

Abstract
CA3–CA1 glutamatergic synapses in the hippocampus exhibit a large heterogeneity in release probability (p) and paired-pulse (PP) plasticity, established already in the early neonatal period when the CA3–CA1 connections consist of only a single release site. At such a site two factors decide initial release probability: the number of immediately releasable vesicles (preprimed pool) and the vesicle release probability (Pves1). Depletion and replenishment of this pool, an alteration in Pves, and desensitization of postsynaptic receptors may contribute to PP plasticity. A model based on data from single neonatal CA3–CA1 synapses has been used to address the relative importance of these factors for the heterogeneity in PP plasticity. At a 20 msec PP interval, the PP ratio (P2/P1) varied from 0.1 to 4.5 among the synapses. At this interval desensitization and replenishment were of little importance. The heterogeneity was explained mostly by the variation inPves1, whereas the preprimed pool size was of minor importance. Pves altered from the first to the second stimulus such thatPves2 was rather uniform among the synapses. Its variation thus contributed little to the heterogeneity in PP ratio. The model also shows that the relationship between alterations in release probability and PP ratio is complex. Thus, an increase in release probability can be associated with an increase, a decrease, or no change at all in PP ratio, depending on the original values ofPves1 and the preprimed pool and on which one of these factors is altered to produce the increase in release probability.
Keywords: paired pulse, release probability, hippocampus, CA1, synaptic plasticity, development
A ubiquitous feature of synapses is that synaptic activity modifies synaptic action. In its simplest form this plasticity can be seen as paired-pulse (PP) facilitation (PPF)/paired-pulse depression (PPD), in which the second afferent stimulation produces more/less synaptic action than the first one (Katz and Miledi, 1968; Zucker, 1989; Thomson, 2000). Mechanisms, both on the presynaptic and postsynaptic side, may contribute to paired-pulse plasticity. These include changes in the number of release-ready vesicles, in the release probability of the individual vesicles, and in the responsiveness of the postsynaptic receptors. Paired-pulse plasticity is of functional interest because it decides the short-term computational properties of the synapse. Moreover, it is also used as a tool for evaluation of possible presynaptic changes after other kind of manipulations of the synapse. For example, manipulations thought to influence presynaptic release are generally assumed to alter paired-pulse plasticity in a predictable manner.
To properly understand what underlies variations in paired-pulse plasticity among synapses, as well as to use it as a tool, one would have to know what factors determine release probability and how these factors may be altered after a first stimulus. For the Schaffer collateral synapses on CA1 pyramidal cells, release probability to the first (initial) stimulus and paired-pulse plasticity vary considerably among the synapses in a correlated manner (Dobrunz and Stevens, 1997). It has been suggested that the initial release probability is primarily decided by the number of readily releasable vesicles (Dobrunz and Stevens, 1997), but how this latter factor could give rise to the correlated variation in paired-pulse behavior is unclear. Recently, examination of release from these synapses in the neonatal rat has indicated that initial release probability also depends to a considerable extent on a variation in vesicle release probability (Pves) (Hanse and Gustafsson, 2001a,b). Moreover, the release-ready pool was found to be very small (on average only one vesicle) and variable among the synapses. Such information would then allow for an evaluation of the manner in which vesicle release probability and the size of the release-ready pool could create a variation in the paired-pulse behavior among these synapses.
In the present article, we have modeled the paired-pulse behavior of single release sites exhibiting the above features. The aim has not been to give a full account of all aspects of paired-pulse behavior. Rather, by explaining a specific instance of paired-pulse behavior our aim is that some general insights into how paired-pulse plasticity is molded and how it can be used as a tool may emerge.
MATERIALS AND METHODS
Hippocampal slice preparation. The experimental data used in the present study were obtained as described in detail previously (Hanse and Gustafsson, 2001a,b). Hippocampal slices were prepared from 1- to 7-d-old Wistar rats. The rats were killed by decapitation in accordance with the guidelines of the local ethical committee for animal research. Whole-cell patch-clamp recordings were performed from visually identified CA1 pyramidal cells using a pipette solution containing (in mm): 95 Cs-gluconate, 20 TEA-Cl, 10 NaCl, 5 QX-314, 4 Mg-ATP, 0.4 Na-GTP, 0.2 EGTA, and 10 HEPES, pH 7.3, adjusted with CsOH. Recordings were performed at 30–32°C and the extracellular solution contained (in mm): 124 NaCl, 3.0 KCl, 4 CaCl2, 4 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, and 0.02 bicuculline methiodide or 0.1 picrotoxin.
Afferents in the stratum radiatum were activated with 10 impulse, 50 Hz trains at 0.2 Hz using minimal extracellular stimulation. Several findings suggested that this minimal stimulation consistently resulted in the activation of a single axon contributing a single synapse to the cell recorded from this synapse containing a single release site (Hanse and Gustafsson, 2001a). They also suggested that this release site releases one vesicle per stimulus at most (Triller and Korn, 1982;Stevens and Wang, 1995; Dobrunz and Stevens, 1997; Liu et al., 1999;Matveev and Wang, 2000). Most importantly, the average EPSC amplitude, excluding failures, was found to be independent of release probability during the burst (Hanse and Gustafsson, 2001a). This result strongly argues against a release of more than one vesicle per action potential, because that would have resulted in larger EPSC amplitudes at positions of higher release probability during the burst.
