

Abstract
The role of limbic-striato-pallidal circuitry in cocaine-induced reinstatement was evaluated. The transient inhibition of brain nuclei associated with motor systems [including the ventral tegmental area (VTA), dorsal prefrontal cortex (dPFC), core of the nucleus accumbens (NAcore), and ventral pallidum (VP)] prevented cocaine-induced reinstatement. However, only the VP proved to be necessary for food reinstatement, suggesting that the identified circuit is specific to drug-related reinstatement. Supporting the possibility that the VTA–dPFC–NAcore–VP is a series circuit mediating reinstatement, simultaneous unilateral microinjection of GABA agonists into the dPFC in one hemisphere and into the VP in the contralateral hemisphere abolished cocaine reinstatement. Although dopamine projections from the VTA innervate all three forebrain nuclei, the blockade of dopamine receptors only in the dPFC antagonized cocaine-induced reinstatement. Furthermore, DA administration into the dPFC was sufficient to elicit a reinstatement in drug-related responding. These data demonstrate that dopamine release in the dPFC initiates a dPFC–NAcore–VP series circuit that mediates cocaine-induced drug-seeking behavior.
Keywords: cocaine, dopamine, glutamate, self-administration, craving, reinstatement
One of the most insidious problems associated with chronic drug use is the tendency for users to relapse even after extended periods of drugs abstinence. Many types of stimuli can increase reports of craving and subsequent relapse in drug addicts, including reexposure to the drug itself, stress, and cocaine-associated cues (Jaffe et al., 1989; Childress et al., 1999), and animal reinstatement models of drug-seeking behavior have been established to evaluate the neurobiology of reinstatement and relapse (Shaham and Stewart, 1995; Erb et al., 1996; McFarland and Ettenberg, 1997; Meil and See, 1997). Although the rewarding effect of acute psychostimulant administration has been successfully linked to limbic circuitry, including the mesolimbic dopamine projection from the ventral tegmental area (VTA) to the nucleus accumbens (Koob et al., 1993; Robbins and Everitt, 1996), the circuitry mediating drug seeking in animal models of reinstatement has not been as clearly delineated.
Along with the VTA and nucleus accumbens, the amygdala, prefrontal cortex (PFC), and ventral pallidum (VP) have been examined for involvement in drug reward (Pierce and Kalivas, 1997; Wolf, 1998;Childress et al., 1999). Topographic analysis of the interconnections among these nuclei has established two subcircuits: one subcircuit comprising the ventral PFC (vPFC), shell of the accumbens (NAshell), medial VP, amygdala, and the VTA, that is more intimately connected with limbic structures, and another subcircuit comprised of the dorsal PFC (dPFC), core of the accumbens (NAcore), dorsolateral VP, and substantia nigra, that is more extensively interconnected with motor structures (Zahm and Brog, 1992; Kalivas et al., 1993; Groenewegen et al., 1996). Traditionally, they have been termed the “limbic” and “motor” subcircuits in deference to this anatomical specificity. It is generally assumed that limbic circuitry underlies behavioral changes associated with chronic drug use (e.g., craving and relapse), an assumption supported by studies showing that inactivation of the amygdala prevents cue-induced reinstatement (Meil and See, 1997; Grimm and See, 2000), as well as numerous data indicating enduring cellular adaptations in limbic nuclei after repeated administration of drugs of abuse (Nestler and Aghajanian, 1997; White and Kalivas, 1998). However, craving and relapse are often described as compulsive or automatic (Grant et al., 1996; Childress et al., 1999), raising the possibility that these behaviors may rely more on activation of motor than limbic circuitry.
The present study evaluated the nuclei outlined above and tested whether the motor or limbic subregion of each nucleus was more critical for drug-induced reinstatement. In an attempt to parallel human addicts who relapse after reexposure to a previously self-administered drug, rats were trained to lever press for cocaine reinforcement and then underwent a period of behavioral extinction during which no cocaine reinforcement was available (de Wit and Stewart, 1981). To identify nuclei that are necessary for drug-seeking behavior, the neuronal activity in individual nuclei was transiently inhibited with an intracranial microinjection of a combination of GABAA and GABAB agonist before reinstatement testing. GABA-mediated inhibition was chosen over other inactivation techniques because it is both reversible and leaves fibers of passage unaffected.
MATERIALS AND METHODS
Animal housing and surgery. All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The subjects were 164 male Sprague Dawley rats (Charles Rivers Laboratories. Wilmington, MA) weighing 250–275 gm on arrival, and they were individually housed in an Association for the Assessment and Accreditation of Laboratory Animal Care-approved facility maintained on a 12 hr reversed light/dark cycle (lights on 7:00 P.M.). Subjects were weighed and handled daily for the duration of the experiment and were given ad libitum access to food and water until 7 d after surgery, when they received a 20 gm daily ration of rat chow for the remainder of the experiment. This feeding regimen aided in the acquisition of the operant lever-press response but allowed rats to gain weight throughout the course of the experiment.
After 1 week of acclimation, animals were anesthetized with ketamine HCl (87.5 mg/kg Ketaset; Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (5 mg/kg Rompum; Bayer, Shawnee Mission, KS) and implanted with indwelling jugular catheters and bilateral guide cannulas (26 gauge; Small Parts Inc., Roanoke, VA) aimed at one of 10 brain regions according to the atlas of Paxinos and Watson (1998)(all coordinates given relative to bregma): dorsal and ventral PFC, +3.0 mm anteroposterior (AP), ±0.7 mm mediolateral (ML), and −2.0 mm dorsoventral (DV); NAcore, +1.2 mm AP, ±1.6 mm ML, and −6.5 mm DV; NAshell, +1.5 mm AP, ±0.6 mm ML, and −6.5 DV; VP, −0.25 mm AP, ±2.4 mm ML, and −7.9 mm DV; central nucleus of the amygdala (CN), −2.1 mm AP, ±4.0 mm ML, and −8.0 mm DV; basolateral amygdala (BLA), −2.5 mm AP, ±4.7 mm ML, and −8.5 mm DV; mediodorsal thalamus (MD), −2.8 mm AP, ±0.7 mm ML, and −5.5 mm DV; VTA, −5.2 mm AP, ±0.85 mm ML, and 8.15 mm DV with the cannulas angled 6° from the midline; and substantia nigra (SN), −5.2 mm AP; ±2.1 mm ML, and −8.0 mm DV). Guide cannulas (cut to 14 mm) were lowered into place and attached to the skull via small stainless steel screws and dental acrylic. Obdurators (33 gauge; Small Parts Inc.), cut to extend 0.5 mm beyond the tip of each cannula, were inserted to prevent obstruction by debris.
For catheter implantation, a guide cannula (C313G; Plastics One Inc., Wallingford, CT), attached to SILASTIC tubing (0.025 inner diameter, 0.047 outer diameter; Bio-Sil; Bio-Rad, Hercules, CA) and Marlex mesh via dental cement, was inserted subcutaneously between the shoulder blades and exited the skin via a dermal biopsy hole (3 mm). The other end of the tubing was threaded under the skin, inserted 3 cm into the right jugular vein, and then sutured securely to the underlying muscle tissue.
