

Abstract
The data presented here demonstrate that sympathetic neurons have the potential to activate two alternative caspase-dependent pathways either of which is capable of mediating death induced by NGF deprivation and that these neurons have the potential to switch from one pathway to the other. The presence of these two alternative pathways to trophic factor deprivation-induced death may have implications for ensuring the correct development of the nervous system. In wild-type neurons, a caspase-2-dependent pathway is required for death, and a caspase-9-dependent pathway appears to be suppressed by endogenous inhibitors of apoptosis proteins (IAPs). In contrast, for caspase-2-null neurons, death is dependent on the caspase-9 pathway. The mechanism underlying the shift is the result of a threefold compensatory elevation of caspase-9 expression and a doubling of levels of direct IAP binding protein with low pI (DIABLO)/second mitochondria-derived activator of caspase (Smac), an IAP inhibitor, both at the mRNA and protein levels. These findings resolve seemingly discrepant findings regarding the roles of various caspases after NGF deprivation and raise a cautionary note regarding the interpretation of findings with caspase-null animals. The choice of the death-mediating caspase pathway in the sympathetic neurons is thus dependent on the regulated relative expression of components of the pathways including those of caspases, IAPs, and IAP inhibitors.
Keywords: trophic factor deprivation, neuronal cell death, caspases, caspase-2, caspase-9, IAPs, DIABLO/Smac, sympathetic neurons
During development, neurons that fail to find appropriate targets and sources of target-derived neurotrophic factors undergo apoptotic cell death (Pettmann and Henderson, 1998). The elimination of improperly connected neurons constitutes a critical step in the formation of specific connections in the nervous system. Data from caspase-null and apoptotic protease activating factor 1 (Apaf1)-null mice support a role for the caspase-9 pathway in mediating death occurring early in the development of the nervous system (Kuida et al., 1996, 1998; Hakem et al., 1998;Yoshida et al., 1998), when mainly neuroblasts are being removed. It is not clear which caspases are necessary for the removal of neurons that occurs later in development. It has been reported that the caspase dependency of a pathway can be altered in caspase-null mice (Zheng et al., 2000), but no mechanism was provided for the observations described. Studies of the caspases required to execute trophic factor deprivation (TFD)-induced death of NGF-dependent neurons have given apparently conflicting results. In the case of cultured rat and mouse sympathetic neurons, as well as of neuronal pheochromocytoma 12 (PC12) cells, there is evidence that caspase-2 is necessary for TFD-induced death. Both acute downregulation of caspase-2 expression with an antisense oligonucleotide (Troy et al., 1997) and chronic downregulation of caspase-2 in PC12 cells by stable transfection with antisense caspase-2 (Haviv et al., 1998) protect these cells from NGF deprivation. In contrast, sympathetic neurons cultured from caspase-2-null mice retain sensitivity to NGF deprivation and die (Bergeron et al., 1998). Other studies have demonstrated a delay in TFD-induced death in sympathetic neurons from caspase-9-null embryos (Deshmukh et al., 2000). In an attempt to reconcile these apparently contradictory data, we have investigated the mechanism of TFD-induced death in sympathetic neurons cultured from both wild-type and caspase-2-null mice with attention to the levels of expression of caspases and caspase regulators in these mice. Caspases are an evolutionarily conserved family of proteins with at least 14 mammalian members (Thornberry and Lazebnik, 1998). Evidence suggests that caspases are activated in cascades in which upstream (activator) caspases lead to activation of downstream (effector) caspases. One of the most studied cascades is that of caspase-9 that leads to activation of caspase-3 and -7. Activation of the caspase-9-dependent apoptotic pathway is tightly regulated by both the regulatory adaptor molecule APAF1, which recruits caspase-9 to the apoptosome, and the inhibitors of apoptosis proteins (IAPs) (Salvesen, 1999;Hengartner, 2000). The human IAP XIAP has been shown to inhibit caspase-9 as well as the downstream caspases caspase-3 and -7 (Deveraux et al., 1997). Recent work has revealed a mammalian inhibitor of IAPs, direct IAP binding protein with low pI DIABLO/second mitochondria-derived activator of caspase (Smac), that inhibits the IAPs and thus promotes caspase-9, -3, and -7 activities (Du et al., 2000; Verhagen et al., 2000). Similar regulation of the caspase-2 pathway has not been found. The death adaptor protein receptor interacting protein-associated ICH-1 homologous protein with a death domain (RAIDD) activates caspase-2 (Duan and Dixit, 1997), but, to date, no IAPs have been found that bind caspase-2 (Deveraux et al., 1999b). The position of caspase-2 in an activation cascade has not been clarified. It has been proposed to act as either an activator or an effector. In any event, it does appear to be independent of the caspase-9 pathway.
The studies we report reveal specific upregulation in brains and sympathetic neurons of caspase-2-null mice of both caspase-9 and DIABLO/Smac. As a consequence of these changes, TFD-induced death, which is normally dependent on caspase-2, switches to an alternative pathway dependent on caspase-9. These results show the tight regulation of caspase activities in neurons. When caspase-2 is removed early in development, as in the null animals, the redundancy of caspases allows the compensations that we have described. The shift from the caspase-2 to the caspase-9 pathway would ensure the elimination of neurons that are superfluous. This regulated expression of caspases and IAP inhibitors is likely to be important in neurodegenerative disorders as well as in neurodevelopment.
