

Abstract
Superoxide is produced as a result of normal energy metabolism within the mitochondria and is scavenged by the mitochondrial form of superoxide dismutase (sod2). Mice with inactivated SOD2 (sod2 nullizygous mice) die prematurely, exhibiting several metabolic and mitochondrial defects and severe tissue pathologies, including a lethal spongiform neurodegenerative disorder (Li et al., 1995; Melov et al., 1998, 1999). We show that treatment ofsod2 nullizygous mice with synthetic superoxide dismutase (SOD)–catalase mimetics extends their lifespan by threefold, rescues the spongiform encephalopathy, and attenuates mitochondrial defects. This class of antioxidant compounds has been shown previously to extend lifespan in the nematode Caenorhabditis elegans (Melov et al., 2000). These new findings in mice suggest novel therapeutic approaches to neurodegenerative diseases associated with oxidative stress such as Friedreich ataxia, spongiform encephalopathies, and Alzheimer's and Parkinson's diseases, in which chronic oxidative damage to the brain has been implicated.
Keywords: mitochondria, oxidative stress, superoxide dismutase, antioxidants, neurodegeneration, spongiform encephalopathy
Approximately 0.4–4% of all oxygen consumed during normal respiration is converted into superoxide within the mitochondrion (Chance et al., 1979; Turrens and Boveris, 1980;Boveris, 1984; Turrens et al., 1985; Hansford et al., 1997), the chief source of reactive oxygen species (ROS) within the cell. ROS have been implicated in a wide range of disorders including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (ALS), prion disease, and Friedreich ataxia (FA), as well as in ischemia–reperfusion injury and aging (Beal, 1996; Rotig et al., 1997;Beckman and Ames, 1998; Choi et al., 1998; Pandolfo, 1998; Schapira, 1998; Smith et al., 1998; Behl, 1999; Brown et al., 1999; Fridovich, 1999; White et al., 1999; Bradley et al., 2000; Cuajungco et al., 2000;White et al., 2001). Consequently, studies have attempted to ameliorate or attenuate the progression of neurodegenerative disease by chronic or acute antioxidant treatment (Haller et al., 1996). Such studies have yielded at best equivocal or mildly beneficial results with regard to slowing disease progression or improving specific indicators (Jama et al., 1996; Perrig et al., 1997; Morris et al., 1998). In theory, efficacy should be improved by using catalytic antioxidants that can destroy multiple toxic mitochondrial ROS without themselves becoming inactivated. The compounds used in this study, synthetic mimetics of superoxide dismutase and catalase, are examples of such agents (Baudry et al., 1993).
To investigate the protective activity of synthetic catalytic antioxidants in oxidative neurodegenerative disease, we used mice lacking SOD2, the mitochondrial form of superoxide dismutase, on a CD1 background (sod2tm1Cje −sod2 nullizygous mice). Sod2 nullizygous mice die within the first week of life and show a dilated cardiomyopathy, hepatic lipid accumulation, metabolic defects, mitochondrial enzymatic defects, DNA oxidative damage, and organic aciduria (Li et al., 1995;Melov et al., 1999). In a previous study we treated sod2nullizygous mice with the catalytic antioxidant manganese 5,10,15,20-tetrakis (4-benzoic acid) porphyrin (MnTBAP) (Melov et al., 1998). We found that the MnTBAP treatment significantly increased the mean lifespan of the mice and ameliorated dilated cardiomyopathy and hepatic lipid accumulation. However, MnTBAP does not cross the blood–brain barrier (Melov et al., 1998). In extending the life ofsod2 nullizygous mice beyond 2 weeks of age through protection of peripheral tissues, MnTBAP treatment uncovered a severe neurological disorder from the endogenous production of free radicals within mitochondria of the brain, attributable to the inability of MnTBAP to protect against ROS produced within the brain. The neurological phenotype seen in the MnTBAP-treated sod2nullizygous mice is characterized by a severe disturbance in motor control, and the underlying neuropathology is that of spongiform change predominantly within the frontal cortex and focally in brainstem nuclei (Melov et al., 1998).