Self-administration and extinction procedures. Seven days after surgery, subjects began behavioral training. All self-administration experiments were conducted in standard operant chambers (ENV-008; Med Associates Inc., E. Fairfield, VT) fitted with two retractable levers. Initially, all subjects were trained in one 15 hr session to lever press for food on an fixed ratio 1 (FR-1) schedule for reinforcement consisting of one food pellet (45 mg; Noyes, Lancaster, NH). The following day, subjects began lever pressing for cocaine reinforcement. Each press of the correct lever resulted in an infusion of cocaine (0.25 mg/kg over 4 sec), followed by a 20 sec time out, in which correct lever presses were counted but resulted in no scheduled consequences. Responses on the incorrect lever never resulted in cocaine delivery and consequently became very infrequent over the course of training. Each training trial lasted for 2 hr or until the subject had self-administered 200 infusions of cocaine. An arbitrary acquisition criterion required that subjects' active lever presses vary by 10% or less over the course of 3 consecutive maintenance days before they were moved on to the extinction phase of the experiment. During maintenance, subjects administered an average of 20.25 mg/kg cocaine across the 2 hr session. This average did not differ significantly between groups of animals implanted with guide cannulas into different brain regions. Once subjects met the maintenance criterion, extinction procedures were instituted. During extinction, subjects experienced 2 hr daily training sessions; however, saline was substituted for cocaine after presses of the active lever. Thus, active lever presses now resulted in no reinforcer delivery. Subjects remained in extinction until responding on the active lever fell to <10% of the level during maintenance.
Reinstatement testing and microinjection of drug. After extinction, subjects were tested for their propensity to reinstate responding on the active lever after a systemic injection of cocaine (10 mg/kg, i.p.). Before reinstatement testing, subjects received an intracranial infusion of either 0.9% saline vehicle or a combination of the GABAA receptor agonist muscimol (mus) and the GABAB agonist baclofen (bac). Before injection, obdurators were removed from the guide cannulas and 33 gauge microinjection cannula were inserted bilaterally to extend 1 mm below the end of the guide (with the exception of the vPFC, in which injectors were inserted to 3 mm below the end of the guide). All infusions were made in a volume of 0.3 μl over 60 sec. After infusion, 1 min was allowed for diffusion, the microinjectors were removed, obdurators were replaced, and subjects were given an injection of cocaine before being placed in the chambers for 2 hr. This injection volume and procedure has been shown previously to functionally distinguish between the NAshell and NAcore and between the VTA and SN (Johnson et al., 1996). All subjects were tested twice with successive test days separated by additional extinction trials, in which subjects were required to again meet the extinction criterion before the second test trial. During reinstatement testing, active lever presses resulted only in a delivery of intravenous saline and not cocaine.
For bac–mus infusions, the drugs were dissolved in saline at a screening dose of 0.3 nmol of baclofen and 0.03 nmol of muscimol (per 0.3 μl injection volume). For any brain area in which there was an effect of these high doses, a dose–response curve was established at half log unit increments of each drug. Each subject was tested only twice (i.e., saline plus a single dose of bac–mus or two different doses of bac–mus). For the ipsilateral versus contralateral test of the integrity of the circuit, all testing was conducted with 0.3 nmol of bac and 0.03 nmol of mus. The combination of GABA agonists was chosen because projection cells in every nucleus examined are inhibited by one or both of the GABAA or GABAB agonists (Kalivas et al., 1993; Mogenson et al., 1993). Fluphenazine was also dissolved in saline in a concentration of 10 nmol per injection (0.3 μl/side). Dopamine was dissolved in saline and administered in a dose of 30 μg.
Food reinstatement. Food reinstatement experiments were conducted after a procedure that paralleled cocaine reinstatement as closely as possible. After 1 week of handling, subjects were stereotaxically implanted with guide cannulas into the dPFC, NAcore, or VP using the procedure and coordinates described previously. One week after surgery, animals were food restricted to 90% of their free-feeding body weights and were maintained at this weight for the duration of the experiment. Two days after the institution of food restriction procedures, subjects began behavioral training. They were trained to lever press on an FR-1 schedule for reinforcement consisting of a single (45 mg) food pellet. On subsequent days, the schedule was increased to an FR-3 and then an FR-5. An intermittent schedule of reinforcement was instituted in animals responding for food reinforcement to help ensure robust reinstatement responding. On each schedule, subjects were required to display stable operant behavior (<10% variation across 3 d), before being moved to the next schedule. Once each rat displayed stable responding on the FR-5 schedule of reinforcement, extinction procedures were instituted so that lever presses no longer resulted in delivery of food reinforcement. When average responding across three consecutive sessions was <10% of FR-5 reinforced responding, subjects were tested for the ability of noncontingent food delivery to reinstate lever-press responding after either saline (0.3 μl) or bac–mus infusion (0.3 and 0.03 nmol, respectively, in 0.3 μl). One food pellet was placed into the hopper before beginning the test session, and an additional five pellets were dropped at 2 min intervals during the first 10 min of the reinstatement session.
Locomotor testing. Motor activity was monitored in clear Plexiglas boxes (22 × 43 × 33 cm). Each box was monitored by a series of 16 photobeams (eight on each horizontal axis) measuring horizontal activity and eight photobeams measuring vertical activity. Beam breaks were detected, counted, and recorded by a personal computer running Digiscan software (AccuScan Instruments Inc., Columbus, OH). Each test period consisted of a 1 hr acclimation period before testing. After habituation, each subject was removed from its cage, given a microinfusion (using the procedure described above), and then returned to the cage for 2 hr of activity monitoring.
Histology and statistics. Rats were overdosed with pentobarbital (120 mg/kg, i.p.) and then perfused transcardially with 0.9% physiological saline followed by 10% formalin. Brains were removed and placed in a 10% formalin solution for at least 24 hr before slicing. The brains were blocked and sliced in coronal sections (50 μm thick) through the site of guide cannula implantation. Sections were mounted on gel-coated slides and then stained with cresyl violet to allow for verification of cannula placement by an individual unaware each subject's behavioral responses. The data were statistically evaluated using a one-way or two-way ANOVA, andpost hoc comparisons between individual treatments were made using a Tukey's test. Because subjects were tested only twice and did not receive all treatment doses, all analyses were done assuming independent groups, because repeated-measures analyses were not possible. When time course data for locomotor activity were evaluated, a two-way ANOVA with repeated measures over time was used.
RESULTS
Neural circuitry mediating cocaine-induced reinstatement
Figure 1 shows the results from nuclei in which infusion of GABA agonists (a combination of the GABAB agonist baclofen and the GABAA agonist muscimol, bac–mus) blocked cocaine-induced reinstatement. An ANOVA conducted on each panel in Figure 1 revealed a significant dose-dependent effect of baclofen–muscimol inactivation of the dPFC, NAcore, VTA, and VP. After microinjection of saline or a very low dose of bac–mus, subjects displayed robust lever-press responding after cocaine challenge. However, pretreatment with higher doses of bac–mus reduced reinstatement responding to extinction levels (minimum effective dose of 0.03 nmol/side bac–0.003 nmol/side mus for each nucleus, except the NAcore in which the dose was 0.1 nmol/side bac–0.01 nmol/side mus).