MATERIALS AND METHODS
Sympathetic neuron cultures. Sympathetic neuron cultures were prepared from 1-d-old wild-type and caspase-2 −/− mouse pups (Bergeron et al., 1998), generous gifts from L. Bergeron and J. Yuan (Harvard University), as described previously (Troy et al., 2000). Cultures were grown in 24-well collagen-coated dishes for survival experiments and in 6-well collagen-coated dishes for RNA and protein extraction in RPMI 1640 medium plus 10% horse serum with mouse NGF (100 ng/ml). One day after plating, uridine and 5-fluorodeoxyuridine (10 μm each) were added to the cultures and allowed to remain for 3 d to eliminate non-neuronal cells (<1% non-neuronal cells remain after 3 d). For survival experiments, on the sixth day after plating, NGF was removed by washing the cultures three times with RPMI 1640 medium plus 10% horse serum, followed by the addition of medium containing anti-mouse NGF (1:200; Sigma, St. Louis, MO). Caspase inhibitors (Enzyme Systems Products, Livermore, CA) were added as indicated. Each culture was scored, as described previously (Troy et al., 1997), for numbers of living, phase-bright neurons present in the same field at various times. Three replicate cultures were assessed for each condition, and data are normalized to numbers of neurons present in each culture at the time of NGF deprivation and reported as the mean ± SEM. For RNA and protein extraction on the sixth day after plating, RNA and protein were extracted using the Trizol reagent according to the manufacturer's protocol.
Synthesis of antisense oligonucleotides. Oligonucleotides containing an SH group at the 5′ end and an NH group at the 3′ end were synthesized by Operon (Alameda, CA). As described previously (Troy et al., 1996), oligonucleotides were resuspended in deionized water, an equimolar ratio of Penetratin1 (Oncor) was added, and the mixture was incubated at 37°C for 1 hr. The yield of the reaction, estimated by SDS-PAGE followed by Coomassie blue staining, was routinely >50%. As a control, a scrambled sequence of the antisense oligonucleotide (same base composition; different order) was used. The antisense sequences used were as follows: antisense caspase-1 (ACasp1), CCTCAGGACCTTGTCGGCCAT; ACasp2, GCTCGGCGCCGCCATTTCCCAG; ACasp3, GTTGTTGTCCATGGTCACTTT; ACasp6, TGTTTCCATCATGCTTTATTG; ACasp7N1, ATCGTCTGTCATCGTTCCCAC; ACasp7N2, CTCGAAGTCCATACGGTACAG; ACasp8, GTGGAAATCCATTCTTACCAA; ACasp9, CTGCCGGTCCGCCTCGTCCAT; ADIABLO, AGAGCCGCCATCCCGCGGCCA; AAPAF1, CTTTGCATCCATTGTGCCTCA; and AMIAP3, GTTAAAAGTCATCTTCTCTGG.
Western blotting. Postnatal day 1 mouse brains were harvested in sample buffer. For antisense downregulation studies, PC12 cells, grown as described previously, were treated with various antisense constructs for 5 hr and harvested in sample buffer. Equal amounts of protein were separated by 15% PAGE, transferred to nitrocellulose, and immunostained as described previously (Troy et al., 2000). Anti-caspase-9 (Medical and Biological Laboratories Co., Nagoya, Japan) was used at 1:1000, anti-APAF1 (StressGen) was used at 1:1000, anti-Smac (a generous gift of X. Wang, Howard Hughes Medical Institute and University of Texas Southwestern Medical Center, Dallas, TX) was used at 1:2000, anti-XIAP (StressGen) was used at 1:1000, anti-RAIDD (StressGen) was used at 1:500, and anti-actin (Sigma) was used at 1:200. Visualization was with ECL, using goat anti-rabbit peroxidase at 1:1000. The relative intensities of the protein bands were quantified using Scion NIH Image 1.55 software.
Quantitative PCR. Primers were designed to amplify a 300–400 base piece of each gene of interest. cDNA from brains of wild-type and caspase-2-null mice or cDNA from cultured sympathetic neurons was added to a reaction mix together with appropriate primers at 0.5 μm each. The reaction mix for the Roche Light Cycler was DNA Master SYBR Green 1 (Roche Molecular Biochemicals, Indianapolis, IN). The reaction mix for the Cepheid Smart Cycler (Fisher Scientific, Houston, TX) was PCR ready-to-go beads (Amersham Pharmacia Biotech, Arlington Heights, IL) with SYBR Green (Molecular Probes, Eugene, OR). Levels of gene transcripts were analyzed using the Roche Light Cycler or the Cepheid Smart Cycler following the manufacturers' specifications. Real-time fluorescence of SYBR green indicated that double-stranded DNA was measured. Melting curve analysis was used for each protocol to characterize and identify the specific amplicon. In each case quantification was made from the linear portion of the amplification curve. Actin was used to normalize input cDNA.
Immunocytochemistry. Sympathetic neurons were grown on collagen-coated eight-well LabTek chamber slides. After indicated treatments, cells were fixed with 4% paraformaldehyde and immunostained as described previously (Troy et al., 1997). Cells were double labeled with anti-actin (Sigma) at 1:250 and anti-activated caspase-3 (New England Biolabs, Beverly, MA) at 1:100. Western blotting showed that the lot of the activated caspase-3 antibody used for these studies detected activated caspase-3 but not caspase-3 zymogen. Secondary antibodies were goat anti-rabbit Alexafluor 546 and goat anti-mouse Alexafluor 488 (Molecular Probes), both at 1:1000. Cells were examined with a PerkinElmer Spinning Disc confocal imaging system mounted on a Nikon inverted microscope.