The present study is focused on the ability of neurologically active synthetic catalytic ROS scavengers to rescue the oxidative neurodegenerative phenotype and further extend the lifespan ofsod2 nullizygous mice. The salen manganese complexes, exemplified by the prototype EUK-8, are mimetics of both SOD and catalase (Baudry et al., 1993; Gonzalez et al., 1995; Baker et al., 1998). The compounds have shown efficacy in a variety of oxidative stress paradigms, including in vivo models for stroke, Parkinson's disease, autoimmune disease, excitotoxic neuronal death, and familial ALS (Doctrow et al., 1997; Malfroy et al., 1997; Rong et al., 1999; Jung et al., 2001). Recently these compounds have proven effective in extending the wild-type lifespan of the invertebrateCaenorhabditis elegans, demonstrating the importance of ROS as a major factor in limiting lifespan (Melov et al., 2000). It was therefore of considerable interest to determine whether the compounds would be effective in a mammalian model involving mitochondrial dysfunction and accelerated oxidative tissue damage, and in particular whether they would rescue an oxidative neurodegenerative process. Therefore, in the present study, sod2 nullizygous mice received salen manganese complexes via daily injection from 3 d of age to assess effects on lifespan as well as oxidative pathologies.
MATERIALS AND METHODS
EUK-8, EUK-134, or EUK-189 was synthesized and assayed for SOD and catalase activity as described previously (Baker et al., 1998). They were assayed for lipophilicity by solvent partitioning that was conducted as follows. A total of 20 nmol of the compound was dissolved in 100 μl of water; next, 500 μl of N-octanol was added. The sample was vortexed and the organic and aqueous phases were allowed to separate. The amount of compound partitioned into each phase was determined by HPLC on an octadecyl–silica column eluted isocratically with a mobile phase of 100 mm NaCl in water/methanol (60:40) with quantitation by UV absorbance.
For animal studies, EUK-8, EUK-134, and EUK-189 were dissolved in sterile water at 5 mg/ml. MnTBAP was prepared and injected as described previously (Melov et al., 1998). Mice were genotyped as described previously (Melov et al., 1998) and then injected intraperitoneally on a daily basis at 1 or 30 mg/kg body weight for EUK compounds or at 5 mg/kg body weight for MnTBAP from 3 d of age until death or until they were killed.
Tissue harvesting, histology, histochemistry, electron microscopy, and immunohistochemistry against glial fibrillary acidic protein (GFAP) were performed as described previously (Melov et al., 1998). Survival analysis was performed using the Kaplan–Meier (product-limit) method with the JMP Statistics program (SAS Institute, Inc., Cary, NC) or PRISM (Graphpad software, San Diego, CA). Differences in survival between treatment groups were assessed by nonparametric analysis (log-rank and Wilcoxon analysis). Animals that became moribund were killed and therefore were counted as dead in the survival analysis, whereas animals killed early for various analyses were counted as censored. Mitochondria were isolated and assayed for aconitase and fumarase from each brain region as described previously (Patel et al., 1996; Melov et al., 1999). Fumarase activity did not vary significantly between control and experimental groups in any comparison, and therefore is not shown.
RESULTS
Three salen manganese complexes, EUK-8, EUK-134, and EUK-189, were used in this study (Fig. 1). They belong to a class of stable organometallic complexes that contain tightly bound manganese, enabling them to mimic the active site of metalloenzymes such as SOD2 (Baudry et al., 1993; Doctrow et al., 1997). These three compounds share similarities but also have some distinctive properties. EUK-134 has an SOD activity equivalent to that of EUK-8 but is a more active catalase and is more effective in a rat stroke model (Baker et al., 1998). EUK-189, a novel analog whosein vivo efficacy has not yet been reported, has SOD and catalase activities equivalent to those of EUK-134 but is more lipophilic, based on solvent partitioning. The catalytic activities and lipophilicities of the three compounds are compared in Table1.
Fig. 1.

Structures of salen manganese complexes. The ring substituents (R) of the three compounds differ as shown. The axial ligand (X) is chloride for EUK-8 and EUK-134 and acetate for EUK-189.
Table 1.
Catalytic activities and lipophilicities of salen manganese complexes
| Compound | SOD activity IC50 (μm) | Catalase rate | Octanol partitioning |
|---|---|---|---|
| EUK-8 | 1.5 ± 0.4 | 103 ± 9 | 8.2 ± 1.8 |
| EUK-134 | 1.3 ± 0.1 | 209 ± 23 | 7.6 ± 0.7 |
| EUK-189 | 1.4 ± 0.5 | 180 ± 9 | 14.2 ± 0.4 |
SOD activity was assayed as described previously (Baker et al., 1998) using oxidized cytochrome c as the electron acceptor. IC50 values (μm) represent the concentration of compound having 1 U of SOD activity. Each value shown is the mean ± SD for IC50 values obtained from eight separate concentration curves, each including seven different concentrations of compound. Catalase activity was assayed by monitoring conversion of hydrogen peroxide to oxygen as described previously (Baker et al., 1998). Initial rate (μmoxygen produced/min) values are the means ± SD for five reactions. Lipophilicity was assessed by solvent partitioning as described in Materials and Methods and is expressed as the percentage of partitioning into octanol (mean ± SD for five samples).