Fig. 1.
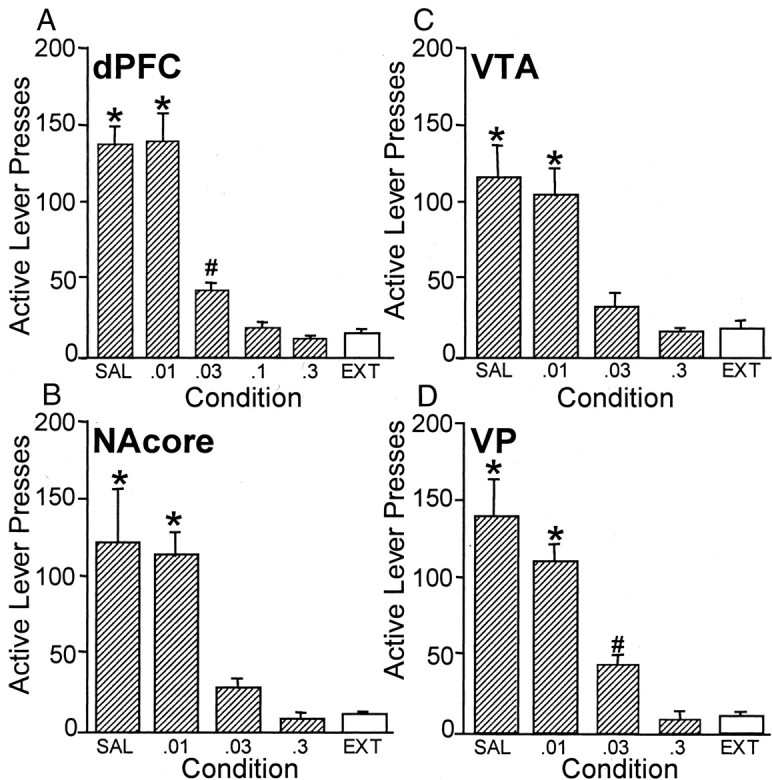
GABA receptor activation prevented cocaine-induced reinstatement. Pretreatment with combinations of the GABAB and GABAA receptor agonists bac–mus into the dPFC (A), ) NAcore (B), VTA (C), and VP (D) blocked the reinstatement elicited by cocaine administration (n = 6–8 for each dose in each nucleus). Subjects pretreated with a screening dose (0.3 and 0.03 nmol/side, respectively) into any of these areas showed no change in active lever presses compared with extinction (EXT) responding, suggesting that neural activation of these areas is critical for the ability of cocaine to elicit reinstatement. Note that, when pretreated with saline vehicle (SAL), all subjects exhibited a robust return to drug seeking. Data depicted as mean + SEM active lever presses. Dose combinations of baclofen and muscimol were 0.3 and 0.03, 0.1 and 0.01, 0.03 and 0.003, and 0.01 and 0.001 nmol/side, respectively. x-Axis labels are designated using the baclofen dose. *p < 0.001, increase in active lever presses compared with EXT. #p< 0.05, responding that was greater than EXT and less than SAL.
Figure 2 reveals that, at the maximum screening dose of bac–mus (0.3 and 0.03 nmol/side, respectively), none of the other regions tested showed any involvement in cocaine-induced reinstatement, including the vPFC, NAshell, SN, CN, BLA, or MD. There was a main effect of condition, indicating that there was a significant reinstatement in animals pretreated with a microinjection of saline vehicle. However, Tukey's honestly significant difference post hoc analyses showed that there was no difference between bac–mus pretreatment and vehicle pretreatment.
Fig. 2.
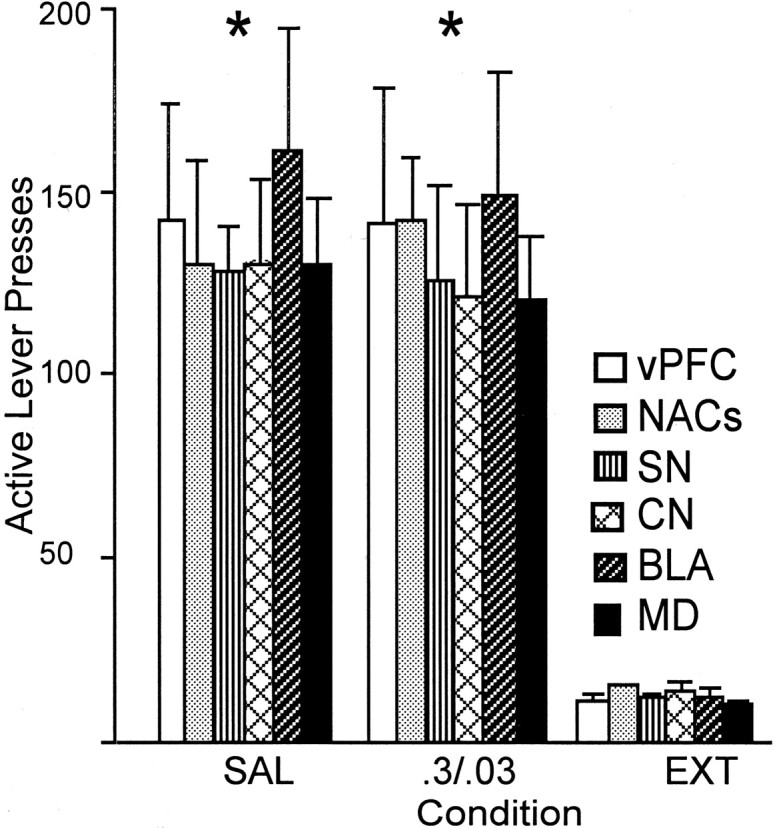
Inactivation of some brain regions tested was ineffective in altering cocaine-induced reinstatement. Subjects pretreated with a high screening dose of bac–mus (0.3 and 0.03 nmol/side, respectively) into the vPFC, NAshell, SN, CN, BLA, or MD (n = 6–8) displayed robust lever-press responding on reinstatement testing equivalent to that seen after saline (SAL) pretreatment. *p < 0.05, comparing cocaine-induced active lever pressing with extinction (EXT) responding.
These data suggest that the VTA, dPFC, NAcore, and VP form a circuit that is critical for cocaine-induced reinstatement of drug-seeking behavior. To test the specificity of the circuit in the control of drug-seeking behavior, each of the targets of VTA dopamine projections implicated above (i.e., the dPFC, NAcore, and VP) were tested for their involvement in the food-induced reinstatement of food-seeking behavior. Figure 3 shows that bac–mus inhibition of the VP, but not the dPFC or NAcore, blocked the ability of food to elicit extinguished food-seeking behavior, suggesting that the identified circuit is specific to the drug-related behavior under examination.
Fig. 3.

Reinstatement of food-seeking behavior is blocked by inhibition of the VP but not the dPFC or NAcore. Pretreatment with the screening dose of bac–mus (0.3 and 0.03 nmol/side, respectively) into the VP inhibited lever-press responding elicited by noncontingent food presentation, whereas infusion of bac–mus into the NAcore or PFC (0.3 and 0.03 nmol/side, respectively) resulted in response rates indistinguishable from saline (SAL) pretreatment (i.e., a robust reinstatement). *p < 0.001, comparing food reinstatement responding with extinction (EXT) responding.