RESULTS
Caspase-2-null neurons use an alternative caspase path to death after trophic factor deprivation
Caspase-2 has been identified as critical for trophic factor death in sympathetic neurons and PC12 cells (Troy et al., 1997; Haviv et al., 1998). However, cultured sympathetic neurons from caspase-2-null mice die when deprived of NGF (Bergeron et al., 1998). To ascertain that TFD-induced death in the caspase-2-null neurons was caspase dependent, both wild-type and caspase-2-null cells were treated with the pseudosubstrate caspase inhibitors BAF and DEVD-fluoromethylketone (DEVD-FMK). Figure1 shows that the broad-spectrum caspase inhibitor BAF protects caspase-2-null neurons as well as wild-type neurons, confirming that the death process is caspase-mediated in both sets of neurons. However, DEVD-FMK, used at a concentration (10 μm) that is relatively specific for caspase-3 family members, provided protection only for caspase-2-null neurons (Fig. 1B). This suggested that although caspase activity was required for death in both cases after removal of NGF, different caspases were used in each case. The rescue of the caspase-2-null neurons from TFD by DEVD-FMK suggested that this was a member of the caspase-3 family.
Fig. 1.

Differential inhibition of TFD-induced death by DEVD-FMK in wild-type and caspase-2-null neurons. Sympathetic neurons from wild-type (A) and caspase-2-null (B) mice were cultured for 5 d and then washed and treated with anti-NGF in the presence and absence of BAF (50 μm) or DEVD-FMK (10 μm). Cultures were counted daily, and survival is reported relative to that in the same cultures before NGF deprivation and is given as the mean ± SEM (n = 3). Error bars are sometimes too small to be visible. This is a representative experiment; similar results were obtained in four independent experiments.
Caspase-2-null mice brains and sympathetic neurons have increased expression of caspase-9 and DIABLO/Smac
The differential effects of DEVD-FMK led us to investigate whether the targeted knock-out of caspase-2 resulted in changes in expression of other caspases or other constituents of cell death pathways that would preserve vulnerability to TFD. Brains, not including cerebella, from wild-type and caspase-2-null postnatal day 1 (P1) mice were harvested for RNA and protein, and the relative expression of various caspases was determined using quantitative PCR and Western blotting. These studies revealed that caspase-2 and -9 transcripts are differentially expressed in the two groups of animals; caspase-2-null animals have no caspase-2 mRNA but have more than three times the levels of caspase-9 mRNA, relative to actin expression (Fig.2A). No significant changes were observed for transcripts encoding caspase-1, -3, -6, -7, -8, -11, or -14. The increase in caspase-9 mRNA was confirmed by Northern blotting (data not shown). Western blotting also revealed changes only in caspase-2 and -9 levels. Caspase-9 protein was increased approximately threefold in the caspase-2-null mouse brain (Fig. 2B), and as expected, caspase-2 protein was absent (data not shown). Other caspases (caspase-1, -3, -6, -7, -8, and -11) were unchanged (caspase-3 levels are shown in Fig.2B). The increase in caspase-9 expression was confirmed in cultured sympathetic neurons as well. Sympathetic neurons from wild-type and caspase-2-null P1 animals were cultured for 5 d and harvested for total cellular RNA and protein assays. Quantitative PCR showed a more than fivefold increase in caspase-9 expression (Fig.2E). Western blotting confirmed the increase in caspase-9 protein in the neurons (data not shown).
Fig. 2.

Differential expression of caspases and caspase regulatory molecules in wild-type and caspase-2-null mice.A, Relative expression of caspase mRNAs in wild-type and caspase-2-null P1 mouse brains. mRNA was prepared from six wild-type and nine caspase-2-null mouse brains. cDNA from each brain was analyzed individually, using serial dilutions in duplicate, with real-time PCR. Each sample was analyzed three times. Results were normalized to actin mRNA levels. For each caspase, expression in wild-type brains was set at a value of 1. Data are the means ± SEM (n= 6 for wild type; n = 9 for caspase-2 null).B, Western blots of caspase-9 and -3. Wild-type and caspase-2-null P1 mouse brains were homogenized in sample buffer, and equal amounts of protein (determined by the Bradford protein assay) were subjected to Western blotting using the indicated antisera. Actin staining confirmed equal loading. These are representative blots; similar results were obtained in six independent blots for caspase-9 and three independent blots for caspase-3. C, Relative expression of DIABLO, Apaf1, RAIDD, and MIAP3 mRNA in wild-type and caspase-2-null mouse brains. mRNA from six wild-type and nine caspase-2 mouse brains was analyzed using real-time PCR as described inA. Results were normalized to actin mRNA levels. For each transcript, results in the wild type are set at a value of 1. Data are the means ± SEM (n = 6 for wild type;n = 9 for caspase-2 null). D, Western blots of DIABLO/Smac, APAF1, and MIAP3. Wild-type and caspase-2-null P1 mouse brains were homogenized in sample buffer, and equal amounts of protein were subjected to Western blotting using the indicated antisera. Actin staining confirmed equal loading. These are representative blots; similar results were obtained in six independent blots for DIABLO/Smac and three independent blots for APAF1 and MIAP3.E, Relative expression of caspase, DIABLO/Smac, and MIAP3 mRNAs in wild-type and caspase-2-null P1 cultured sympathetic neurons. mRNA was prepared from wild-type and caspase-2-null sympathetic neurons grown in culture for 6 d. cDNA was analyzed with real-time PCR using serial dilutions in duplicate. Each sample was analyzed three times. Results were normalized to actin mRNA levels. For each mRNA, expression in wild-type brains was set at a value of 1. Data are the means ± SEM (n = 3 for wild type;n = 3 for caspase-2 null).
