (F(1,61) = 4.02;p = 0.0492), in which amounts were decreased by 43% (Fig. 4C). Soluble Aβ levels were unchanged with high-dose curcumin treatment (F(1,48) = 0.192;p = 0.66).
Low doses of curcumin reduces plaque burden in APPSw brains
To evaluate whether curcumin treatment affected plaque pathology, cryostat hemibrain sections from Tg+ control and Tg+ low-dose curcumin-treated mice were immunostained with an antibody against Aβ1–13 (DAE). Two-factor ANOVA (treatment × region) revealed a significant reduction in plaque burden in curcumin-treated animals (F(1,60) = 4.74; p = 0.03), in which amyloid burden was decreased by 43.6% in treated animals compared with untreated animals (Fig. 4B). Additional analysis of the data showed that total area of plaques was also decreased by 42.7% in curcumin-treated animals (with square root transformation to achieve the homogeneity of variance required for ANOVA; p = 0.01). The mean number of plaques was reduced 32.6% (log transformation for homogeneity of variance;p = 0.045). Although there was a mean 14% decrease in the size of the plaque with curcumin treatment, this observation was not statistically significant (p = 0.33). The mean diameter of the plaque was also not statistically reduced (p = 0.70).
DISCUSSION
In this study, we report that the Indian spice curcumin suppresses indices of inflammation and oxidative damage in the brains of APPSw mice, factors that have been implicated in AD pathogenesis. Furthermore, low, nontoxic doses of curcumin decrease levels of insoluble and soluble amyloid and plaque burden in many affected brain regions.
Curcumin is a potent anti-inflammatory compound. Part of its NSAID-like activity is based on the inhibition of nuclear factor κB (NFκB)-mediated transcription of inflammatory cytokines (Xu et al., 1998), inducible nitric oxide synthase (iNOS) (Chan et al., 1998), and cyclooxygenase 2 (Zhang et al., 1999). Elevated IL-1 has been linked to neuroinflammatory cascades involved in neuritic plaque pathogenesis (Sheng et al., 1996; Griffin et al., 1998) and in age-related LTP deficits (Murray and Lynch, 1998). Both low and high doses of curcumin were effective in significantly lowering our principal index of inflammation, the proinflammatory cytokine IL-1β by 57–61.8%, suggesting that inflammation was reduced in curcumin-treated animals. GFAP, an astrocytic marker associated with injury and inflammatory processes, was also significantly reduced with low-dose curcumin treatment. Similarly, another index of inflammation, PT immunolabeling of microglia in cortical and hippocampal neuronal layers, was significantly reduced by curcumin treatment.
Extensive evidence of oxidative damage has been reported in AD (Conrad et al., 2000; Praticò and Delanty, 2000; Varadarajan et al., 2000) and results in lipid peroxidation products such as 4-hydroxynonenal and isoprostanes (Montine et al., 1998; Praticòet al., 1998). Immunohistochemical evidence of peri-plaque oxidative damage has been found in aged APPSw mice (Perry and Smith, 1997;Pappolla et al., 1998; Smith et al., 1998). In Tg+ animals, we detected markedly elevated protein carbonyl formation using a convenient quantifiable Western blot analysis of DNPH derivatized carbonyls. Low and high doses of curcumin clearly suppressed the level of elevated protein carbonyls by 46–61.5%, which is consistent with its known antioxidant activity in brain (Rajakumar and Rao, 1994; Sreejayan and Rao, 1994; Kaul and Krishnakantha, 1997). Oxidized protein levels were not reduced in ibuprofen-treated Tg2576 mice (Lim et al., 2001). Because ibuprofen reduces inflammation indexed by IL-1β and plaque-associated microgliosis, this result suggests that reactive oxygen species (ROS) secondary to inflammation in plaque-associated reactive glia (Fig. 5) are not the primary source of increased ROS-driven oxidative damage in this β-amyloidosis model. This conclusion is consistent with a recent report in which isoprostanes were elevated in young mice before plaque formation and associated reactive glia (Praticò et al., 2001). These results suggest a combined antioxidant and NSAID approach to AD prevention or therapeutics.
Fig. 5.