Determination of vesicle release probability and size/variation of preprimed pool. The release probability (p) of a single release site is thought to be determined by the number of release-ready, or primed, vesicles (npool) and by thePves, such that
| Equation 1 |
Pves varies among the release sites but is considered to be the same within a release site (Hanse and Gustafsson, 2001b). We refer to the pool of vesicles primed at the arrival of the first stimulus of the 10 impulse train as the preprimed pool. The analysis to determine Pves1(Pves at the first stimulus position of the train) and the size of the preprimed pool for individual synapses have been described previously (Hanse and Gustafsson, 2001b). Briefly, all of the 10 impulse trials, ∼100 per synapse, were also considered as 9 impulse trials (disregarding the last response), and so on, down to 2 impulse trials. Then, for each of thesen-impulse trials, two groups of trials were selected. One group contained trials in which only a single release event occurred during the n stimuli. The other one contained trials in which two release events occurred during the n stimuli, with the condition that one of the events occurred at the last (nth) stimulus position for the given length. For each trial length the probability that a release event occurred at the first stimulus position was then compared between the one release and two release event groups. When the release probabilities in the first stimulus position in the train were the same for the two groups, the release event at the nth position in the train could not have originated from a vesicle that was primed at the onset of train stimulation. However, when the release probability was higher for the two release event group, this second vesicle must have belonged to the preprimed pool. This analysis then defined, for each synapse, the stimulus position at which release no longer originates from preprimed vesicles but rather originates from vesicles recruited to a primed state during the train (refilling point). The average preprimed pool for each synapse was then estimated as the cumulative release occurring before the refilling point was reached.
This analysis also allows for an estimation ofPves1, because the trials in which the preprimed pool consisted of only one vesicle are the trials in which only one release event occurred before the refilling point. Thus,Pves1 was determined as the release probability in the first stimulus position for those single release trials. This method for determiningPves1 is thus based on a subset of the trials (∼100) to which each synapse was subjected. To estimate the sampling error in the estimation ofPves1 we performed a Monte Carlo simulation of release from a preprimed pool according to Equation 1 and a binomial distribution of the pool size (see Fig.2B). These simulations, each with 100 trials, gave an estimated Pves1 that on average was equal to the predefined one with a SD of ∼0.06. This can be compared with the SD of 0.28 for the variation between synapses (Hanse and Gustafsson, 2001b). Because there was an intersimulation variation in the release probability at position 1 (P1) (SD = 0.04) that correlated with that of the estimated Pves1(r ≈ 0.7), the error with respect to the calculatedP1 (for a given simulation or experiment) will thus only be approximately one-half the above estimated error in Pves1. ThePves value at the second stimulus position (Pves2) was calculated as follows:
| Equation 2 |
where P2 is the release probability at position 2 (using all trials) andnpool is the average size of the preprimed pool.
Fig. 2.
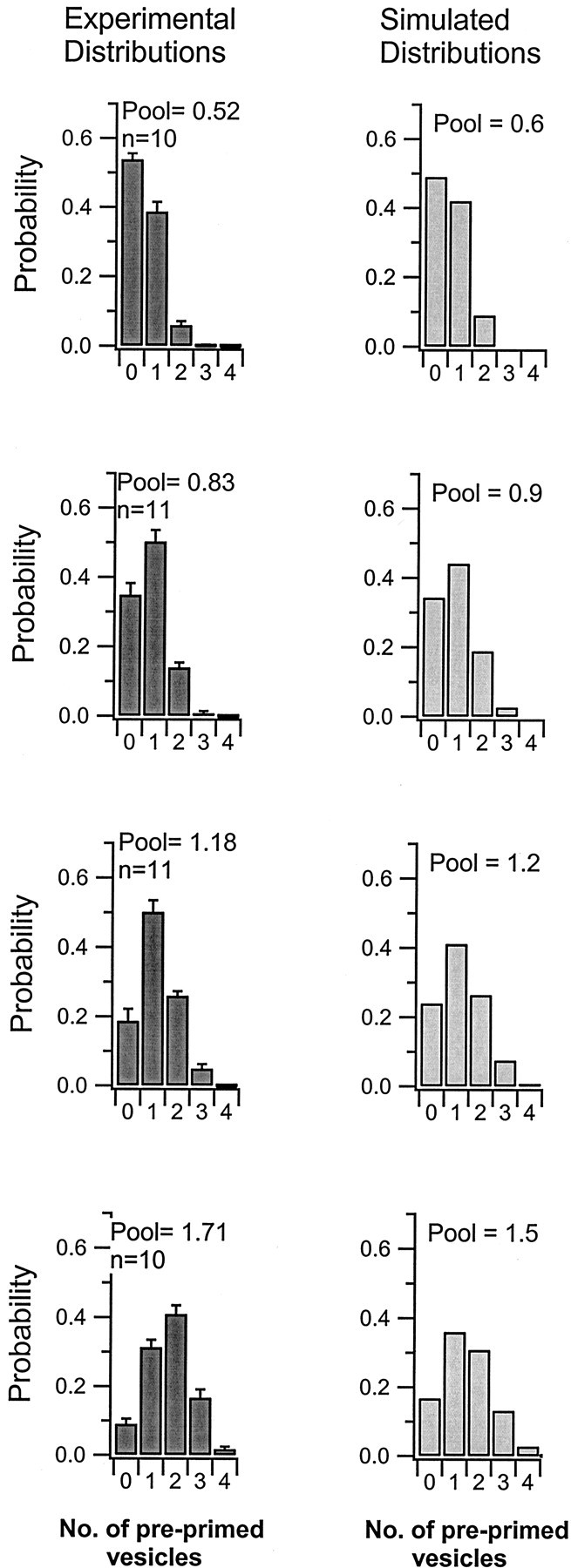
Distribution of the trial-to-trial variation in the preprimed pool size. The left column shows the trial-to-trial variability in the number of preprimed vesicles. Results from different synapses were pooled together in four groups according to the average size of their preprimed pool. The average pool size within each group is indicated in the graphs together with the number of synapses included. The right column shows binomial distributions using two, three, four, and five sites, respectively; each site had an occupancy probability of 0.3.