We next investigated whether loss of caspase-2 resulted in changes in other molecules known to modulate the caspase-2 or caspase-9 pathways. These include RAIDD for caspase-2 and APAF1, MIAP3, and DIABLO/Smac for caspase-9. There were no changes in expression of either message or protein for RAIDD, the death adaptor protein for caspase-2 (Duan and Dixit, 1997), or APAF1, the mammalian ced-4 homolog that activates caspase-9 (Zou et al., 1997) (Fig. 2C,D). However, in the case of the recently discovered DIABLO/Smac, an inhibitor of IAPs that is permissive for caspase-9 activation (Chai et al., 2000; Du et al., 2000; Verhagen et al., 2000), both mRNA and protein were increased by approximately twofold in brain as well as in sympathetic neurons (Fig. 2C–E). MIAP3, a mouse homolog of XIAP (Farahani et al., 1997), an IAP that has been shown to inhibit the activity of caspase-3, -7, and -9 (Deveraux et al., 1997, 1999a;Takahashi et al., 1998), was unchanged (Fig. 2C–E).
Specific inhibition of the caspase-9 pathway protects caspase-2-null neurons but not wild-type neurons from TFD
We next assessed whether, as indicated by the above findings, TFD-induced death of caspase-2-null and wild-type sympathetic neurons uses different sets of caspases. Because none of the available pharmacologic caspase inhibitors are completely specific for an individual caspase, we turned to antisense oligonucleotides to decrease expression of specific caspases. This was achieved by using antennapedia peptide (Penetratin1)-mediated intracellular delivery of antisense oligonucleotides, which is a technique that has been widely and successfully used for such purposes (Allinquant et al., 1995; Troy et al., 1996; Pooga et al., 1998; Nakagawa et al., 2000). By this means, we have been able to achieve 50–80% downregulation of individual caspases in cultured neuronal cells without affecting levels of other caspases (Troy et al., 1997, 2000) (Fig. 3A, representative blots). Control (scrambled) oligonucleotides had no discernable effect on expression. Antisense oligonucleotides were designed to downregulate caspase-1, -2, -3, -6, -7, -8, and -9 and linked to Penetratin1. The specificity of sequences was verified by BLAST search (National Center for Biotechnology Information, Bethesda, MD). The efficacy of downregulation was evaluated by Western blotting of the target caspase in PC12 cell cultures with or without exposure to the Penetratin1-linked oligonucleotides [Fig. 3A for caspase-6, -7, -8, and -9; Troy et al. (1997) for caspase-2; Troy et al. (2000) for caspase-1 and -3]. PC12 cell cultures were used because of the greater amount of material available for the biochemical measurements. Our previous work supports the concurrence of mechanisms in PC12 cells and sympathetic neurons (Farinelli et al., 1996; Troy et al., 1997, 2000; Park et al., 1998; Stefanis et al., 1998). All antisense oligonucleotides provided >50% downregulation of the targeted caspase within 5 hr of treatment. Levels of the other nontargeted caspases were not affected (data not shown). Sympathetic neurons from wild-type and caspase-2-null mice were deprived of NGF in the presence and absence of each of these antisense oligonucleotides, and survival was assessed daily for 3 d (Fig. 3B,C). As we have shown previously, the Penetratin1-linked antisense oligonucleotide to caspase-2 (V-ACasp2, previously called V-ANedd) protected wild-type neurons from NGF withdrawal. As anticipated, there was no protection of caspase-2-null neurons by this construct. In contrast, caspase-2-null neurons were protected by V-ACasp3, V-ACasp7, and V-ACasp9. These antisense constructs, however, provided no protection for wild-type neurons. No protection was afforded for either wild-type or caspase-2-null neurons by control (scrambled) oligonucleotides (data not shown) or by downregulation of caspase-1, -6, or -8.