As shown in Figure 2, EUK-8, EUK-134, or EUK-189 (30 mg/kg), significantly extended the lifespan ofsod2 nullizygous mice beyond that of untreated mice and mice treated with MnTBAP (5 mg/kg). A prominent feature of the survival curves is that all salen manganese complexes dramatically increased the percentage of sod2 nullizygous mice living beyond 3 weeks of age. This is the approximate age at which MnTBAP-treatedsod2 nullizygous mice succumb to the previously described neurodegenerative phenotype (Melov et al., 1998). Key parameters of the survival data obtained with all treatment groups are summarized in Table 2. The effect of EUK-8 and EUK-134 was dose dependent, with 30 mg/kg enhancing survival significantly more than 1 mg/kg. EUK-189 at 30 mg/kg significantly enhanced survival beyond that observed with EUK-134 at the same dose. EUK-8 at 30 mg/kg appeared to have an intermediate effect compared with the other two analogs, with survival differences not achieving statistical significance with either EUK-189 or EUK-134. The percentage of mice surviving beyond 3 weeks of age was greater with EUK-189 treatment (80%), compared with 54% and 49% for EUK-8 and EUK-134, respectively (Table 2).
Fig. 2.

Survival analysis of sod2nullizygous mice treated with synthetic catalytic antioxidants. Kaplan–Meier survival analysis demonstrates enhanced survival ofsod2 nullizygous mice treated with EUK-8, EUK-134, or EUK-189 (30 mg/kg) versus untreated sod2 nullizygous mice. MnTBAP was administered at the limiting dose of 5 mg/kg as described previously (Melov et al., 1998).
Table 2.
Survival data for sod2 nullizygous mice treated with SOD–catalase mimetics
| Catalytic antioxidant treatment | Total number in survival | Percentage surviving to 21 days | Mean lifespan ± SD (days) | Versus untreated | Versus EUK-8 (30 mg/kg) |
|---|---|---|---|---|---|
| Untreated | 150 | 2% | 9 ± 4 | — | * |
| MnTBAP (5 mg/kg) | 193 | 24% | 14 ± 6 | * | * |
| EUK-8 (1 mg/kg) | 98 | 49% | 17 ± 7 | * | * |
| EUK-8 (30 mg/kg) | 144 | 54% | 24 ± 9 | * | — |
| EUK-134 (1 mg/kg) | 26 | 29% | 14 ± 7 | * | * |
| EUK-134 (30 mg/kg) | 26 | 47% | 19 ± 9** | * | NS |
| EUK-189 (30 mg/kg) | 24 | 80% | 28 ± 4 | * | NS |
These data summarize the results of the Kaplan–Meier survival analysis for all groups. Statistical tests were performed between indicated groups, as described in Materials and Methods, with significant differences (p < 0.05) between groups as noted by an asterisk. Additional tests compared the EUK-189 (30 mg/kg) treatment group with those treated with EUK-134 or EUK-8 (both at 30 mg/kg); a significant difference (p < 0.05) is denoted by a double asterisk.
At 3 weeks of age, mice treated with EUK-8, EUK-134, or EUK-189 (30 mg/kg) showed no clinical evidence of the severe brain disorder uncovered in the MnTBAP study. This is consistent with the salen manganese complexes crossing the blood–brain barrier and rescuing the neurological phenotype that is the likely cause of death in MnTBAP-treated animals. The neuropathological observations discussed below are consistent with this interpretation. Histopathological analysis revealed no spongiform change in the brains of sod2nullizygous mice treated with 30 mg/kg EUK-8, 30 mg/kg EUK-134, or 30 mg/kg EUK-189 at any age (19–44 d of age, n = 12). Electron microscopic analysis of the frontal cortex from the brains of two mice treated with EUK-8 at 30 mg/kg at 42 and 44 d of age revealed no significant differences compared with EUK-8-treated wild-type controls. In combination with the neuropathological findings at the light level, this is consistent with a rescue of the spongiform encephalopathy observed previously in MnTBAP-treated sod2nullizygous mice by EUK-8, EUK-134, and EUK-189 at 30 mg/kg.