If release events occurring up to the refilling point were considered, any given synapse displayed a substantial trial-to-trial variation in the number of such events. This variation was used to estimate the size distribution of the preprimed pool across all trials (see Fig. 2). Although a fixed refilling point provides for an accurate estimation of the average preprimed pool (Hanse and Gustafsson, 2001b), it may introduce some bias in the estimation of the size distribution. First, a vesicle that is recruited during the train may be released before the refilling point and, if so, bias the distribution toward larger pool contents. Second, if there are more than two preprimed vesicles (the refilling point analysis is based on a preprimed pool of two vesicles), a preprimed vesicle may be released after that position and, if so, bias the distribution toward smaller pool contents. These putative biasing effects are partially opposing each other and their influence should be small as long as the preprimed pool is small. A small (0.5–2 vesicles) average preprimed pool was also found among these synapses (Hanse and Gustafsson, 2001b). Nevertheless, because of these considerations, the distributions of the trial-to-trial variation of the preprimed vesicle pool should be considered provisional.
Model computations. Based on the range of the experimentally estimated values of Pves1 andPves2 and the size and variation of the preprimed pool, release probabilities in response to a first (P1) and a second (P2) stimulus were calculated using Equation 1. Pves1 was varied between 0.1 and 1.0 and the (average) preprimed pool was varied between 0.6 and 1.8. Pves2 was set to a fixed value of 0.35. The trial-to-trial variation in the preprimed pool size was simulated by a binomial distribution. The paired-pulse ratio was calculated asP2/P1. The analysis and the calculations were performed using custom software written in Igor Pro (Wavemetrics, Lake Oswego, OR).
Unless otherwise indicated, data are presented as mean ± SEM. Student's t test was used to determine statistical significance.
RESULTS
Figure 1Aillustrates unitary quantal release from a single terminal of a Schaffer collateral axon onto a CA1 pyramidal cell in response to train activation (20 msec interval). Such activation of synapses in 1- to 7-d-old rats resulted in release that varied considerably among the synapses with respect to initial release probability (P1), and facilitation/depression behavior (Hanse and Gustafsson, 2001b). TheP1 values varied from almost 0 up to almost 1.0, whereas release probability to the second stimulus (P2) had a somewhat more narrow distribution, mostly between 0.1 and 0.4 (Fig. 1B). No correlation was observed between theP1 and theP2 values (Fig. 1B). The paired-pulse ratio (P2/P1) varied widely among the synapses, with most synapses demonstrating PPD (Fig. 1C). Synapses with lowP1 produced PPF, and the larger the P1 the more the PPD. However, the scatter was considerable, indicating a rather poor predictive ability of P1 with respect to paired-pulse plasticity. TheP2/P1ratio did not correlate with the age of the animal during the first postnatal week (r = 0.03; p > 0.05).
Fig. 1.
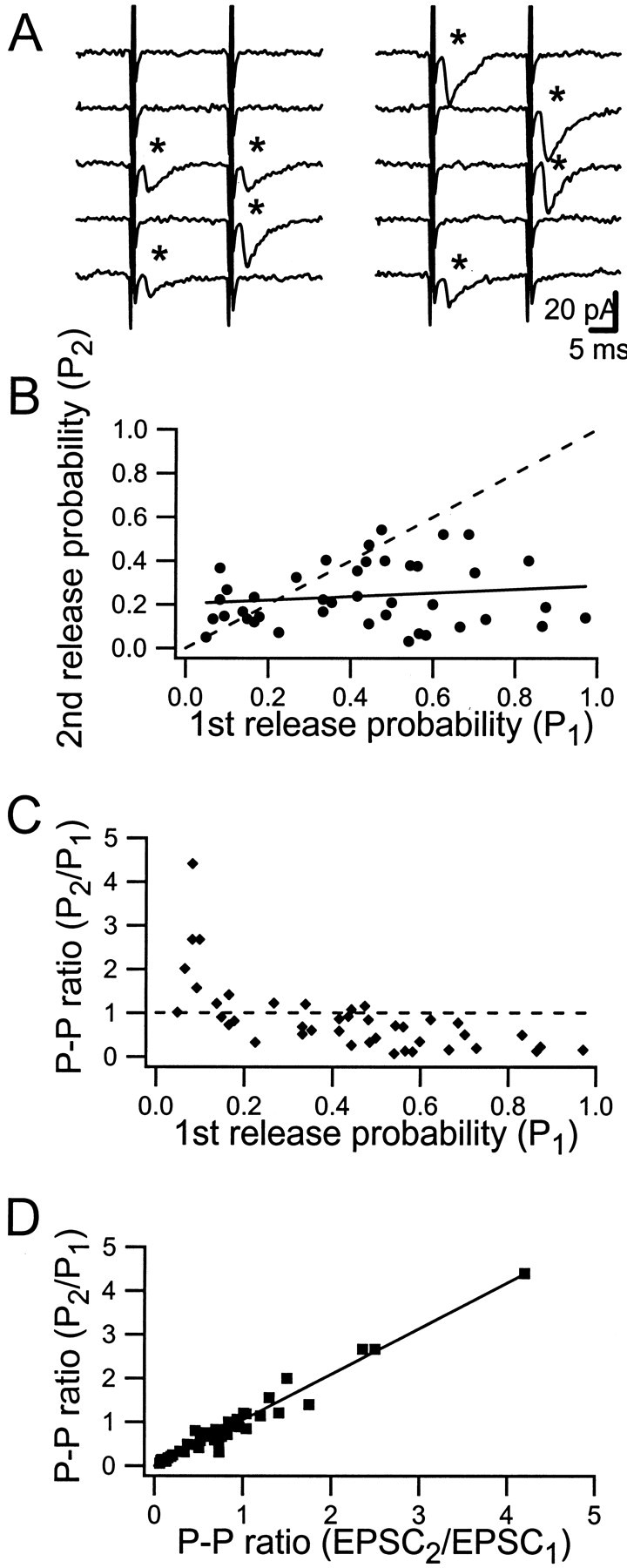
Release probabilities and paired-pulse (20 msec) ratio among synapses. A, Ten consecutive sweeps (0.2 Hz) from one synapse in response to paired stimuli. EPSCs are indicated byasterisks. B, Relationship betweenP1 and P2 among synapses (n = 42). The solid line is a linear regression line (r = 0.14;p > 0.05), and the dashed lineindicates equality between P1 andP2. Nine synapses with no first-release probability (low-frequency mute synapses) are not included in the graph. C, Relationship between the paired-pulse ratio (P2/P1) and P1 among the synapses (n = 42). The dashed line indicates equality between P1 andP2. D, Relationship between paired-pulse ratios measured using release probabilities and EPSC areas. The solid line is a linear regression line (r = 0.96; p < 0.001).