An implication of these findings is that the VTA–dPFC–NAcore–VP connections form a functional series circuit that is critical for cocaine-induced reinstatement of drug-seeking behavior in rats (i.e., information flows sequentially from one nucleus to the next and not in parallel pathways) (Fig.4A). To directly test this possibility, rats were implanted with guide cannulas into both the dPFC and the VP. Before reinstatement testing, each animal received a bac–mus infusion (0.3 and 0.03 nmol, respectively) into the dPFC and VP on either ipsilateral or contralateral sides of the brain. Figure4B reveals that animals having the VP and the dPFC in the same hemisphere inactivated showed normal reinstatement after cocaine challenge. However, if the inactivated VP and dPFC were on alternate sides, animals showed no reliable cocaine-induced reinstatement. This experiment indicates that a flow of information in series between the dPFC and VP is required, presumably via a synapse in the NAcore, because the primary conduit whereby the dPFC gains access to the VP is via a synapse in the NAcore (Sesack et al., 1989; Zahm and Brog, 1992). The VP and dPFC were chosen as targets for the disconnection experiment because there are no direct connections between them. Thus, a positive disconnection implicates a serial circuit flowing from the dPFC through the NAcore to the VP in the production of cocaine-induced reinstatement. Selection of any other pair of nuclei (e.g., the dPFC and the NAcore) would implicate only two of these brain regions in this serial circuitry.
Fig. 4.

A series, rather than parallel, circuit is involved in cocaine-primed reinstatement. A, Circuits can be organized in either parallel or series. Parallel circuits would suggest that information could flow simultaneously along multiple pathways, whereas a series circuit would suggest that information flows sequentially from one nucleus to the next. B, Subjects receiving bac–mus (0.3 and 0.03 nmol/side, respectively) infusion into the dPFC and VP on the same side (ipsilateral;n = 5) of the brain displayed vigorous active lever-press responding after cocaine challenge. However, when subjects received bac–mus treatment into one dPFC and the contralateral VP (contralateral; n = 6), responding on the active lever did not differ from extinction (EXT) responding, suggesting these brain regions are in series circuit essential for the ability of cocaine to elicit a reinstatement of drug-seeking behavior. *p < 0.001, comparing ipsilateral with extinction (EXT) responding.
Role of dopamine in response initiation
Acute doses of cocaine increase extracellular levels of dopamine, which presumably serves as the intereoceptive cue to initiate reinstatement because systemically administered dopamine antagonists (De Vries et al., 1999) or GABAergic inhibition of dopamine cells in the VTA (Fig. 1) inhibit cocaine-induced reinstatement. Dopaminergic projections from the VTA innervate all three forebrain regions found sensitive to GABA agonist-induced inhibition of cocaine reinstatement (Kalivas et al., 1993). Figure5A shows that microinjection of the D1/D2 antagonist fluphenazine (10 nmol/side) into the dPFC, but not into the NAcore or VP, blocked cocaine-induced reinstatement.
Fig. 5.

Role of dopamine in the dPFC in cocaine reinstatement. A, Fluphenazine (FLU) infusion into the dPFC, but not the NAcore or VP (n = 6 in each condition), before reinstatement testing abolished the increase in active lever pressing observed after pretreatment with saline vehicle (SAL).B, After bac–mus (0.3 and 0.03 nmol/side, respectively) activation of the VTA, subjects received either saline (0 nmol;n = 5) or dopamine (30 nmol/side;n = 7) infusions into the dPFC before a cocaine (COC) reinstatement challenge. When no dopamine was infused into the dPFC, subjects exhibited the expected blockade of cocaine-induced reinstatement, similar to that seen after inactivation of the VTA in Figure 1. However, dopamine replacement into the dPFC resulted in a highly significant reinstatement of self-administration behavior. Similarly, dopamine alone into the dPFC was sufficient to induce robust responding. *p < 0.001, comparing extinction (EXT) responding with other treatments.
These data implicate the dopaminergic projection from the VTA to the dPFC as the initial step in producing cocaine-induced reinstatement. Given that the increase in extracellular dopamine after cocaine is predominantly action potential dependent (Seiden et al., 1993), it is possible that cocaine is not inducing reinstatement by activating dopamine cells in the VTA but rather is relying on ongoing activity in these cells to increase dopamine release in the dPFC. To examine this possibility, animals were prepared with bilateral guide cannulas aimed at both the VTA and the dPFC. Before reinstatement testing, subjects received bac–mus infusions (0.3 and 0.03 nmol/side, respectively) into the VTA, followed by either saline or dopamine (30 μg/side) into the dPFC. When saline was infused into the dPFC, cocaine failed to produce reinstatement when the VTA was inactivated by GABA agonists. However, when dopamine tone was returned to the dPFC, cocaine once again elicited a robust reinstatement (Fig. 5B). Furthermore, dopamine (30 μg/side) infused into the dPFC in the absence of systemic cocaine was sufficient to elicit a reinstatement of lever-pressing responding. Together, these findings support a permissive role for the VTA rather than an activation of dopamine cells by the priming injection of cocaine. Indeed, electrophysiological studies indicate that acute cocaine administration decreases spontaneous action potential generation in dopamine cells (Henry et al., 1989).
Role of locomotor activity
The data presented thus far implicate a VTA–dPFC–NAcore–VP series circuit in the production of cocaine-induced reinstatement, with the projection from the VTA to the dPFC being the dopamine-dependent initiating step in the process. However, it is possible that the inhibitory effects of GABA agonists or fluphenazine are not specific to the goal-directed behavior under examination and may arise from a generalized motoric incapacity. A series of experiments were conducted examining the effect of intracranial bac–mus or fluphenzine on spontaneous and cocaine-induced locomotor activity. bac–mus (at the minimum dose effective for blocking cocaine-induced reinstatement) was infused into either the dPFC, NAcore, VTA, or VP before a systemic injection of either saline or cocaine (10 mg/kg, i.p.). Thus, there were four treatment conditions for each of the target brain nuclei: saline plus saline, bac–mus plus saline, saline plus cocaine, and bac–mus plus cocaine. Two-way ANOVAs were conducted comparing saline with bac–mus pretreatment across the 2 hr test session for both spontaneous and cocaine-induced activity. Figure6A shows that microinjection of bac–mus did not significantly reduce total photocell counts in any brain region examined. However, an analysis of the time course revealed that GABA receptor activation produced a decrement in cocaine-induced locomotion in the VP over the first 20 min of the test session (data not shown). No other brain region tested showed any effect of bac–mus pretreatment at any time point.
Fig. 6.