As with survival, rescue of the spongiform encephalopathy by EUK-8 and EUK-134 was dose dependent, with pathological observations indicating complete prevention of the spongiform changes at 30 mg/kg and lesser protection at 1 mg/kg (Fig. 3). Histopathological analysis of the brains of sod2 nullizygous mice treated with 1 mg/kg of either compound at 3 weeks of age demonstrated a regional specific spongiform encephalopathy in six of six mice that was similar to that seen in MnTBAP-treated mice (Melov et al., 1998) (Fig. 3). Vacuoles ranged from 4 to >40 μm (Fig. 3A), impinged on neighboring structures such as neurons and blood vessels, and were occasionally seen within neuronal perikaryon. Vacuoles showed occasional septa, and changes were noted focally within the cerebral cortex in all cases, predominately involving frontal regions, although the parietal and occipital regions were also involved. All but two cases showed moderate to severe involvement of the motor nucleus of the trigeminal nerve. Four of the six cases showed a mild to moderate degree of involvement in the retrosplenial granular and agranular cortex, the somatosensory cortex, and the motor cortex. Occasional vacuoles were also observed in the secondary sensory and motor cortex, cingulate, entorhinal cortex, subiculum, perirhinal cortex, auditory and visual cortices, lateral orbital cortex, periolivary nucleus, and lateral superior olive. There was preferential involvement of the midlayers of the cortex except in the cingulate and in the retrosplenial cortex and M1 and M2, where the molecular layers were predominately affected. There was general sparing of the white matter and subcortical nuclei. Immunohistochemistry using antibodies specific for GFAP, showed some positivity in areas of vacuolization and in areas not affected by vacuoles. However, this was not considered different from the GFAP positivity in the brains of control animals, suggesting a histologic absence of reactive gliosis in affected animals (data not shown). The prominent GFAP positivity (reactive gliosis) in association with vacuolization observed previously in MnTBAP-treated mice (Melov et al., 1998) was not observed in sod2 nullizygous mice treated with either dose of EUK-8 or EUK-134. These observations, in conjunction with the lesser severity of the vacuolar changes, are consistent with a partially neuroprotective effect of the lower dose of EUK-8 and EUK-134. Figure3B shows the equivalent region of a 3-week-oldsod2 nullizygous mouse treated with EUK-8 at 30 mg/kg, demonstrating an absence of spongiform pathology.
Fig. 3.
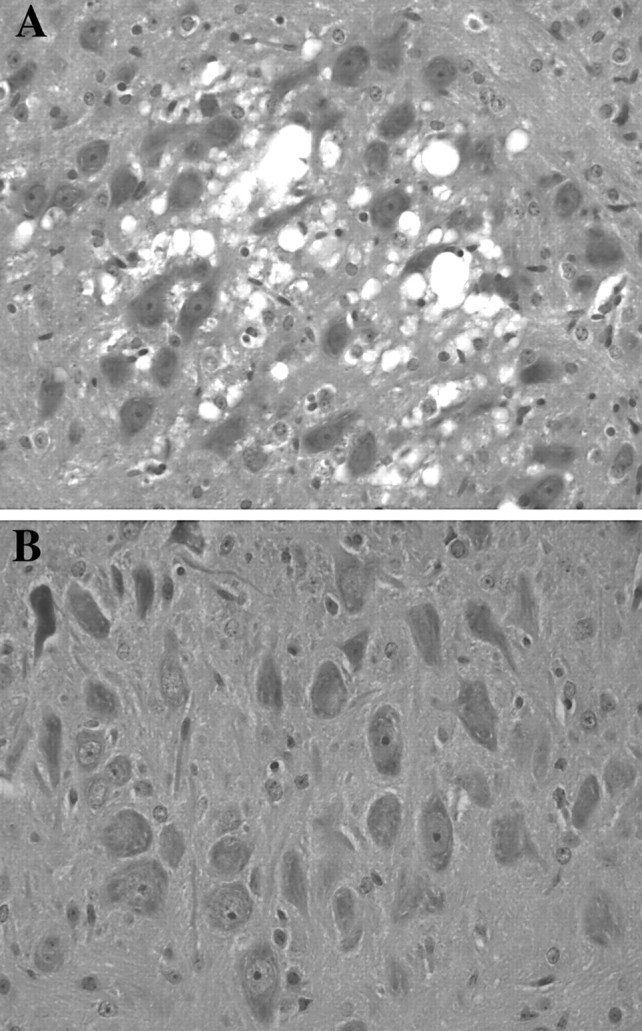
Dose-dependent rescue of spongiform encephalopathy in sod2 nullizygous mice. A shows the trigeminal motor nuclei (hematoxylin and eosin) (400× magnification) of a 3-week-old sod2 nullizygous mouse treated with EUK-8 at 1 mg/kg; prominent spongiform changes are seen that are consistent with those reported previously in MnTBAP-treated sod2 nullizygous mice (Melov et al., 1998). B shows the equivalent regions from a 3-week-oldsod2 nullizygous mouse treated with EUK-8 at 30 mg/kg, demonstrating the rescue of spongiform changes.