Paired-pulse plasticity was also evaluated as EPSC2/EPSC1, where EPSC1 and EPSC2 are the average EPSC magnitudes (including failures). Discrepancies between this ratio and that ofP2/P1would indicate the influence of changes in quantal size on paired-pulse plasticity. On average, both the magnitude and probability ratio were 0.84 ± 0.12 (n = 43), and the two ratios correlated well (slope = 0.98) throughout the range of paired-pulse plasticity (Fig. 1D). That is, postsynaptic factors such as desensitization do not participate in paired-pulse plasticity under these conditions.
To analyze what may explain the above paired-pulse behavior,P1 andP2 were calculated using Equation 1 as described in Materials and Methods.
Binomial trial-to-trial distribution of preprimed vesicles
As described in Materials and Methods, the size of the preprimed pool at a given release site varies from trial to trial. To account in the model for this variation, the nature of this variation was examined. For each synapse the number of preprimed vesicles at each stimulus trial was determined (see Materials and Methods), and a distribution of these values was obtained. Synapses were then subdivided arbitrarily into four groups with respect to their average pool size, and the distributions within each of these groups were pooled together (Fig. 2, left column). In the group with the lowest average size of preprimed pool (Fig. 2, left, uppermost graph), the pool was zero in more than one-half of the trials, and trials with two preprimed vesicles were rare. However, in the group with the largest average pool (Fig. 2, left, lowermost graph) preprimed vesicles were absent in <10% of the trials, and up to four preprimed vesicles could occur.
The fact that on some trials preprimed vesicles were absent suggests that priming is reversible. Otherwise, it seems difficult to understand why during some of the 5 sec intertrial intervals two to three vesicles have entered a primed state whereas during many other such intervals no vesicle has entered such a state. It may then be conceived that docked vesicles are in equilibrium between a primed and a nonprimed state. The simplest model is to assume that each synapse has a number of docking/priming sites equal to the largest number of preprimed vesicles observed for that synapse. We thus computed the binomial distribution of site occupancy by varying the number of sites from two to five and keeping the probability of site occupancy (at each site) constant. Theright column of Figure 2 shows that for matching average preprimed pool sizes, a p value of 0.3 gave distributions closely resembling the experimental ones.
Such a value can then used to model the trial-to-trial variability of the preprimed pool.
Computed paired-pulse ratio: Pves1 and preprimed pool
The experimentally determined values ofPves for the first stimulus (Pves1) vary among the synapses from close to 0 up to almost 1 (Fig. 3) (Hanse and Gustafsson, 2001b). For the calculations,Pves1 was thus allowed to vary from 0.1 to 1.0 in steps of 0.1. Experimentally,Pves for the second stimulus (Pves2) was found to vary mostly between 0.2 and 0.5, independently ofPves1 (Fig. 3). In the model we therefore chose to use a fixed value of 0.35 forPves2. The average size of the preprimed pool was varied between 0.6 and 1.8 in steps of 0.3, with each average size represented by a binomial distribution ofnpool sizes (compare Fig. 2,right column). The calculated values ofP1 andP2 are plotted against each other in Figure 4A. In agreement with the experimental values (Fig. 1B), the modeledP1 values vary from close to 0 up to 0.9, and the P2 values are more narrowly distributed, mostly within 0.1–0.4. It can be noted that whenP1 increases because of an increase in Pves1 (for a given pool size) P2 decreases, but this decrease is relatively less the larger the pool size. However, whenP1 increases because of an increase in pool size (for a given Pves1) thenP2 increases, relatively more the larger the Pves1. These two contrasting effects indicate an overall lack of a relationship betweenP1 andP2, as also observed experimentally (Fig. 1B).
Fig. 3.

Experimentally obtained relationship betweenPves2 and Pves1among the synapses (n = 42). The dashed line is the regression line for these data (r = 0.09; p > 0.05).Pves2 was calculated using Equation 2, whereP2 is the release probability at stimulus position 2, npool is the average size of the preprimed pool, and P1 is the release probability at stimulus position 1.
Fig. 4.