Fig. 3.
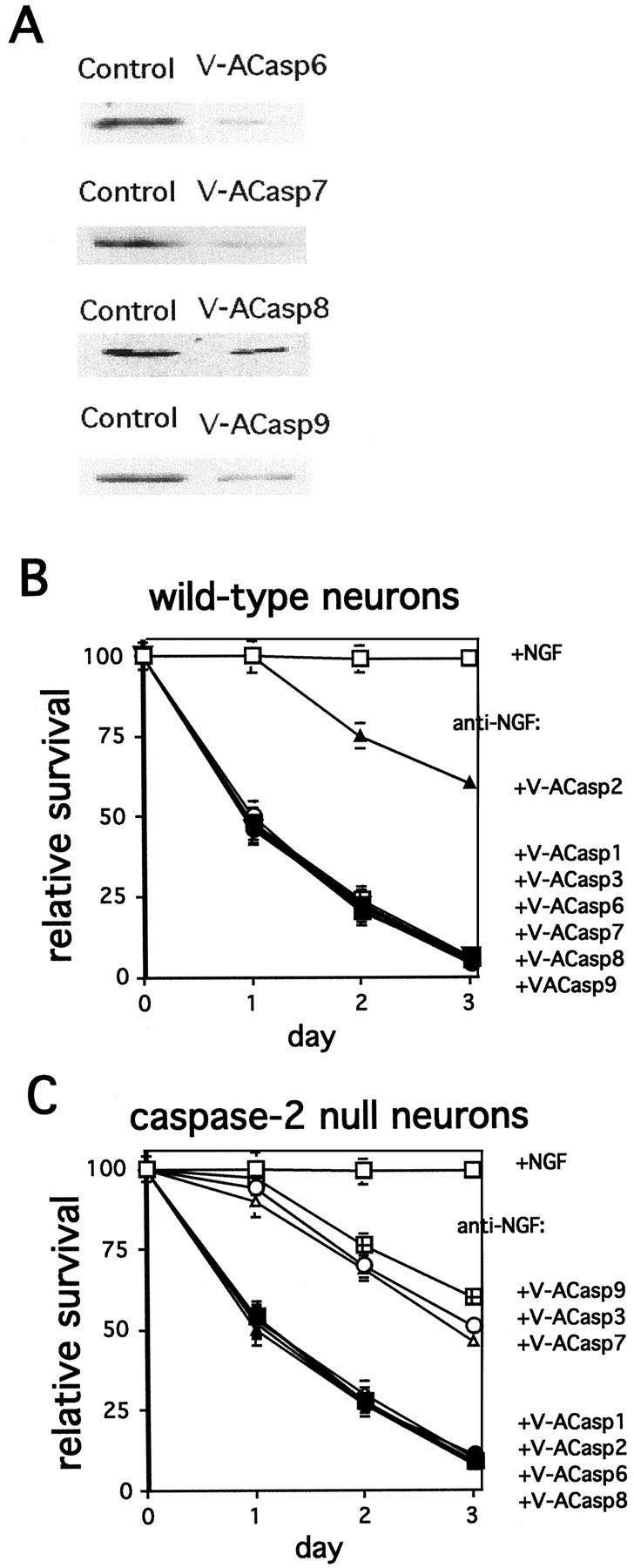
Differential effects of downregulation of specific caspases on TFD-induced death of wild-type and caspase-2-null sympathetic neurons. A, Penetratin1-linked antisense oligonucleotides specifically downregulate targeted caspases. PC12 cells were treated with the indicated antisense oligonucleotides (240 nm) for 6 hr. Cells lysates containing equal amounts of protein were subjected to Western blotting using the corresponding antisera. Actin staining confirmed equal loading. These are representative blots; similar results were obtained in two independent experiments. B, C, Sympathetic neurons from P1 wild-type (B) and caspase-2-null (C) mice were cultured for 5 d. Cultures were then washed and treated with anti-NGF in the presence and absence of the indicated antisense oligonucleotides (open squares, control; closed squares, anti-NGF;open diamonds, anti-NGF + V-ACasp1; closed triangles, anti-NGF + V-ACasp2; open circles, anti-NGF + V-ACasp3; closed circles, anti-NGF + V-ACasp6; open triangles, anti-NGF + V-ACasp7;closed inverted triangles, anti-NGF + V-ACasp8;squares with crosses, anti-NGF + V-ACasp9). Cultures were scored daily, and survival is reported relative to the numbers of living neurons in the same cultures before NGF deprivation and is given as the mean ± SEM (n = 3). Error bars are sometimes too small to be visible. This is a representative experiment; similar results were obtained in six independent experiments.
Downregulation of DIABLO/Smac or APAF1 selectively protects caspase-2-null neurons from TFD
We next tested whether downregulation of additional components of the caspase-9 pathway would bring about differential protection. Antisense-mediated downregulation of either DIABLO/Smac (Fig. 4A,B) or APAF1 (Fig. 4D,E) provided complete protection against NGF withdrawal for caspase-2-null neurons but had no effect on the survival of wild-type neurons. Figure 4C shows the efficacy of downregulation by these constructs. The photomicrographs in Figure 5 show that inhibition of APAF1 (Fig. 5D), DIABLO/Smac (Fig.5C), caspase-9 (Fig. 5E), or caspase-3 (Fig. 5F) expression in caspase-2-null neurons protected not only cell bodies but also neurites.
Fig. 4.

Caspase-2-null neurons use an alternative pathway to TFD-induced death. A, B, D, E, Sympathetic neurons from P1 wild-type (A, D) and caspase-2-null (B, E) mice were cultured for 5 d. Cultures were then washed and treated with anti-NGF in the presence and absence of V-ADIABLO (A, B) or V-AAPAF1 (D, E). Cultures were scored daily, and survival is reported relative to that in the same cultures before NGF deprivation and is given as the mean ± SEM (n = 3). Error bars are sometimes too small to be visible. This is a representative experiment; similar results were obtained in three independent experiments.C, Specific downregulation of DIABLO/Smac and APAF1 by antisense oligonucleotides is shown. PC12 cells were treated with the indicated antisense oligonucleotides (240 nm) for 6 hr. Cells lysates containing equal amounts of protein were subjected to Western blotting using the corresponding antisera. Actin staining confirmed equal loading. These are representative blots; similar results were obtained in two independent experiments.
Fig. 5.
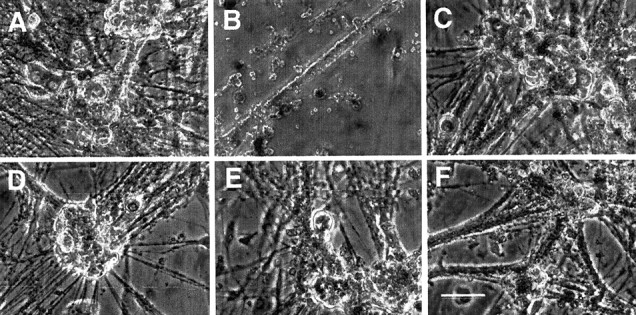
Photomicrographs of SCGs from caspase-2-null mice rescued by downregulation of various components of the caspase-9 pathway. Sympathetic neurons of caspase-2-null mice were cultured for 5 d. Cultures were then washed and treated with anti-NGF in the presence or absence of various antisense oligonucleotides. The photomicrographs were taken after 2 d of treatment.A, +NGF. B, Anti-NGF. C, Anti-NGF + V-ADIABLO. D, Anti-NGF + V-AAPAF1.E, Anti-NGF + V-ACasp9. F, Anti-NGF + V-ACasp3. Scale bar, 100 μm.