Curcumin blocks AD pathogenesis at multiple sites. Curcumin can act as a scavenger of ROS, including NO and peroxynitrite generated by reactive glia and hydroxyl radicals generated by neurons as a result of direct Aβ toxicity. Ibuprofen (NSAID action at site 1) can inhibit microglial activation and cytokine production but was not sufficient to reduce oxidative damage. Antioxidants that can block ROS at multiple sites may be required. Curcumin can also limit damage by inhibiting NFκB-induced iNOS, cyclooxygenase 2, and inflammatory cytokine production by reactive glia. By blocking NFkB and reducing IL-1β, IL-6, and ApoE, curcurmin should reduce proamyloidogenic factors (APOE, α1ACT). Finally, curcumin can lower plasma and tissue cholesterol, potentially lowering Aβ production. LOX, Lipoxygenase; Cox-2, cyclooxygenase 2; SCR, Scavenger receptors;Fc, Fc Ig receptors.
Studies indicate that there is a reduced age-adjusted prevalence of AD in India (Ganguli et al., 2000), as well as a lower prevalence of Parkinson's disease (Muthane et al., 1998). Both diseases are linked to increased oxidative damage, including NO-based damage to a specific protein, synuclein (Glasson et al., 2000). Curcumin may effectively inhibit this damage.
Low-dose curcumin treatment significantly lowered both overall insoluble amyloid and plaque burden by 39 and 43%, respectively. These Aβ-lowering effects were not mediated by reductions in APP expression because we did not see any decrease in APP production in the SDS fraction of curcumin-treated mice using Western blots with C-terminal anti-APP681–695 antibody (Cole et al., 1992) (data not shown). Although we did not see any consistent effect on APP levels, additional experiments are required to determine whether curcumin influences APP processing. These findings are consistent with previous observations seen in ibuprofen-treated animals (10–16 months old), in which amyloid levels and plaque pathology were decreased by ∼50% (Lim et al., 2000). High-dose curcumin treatment, however, did not affect the amount of insoluble amyloid. Soluble Aβ levels were also significantly lowered by 43% in animals treated with low doses of curcumin, which is consistent with previous observations seen in mice treated with ibuprofen (Lim et al., 2000). Because “soluble Aβ” fractions may be involved with neurotoxicity, our data suggest that NSAIDs such as curcumin and ibuprofen can protect against neurotoxicity by reducing the level of these fractions.
Potential mechanisms underlying these curcumin treatment effects are multifactorial, as illustrated in a schematic diagram (Fig. 5). Curcumin suppressed microgliosis in neuronal layers but failed to reduce microgliosis within plaques and even significantly increased microgliosis immediately adjacent to plaques, raising the possibility that it may stimulate microglial phagocytosis of amyloid. Other possible mechanisms include inhibition of IL-1-induced increases in alpha-1-antichymotrypsin (α1ACT) and NFκB-mediated transcription of apolipoprotein E (ApoE). Both α1ACT (Rozemuller et al., 1990; Shoji et al., 1991; Aksenova et al., 1996) and ApoE (Wisniewski and Frangione, 1992;Wisniewski et al., 1994; Weisgraber and Mahley, 1996; Beffert et al., 1999) have been shown to be proamyloidogenic in APP transgenic mice. Curcumin can reduce two other proamyloidogenic pathways: oxidative damage (Bush et al., 1994; Friedlich and Butcher, 1994; Hensley et al., 1994) and cholesterol levels (Soudamini et al., 1992; Ramirez-Tortosa et al., 1999; Kamal-Eldin et al., 2000). Cholesterol could promote amyloidogenesis by regulating α- and β-secretase activity (Bodovitz and Klein, 1996; Frears et al., 1999; Wolozin, 2001).
In addition to the illustrated inflammation-related targets, curcumin is also reported to inhibit lipoxygenases and phospholipase D (Yamamoto et al., 1997; Began and Sudharshan, 1998; Skrzypczak-Jankun et al., 2000), which could contribute to overall NSAID or neuroprotective function. Given that there are multiple curcumin targets with varying dose–response curves, it is not surprising that some effects are dose related. For example, the amyloid reduction effect was dose-dependent and lost at 5000 ppm. One explanation for this effect may be that high doses of curcumin appear to suppress glial amyloid clearance in organotypic hippocampal slice cultures (T. Chu, G. P. Lim, and G. M. Cole, our unpublished observations), which could counterbalance any anti-amyloidogenic effects. Ongoing efforts at dissection of the relative importance of the different possible pathways for the amyloid reduction effects by curcumin may or may not reveal a single essential mode of action. In fact, the ability of curcumin to partially block multiple pathways implicated in AD pathogenesis may potentially provide greater in vivo efficacy than more potent but specific inhibitors of any of the individual targets.