Mice in the 30 mg/kg treatment groups that survived to >25 d of age developed a progressive movement disorder (n = 67) concurrent with a loss of weight, at which point they were killed. However, this was not associated with gross spongiform changes because, as noted above, brains from sod2 nullizygous mice at any age in the 30 mg/kg dose treatment groups did not exhibit this pathology.Sod2 nullizygous mice treated with EUK-8 or EUK-134 at 1 mg/kg became ataxic at an earlier age (between 15 and 21 d of age) (n = 69). Wild-type mice treated chronically with the compounds at either dose showed no ill effects with up to 41 d of continuous treatment, and there were no mortalities in any wild-type groups.
Previous studies indicate that untreated sod2 nullizygous mice suffer mitochondrial damage, as indicated by a loss of activity of certain oxidation-sensitive mitochondrial enzymes in the brain and peripheral tissues. For example, mitochondrial aconitase is decreased by 70–80% in striatum, frontal cortex, cerebellum, and brain stem ofsod2 nullizygous mice, whereas another tricarboxylic acid cycle enzyme, fumarase, is unaffected (Melov et al., 1999). Because EUK-8 rescues the neuropathology in these animals, consistent with penetrating the blood–brain barrier, we investigated whether it concomitantly prevented the loss of mitochondrial aconitase activity in the brains of sod2 nullizygous mice. For this experiment,sod2 nullizygous mice treated with EUK-8 (30 mg/kg) were compared with those treated with MnTBAP (5 mg/kg) at 3 weeks of age. Untreated sod2 nullizygous mice could not serve as controls in this study because they do not live long enough in sufficient numbers to exhibit the neuropathology. The mitochondrial aconitase activity in the brains of EUK-8-treated sod2 nullizygous mice, although not achieving the levels observed in mitochondria from wild-type and other control mice, was 2.5-fold to threefold higher than that of MnTBAP-treated mice (Fig. 4). This is consistent with EUK-8 protecting mitochondrial aconitase from inactivation by endogenously generated mitochondrial superoxide.
Fig. 4.

Increase in mitochondrial aconitase activity in the brains of sod2 nullizygous mice at 3 weeks of age.Open bars, MnTBAP-treated sod2−/− mice (n = 11); graybars, EUK-8-treated sod2−/− mice (n = 6); closed bars, EUK-8- or MnTBAP-treated sod2+/+ mice or untreatedsod2+/+ mice. BS, Brain stem;Str, striatum; Ctx, cortex;Cb, cerebellum. Mitochondrial aconitase activity in the brains of sod2 nullizygous mice treated with EUK-8 compared with mitochondria isolated from MnTBAP-treated mice at 3 weeks of age via nonparametric t tests reveals that EUK-8 treatment results in statistically significant increases in the level of mitochondrial aconitase activity (*p < 0.05; **p < 0.01). No statistically significant difference was observed in the activity of brain mitochondrial aconitase from sod2+/+ mice treated with either EUK-8 or MnTBAP compared with untreated controls; therefore, these mice were grouped together. Error bars represent the SEM.