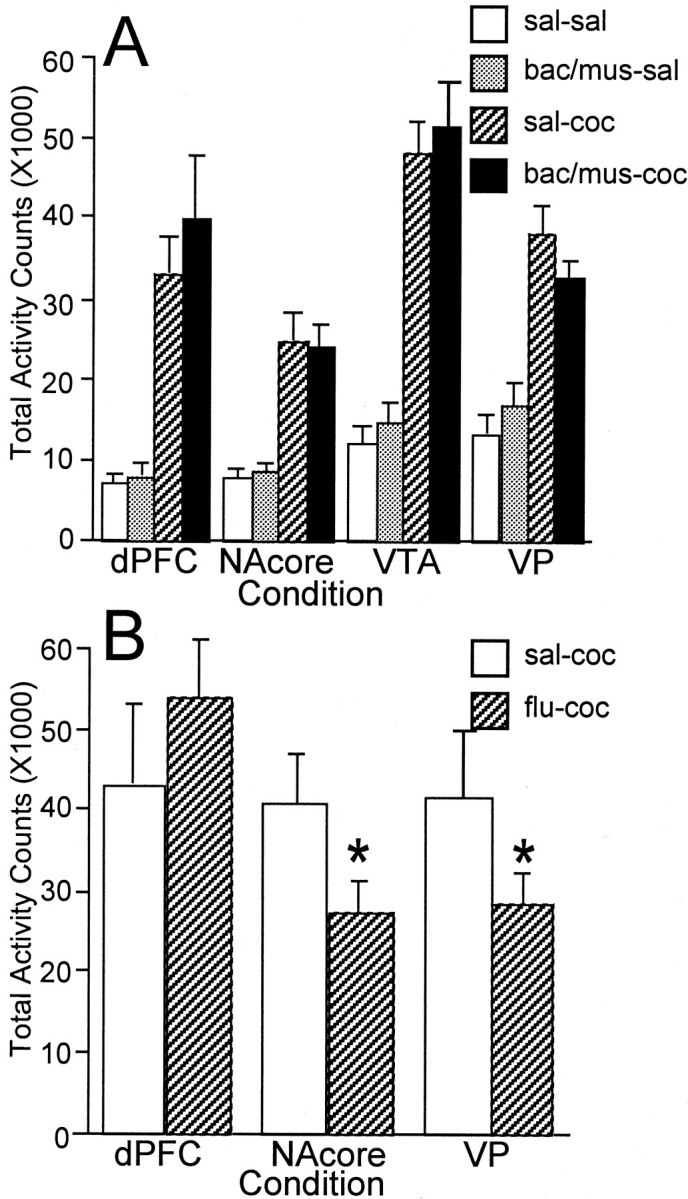
Locomotor activity. Mean ± SEM photocell counts are shown for the 2 hr test sessions for both spontaneous and cocaine-induced motor activity studies. A, Subjects received two treatments before each test: an intracranial infusion of saline (sal) or bac–mus followed by an intraperitoneal injection of either saline (sal) or cocaine (coc) (10 mg/kg; n = 8 in each condition). bac–mus pretreatment did not block either spontaneous or cocaine-induced locomotor activity when infused into the dPFC, NAcore, VTA, or VP. B, Fluphenazine (flu), when infused into the VP or NAcore but not the dPFC, produced a significant reduction in cocaine-induced locomotor activity. *p < 0.01, comparing bac–mus or fluphenazine with saline.
The effect of fluphenazine pretreatment on cocaine-induced locomotor activity was also examined (Fig. 6B). Fluphenazine pretreatment into the NAcore or into the VP produced a reliable decrement in locomotor activity across the first 40 min of the test session. However, fluphenazine did not produce any decrement in cocaine-induced activity when microinfused into the dPFC (the only brain site in which fluphenazine antagonized cocaine reinstatement) (Fig. 5A). Together, these data demonstrate that the effects of GABA agonists and dopamine antagonists on self-administration are separable from effects on motoric capacity.
Histology
Figure 7 shows the location of cannula tips from animals used in the self-administration experiments. Placements in the dPFC were at the interface between the anterior cingulate cortex and the prelimbic cortex (as depicted by Paxinos and Watson, 1998), whereas cannula tips designated vPFC were ∼2 mm more ventral at the border of the prelimbic and infralimbic cortices (Fig.7A). Cannula placements in the NAshell were medial to the anterior commissure and were not in the ventrolateral limb of the nucleus, whereas placements in the NAcore were clustered around the anterior commissure (Fig. 7B). Placements in the VP were generally in the dorsal half of the nucleus in both the subcommissural and sublenticular compartments (Fig. 7C). Cannulas in the MD were located primarily in the ventral half of the nucleus, and placements in the amygdala were clustered at the medial and lateral edges of the CN and BLA, respectively, to maximize the distinction between injections into these near adjacent nuclei (Fig.7D). Similarly, distinctions between the VTA and SN were maximized by having cannula placements in the VTA located in the medial half of the nucleus paranigralis and parabrachialis pigmentosus and injections into the SN made into the middle of the pars reticulata (Fig. 7E).
Fig. 7.
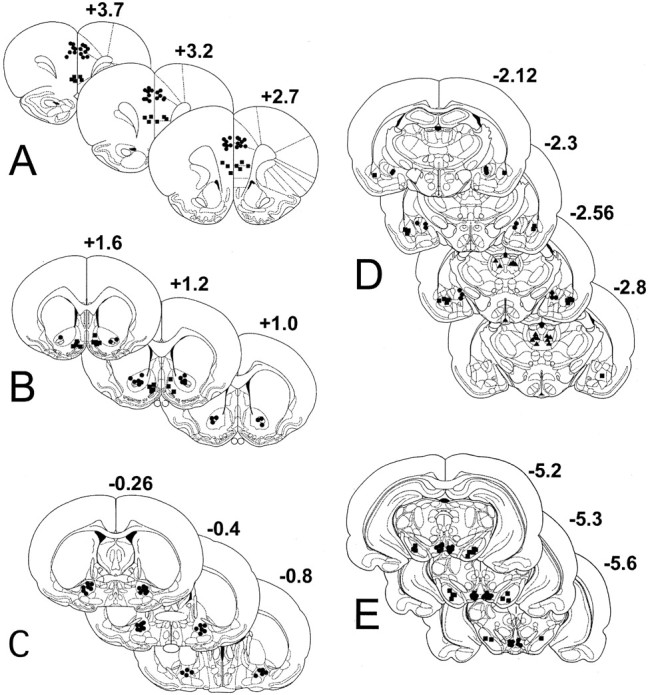
Location of microinjection cannula tips. This figure depicts the location of the microinjection cannula tips in coronal section based on the atlas of Paxinos and Watson (1998).A, Dorsal (●) and ventral (▪) PFC; B, NAcore (●) and NAshell (▪); C, ventral pallidum (●); D, basolateral amygdala (▴), central nucleus of the amygdala (✚), and mediodorsal nucleus of the thalamus (▪);E, VTA (●) and substantia nigra (▪). Note that the ● was used in brain regions critical for the normal production of cocaine-induced reinstatement in the inactivation studies. Numbers indicate the distance from bregma in the anteroposterior plane.