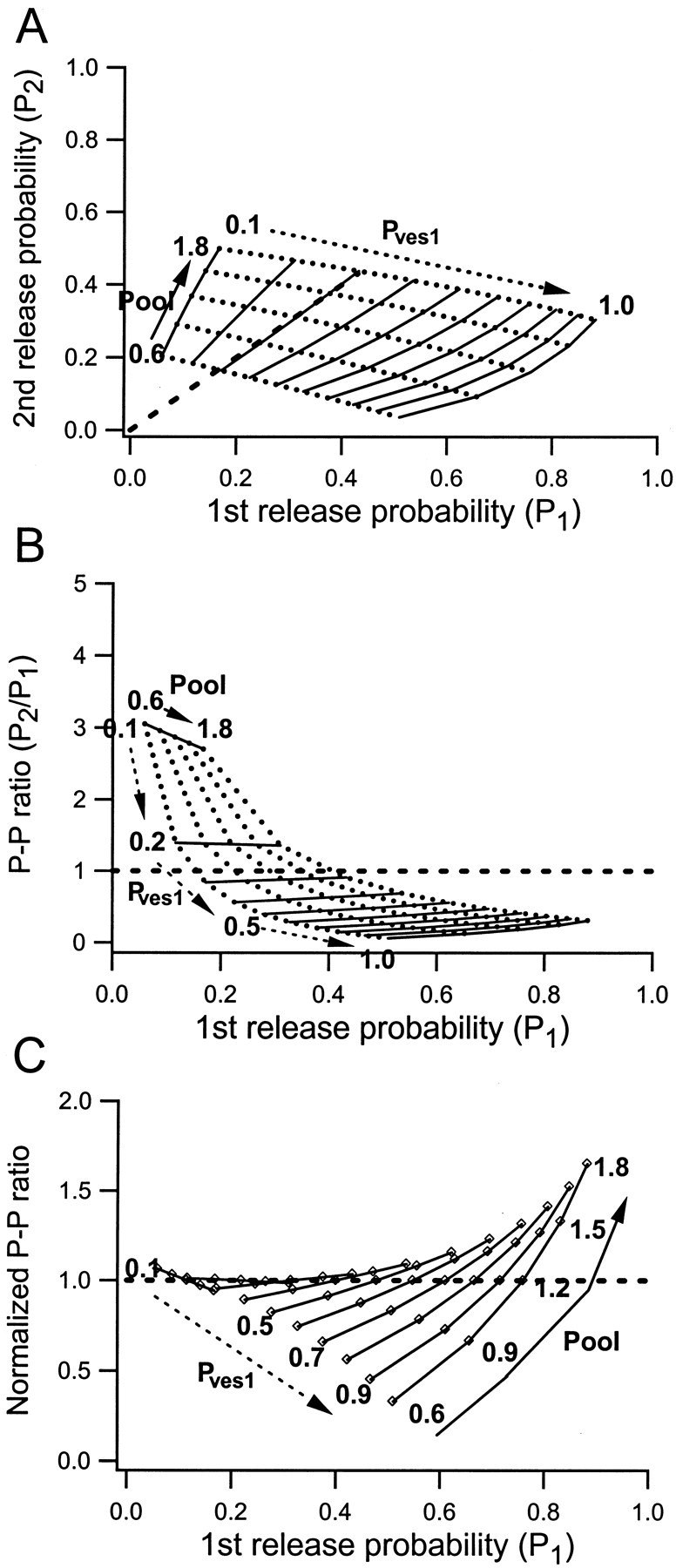
Calculated relationships betweenP1,P2, and the PP ratio at different magnitudes of Pves1 and the preprimed pool. The release probabilities in response to the first (P1) and second (P2) stimulus were calculated using Equation 1 as described in Materials and Methods.Pves1 was varied between 0.1 and 1 in steps of 0.1. The average preprimed pool was varied between 0.6 and 1.8 vesicles in steps of 0.3. Pves2 was set to a fixed value of 0.35. A, Relationship betweenP1 and P2. Thedashed line indicates equality betweenP1 and P2.B, Relationship between the paired-pulse ratio (P2/P1) and P1. The dashed lineindicates a PP ratio of 1. C, Same as inB, but each PP–Pves1 curve is normalized with respect to the paired-pulse ratio value obtained at a pool of 1.2.
Figure 4A shows that most calculated values fall below the “P1 =P2 line” (dashed line), indicating a predominance of PPD over PPF. When theP2/P1ratio is plotted against P1 (Fig.4B), there is an overall but broad relationship between the paired-pulse ratio and P1, in agreement with the experimentally observed relationship (Fig.1C). The broadness is again related to the contrasting actions of Pves and preprimed pool variations, respectively. When P1increases because of an increase inPves1, the paired-pulse ratio decreases, in line with the common notion that an increase in release probability should be associated with a substantial decrease in paired-pulse ratio. However, the horizontal nature of the lines drawn through the iso-Pves values in Figure4B shows that whenP1 increases because of an increase in the pool size, the paired-pulse ratio changes little or, for largePves1 values, actually increases. To better illustrate the relative change in the paired-pulse ratio introduced by preprimed pool variations, the same values have been replotted in Figure 4C after normalization to the PP ratio calculated for the median (average) preprimed pool (1.2). It can be noted that for Pves1 values of >0.5, an increase in the preprimed pool causes an increase inP1 associated with a substantial relative increase in the paired-pulse ratio.
Nevertheless, the above calculations suggest that the variation in paired-pulse ratio is dominated by the variation inPves1. This effect ofPves1 depends on two factors. The first factor is that a change in Pves1alters thePves2/Pves1ratio, thereby changing theP2/P1ratio. The second factor is that a largerPves1 will lead to a larger depletion after the first stimulus. In Figure5A, the calculated values ofP2/P1(solid lines) are plotted against thePves1 value for the five different pool sizes. It can be noted that whenPves1 is small (0.1–0.2), there is a large PPF of 100–200%. When Pves1 is large (>0.7) there is a large PPD, with the second response being only some 10–30% of the first one. To separate the above two effects ofPves1, the depletion factor was annulled by using the same pool size for the calculation ofP2 as forP1. TheseP2/P1ratios are shown in the same graph (Fig. 5A, dashed lines). Comparison between the two sets ofP2/P1calculations indicates that most of the variation in paired-pulse ratio depends on changes in thePves2/Pves1ratio. That is, depletion does not cause large changes in the absolute values of the paired-pulse ratio. Nevertheless, depletion appears to cause considerable changes in the normalized paired-pulse ratio, in particular for larger values of Pves1. To illustrate that effect more directly, the calculated values (Fig.5A, solid lines) were, for each size of (average) preprimed pool, normalized with respect to those calculated using the same pool size for P2 as forP1 (Fig. 5A, dashed lines). This procedure demonstrates (Fig. 5B) that depletion causes a decrease in the normalized paired-pulse ratio that depends strongly on the value of Pves1as well as on the preprimed pool size.