DISCUSSION
Mice lacking SOD2, the mitochondrial form of the enzyme, generally die within a few days of birth, exhibiting mitochondrial enzyme deficiencies and numerous severe pathologies resulting from mitochondrial oxidative stress (Li et al., 1995; Melov et al., 1998,1999). This is in dramatic contrast to mice lacking the far more abundant cellular enzyme SOD1, which show a very mild phenotype and normal lifespan (Reaume et al., 1996). This difference alone illustrates the profound impact that mitochondrial oxidative stress can have on tissue function and, therefore, on disease. Thus, it is of considerable interest to examine the pathologies manifested bysod2 nullizygous mice as well as the potential effects of specific pharmacological interventions on these pathologies. For example, we have reported previously that when sod2nullizygous mice, which normally die of a dilated cardiomyopathy, are treated with a synthetic antioxidant that does not cross the blood–brain barrier, they live longer and develop a lethal develop a spongiform encephalopathy (Melov et al., 1998). This disorder, resulting from mitochondrial oxidative damage to the brain, was unmasked when earlier-onset peripheral pathologies were alleviated. As will be discussed further below, this phenotype is remarkably similar to that observed in a recently reported mouse model for FA, a severe neurodegenerative mitochondrial disease (Puccio et al., 2001). More broadly, mitochondrial dysfunction and oxidative stress have been implicated in such prevalent neurodegenerative disorders as Alzheimer's and Parkinson's diseases (Cherny et al., 2001; Conn et al., 2001). Thus, insofar as the spongiform encephalopathy represents a model for pathological consequences of brain mitochondrial dysfunction, our studies with the sod2 nullizygous mouse may provide some insight into potential therapeutic approaches to human neurodegenerative disease.
Consequently, the current study was focused on investigating whether the spongiform encephalopathy in sod2 nullizygous mice could be rescued by treatment with a class of synthetic catalytic antioxidants that have been shown previously to be neuroprotective (Baker et al., 1998; Rong et al., 1999; Jung et al., 2001). Our data show that the compounds, administered daily at 30 mg/kg, extended the lifespan of sod2 nullizygous mice approximately threefold, beyond the age (∼2–3 weeks) at which, with MnTBAP treatment, they succumb to the spongiform encephalopathy. In addition, at 2–3 weeks of age, the treated mice showed no clinical signs of the associated neurobehavioral phenotype described previously, which consists of severe motor disturbances (Melov et al., 1998). Finally, analysis of the brains of the treated mice showed an absence of spongiform pathology at all ages examined (up to 44 d). Together, these data indicate that the compounds, unlike MnTBAP, can cross the blood–brain barrier and rescue the spongiform encephalopathy. This protection was dose dependent, as evidenced by the partial protection, with respect to survival as well as brain pathology, observed with the lower dose (1 mg/kg EUK-8 or EUK-134) treated groups.
The eventual cause of death in sod2 nullizygous mice treated with the compounds is not yet known. Mice surviving for >25 d exhibited a progressive movement disorder, but this was not associated with gross spongiform changes as is the phenotype seen in the MnTBAP-treated mice. Hence, the mechanistic basis for the later-onset neurological disorder is likely to be different from that unmasked by MnTBAP treatment (Melov et al., 1998) and remains to be further characterized.
The comparative efficacies of the three analogs in the sod2nullizygous mice are of interest. EUK-8 and EUK-134 were approximately equipotent, as indicated by their comparable effects at both doses (Table 2). Thus, in this model, the enhanced catalase activity of EUK-134 did not confer a greater protectiveness than that seen with EUK-8. EUK-8 appeared to be slightly more effective than EUK-134, although this difference was not statistically significant. This observation is consistent with SOD activity, rather than catalase activity, being of primary importance for an agent to rescue thesod2 nullizygous phenotype, because EUK-8 and EUK-134 have equivalent SOD activities. This is in contrast to certain other models, for example a rat stroke model, in which EUK-134 was significantly more effective than EUK-8 (Baker et al., 1998). EUK-189 has SOD and catalase activities equivalent to those of EUK-134 but is significantly more lipophilic than EUK-8 or EUK-134 (Table 1). Thus, the increased effectiveness of EUK-189 compared with EUK-134 in the sod2nullizygous mice might be attributable to an enhanced ability to cross the blood–brain barrier and/or gain access to the mitochondria. In support of the latter concept, EUK-189 has been shown recently to be substantially more potent than EUK-134 in protecting neuronal cultures against staurosporine-induced apoptosis but equipotent in protecting cells against an extracellular hydrogen peroxide insult (Pong et al., 2001). Interestingly, the maximum lifespan of the treatedsod2 nullizygous mice remained approximately the same with EUK-189 treatment as it did with the other compounds (Fig. 1). This suggests a late-onset (>25 d) phenotype that is not amenable to treatment with these compounds, at least under the protocol used in this study.