Downregulation of DIABLO/Smac, APAF1, caspase-9, or caspase-3 suppresses elevation of activated caspase-3 in NGF-deprived neurons from caspase-2-null mice
The preceding findings point to the activation of the caspase-9 pathway in caspase-2-null neurons, with consequent activation of caspase-3 and -7. Using an antibody that specifically recognizes activated caspase-3 (see Materials and Methods), we assessed the cellular localization of this enzyme in caspase-2-null neurons after various treatments. The confocal micrographs in Figure6 depict cultures of sympathetic neurons from caspase-2-null mice double-labeled for actin (green) and activated caspase-3 (red). Control cells show only minimal staining for activated caspase-3 in either cell bodies or neurites (Fig. 6A). After 5 hr of TFD, there is substantial activation of caspase-3. In the two cells shown in Figure 6B, it is clear that, as activation of caspase-3 increases, actin immunostaining decreases, likely because of actin degradation during the death process. The induction of activated caspase-3 seen in caspase-2-null neurons after TFD is blocked by downregulation of either DIABLO or APAF1 with the appropriate antisense oligonucleotide. Downregulation of caspase-9 or -3 (Fig.6E,F) substantially decreased the amount of activated caspase-3 detectable by immunostaining but did not completely block it.
Fig. 6.

Downregulation of various components of the caspase-9 pathway suppress caspase-3 activation in NGF-deprived sympathetic neurons from caspase-2-null mice. Sympathetic neurons from caspase-2-null mice were cultured on chamber cover glass slides for 5 d. Cultures were then washed and treated with anti-NGF in the presence and absence of various oligonucleotides. After 5 hr, cells were fixed, immunostained for actin (green) and activated caspase-3 (red), and examined by confocal microscopy. A, +NGF. B, Anti-NGF.C, Anti-NGF + V-ADIABLO. D, Anti-NGF + V-AAPAF1. E, Anti-NGF + V-ACasp9. F, Anti-NGF + V-ACasp3. Scale bar, 50 μm.
The caspase-9 pathway is suppressed in wild-type neurons by IAPs
Our previous findings have indicated that although NGF deprivation induces DEVDase activity in wild-type sympathetic neurons and PC12 cells, this is neither necessary nor sufficient to induce death (Troy et al., 1997; Stefanis et al., 1998). In addition, endogenous suppressors of caspases are likely to play an important role in the regulation of caspase activity and death. The IAP family of caspase inhibitors has been shown to block caspase-3, -7, and -9 activities (Deveraux et al., 1997, 1999b). To reduce IAP activity in cultured sympathetic neurons, we designed a Penetratin1-linked antisense oligonucleotide (V-AMIAP3) to MIAP3. MIAP3 was chosen because it is the mouse homolog of XIAP, the IAP that has been most closely linked with the caspase-9 pathway. V-AMIAP3 promotes 70% downregulation of MIAP3 within 6 hr (Fig. 7A). To determine whether in vivo activation of the caspase-9 pathway might be suppressed by IAPs in wild-type neurons, we withdrew NGF in the presence of V-ACasp2 and V-AMIAP3. We have shown previously that simultaneous treatment with multiple Penetratin1-linked antisense oligonucleotides does not alter the effects of the individual oligonucleotides (Troy et al., 1996). As shown in Figure 7B, the protection conferred by caspase-2 downregulation (by V-ACasp2) was reversed by downregulation of MIAP3 (by cotreatment with V-AMIAP3). This suggests that reduction of MIAP3 levels permits death by an otherwise suppressed caspase-9-dependent pathway. Consistent with this, the death induced by V-ACasp2 plus V-AMIAP3 was prevented by downregulation of either APAF1 or caspase-9 (Fig. 7B). This suggestion was further supported when we examined immunostaining of activated caspase-3 in these neurons. Withdrawal of NGF in the presence of V-ACasp2 plus V-AMIAP3 induced a strong signal in both cell bodies and neurites (Fig. 7C,D). This activated caspase-3 immunostaining was suppressed by the downregulation of caspase-9 (Fig.7E).
Fig. 7.

Downregulation of MIAP3 permits caspase-9-dependent TFD-induced death of wild-type SCGs.A, Specific downregulation of MIAP3 is shown. PC12 cells were treated with V-AMIAP3 (240 nm) for 6 hr. Cells lysates containing equal amounts of protein were subjected to Western blotting using the corresponding antisera. Actin staining confirmed equal loading. These are representative blots; similar results were obtained in two independent experiments. B, Sympathetic neurons from wild-type mice were cultured for 5 d. Cultures were then washed and treated with anti-NGF in the presence and absence of the indicated antisense oligonucleotides. Cultures were scored daily, and survival is reported relative to that in the same cultures before NGF deprivation and is given as the mean ± SEM (n= 3). Error bars are sometimes too small to be visible. This is a representative experiment; similar results were obtained in three independent experiments. C–E, Activation of caspase-3 in NGF-deprived sympathetic neurons is dependent on caspase-9. Sympathetic neurons from wild-type mice were cultured on chamber cover glass slides for 5 d. Cultures were then washed and treated with anti-NGF in the presence and absence of various oligonucleotides. After 5 hr, cells were fixed, immunostained for actin (green) and activated caspase-3 (red), and examined by confocal microscopy.C, +NGF. D, Anti-NGF + V-ACasp2 + V-AMIAP3. E, Anti-NGF + V-ACasp2 + V-AMIAP3 + V-ACasp9. Scale bar, 50 μm.
DISCUSSION
In an attempt to reconcile the apparently conflicting observations that NGF withdrawal induces apoptosis in caspase-2-null sympathetic neurons (Bergeron et al., 1998) but is unable to cause death in neurons in which caspase-2 has been downregulated (Troy et al., 1997), we examined the ability of caspase inhibitors to rescue each of these cell types from death. Both cell types were rescued by the broad-spectrum inhibitor BAF, but only the caspase-2-null neurons were rescued by DEVD-FMK, which is relatively selective for caspase-3-like activities when used at 10 μm. This differential sensitivity to the caspase inhibitors led us to question whether there were differences in the expression of caspases or the regulators of caspase activity in these two cell types. We found that caspase-9 mRNA and protein are selectively increased by approximately threefold in the newborn caspase-2-null mouse brain and more than fivefold in cultured sympathetic neurons. Expression of the proapoptotic death regulator DIABLO/Smac was also elevated.