Fig. 5.
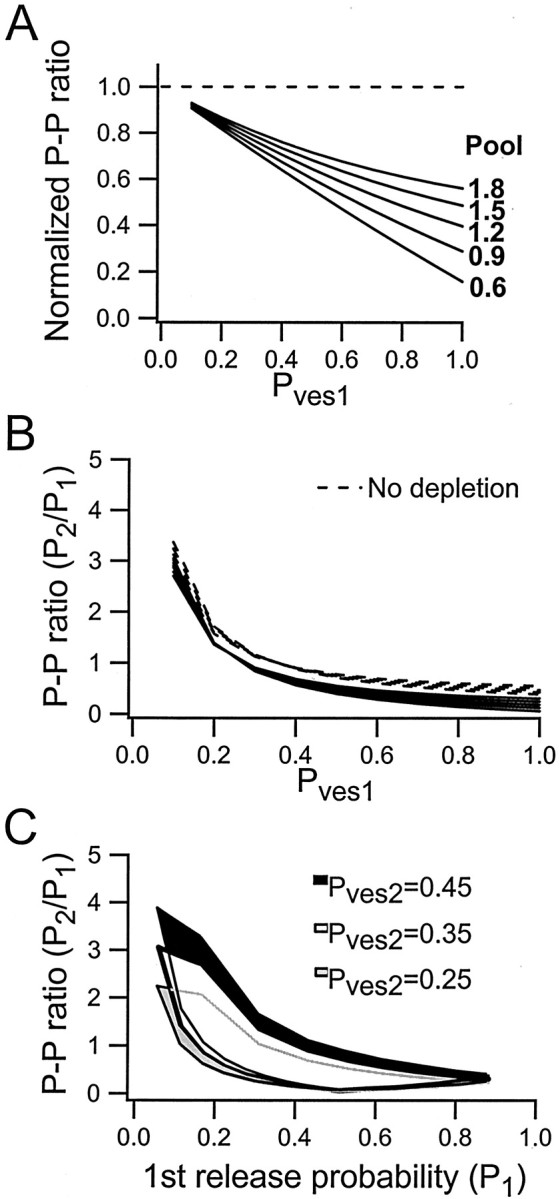
Paired-pulse ratio (P2/P1) against Pves1: effects ofPves2 variations and of depletion. These calculations were performed as described in the legend to Figure4. A, Relationship betweenPves1 and the paired-pulse ratio.Solid lines represent the relationship for five different pool sizes (0.6, 0.9, 1.2, 1.5, and 1.8, respectively). Thedashed line represents calculations using the same five pool sizes, but in this case the pool size was the same for the first and second stimulus (i.e., there was no depletion of vesicles).B, Relative effect of vesicle depletion for paired-pulse ratio for different pool sizes (0.6–1.8) and for differentPves1 values. The plot is derived fromA such that for each pool size, the ratio between the curves with (solid line in A) and without (dashed line in A) depletion was constructed. C, Effect of differentPves2 values on the relationship between the paired-pulse ratio and first-release probability. The white area, corresponding to a Pves2 value of 0.35, is the same as the plot shown in Figure4B. The gray andblack areas correspond toPves2 values of 0.25 and 0.45, respectively. These Pves2 values correspond approximately to the mean ± SD of the experimentally obtainedPves2 (0.33 ± 0.11).
Computed paired-pulse ratio: Pves2
Figure 3 shows that the experimentally estimated values ofPves2 varied from ∼0.2–0.5 independently of Pves1. The variation in Pves2 will then not be expected to affect the paired-pulse ratio in a manner that is correlated with initial release probability but rather to introduce scatter in the PPF/PPD–P1 relationship. The influence of Pves1 (and of preprimed pool) variations on paired-pulse ratio was thus examined using a fixedPves2 value of 0.35. However, to indicate the effect of Pves2 on the paired-pulse ratio, the calculations ofP2 andP1 illustrated in Figure4B were also performed usingPves2 values of 0.25 and 0.45. Figure5C shows that this variation ofPves2 introduces some shift of the relationship between paired-pulse ratio and initial release probability without altering its overall shape.
Paired-pulse ratio versus Pves1 and preprimed pool size
Heterogeneity in P1 among the synapses is explained about equally by variations inPves1 and in preprimed pool size (Hanse and Gustafsson, 2001b). The above calculations suggest that it is mostly P1 differences attributed to variation in Pves1 that should cause differences in the paired-pulse ratio. Figure6 shows the experimentally observed paired-pulse ratios plotted againstPves1 (Fig. 6A) and the preprimed pool size (Fig. 6B) estimated for these synapses. As can be noted from the individual values (Fig. 6,filled circles) as well as from the binned averages (Fig.6A, filled squares) and the regression line (Fig. 6B, dashed line), these paired-pulse ratio values covaried withPves1 and not with pool size. Calculated relationships are also included in these graphs (assolid lines).
Fig. 6.

Pves1, but not the preprimed pool, determines the paired-pulse ratio.A, Relationship betweenP2/P1 andPves1. Filled circles are experimental data obtained from 42 synapses. Filled squares represent binned averages (bin size is 0.2Pves units) of the experimental data. Error bars are ±SEM and are only seen outside thesymbol for the lowest Pves1value. Lines are connecting calculated values using the parameter values given in the legend to Figure 4.B, Relationship betweenP2/P1 and the preprimed pool. Filled circles are experimental data obtained from 42 synapses. The dashed line is the regression line for these data (r = −0.14;p > 0.05). Solid lines are connecting calculated values using the parameter values given in the legend to Figure 4.