As noted above, the pathologies observed in the sod2nullizygous mice are consistent with damage from mitochondrial oxidative stress, and indeed, the mice have been shown to have numerous defects in mitochondrial enzymes (Melov et al., 1999). Analysis of isolated brain mitochondria from treated sod2 nullizygous mice (Fig. 4) strongly supports our hypothesis that the compounds investigated in this study exert their neuroprotective function directly through protection of brain mitochondria from endogenously generated ROS. Essentially, these data demonstrate that EUK-8-treatedsod2 nullizygous mice showed a significant preservation of active cis-aconitase, a known target of mitochondrial oxidative damage, compared with age-matched MnTBAP-treated mice exhibiting the spongiform neuropathy.
Overall these observations in sod2 nullizygous mice suggest that the class of synthetic catalytic antioxidants exemplified by EUK-8 and its analogs can permeate the brain, gain access to the mitochondria, and attenuate the mitochondrial damage attributable to oxidative stress, as well as its resulting pathologies. This may have important clinical implications in the design and implementation of therapeutic approaches to several diseases. Interestingly, a recent report describing a mouse model of FA (Puccio et al., 2001) shows a highly similar phenotype to the sod2 nullizygous mouse treated with MnTBAP (Melov et al., 1998). Commonalities between these two mouse models include a regional spongiform encephalopathy, cardiomyopathy, and mitochondrial enzymatic abnormalities (Melov et al., 1998; Puccio et al., 2001). This implies that mitochondrial oxidative stress may be a central feature of the pathology of FA and that, therefore, “mito-protective” antioxidants such as the ones described in this report would be efficacious for the human disease. In addition, a recent report demonstrated that oxidative stress precedes amyloid formation in the brains of Tg2576 Alzheimer mice, indicating that effective antioxidant therapy in this model might prevent the development of the pathology (Pratico et al., 2001). More generally, the observations of a prolonged lifespan in the sod2nullizygous mice by threefold and the concomitant rescue of the spongiform neurodegenerative disorder imply that such mito-protective SOD–catalase mimetics might retard the pathogenesis of other neurodegenerative disorders with reactive oxygen species implicated in their etiology, such as Parkinson's and Alzheimer's diseases and, perhaps, the degenerative processes of aging.
Footnotes
This work was supported by National Institutes of Health Grants DCW-AG13154, HL45572, NS21328, and AG18679 (S.M.). We thank B. A. Day and J. D. Crapo for their kind gift of MnTBAP, T.-T. Huang and C. J. Epstein for the gift of sod2 nullizygous mice, and Tamara Golden for helpful comments. All procedures with animals were performed under approved Buck Institute or Emory University Institutional Animal Use and Care Committee protocols.
Correspondence should be addressed to Simon Melov, Buck Institute for Age Research, 8001 Redwood Boulevard, Novato, CA 94945 E-mail: smelov@buckinstitute.org.
REFERENCES
- 1.Baker K, Marcus CB, Huffman K, Kruk H, Malfroy B, Doctrow SR. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J Pharmacol Exp Ther. 1998;284:215–221. [PubMed] [Google Scholar]
- 2.Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E, Malfroy B. Salen-manganese complexes are superoxide dismutase-mimics. Biochem Biophys Res Commun. 1993;192:964–968. doi: 10.1006/bbrc.1993.1509. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 4.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 5.Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 6.Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- 7.Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical, and molecular genetic correlations in Friedreich's ataxia. Hum Mol Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- 8.Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochem J. 1999;344:1–5. [PMC free article] [PubMed] [Google Scholar]
- 9.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 10.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 11.Choi SI, Ju WK, Choi EK, Kim J, Lea HZ, Carp RI, Wisniewski HM, Kim YS. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol (Berl) 1998;96:279–286. doi: 10.1007/s004010050895. [DOI] [PubMed] [Google Scholar]