Curcumin has an extensive history as a food preservative and medicinal herb in India (Ammon and Wahl, 1991), and it is possible that widespread curcumin use may contribute to the reduced age-adjusted prevalence of AD in India (Ganguli et al., 2000). Studies have consistently shown that curcumin is relatively nontoxic and has few side effects at doses greater than the low dose tested in our mice. (Kelloff et al., 1991, 2000). Toxicity studies performed at 2000 mg/kg, which is 83-fold greater than our low-dose curcumin treatment (∼24 mg/kg), revealed no mortalities in any group of mice tested; the compound also had a low ulcerogenic index (Srimal and Dhawan, 1972). Even with the high-dose curcumin treatment (5000 ppm), which is 31-fold greater than our low dose of curcumin, there was no impact on presynaptic markers and no increase in GFAP in any region, consistent with the absence of overt CNS toxicity (data not shown). Although side effects have been limited in chronic animal and short-term clinical studies, sustained clinical trials are needed to establish the safety of chronic curcumin at antioxidant and anti-inflammatory doses.
In summary, at the relatively low, 160 ppm dose, curcumin significantly suppressed the inflammatory cytokine IL-1β and the astrocytic inflammatory marker GFAP, reduced oxidative damage, and decreased overall insoluble amyloid, soluble amyloid, and plaque burden. In a rat intraventricular Aβ infusion model, a similar dose of dietary curcumin reduced an isoprostane index of oxidative damage, amyloid plaque burden, and Aβ-induced spatial memory deficits in the Morris water maze (Frautschy et al., 2001). Hence, curcumin is not only efficacious at multiple levels but may have fewer side effects and toxicity issues than many other NSAIDs, including ibuprofen. Together, the multiple beneficial effects of curcumin make it a promising agent for controlled clinical trials to establish its safety and efficacy as a chronic antioxidant and NSAID prophylactic for prevention or treatment of Alzheimer's and possibly other neurodegenerative diseases of aging, such as Parkinson's disease.
Footnotes
This work was supported by National Institute on Aging Grant AG13471 (G.M.C.), Veterans Affairs Merit, the Alzheimer Association, and The Elizabeth and Thomas Plott Family Foundation. We thank Dr. Karen Hsiao Ashe for her continued support and collaboration in studies pertaining to the Tg2576 mouse. We acknowledge Boris Oks, John Alcantara, Veronica Talamantes, and Ulises Garcia for their excellent work genotyping the transgenic mice. We are also grateful to Dr. Judith Harker for her help with the statistical analyses.
Correspondence should be addressed to Dr. Gregory M. Cole, Greater Los Angeles Veterans Affairs Healthcare System, GRECC11E, University of Los Angeles, Departments of Medicine and Neurology (San Fernando Valley Program), 16111 Plummer Street, Sepulveda, CA 91343. E-mail: gmcole@ucla.edu.
REFERENCES
- 1.Adams JD, Klaidman LK, Odunze IN, Shen HC, Miller CA. Alzheimer's and Parkinson's disease. Brain levels of glutathione, glutathione disulfide, and vitamin E. Mol Chem Neuropathol. 1991;14:213–226. doi: 10.1007/BF03159937. [DOI] [PubMed] [Google Scholar]
- 2.Aksenova MV, Aksenov MY, Butterfield DA, Carney JM. Alpha-1-antichymotrypsin interaction with a beta (1–40) inhibits fibril formation but does not affect the peptide toxicity. Neurosci Lett. 1996;211:45–48. doi: 10.1016/0304-3940(96)12717-8. [DOI] [PubMed] [Google Scholar]
- 3.Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta Med. 1991;57:1–7. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- 4.Beffert U, Aumont N, Dea D, Lussier-Cacan S, Davignon J, Poirier J. Apolipoprotein E isoform-specific reduction of extracellular amyloid in neuronal cultures. Brain Res Mol Brain Res. 1999;68:181–185. doi: 10.1016/s0169-328x(99)00073-x. [DOI] [PubMed] [Google Scholar]
- 5.Began G, Sudharshan E, Appu Rao AG. Inhibition of lipoxygenase 1 by phosphatidylcholine micelles-bound curcumin. Lipids. 1998;33:1223–1228. doi: 10.1007/s11745-998-0327-2. [DOI] [PubMed] [Google Scholar]