DISCUSSION
The present study demonstrates that neural activity in the VTA, dPFC, NAcore, and VP is necessary for cocaine-induced reinstatement of drug-seeking behavior. Inactivation of these nuclei using the GABA agonists baclofen and muscimol prevented cocaine-primed drug-seeking behavior in an animal reinstatement model of relapse. The same treatment into the vPFC, NAshell, MD, SN, BLA, and CN produced no effect. These data strongly suggest that a limbic–cortico–striato–pallidal circuit created by VTA–dPFC–NAcore–VP connections form a functional series circuit that is crucial for the ability of cocaine to elicit renewed drug self-administration. Supporting the presence of a series circuit, simultaneous contralateral (but not ipsilateral) inactivation of the dPFC and the VP blocked the reinstatement of self-administration. Because the dPFC and VP have few if any direct connections (Sesack et al., 1989; Haber et al., 1995), a multisynaptic connection through the NAcore is likely required for the behavior.
Notably, inhibition of the dPFC and NAcore did not block the ability of noncontingent food delivery to reinstate food-seeking behavior, indicating that the circuitry implicated in cocaine-induced reinstatement does not generalize to all goal-directed responding. This finding strongly suggests that chronic cocaine elicits neuroadaptations in prefrontal and accumbal motor circuitry in the control of drug-seeking behavior, a notion consistent with the recent suggestion that chronic cocaine causes behavioral impulsivity resulting from disinhibition of frontostriatal function (Jentsch and Taylor, 1999). Inhibition of the VP, however, blocked both food- and cocaine-seeking behavior, suggesting that outflow of information through the VP might serve as a common pathway by which motivationally relevant information is transmitted to motor circuitry.
The effect of drug treatment on reinstatement cannot be explained as a consequence of motoric incapacity, because infusions of bac–mus into the dPFC, NAcore, and VTA produced no decrement in either spontaneous or cocaine-induced locomotor activity and inhibited cocaine-induced locomotion only in the first 20 min after infusion into the VP. Notably, microinfusion of fluphenazine into the NAcore and VP reliably diminished cocaine-induced locomotion, although it had no effect when infused into the dPFC, the only location in which it prevented cocaine-primed drug-seeking behavior. These data suggest that circuitry mediating cocaine-induced reinstatement is distinct from that mediating spontaneous or cocaine-induced locomotion.
Mesocortical versus mesoaccumbens dopamine distinguishes reinstatement from reinforcement
Because the primary molecular mechanism of cocaine is blocking dopamine transporters (Povlock and Schenk, 1997), enhanced extracellular dopamine is likely the priming neurochemical signal initiating reinstatement. Consistent with this possibility, inhibition of neural activity in the VTA antagonized cocaine reinstatement. Similarly, systemic administration of dopamine antagonists prevents psychostimulant-induced reinstatement (De Vries et al., 1999). Although systemic cocaine administration enhances the extracellular levels of dopamine in all dopamine axon terminal fields, dopamine antagonist administration in the dPFC, and not in the NAcore or VP, prevented reinstatement after systemic cocaine. These data argue that enhanced dopamine transmission only in the dPFC is necessary for cocaine-induced reinstatement. This possibility is strengthened by the findings that (1) inhibition of cocaine-induced reinstatement after GABA agonist administration into the VTA was reversed by dopamine administration directly into the dPFC and (2) infusion of DA into the dPFC is sufficient to cause reinstatement of cocaine-seeking behavior in the absence of systemic cocaine.
In contrast to the findings regarding cocaine-primed reinstatement of drug seeking, studies examining the reinforcing properties of psychostimulants clearly implicate enhanced mesoaccumbens dopamine release as a critical substrate (Wise and Rompre, 1989; Koob et al., 1993; Robbins and Everitt, 1996). Thus, dopamine release in the nucleus accumbens is postulated to be critical for the reinforcing capacity of psychostimulants and consequently for establishing drug-seeking behavior (Robinson and Berridge, 1993). However, some evidence suggests that, once the behavior is well-learned and automatic, it can become independent of dopamine release in the nucleus accumbens. For example, along with the present evidence indicating that cocaine-induced reinstatement of drug seeking proceeds after DA receptor antagonist treatment into the NA, the same treatment fails to block the reinstatement of food-seeking behavior in a straight-arm runway (Chausmer and Ettenberg, 1999).
A cortico–striato–pallidal circuit mediates drug-seeking behavior
Pyramidal cells in the PFC provide a dense glutamatergic projection to the nucleus accumbens that is topographically organized such that the dPFC projects most densely to the NAcore and the vPFC selectively innervates the NAshell (Gorelova and Yang, 1997; Pinto and Sesack, 2000). Dopaminergic afferents from the VTA innervate both interneurons and pyramidal cells in the PFC (Krimer et al., 1997), resulting in a complex electrophysiological impact on corticofugal projections to the nucleus accumbens. The emerging picture reveals that pyramidal projection neurons within the PFC display biphasic states, fluctuating between a membrane potential that is relatively hyperpolarized “down state” and an “up state” in which the membrane potential is relatively depolarized (Yang et al., 1996), and that the increase in dopamine caused by cocaine supports the up state (Lewis and O'Donnell, 2000). Enhanced residence in the up state would be expected to increase corticofugal excitatory transmission and is consistent with observations that repeated cocaine results in enhanced releasability of glutamate in the NAcore (Pierce et al., 1996; Reid and Berger, 1996) and that such release is partly prevented by lesioning the dPFC (Pierce et al., 1998). Moreover, intra-accumbens administration of ionotropic glutamate receptor antagonist abolishes cocaine-induced reinstatement (Cornish and Kalivas, 2000).
Increased glutamate release augments the firing frequency of medium spiny cells in the NAcore (O'Donnell and Grace, 1995; You et al., 1998), which have a dense GABAergic projection to the VP (Heimer et al., 1991). Activation of these cells seems critical for cocaine-primed drug-seeking behavior, because stimulation of GABA receptors in the VP inhibited cocaine-primed drug seeking. It appears contradictory to the postulate that dPFC-induced activation of medium spiny GABAergic cells projecting from the NAcore to the VP mediates drug-seeking behavior when GABA-mediated inhibition of VP cells blocks reinstatement. This incongruity may be attributable to a neuromodulatory role for GABA within the VP, such that it can excite or inhibit behavioral output depending on the balance of neurotransmission within the nucleus. Consistent with this argument, GABA agonists have been shown to both inhibit and stimulate locomotor activity when infused into the VP (Baud et al., 1989; Napier, 1993). Moreover, GABA is colocalized and coreleased with a variety of neuropeptides in efferents from the nucleus accumbens that can differentially modulate the effect of GABA on VP neurons (Napier, 1993). For example, μ-opioid receptor stimulation in the VP modulates the firing activity of VP neurons, inhibits GABA release, initiates locomotor activity, and facilitates motivated behavior (Koob, 1992; Johnson and Stellar, 1994; Johnson and Napier, 1997; Kalivas et al., 2001).
Another equally viable explanation of the fact that activation of GABAergic projection neurons from the NAcore to the VP seems to mediate drug-seeking behavior whereas GABA mediated inhibition of the VP blocks reinstatement relies on the concept of neural ensembles (Pennartz et al., 1994). According to this idea, inputs from the dPFC activate only a particular subset (or ensemble) of neurons in the NAcore, which in turn inhibit only a particular subset of VP neurons. Thus, whereas selective inhibition of this ensemble of neurons, mediated by specific spatiotemporal firing patterns within the NAcore, initiates reinstatement, broad-range inhibition of all VP neurons (such as an infusion of bac–mus) would disrupt these specific patterns of activity within the circuit, and thereby disrupt behavior.