- 12.Conn KJ, Ullman MD, Eisenhauer PB, Fine RE, Wells JM. Decreased expression of the NADH:ubiquinone oxidoreductase (complex I) subunit 4 in 1-methyl-4-phenylpyridinium-treated human neuroblastoma SH-SY5Y cells. Neurosci Lett. 2001;306:145–148. doi: 10.1016/s0304-3940(01)01888-2. [DOI] [PubMed] [Google Scholar]
- 13.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the β-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 14.Doctrow SR, Huffman K, Marcus CB, Musleh W, Bruce A, Baudry M, Malfroy B. Salen-manganese complexes: combined superoxide dismutase/catalase mimics with broad pharmacological efficacy. Adv Pharmacol. 1997;38:247–269. doi: 10.1016/s1054-3589(08)60987-4. [DOI] [PubMed] [Google Scholar]
- 15.Fridovich I. Fundamental aspects of reactive oxygen species, or what's the matter with oxygen? Ann NY Acad Sci. 1999;893:13–18. doi: 10.1111/j.1749-6632.1999.tb07814.x. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez PK, Zhuang J, Doctrow SR, Malfroy B, Benson PF, Menconi MJ, Fink MP. EUK-8, a synthetic superoxide dismutase and catalase mimetic, ameliorates acute lung injury in endotoxemic swine. J Pharmacol Exp Ther. 1995;275:798–806. [PubMed] [Google Scholar]
- 17.Haller J, Weggemans RM, Ferry M, Guigoz Y. Mental health: minimental state examination and geriatric depression score of elderly Europeans in the SENECA study of 1993. Eur J Clin Nutr. 1996;50 [Suppl 2]:S112–S116. [PubMed] [Google Scholar]
- 18.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 19.Jama JW, Launer LJ, Witteman JC, den Breeijen JH, Breteler MM, Grobbee DE, Hofman A. Dietary antioxidants and cognitive function in a population-based sample of older persons. The Rotterdam Study. Am J Epidemiol. 1996;144:275–280. doi: 10.1093/oxfordjournals.aje.a008922. [DOI] [PubMed] [Google Scholar]
- 20.Jung C, Rong Y, Doctrow S, Baudry M, Malfroy B, Xu Z. Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett. 2001;304:157–160. doi: 10.1016/s0304-3940(01)01784-0. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 22.Malfroy B, Doctrow SR, Orr PL, Tocco G, Fedoseyeva EV, Benichou G. Prevention and suppression of autoimmune encephalomyelitis by EUK-8, a synthetic catalytic scavenger of oxygen-reactive metabolites. Cell Immunol. 1997;177:62–68. doi: 10.1006/cimm.1997.1091. [DOI] [PubMed] [Google Scholar]
- 23.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 24.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang T-T, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 26.Morris MC, Beckett LA, Scherr PA, Hebert LE, Bennett DA, Field TS, Evans DA. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–126. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Pandolfo M. Molecular genetics and pathogenesis of Friedreich ataxia. Neuromuscul Disord. 1998;8:409–415. doi: 10.1016/s0960-8966(98)00039-x. [DOI] [PubMed] [Google Scholar]
- 28.Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 29.Perrig WJ, Perrig P, Stahelin HB. The relation between antioxidants and memory performance in the old and very old. J Am Geriatr Soc. 1997;45:718–724. doi: 10.1111/j.1532-5415.1997.tb01476.x. [DOI] [PubMed] [Google Scholar]
- 30.Pong K, Doctrow SR, Huffman K, Baudry M. Attenuation of staurosporine-induced apoptosis, oxidative stress, and mitochondrial dysfunction by synthetic superoxide dismutase and catalase mimetics in cultured cortical neurons. Exp Neurol. 2001;171:84–97. doi: 10.1006/exnr.2001.7747. [DOI] [PubMed] [Google Scholar]
- 31.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect, and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 33.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 34.Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci USA. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 36.Schapira AHV. Mitochondrial dysfunction in neurodegenerative disorders. Biochim Biophys Acta. 1998;1366:225–233. doi: 10.1016/s0005-2728(98)00115-7. [DOI] [PubMed] [Google Scholar]
- 37.Smith MA, Sayre LM, Anderson VE, Harris PL, Beal MF, Kowall N, Perry G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 38.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 40.White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol. 1999;155:1723–1730. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White AR, Huang X, Jobling MF, Barrow CJ, Beyreuther K, Masters CL, Bush AI, Cappai R. Homocysteine potentiates copper- and amyloid β peptide-mediated toxicity in primary neuronal cultures: possible risk factors in the Alzheimer's-type neurodegenerative pathways. J Neurochem. 2001;76:1509–1520. doi: 10.1046/j.1471-4159.2001.00178.x. [DOI] [PubMed] [Google Scholar]