- 6.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 7.Bjorkman D. Nonsteroidal anti-inflammatory drug-associated toxicity of the liver, lower gastrointestinal tract, and esophagus. Am J Med. 1998;105:17S–21S. doi: 10.1016/s0002-9343(98)00276-9. [DOI] [PubMed] [Google Scholar]
- 8.Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 9.Breitner JCS, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, Pericak-Vance MA, Saunders AM. Delayed onset of Alzheimer's disease with nonsteroidal anti-inflammatory and Histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–530. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- 10.Bush AI, Pettingell WH, Jr, de Paradis M, Tanzi RE, Wasco W. The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J Biol Chem. 1994;269:26618–26621. [PubMed] [Google Scholar]
- 11.Cappell MS, Schein JR. Diagnosis and treatment of nonsteroidal anti-inflammatory drug-associated upper gastrointestinal toxicity. Gastroenterol Clin North Am. 2000;29:97–124. doi: 10.1016/s0889-8553(05)70109-6. [DOI] [PubMed] [Google Scholar]
- 12.Chan MM, Huang HI, Fenton MR, Fong D. In vivo inhibition of nitric oxide synthase gene expression by curcumin, a cancer preventive natural product with anti-inflammatory properties. Biochem Pharmacol. 1998;55:1955–1962. doi: 10.1016/s0006-2952(98)00114-2. [DOI] [PubMed] [Google Scholar]
- 13.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Hsiao K. Impaired synaptic plasticity and learning in amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 14.Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN. Gamma-tocopherol traps mutagenic electrophiles such as NOx and complements alpha-tocopherol: physiological implications. Proc Natl Acad Sci USA. 1997;94:3217–3222. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole GM, Bell L, Truong QB, Saitoh T. An endosomal lysosomal pathway for degradation of amyloid precursor protein. Ann NY Acad Sci. 1992;674:103–117. doi: 10.1111/j.1749-6632.1992.tb27480.x. [DOI] [PubMed] [Google Scholar]
- 16.Conrad CC, Marshall PL, Talent JM, Malakowsky CA, Choi J, Gracy RW. Oxidized proteins in Alzheimer's plasma. Biochem Biophys Res Commun. 2000;275:678–681. doi: 10.1006/bbrc.2000.3356. [DOI] [PubMed] [Google Scholar]
- 17.Franklin KBJ, Paxinos G. The mouse brain in stereotaxis coordinates, Ed 1. Academic; London: 1997. [Google Scholar]
- 18.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 19.Frautschy SA, Hu W, Miller SA, Kim P, Harris-White ME, Cole GM (2001) Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiol Aging, in press. [DOI] [PubMed]
- 20.Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. NeuroReport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- 21.Friedlich AL, Butcher LL. Involvement of free oxygen radicals in β-amyloidosis: an hypothesis. Neurobiol Aging. 1994;15:443–455. doi: 10.1016/0197-4580(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 22.Ganguli M, Chandra V, Kamboh MI, Johnston JM, Dodge HH, Thelma BK, Juyal RC, Pandav R, Belle SH, DeKosky ST. Apolipoprotein E polymorphism and Alzheimer disease: the Indo-US Cross-National Dementia Study. Arch Neurol. 2000;57:824–830. doi: 10.1001/archneur.57.6.824. [DOI] [PubMed] [Google Scholar]
- 23.Glasson BT, Dude JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 24.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin WS, Nicoll JA, Grimaldi LM, Sheng JG, Mrak RE. The pervasiveness of interleukin-1 in Alzheimer's pathogenesis: a role for specific polymorphisms in disease risk. Exp Gerontol. 2000;35:481–487. doi: 10.1016/s0531-5565(00)00110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 28.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Kamal-Eldin A, Frank J, Razdan A, Tengblad S, Basu S, Vessby B. Effects of dietary phenolic compounds on tocopherol, cholesterol, and fatty acids in rats. Lipids. 2000;35:427–435. doi: 10.1007/s11745-000-541-y. [DOI] [PubMed] [Google Scholar]
- 30.Kaul S, Krishnakantha TP. Influence of retinol deficiency and curcumin/turmeric feeding on tissue microsomal membrane lipid peroxidation and fatty acids in rats. Mol Cell Biochem. 1997;175:43–48. doi: 10.1023/a:1006829010327. [DOI] [PubMed] [Google Scholar]