Motor versus limbic subcircuits
Figure 8 illustrates the circuit containing the nuclei examined in this study and highlights topographic organization according to associations with limbic or motor subcircuits (Heimer and Alheid, 1991; Mogenson et al., 1993). The interconnected nuclei associated with motor output were most sensitive to GABA agonist-induced inhibition of cocaine reinstatement, whereas nuclei generally described as providing integration of motivational stimuli were not affected. The VTA is the only brain region typically associated with limbic integration that was sensitive to GABA agonists. However, this involvement may be permissive as a source of dopamine to the cortex and not necessarily as an active component of the reinstatement because increasing dopamine tone in the dPFC elicits cocaine-induced reinstatement (Fig. 5B).
Fig. 8.

Circuit containing the nuclei injected with GABA agonists. The circuit illustrates the connectivity of the limbic and motor regions that have been implicated in the production of goal-directed behavior. The limbic subcircuit is more closely associated with limbic structures, and the motor subcircuit is more intimately connected with motor structures (Heimer et al., 1991; Zahm and Brog, 1992). Arrowheads indicate the direction of the projection, and bidirectional arrowsindicate reciprocal connections. Larger circles andarrows indicate nuclei identified as critical for drug-seeking behavior.
Some nuclei in the circuit, such as the BLA and MD, have been implicated previously in drug reward and cue-induced reinstatement or craving but were found to be not involved in cocaine-induced reinstatement (Weissenborn et al., 1998; Grimm and See, 2000). This is most readily explained by the fact that different types of eliciting stimuli (e.g., cocaine versus cocaine-paired cues) depend on differential processing within the motive circuit to produce their behavioral activating effects. Thus, different stimuli may initiate reinstatement behavior by recruiting nuclei in the limbic subcircuit, although the execution of the behavior may rely on the motor subcircuit consisting of the series connection between the dPFC, NAcore, and VP.
The fact that the nuclei shown to be critical for cocaine-induced reinstatement are part of the motor subcircuit argues that drug-seeking behavior in a drug-experienced subject requires little limbic integration. Thus, in parallel with reports from cocaine addicts (Childress et al., 1999), the motor behaviors mediating drug-seeking behavior are easily primed and relatively automatic. In this sense, drug-seeking behavior resembles other compulsive motor disorders (Jog et al., 1999) known to involve cortico–striato–pallidal circuitry.
Footnotes
This research was supported in part by United States Public Health Service Grants DA12513, MH40817, and DA03906 and postdoctoral National Research Service Award DA05978 (K.M.). We thank Christopher Lapish for his outstanding technical assistance.
Correspondence should be addressed to Krista McFarland, Department of Physiology and Neuroscience, Medical University of South Carolina, 173 Ashley Avenue, Room 403, Charleston, SC 29425. E-mail:mcfarlk@musc.edu.
REFERENCES
- 1.Baud P, Mayo W, le Moal M, Simon H. Locomotor hyperactivity in the rat after infusion of muscimol and [d-Ala2]Met-enkephalin into the nucleus basalis magnocellularis. Possible interaction with cortical cholinergic projections. Brain Res. 1989;452:203–211. doi: 10.1016/0006-8993(88)90024-8. [DOI] [PubMed] [Google Scholar]
- 2.Chausmer A, Ettenberg A. Intraaccumbens raclopride attenuates amphetamine-induced locomotion, but fails to prevent the response-reinstating properties of food reinforcement. Pharmacol Biochem Behav. 1999;62:299–305. doi: 10.1016/s0091-3057(98)00165-8. [DOI] [PubMed] [Google Scholar]
- 3.Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O'Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–18. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cornish J, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci 20 2000. RC89(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Vries T, Schoffelmeer A, Binnekade R, Vanderschuren L. Dopaminergic mechanisms mediating the incentive to seek cocaine and heroin following long-term withdrawal of IV drug self-administration. Psychpharmacology. 1999;143:254–260. doi: 10.1007/s002130050944. [DOI] [PubMed] [Google Scholar]
- 6.de Wit H, Stewart J. Reinstatement of cocaine-reinforced responding in the rat. Psychopharmacology. 1981;75:134–143. doi: 10.1007/BF00432175. [DOI] [PubMed] [Google Scholar]
- 7.Erb S, Shaham Y, Stewart J. Stress reinstates cocaine-seeking behavior after prolonged extinction and a drug-free period. Psychopharmacology. 1996;128:408–412. doi: 10.1007/s002130050150. [DOI] [PubMed] [Google Scholar]
- 8.Gorelova N, Yang CR. The course of neural projection from the prefrontal cortex to the nucleus accumbens in the rat. Neuroscience. 1997;76:689–706. doi: 10.1016/s0306-4522(96)00380-6. [DOI] [PubMed] [Google Scholar]
- 9.Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C, Phillips RL, Kimes AS, Margolin A. Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci USA. 1996;93:12040–12045. doi: 10.1073/pnas.93.21.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grimm J, See R. Dissociation of primary and secondary reward-relevant limbic nuclei in an animal model of relapse. Neuropsychopharmacology. 2000;22:473–479. doi: 10.1016/S0893-133X(99)00157-8. [DOI] [PubMed] [Google Scholar]
- 11.Groenewegen HJ, Wright CI, Beijer VJ. The nucleus accumbens: gateway for limbic structures to reach the motor system? Prog Brain Res. 1996;107:485–551. doi: 10.1016/s0079-6123(08)61883-x. [DOI] [PubMed] [Google Scholar]
- 12.Haber SN, Kunishio K, Mizobuchi M, Lynd-balta E. The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci. 1995;15:4851–4867. doi: 10.1523/JNEUROSCI.15-07-04851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heimer L, Alheid GF. Piecing together the puzzle of basal forebrain anatomy. In: Napier TC, Kalivas PW, Hanin I, editors. The basal forebrain: anatomy to function. Plenum; New York: 1991. pp. 1–42. [DOI] [PubMed] [Google Scholar]
- 14.Heimer L, Zahm DS, Churchill L, Kalivas PW, Wohltmann C. Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience. 1991;41:89–125. doi: 10.1016/0306-4522(91)90202-y. [DOI] [PubMed] [Google Scholar]
- 15.Henry DJ, Greene MA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: repeated administration. J Pharmacol Exp Ther. 1989;251:833–839. [PubMed] [Google Scholar]
- 16.Jaffe JH, Cascella NG, Kumor KM, Sherer MA. Cocaine-induced cocaine craving. Psychopharmacology. 1989;97:59–64. doi: 10.1007/BF00443414. [DOI] [PubMed] [Google Scholar]