The increases in the expression of these two proapoptotic molecules suggest that they might compensate for the loss of caspase-2 and enable neurons from caspase-2-null mice to die by an alternative pathway mediated by caspase-9 and its downstream targets such as caspase-3 and -7. This was confirmed in the series of experiments showing that downregulation of caspase-2 suppresses TFD in wild-type neurons, but not in caspase-2-null neurons. In contrast, downregulation of caspase-3, -7, and -9 rescues caspase-2-null neurons, but not wild-type neurons, from NGF deprivation. Furthermore, interference with caspase-9 activation by downregulation of APAF1 provided protection for caspase-2-null neurons, but not for wild-type neurons.
The compensatory switch to the caspase-9 pathway that we observed in caspase-2-null mice appears to involve more than simply elevation of caspase-9 levels. DIABLO/Smac is a recently identified protein that enables activation of caspase-9 (and most likely, caspase-3 and -7) by binding to members of the IAP family (Chai et al., 2000; Du et al., 2000; Verhagen et al., 2000). We observed that DIABLO/Smac levels are doubled in caspase-2-null brains and sympathetic neurons and that downregulation of DIABLO/Smac protects caspase-2-null, but not wild-type, sympathetic neurons from NGF deprivation. Thus, the availability of the caspase-9 pathway for induction of death in NGF-deprived neurons may be at least in part dependent on its regulation by the competing activities of IAPs and DIABLO/Smac. In caspase-2-null neurons, the elevated levels of DIABLO/Smac might help swing the balance in favor of enhanced activation of the caspase-9 pathway.
The apparent involvement of DIABLO/Smac in promoting death of caspase-2-null neurons raised the issue of whether IAPs may play a role in the repression of the caspase-9 pathway in wild-type neurons. The IAPs effectively suppress activity of caspase-3, -7, and -9 (Deveraux et al., 1997, 1998, 1999b). We chose to investigate the role of MIAP3, the mouse homolog of XIAP, because it is expressed in sympathetic neurons and can inhibit all three of the above caspases (Farahani et al., 1997). We found that although downregulation of caspase-2 protects wild-type neurons from NGF deprivation, the simultaneous downregulation of MIAP3 results in death. This death appeared to involve the caspase-9 pathway because it was inhibited in turn by the additional downregulation of either caspase-9 or APAF1. The simplest interpretation of these observations is that, when NGF is removed from wild-type neurons in our system, caspase-2 is activated and mediates death and that the caspase-9 pathway is held in check by MIAP3. When MIAP3 is downregulated in such neurons, the caspase-9 pathway is no longer suppressed, and when caspase-2 is also downregulated, the caspase-9 pathway mediates death. A schematic depiction of the alternative death pathways is shown in Figure8.
Fig. 8.

Schematic representation of the trophic factor deprivation death pathways in sympathetic neurons.
Taken together, our findings support the idea that wild-type sympathetic neurons possess two alternative caspase pathways that have the potential to mediate TFD-induced death. Under the conditions of our experiments, the caspase-2 pathway is predominant, and the caspase-9 pathway is held in check by IAPs. One result of this arrangement is that various circumstances may switch use of the two pathways. For instance, as we observed, knock-out of caspase-2 results in compensatory enablement of the caspase-9 pathway because, at least in part, of upregulation of caspase-9 and of DIABLO/Smac. Another variable is developmental stage. Although the existing literature is incomplete, it appears that caspase 9 is highly expressed in the developing mouse brain at embryonic day 7 and declines after that (Kuida et al., 1998). In contrast, mouse caspase-2 [originally identified because of its downregulation in brain during development (Kumar et al., 1994)] is barely expressed at embryonic day 8 and has peak expression in the brain at embryonic day 12. However, rodent sympathetic neurons show high expression of caspase-2 in P1 animals and a subsequent decrease so that expression is minimal by P11 (Savitz and Kessler, 2000). Developmental expression patterns for other elements of either the caspase-2 or caspase-9 pathways have yet to be established in sympathetic neurons, but such time-dependent changes represent potentially important variables in the choice of caspase death mechanisms. Similarly, it is likely that additional factors can influence the expression of specific caspases and caspase regulatory molecules and, thereby, switch cells from one death pathway to another.
Because of our findings, it is not surprising that circumstances may occur in which TFD-induced death of sympathetic neurons is dependent on the caspase-9 pathway. It was reported recently that sympathetic neurons from caspase-9-null embryos undergo delayed TFD-induced death (Deshmukh et al., 2000), and on this basis, it was suggested that caspase-9 plays a critical role in death caused by NGF deprivation. These studies used E17 embryos, a developmental stage at which the expression of caspase-9 may normally be higher and that of caspase-2 may be lower than that in the postnatal (P1) neurons used in our studies. It is also possible that the knock-out of caspase-9 also leads to depression of the caspase-2 pathway and delay of death.
The failure of caspase 3 activation to cause death in the wild-type neurons in which caspase-2 has been downregulated suggests that the level of activation falls below a critical level for inducing apoptosis. This possibility is supported by the observation that increasing the activity of the caspase-9 pathway further by downregulation of MIAP3 leads to death and by the increased concentrations of both caspase-9 and DIABLO/Smac in the caspase-2-null mouse brains and neurons. It is possible that “subapoptotic” activation of the caspases in the caspase-9 pathway serves one or more important functions, such as mediating cytoskeletal breakdown.