- 31.Kelloff GJ, Crowell JA, Hawk ET, Steele VE, Lubet RA, Boone CW, Covey JM, Doody LA, Omenn GS, Greenwald P, Hong WK, Parkinson DR, Bagheri D, Baxter GT, Blunden M, Doeltz MK, Eisenhauer KM, Johnson K, Knapp GG, Longfellow DG, Malone WF, Nayfield SG, Seifried HE, Swall LM, Sigman CC. Strategy and planning for chemopreventive drug development: clinical development plans II. J Cell Biochem Suppl. 1991;26:54–71. doi: 10.1002/jcb.240630705. [DOI] [PubMed] [Google Scholar]
- 32.Kelloff GJ, Crowell JA, Steele VE, Lubet RA, Malone WA, Boone CW, Kopelovich L, Hawk ET, Lieberman R, Lawrence JA, Ali I, Viner JL, Sigman CC. Progress in cancer chemoprevention: development of diet-derived chemopreventive agents. J Nutr. 2000;130:467S–471S. doi: 10.1093/jn/130.2.467S. [DOI] [PubMed] [Google Scholar]
- 33.Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Hsiao Ashe K, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao Ashe K, Frautschy SA, Cole GM (2001) Ibuprofen effects of Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging, in press. [DOI] [PubMed]
- 35.Martin-Aragon S, Benedi JM, Villar AM. Modifications on antioxidant capacity and lipid peroxidation in mice under fraxetin treatment. J Pharm Pharmacol. 1997;49:49–52. doi: 10.1111/j.2042-7158.1997.tb06751.x. [DOI] [PubMed] [Google Scholar]
- 36.Mattson MP, Goodman Y. Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res. 1995;676:219–224. doi: 10.1016/0006-8993(95)00148-j. [DOI] [PubMed] [Google Scholar]
- 37.McGettigan P, Henry D. Current problems with non-specific COX inhibitors. Curr Pharm Des. 2000;6:1693–1724. doi: 10.2174/1381612003398690. [DOI] [PubMed] [Google Scholar]
- 38.Montine TJ, Markesbery WR, Roberts RJI, Morrow JD. Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer's disease. Ann Neurol. 1998;44:410–413. doi: 10.1002/ana.410440322. [DOI] [PubMed] [Google Scholar]
- 39.Montine TJ, Markesbery WR, Zackert W, Sanchez SC, Roberts LJ, Morrow JD. The magnitude of brain lipid peroxidation correlates with the extent of degeneration but not with density of neuritic plaques or neurofibrillary tangles or with ApoE genotype in Alzheimer's disease patients. Am J Pathol. 1999;155:863–868. doi: 10.1016/S0002-9440(10)65185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muthane U, Yasha TC, Shankar SK. Low numbers and no loss of melanized nigral neurons with increasing age in normal human brains from India. Ann Neurol. 1998;43:283–287. doi: 10.1002/ana.410430304. [DOI] [PubMed] [Google Scholar]
- 42.Pappolla MA, Chyan Y-J, Omar RA, Hsiao K, Perry G, Smith MA, Bozner P. Evidence of oxidative stress and in vivo neurotoxicity of β-amyloid in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 1998;152:871–877. [PMC free article] [PubMed] [Google Scholar]
- 43.Perry G, Smith MA. A central role for oxidative damage in the pathogenesis and therapeutics of Alzheimer's disease. Alzheimer's Res. 1997;2:319–324. [Google Scholar]
- 44.Praticò D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer's disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 45.Praticò D, Lee VM-Y, Trojanowski JQ, Rokach J, FitzGerald GA. Increased F2-isoprostanes in Alzheimer's disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 46.Praticò D, Uryu K, Leight S, Trojanowski JQ, Lee VM. Increased lipid peroxidation preces amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajakumar DV, Rao MN. Antioxidant properties of dehydrozingerone and curcumin in rat brain homogenates. Mol Cell Biochem. 1994;140:73–79. doi: 10.1007/BF00928368. [DOI] [PubMed] [Google Scholar]
- 48.Ramirez-Tortosa MC, Mesa MD, Aguilera MC, Quiles JL, Baro L, Ramirez-Tortosa CL, Martinez-Victoria E, Gil A. Oral administration of a turmeric extract inhibits LDL oxidation and has hypocholesterolemic effects in rabbits with experimental atherosclerosis. Atherosclerosis. 1999;147:371–378. doi: 10.1016/s0021-9150(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 49.Rogers J, Webster S, Lih-Fen L, Brachova L, Civin WH, Emmerling M, Brenda S, Walker D, McGeer P. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. 1996;17:681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- 50.Rozemuller JM, Stam FC, Eikelenboom P. Acute phase proteins are present in amorphous plaques in the cerebral but not in the cerebellar cortex of patients with Alzheimer's disease. Neurosci Lett. 1990;119:75–78. doi: 10.1016/0304-3940(90)90759-3. [DOI] [PubMed] [Google Scholar]