DISCUSSION
The present experimental results and calculations suggest that heterogeneity in paired-pulse plasticity among the Schaffer collateral synapses in the neonatal rat is primarily explained by a variation inPves1. Variations inPves2 and in the size of the release-ready vesicle pool were found to be of secondary importance, and desensitization was of no importance. The present model also suggests that an increase in initial release probability is not necessarily associated with a decrease in paired-pulse ratio but can also be associated with no significant alteration or even with a substantial increase.
Paired-pulse plasticity: Pves changes, pool changes, and desensitization
Paired-pulse plasticity is commonly seen as resulting from the interaction of three factors: two presynaptic ones and a postsynaptic one. Facilitation results from an increase inPves, based for example on “residual” calcium from the first stimulus. Depression can result from a decrease in Pves, a decrease in vesicle pool size (depletion), and desensitization of postsynaptic receptors. In the present calculations, desensitization was not taken into account because, at least with the paired-pulse intervals used (20 msec), paired-pulse plasticity was found experimentally to be the same regardless of whether it was evaluated by EPSC magnitude or by release probability. That is, postsynaptic factors such as desensitization do not play any role (Hjelmstad et al., 1999).
The importance of vesicle depletion was evaluated using model calculations. Depletion was found to have an effect on paired-pulse plasticity that was strongly dependent onPves1 and on the preprimed pool. For lower Pves1 values, depletion gave ∼20% lower values of paired-pulse plasticity that were independent of preprimed pool size. For largePves1 values, corresponding values were 40–80%, with the effect being more accentuated for the smaller preprimed pool sizes. Vesicle depletion can thus be a significant factor in the generation of PPD under certain conditions. These calculations did not take into account possible recruitment of newly primed vesicles. Approximately one-sixth of the hippocampal neonatal synapses lack initial release but display a low release probability (0–0.1) to the second stimulus (“low-frequency mute” synapses) (Hanse and Gustafsson, 2001b). This behavior was explained by a lack of a preprimed pool and by a fast onset of recruitment. We have no evidence that such a fast recruitment occurs in synapses exhibiting a preprimed pool. However, if so, such recruitment should act to diminish the depletion effect.
Our model calculations suggest that the variation inPves1 among the synapses creates the large heterogeneity in paired-pulse plasticity. This effect ofPves1 depends on two factors. The first is that a change in Pves1 alters thePves2/Pves1ratio, thereby changing theP2/P1ratio. The second is that a largerPves1 will lead to a larger depletion effect. A key model factor is thatPves2 is kept constant at an intermediate value (0.35). At synapses with higher/lowerPves1 values,Pves is thus supposed to decrease/increase during the paired activation and lead to PPD/PPF (see also Matveev and Wang, 2000). A decrease inPves with paired activation does not conform to the residual calcium hypothesis. However, such a reduction in Pves has been described in other synapses (Bellingham and Walmsley, 1999; Thomson and Bannister, 1999;Wu and Borst, 1999; Waldeck et al., 2000). Thus, in agreement with these other recent studies (Bellingham and Walmsley, 1999; Thomson and Bannister, 1999; Wu and Borst, 1999; Matveev and Wang, 2000; Waldeck et al., 2000), our results suggest that depression is not explained solely by vesicle depletion. In the presently analyzed synapses,Pves2 was not found to be constant, but its variation was considerably less than that ofPves1, and was independent ofPves1. The model calculations indicated that this Pves2 variation created some broadening of the relationship between paired-pulse ratio and P1.
Temporal aspects of paired-pulse plasticity
Experimental data have been limited to a single paired-pulse interval (20 msec). This is an interval at which the initial release-dependent release inhibition should have vanished (Stevens and Wang, 1995; Dobrunz et al., 1997; Hjelmstad et al., 1997) and at which possible recruitment of newly primed vesicles is likely kept at a minimum. An understanding of paired-pulse plasticity along the time axis would then in addition require knowledge of thePves2 dynamics, both from a value higher than Pves1 as from a lower one, as well as of vesicle recruitment dynamics.
Dissociation between initial release probability and paired-pulse plasticity
It is commonly assumed that an increase in initial release probability is associated with a decrease in paired-pulse ratio. However, the calculations show that whenP1 increases because of an increase in preprimed pool size there is either little change, or, for larger values of Pves1, a substantial increase in the paired-pulse ratio. Because of the small size of the preprimed pool, its average size had a large effect on release probability. For example, a doubling of pool size could cause an ∼50% increase in P1 associated with anything from no change to a 100% increase in paired-pulse ratio. In the model, a pool-size change can be achieved by an increase either in the probability for a docked vesicle to be in a primed state or in the number of docking/priming sites. The observation of a phorbol ester-induced change in release probability unaccompanied by changes in paired-pulse ratio (Honda et al., 2000) may then be explained in this manner. Conversely, the finding of a change in paired-pulse ratio unaccompanied by any change in release probability after BDNF treatment (Sherwood and Lo, 1999) in NT-3-deficient mice (Kokaia et al., 1998) and in Rab3A-deficient mice (Geppert et al., 1997) can be explained either by a selective change in Pves2or by a reciprocal change in Pves1 and the size of the preprimed pool.
Preprimed pool distribution
The experimental analysis suggested that the preprimed pool of vesicles fluctuated in size from trial to trial. A binomial model for this fluctuation was constructed for usage in the paired-pulse calculations. This binomial model was based on the assumption that the final release-ready stage is reversible, with docked vesicles being in equilibrium between a nonprimed and a primed state. Such reversibility in the final release-ready stage appears compatible with current molecular models of the release process (Murthy and Stevens, 1999;Matveev and Wang, 2000; Voets, 2000; Zenisek et al., 2000). To vary the average preprimed pool in the model, the number of docking sites was varied (two to five) and the probability of a vesicle to be in the primed state was kept constant at 0.3. Experimentally estimated distributions of preprimed pool fluctuations from synapses with different average preprimed pool sizes were quite similar to the simulated ones, which lends support to this kind of binomial model. Moreover, the number of docking/priming sites used (two to five) is in good agreement with the number of such sites contained within a region destined to be a release site (Zhai et al., 2001). However, the experimental data would also be compatible with other parameters of such a model. Nevertheless, this binomial description may be helpful for calculations of release probabilities and may serve as an analytical tool for the evaluation of release under experimental circumstances.