In contrast to the caspase 9 pathway, relatively little is known about the mechanisms by which caspase-2 is activated and how such activation leads to death. Our previous work indicates that caspase-2 is not downstream of caspase-3-like activity in NGF-deprived sympathetic neurons and visa versa (Stefanis et al., 1998). Caspase-2 possesses a long caspase recruitment domain (CARD)-containing prodomain that appears important for activation via specific association with CARD-containing adapter proteins such as RAIDD (Duan and Dixit, 1997). However, little is known about how NGF deprivation might trigger interaction between caspase-2 and RAIDD and/or other activators. Several types of evidence indicate that cytochrome C release from mitochondria is required for TFD-induced death of sympathetic neurons (Deshmukh and Johnson, 1998; Neame et al., 1998). Because of the importance of caspase-2 in TFD-induced death, this raises the yet untested possibility that activation of caspase-2 lies downstream of mitochondrial perturbation. In this regard, it may be relevant that procaspase-2 has been reported to be present within mitochondria and to be released during the apoptotic process (Susin et al., 1999). Also in contrast to the caspase-9 pathway, there are no currently known negative regulators of caspase-2 activity. Thus it is possible that, unlike caspase-3, -7, and -9 that are subject to inhibition by IAPs, caspase-2, after it is activated, inevitably leads to death.
Our findings raise a note of caution regarding interpretation of data from knock-out animals and emphasize the flexibility and redundancy of apoptotic pathways. We uncovered two compensatory changes in expression of apoptosis-related proteins that occur in response to loss of caspase-2, and these appear to contribute to enablement of an alternative apoptotic caspase pathway in sympathetic neurons. It is conceivable that similar or additional changes also occurred in other tissues and that these may affect other aspects of the phenotype of caspase-2-null mice. It has been reported recently that compensatory changes in caspase activation occur in caspase-9- and caspase-3-null animals (Zheng et al., 2000). Although the latter study did not identify specific molecular changes that underlie the compensatory activation of alternative caspases, it does underscore the point raised here that cells possess multiple apoptotic caspase pathways and the means to switch from one to another.
In summary, the data presented here show that TFD-induced death of sympathetic neurons has the potential to proceed by either of two distinct pathways and that the decision concerning which pathway to use in a given situation can be regulated by alterations in the relative levels of the components of each of the pathways. It is of particular interest that such regulation included both caspases and an IAP inhibitor. Although we have manipulated these levels by genetic and antisense approaches, it is likely that they are regulated to similar effect both during development and in neurodegenerative disorders.
Footnotes
This work was supported by grants from the National Institutes of Health (C.M.T., L.A.G., and M.L.S.), the Muscular Dystrophy Association (C.M.T.), and the Blanchette Rockefeller Foundation (L.A.G.). We thank Seonia Hutchinson for technical assistance.
Correspondence should be addressed to Dr. Carol M. Troy, Columbia University College of Physicians and Surgeons, Department of Pathology, 630 West 168th Street, New York, NY 10032. E-mail:cmt2@columbia.edu.
S. A. Rabacchi's present address: Biogen, 14 Cambridge Center, Cambridge, MA 02142.
REFERENCES
- 1.Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol. 1995;128:919–927. doi: 10.1083/jcb.128.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 4.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 5.Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 7.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999a;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deveraux QL, Stennicke HR, Salvesen GS, Reed JC. Endogenous inhibitors of caspases. J Clin Immunol. 1999b;19:388–398. doi: 10.1023/a:1020502800208. [DOI] [PubMed] [Google Scholar]
- 10.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Duan H, Dixit VM. RAIDD is a new “death” adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 12.Farahani R, Fong WG, Korneluk RG, MacKenzie AE. Genomic organization and primary characterization of miap-3: the murine homologue of human X-linked IAP. Genomics. 1997;42:514–518. doi: 10.1006/geno.1997.4742. [DOI] [PubMed] [Google Scholar]
- 13.Farinelli SE, Park DS, Greene LA. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J Neurosci. 1996;16:2325–2334. doi: 10.1523/JNEUROSCI.16-07-02325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 15.Haviv R, Lindenboim L, Yuan J, Stein R. Need for caspase-2 in apoptosis of growth-factor-deprived PC12 cells. J Neurosci Res. 1998;52:491–497. doi: 10.1002/(SICI)1097-4547(19980601)52:5<491::AID-JNR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 16.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 17.Kuida K, Zheng TS, Na S, Kuan Na, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 18.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 21.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 24.Pooga M, Soomets U, Hallbrink M, Valkna A, Saar K, Rezaei K, Kahl U, Hao JX, Xu XJ, Wiesenfeld-Hallin Z, Hokfelt T, Bartfai T, Langel U. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol. 1998;16:857–861. doi: 10.1038/nbt0998-857. [DOI] [PubMed] [Google Scholar]
- 25.Salvesen GS. Programmed cell death and the caspases. APMIS. 1999;107:73–79. doi: 10.1111/j.1699-0463.1999.tb01528.x. [DOI] [PubMed] [Google Scholar]
- 26.Savitz SI, Kessler JA. Leukemia inhibitory factor requires concurrent p75LNTR signaling to induce apoptosis of cultured sympathetic neurons. J Neurosci. 2000;20:4198–4205. doi: 10.1523/JNEUROSCI.20-11-04198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA. Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J Neurosci. 1998;18:9204–9215. doi: 10.1523/JNEUROSCI.18-22-09204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem. 1998;273:7787–7790. doi: 10.1074/jbc.273.14.7787. [DOI] [PubMed] [Google Scholar]
- 30.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 31.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci. 1996;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Troy CM, Stefanis L, Greene LA, Shelanski ML. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 36.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- 37.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]