- 51.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's disease cooperative study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 52.Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin ST. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17:761–766. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoji M, Hirai S, Yamaguchi H, Harigaya Y, Ishiguro K, Matsubara E. Alpha 1-antichymotrypsin is present in diffuse senile plaques. A comparative study of beta-protein and alpha 1-antichymotrypsin immunostaining in the Alzheimer brain. Am J Pathol. 1991;138:247–257. [PMC free article] [PubMed] [Google Scholar]
- 54.Skrzypczak-Jankun E, McCabe NP, Selman SH, Jankun J. Curcumin inhibits lipoxygenase by binding to its central cavity: theoretical and X-ray evidence. Int J Mol Med. 2000;6:521–526. doi: 10.3892/ijmm.6.5.521. [DOI] [PubMed] [Google Scholar]
- 55.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PLR, Siedlank SL, Tabaton M, Perry G. Amyloid-β deposition is Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 58.Soudamini KK, Unnikrishnan MC, Soni KB, Kuttan R. Inhibition of lipid peroxidation and cholesterol levels in mice by curcumin. Indian J Physiol Pharmacol. 1992;36:239–243. [PubMed] [Google Scholar]
- 59.Sreejayan N, Rao MN. Curcuminoids as potent inhibitors of lipid peroxidation. J Pharm Pharmacol. 1994;46:1013–1016. doi: 10.1111/j.2042-7158.1994.tb03258.x. [DOI] [PubMed] [Google Scholar]
- 60.Sreejayan N, Rao MNA. Nitric oxide scavenging by curcuminoids. J Pharm Pharmacol. 1997;49:105–107. doi: 10.1111/j.2042-7158.1997.tb06761.x. [DOI] [PubMed] [Google Scholar]
- 61.Srimal RC, Dhawan BN. Pharmacology of diferuloyl methane (curcumin), a non-steroidal anti-inflammatory agent. J Pharm Pharmacol. 1972;25:447–452. doi: 10.1111/j.2042-7158.1973.tb09131.x. [DOI] [PubMed] [Google Scholar]
- 62.Stewart WF, Kawas C, Corrada M, Meter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 63.Sung J, Russell RI, Nyeomans, Chan FK, Chen S, Fock K, Goh KL, Kullavanijaya P, Kimura K, Lau C, Louw J, Sollano J, Triadiafalopulos G, Xiao S, Brooks P. Non-steroidal anti-inflammatory drug toxicity in the upper gastrointestinal tract. J Gastroenterol Hepatol [Suppl] 2000;15:G58–G68. doi: 10.1046/j.1440-1746.2000.02267.x. [DOI] [PubMed] [Google Scholar]
- 64.Tomoda T, Kurashige T, Hayashi Y, Enzan H. Primary changes in liver damage by aspirin in rats. Acta Paediatr Jpn. 1998;40:593–596. doi: 10.1111/j.1442-200x.1998.tb01996.x. [DOI] [PubMed] [Google Scholar]
- 65.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 66.Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer's disease connection. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 67.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 68.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- 69.Wolozin B. A fluid connection: cholesterol and Abeta. Proc Natl Acad Sci USA. 2001;98:5371–5373. doi: 10.1073/pnas.101123198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu YX, Pindolia KR, Janakiraman N, Chapman RA, Gautam SC. Curcumin inhibits IL-1 alpha and TNF-alpha induction of AP-1 and NK-kB DNA-binding activity in bone marrow stromal cells. Hematopathol Mol Hematol. 1998;11:49–62. [PubMed] [Google Scholar]
- 71.Yamamoto H, Hanada K, Kawasaki K, Nishijima M. Inhibitory effect on curcumin on mammalian phospholipase D activity. FEBS Lett. 1997;417:196–198. doi: 10.1016/s0014-5793(97)01280-5. [DOI] [PubMed] [Google Scholar]
- 72.Zhang F, Altorki NK, Mestre JR, Subbaramaiah K, Dannenberg AJ. Curcumin inhibits cyclooxygenase-2 transcription in bile acid- and phorbol ester-treated human gastrointestinal epithelial cells. Carcinogenesis. 1999;20:445–451. doi: 10.1093/carcin/20.3.445. [DOI] [PubMed] [Google Scholar]
- 73.Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989;14:175–185. doi: 10.1007/BF02797132. [DOI] [PubMed] [Google Scholar]