- 17.Jentsch K, Taylor J. Impulsivity resulting form frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology. 1999;146:373–390. doi: 10.1007/pl00005483. [DOI] [PubMed] [Google Scholar]
- 18.Jog M, Kubota Y, Connolly C, Hillegaart V, Graybiel A. Building neural representations of habits. Science. 1999;286:1745–1749. doi: 10.1126/science.286.5445.1745. [DOI] [PubMed] [Google Scholar]
- 19.Johnson K, Churchill L, Klitenick MA, Hooks MS, Kalivas PW. Involvement of the ventral tegmental area in locomotion elicited from the nucleus accumbens or ventral pallidum. J Pharmacol Exp Ther. 1996;277:1122–1131. [PubMed] [Google Scholar]
- 20.Johnson PI, Napier TC. Morphine modulation of GABA- and glutamate-induced changes of ventral pallidal neuronal activity. Neuroscience. 1997;77:187–197. doi: 10.1016/s0306-4522(96)00482-4. [DOI] [PubMed] [Google Scholar]
- 21.Johnson PI, Stellar JR. Comparison of delta opiate receptor agonist induced reward and motor effects between the ventral pallidum and dorsal striatum. Neuropharmacology. 1994;33:1171–1182. doi: 10.1016/s0028-3908(05)80007-3. [DOI] [PubMed] [Google Scholar]
- 22.Kalivas PW, Churchill L, Klitenick MA. The circuitry mediating the translation of motivational stimuli into adaptive motor responses. In: Kalivas PW, Barnes CD, editors. Limbic motor circuits and neuropsychiatry. CRC; Boca Raton. FL: 1993. pp. 237–287. [Google Scholar]
- 23.Kalivas PW, Jackson D, Romanidies L, Wyndham L, Duffy P. Involvement of pallidothalamic circuitry in working memory. Neuroscience. 2001;104:129–136. doi: 10.1016/s0306-4522(01)00054-9. [DOI] [PubMed] [Google Scholar]
- 24.Koob GF. Neuronal mechanisms of drug reinforcement. Ann NY Acad Sci. 1992;654:171–191. doi: 10.1111/j.1749-6632.1992.tb25966.x. [DOI] [PubMed] [Google Scholar]
- 25.Koob GF, Robledo P, Markou A, Caine SB. The mescorticolimbic circuit in drug dependence and reward: a role for the extended amygdala? In: Kalivas PW, Barnes CD, editors. Limbic motor circuits and neuropsychiatry. CRC; Boca Raton, FL: 1993. pp. 289–310. [Google Scholar]
- 26.Krimer LS, Jakab RL, Goldman-Rakic PS. Quantitative three-dimensional analysis of the catecholaminergic innervation of identified neurons in the macaque prefrontal cortex. J Neurosci. 1997;17:7450–7461. doi: 10.1523/JNEUROSCI.17-19-07450.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis B, O'Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D1 dopamine receptors. Cereb Cortex. 2000;10:1168–1175. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- 28.McFarland K, Ettenberg A. Reinstatement of drug-seeking behavior produced by heroin-predictive environmental stimuli. Psychopharmacology. 1997;131:86–92. doi: 10.1007/s002130050269. [DOI] [PubMed] [Google Scholar]
- 29.Meil WM, See RE. Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav Brain Res. 1997;87:139–148. doi: 10.1016/s0166-4328(96)02270-x. [DOI] [PubMed] [Google Scholar]
- 30.Mogenson GJ, Brudzynski SM, Wu M, Yang CR, Yim CCY. From motivation to action: a review of dopaminergic regulation of limbic-nucleus accumbens-pedunculopontine nucleus circuitries involved in limbic-motor integration. In: Kalivas PW, Barnes CD, editors. Limbic motor circuits and neuropsychiatry. CRC; Boca Raton, FL: 1993. pp. 193–236. [Google Scholar]
- 31.Napier TC. Transmitter actions and interactions on pallidal neuronal function. In: Kalivas PW, Barnes CD, editors. Limbic motor circuits and neuropsychiatry. CRC; Boca Raton, FL: 1993. pp. 125–154. [Google Scholar]
- 32.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 33.O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 4. Academic; New York: 1998. [Google Scholar]
- 35.Pennartz CMA, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbensas a complex of functionally distinct neuronal ensembles: an integration of behavioral, electrophysiological and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 36.Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Rev. 1997;25:192–216. doi: 10.1016/s0165-0173(97)00021-0. [DOI] [PubMed] [Google Scholar]
- 37.Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce RC, Reeder DC, Hicks J, Morgan ZR, Kalivas PW. Ibotenic acid lesions of the dorsal prefrontal cortex disrupt the expression of behavioral sensitization to cocaine. Neuroscience. 1998;82:1103–1114. doi: 10.1016/s0306-4522(97)00366-7. [DOI] [PubMed] [Google Scholar]
- 39.Pinto A, Sesack S. Limited collateralization of neurons in the rat prefrontal cortex that project to the nucleus accumbens. Neuroscience. 2000;97:635–642. doi: 10.1016/s0306-4522(00)00042-7. [DOI] [PubMed] [Google Scholar]
- 40.Povlock SL, Schenk JO. A multisubstrate kinetic mechanism of dopamine transport in the nucleus accumbens and its inhibition by cocaine. J Neurochem. 1997;69:1093–1105. doi: 10.1046/j.1471-4159.1997.69031093.x. [DOI] [PubMed] [Google Scholar]
- 41.Reid MS, Berger SP. Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. NeuroReport. 1996;7:1325–1329. doi: 10.1097/00001756-199605170-00022. [DOI] [PubMed] [Google Scholar]
- 42.Robbins TW, Everitt BJ. Neurobehavioral mechanisms of reward and motivation. Curr Opin Neurol. 1996;6:228–236. doi: 10.1016/s0959-4388(96)80077-8. [DOI] [PubMed] [Google Scholar]
- 43.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 44.Seiden LS, Sabol KE, Ricuarte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 45.Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- 46.Shaham Y, Stewart J. Stress reinstates heroin-seeking in drug-free animals: an effect mimicking heroin, not withdrawal. Psychopharmacology. 1995;119:334–341. doi: 10.1007/BF02246300. [DOI] [PubMed] [Google Scholar]
- 47.Weissenborn R, Whitelaw RB, Robbins TW, Everitt BJ. Excitotoxic lesions of the mediodorsal thalamic nucleus attenuate intravenous cocaine self-administration. Psychopharmacology. 1998;140:225–232. doi: 10.1007/s002130050761. [DOI] [PubMed] [Google Scholar]
- 48.White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 49.Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–215. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 50.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 51.Yang CR, Seamans JK, Gorelova N. Electrophysiological and morphological properties of layers V–VI principal pyramidal cells in rat prefrontal cortex in vitro. J Neurosci. 1996;16:1904–1921. doi: 10.1523/JNEUROSCI.16-05-01904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You Z-B, Tzschentke TM, Brodin E, Wise RA. Electrical stimulation of the prefrontal cortex increases cholecystokinin, glutamate, and dopamine release in the nucleus accumbens: an in vivo microdialysis study in freely moving rats. Neuroscience. 1998;18:6492–6500. doi: 10.1523/JNEUROSCI.18-16-06492.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zahm DS, Brog JS. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience. 1992;50:751–767. doi: 10.1016/0306-4522(92)90202-d. [DOI] [PubMed] [Google Scholar]