Dependence on age
The experimentally observed paired-pulse ratio did not correlate with the age of the animal (within the first postnatal week), indicating that its variability among the synapses was not a developmental feature. The presently found relationship between paired-pulse ratio and initial release probability also agrees in its overall variability and shape with that obtained from putative single release sites of the same hippocampal connections from older animals (Dobrunz and Stevens, 1997). However, the present relationship is substantially shifted toward more PPD/less PPF compared with that previous study. This shift may be related to differences in experimental conditions that may affect release conditions, such as temperature and divalent ion concentrations. However, examination of paired-pulse plasticity using population EPSP recordings has indicated that under the same experimental conditions, a substantial shift occurs in a facilitating direction at approximately days 8–12 (P. Wasling, E. Hanse, B. Gustafsson., unpublished observations). Although not excluding other possibilities, these data point to age difference as a major explanation. It will be of interest to find out in future studies which of the factors that decides paired-pulse plasticity undergoes such developmental change that explains such a shift in paired-pulse ratio.
Paired-pulse plasticity within a synapse
Can the present calculations also explain what happens to the paired-pulse ratio when Pves1 and the preprimed pool change within a given synapse? With respect to changes in pool size, there seems no reason to believe otherwise. However, with respect to Pves1, the situation is more complicated. Recent studies, albeit in neocortical axons (Koester and Sakmann, 2000) and hippocampal cultures (Prakriya and Mennerick, 2000), have indicated, directly or indirectly, a wide variation in action potential-induced calcium influx among synaptic boutons. The variations in Pves1 among synapses may then be explained by a variation in calcium influx. Because manipulation of release probability often concerns a modulation of the amount of presynaptic calcium influx, one may also conjecture that in this situation Pves1 may vary whereasPves2 does not change much at all. However, there is at present no experimental support for such a notion. Moreover, Pves may be altered in a manner unrelated to calcium influx. Thus, to increase the usefulness of the present specific model to understand induced paired-pulse plasticity variations, an extension to incorporate changes in Pves2 may be needed.
Footnotes
This study was supported by grants from the Swedish Medical Research Council (project numbers 12600 and 05180), the Swedish Society of Medicine, and Harald Jeanson's Foundation. We thank F. Asztely, J. Eilers, L. Groc, and P. Wasling for critically reading this manuscript.
Correspondence should be addressed to Eric Hanse, Institute of Physiology and Pharmacology, Göteborg University, Box 432, SE-405 30 Göteborg, Sweden. E-mail: eric.hanse@physiol.gu.se.
REFERENCES
- 1.Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 2.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 3.Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci USA. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geppert M, Goda Y, Stevens CF, Sudhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- 5.Hanse E, Gustafsson B. Quantal variability at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol (Lond) 2001a;531:467–480. doi: 10.1111/j.1469-7793.2001.0467i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanse E, Gustafsson B. Vesicle release probability and pre-primed pool at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol (Lond) 2001b;531:481–493. doi: 10.1111/j.1469-7793.2001.0481i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hjelmstad GO, Nicoll RA, Malenka RC. Synaptic refractory period provides a measure of probability of release in the hippocampus. Neuron. 1997;19:1309–1318. doi: 10.1016/s0896-6273(00)80421-3. [DOI] [PubMed] [Google Scholar]
- 8.Hjelmstad GO, Isaac JT, Nicoll RA, Malenka RC. Lack of AMPA receptor desensitization during basal synaptic transmission in the hippocampal slice. J Neurophysiol. 1999;81:3096–3099. doi: 10.1152/jn.1999.81.6.3096. [DOI] [PubMed] [Google Scholar]
- 9.Honda I, Kamiya H, Yawo H. Re-evaluation of phorbol ester-induced potentiation of transmitter release from mossy fibre terminals of the mouse hippocampus. J Physiol (Lond) 2000;529:763–776. doi: 10.1111/j.1469-7793.2000.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol (Lond) 2000;529:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokaia M, Asztely F, Olofsdotter K, Sindreu CB, Kullmann DM, Lindvall O. Endogenous neurotrophin-3 regulates short-term plasticity at lateral perforant path-granule cell synapses. J Neurosci. 1998;18:8730–8739. doi: 10.1523/JNEUROSCI.18-21-08730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 14.Matveev V, Wang XJ. Implications of all-or-none synaptic transmission and short-term depression beyond vesicle depletion: a computational study. J Neurosci. 2000;20:1575–1588. doi: 10.1523/JNEUROSCI.20-04-01575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 16.Prakriya M, Mennerick S. Selective depression of low-release probability excitatory synapses by sodium channel blockers. Neuron. 2000;26:671–682. doi: 10.1016/s0896-6273(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 17.Sherwood NT, Lo DC. Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J Neurosci. 1999;19:7025–7036. doi: 10.1523/JNEUROSCI.19-16-07025.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 19.Thomson AM. Facilitation, augmentation, and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- 20.Thomson AM, Bannister AP. Release-independent depression at pyramidal inputs onto specific cell targets: dual recordings in slices of rat cortex. J Physiol (Lond) 1999;519:57–70. doi: 10.1111/j.1469-7793.1999.0057o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Triller A, Korn H. Transmission at a central inhibitory synapse. III. Ultrastructure of physiologically identified and stained terminals. J Neurophysiol. 1982;48:708–736. doi: 10.1152/jn.1982.48.3.708. [DOI] [PubMed] [Google Scholar]
- 22.Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/s0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- 23.Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 25.Zenisek D, Steyer JA, Almers W. Transport, capture, and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- 26.Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- 27.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]