

Abstract
Abnormal involuntary movements and cognitive impairment represent the classical clinical symptoms of Huntington's disease (HD). This genetic disorder involves degeneration of striatal spiny neurons, but not striatal large cholinergic interneurons, and corresponds to a marked decrease in the activity of mitochondrial complex II [succinate dehydrogenase (SD)] in the brains of HD patients. Here we have examined the possibility that SD inhibitors exert their toxic action by increasing glutamatergic transmission. We report that SD inhibitors such as 3-nitroproprionic acid (3-NP), but not an inhibitor of mitochondrial complex I, produce a long-term potentiation of the NMDA-mediated synaptic excitation (3-NP-LTP) in striatal spiny neurons. In contrast, these inhibitors had no effect on excitatory synaptic transmission in striatal cholinergic interneurons and pyramidal cortical neurons. 3-NP-LTP involves increased intracellular calcium and activation of the mitogen-activated protein kinase extracellular signal-regulated kinase and is critically dependent on endogenous dopamine acting via D2 receptors, whereas it is negatively regulated by D1 receptors. Thus 3-NP-LTP might play a key role in the regional and cell type-specific neuronal death observed in HD.
Keywords: D2 dopamine receptors, Huntington's disease, striatum, succinate dehydrogenase, synaptic plasticity, excitotoxicity
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder that typically afflicts individuals in midlife (Price et al., 1998; Reddy et al., 1999). The symptoms include involuntary choreiform movements, psychiatric disturbances, and dementia. The primary brain region affected in HD is the striatum: neuronal loss and glial proliferation are seen at early stages of this disease, and they progress during the course of illness. In this pathological condition striatal spiny neurons are selectively lost, whereas large cholinergic interneurons are spared (Ferrante et al., 1985).
Recently, electrophysiological experiments have demonstrated that striatal spiny neurons show an enhanced sensitivity to the NMDA receptor activation in transgenic and knock-in mouse models of HD (Levine et al., 1999). Interestingly, these mice did not show differences in sensitivity to AMPA or kainate. These observations suggest a selective relationship between the genetic alteration underlying HD and NMDA receptor function. Accordingly, an abnormal NMDA-mediated synaptic function has been observed in hippocampal slices obtained from a YAC mouse model for HD (Hodgson et al., 1999).
One characteristic of HD is an expanded glutamine stretch in the protein encoded by the HD locus. However, mutated Huntingtin protein has yet to be directly linked to bioenergetic defects and excitotoxic mechanisms, two pathological events that seem to play a major role in HD (Browne et al., 1997). In fact, the corticostriatal projection represents one of the major glutamatergic pathways in the brain (Graybiel, 1995; Calabresi et al., 1996a), and an abnormal release of glutamate from this pathway seems to play a pathogenic role in HD (Greene and Greenamyre, 1996; Greene et al., 1998). Interestingly, an impaired complex II mitochondrial activity is a prominent metabolic alteration in HD (Gu et al., 1996). Accordingly, accidental or experimental systemic administration of the irreversible SD inhibitor 3-nitropropionic acid (3-NP) mimics the pathology of this genetic disorder in rats, nonhuman primates, and humans (Ludolph et al., 1991;Wullner et al., 1994; Palfi et al., 1996). Furthermore, the reversible complex II inhibitor malonic acid produces similar effects (Greene et al., 1993). Thus, enhanced glutamatergic transmission may trigger neurodegeneration in postsynaptic neurons, the energy metabolism of which is compromised because of impaired SD activity (Greene and Greenamyre, 1996).
Using corticostriatal brain slice preparations (Calabresi et al., 1998), we have studied the electrophysiological effects of the pharmacological blockade of SD by either 3-NP or methylmalonic acid (MMA), another rather selective inhibitor of SD (McLaughlin et al., 1998), on glutamatergic EPSPs. Here we demonstrate that endogenous dopamine (DA) is required to produce a long-term enhancement of NMDA-mediated glutamatergic transmission in striatal spiny neurons but not in striatal cholinergic interneurons or cortical pyramidal neurons. Our data suggest that a synaptic mechanism might possibly underlie cell type-specific neuronal vulnerability in HD.
MATERIALS AND METHODS
Electrophysiological experiments. Corticostriatal coronal slices 270 μm thick were prepared from adult Wistar rats and mice and kept in saline solution containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3) at 35°C gassed with 95% O2/5% CO2. The pH of the bathing solution was 7.4. Intracellular recording electrodes (30–60 MΩ) were filled with 2 m KCl. An AxoClamp 2B amplifier was used to monitor the cellular activity, which was displayed and stored by a digital system. In single-electrode voltage-clamp mode, the switching frequency was 3 kHz. The head-stage signal was monitored continuously on a separate oscilloscope. In some experiments, 100 mm BAPTA was added to the pipette solution to buffer intracellular calcium. In other experiments, 50 mm QX-314, a lidocaine derivative that blocks action potential discharge without affecting excitatory synaptic potentials (Calabresi et al., 1994), was used to block action potential discharge during current-induced membrane depolarization of the recorded neurons. Striatal spiny neurons and striatal cholinergic interneurons were distinguished by their electrophysiological, morphological, and immunohistochemical characteristics as described previously (Kawaguchi et al., 1995; Calabresi et al., 2000). The electrophysiological characteristics of prefrontal and frontal cortical neurons have been described previously (Siniscalchi et al., 1997).
For synaptic stimulation, bipolar electrodes were used and were located in the white matter between cortex and striatum, when recordings were performed from striatal neurons. Conversely, the stimulating electrode was placed close (0.5–3 mm) to the recording electrode in the direction of deep cortical layers when recordings were made from cortical pyramidal neurons (Calabresi et al., 1996b). The stimulation frequency was 0.1 Hz. Usually the intensity of stimulation to monitor EPSP amplitude was adjusted to produce synaptic potentials with amplitudes ranging between 5 and 10 mV. Quantitative data on EPSP modifications are means ± SEM expressed as percentage of EPSP amplitude at the onset of drug administration (t = 0). Student's t test (for paired and unpaired observations) was used to compare EPSP amplitude before and after pharmacological treatments.
Combined electrophysiological and optical recordings. A single slice was transferred into a recording chamber, mounted on the stage of an upright microscope (Axioskop FS, Zeiss) equipped with a 60×, 0.90 numerical aperture (NA) water immersion objective (LUMPlan FI, Olympus), and fully submerged in a continuously flowing Krebs' solution (33°C, 3 ml/min) gassed with 95% O2/5% CO2. The tip of sharp microelectrodes was filled with 1 mmbis-fura-2 (hexapotassium salt; Molecular Probes, The Netherlands) in 200 mm KCl. The shank of the electrode was backfilled with a 2 m KCl solution. After cell impalement, cells were loaded with bis-fura-2 by injecting 0.1–0.5 nA negative current through the recording pipette. Electrode resistance dropped from initial values of 120–150 to 35–45 MΩ. Fluorescence of bis-fura-2 was elicited by a 75 W Xenon lamp bandpass-filtered at 340 and 380 nm. Emission light was filtered (500 nm) and then detected by a charge-coupled device camera (Photonic Science). Pairs of 340 and 380 nm images were acquired at intervals of 3–6 sec. Analysis of the data was performed off-line [IonVision (ImproVision) and Microcal Origin 4.1 (Microcal Software), running on PowerMac 8100 and on a PC, respectively]. Pairs of 340 and 380 nm images were background subtracted where backgrounds were regions free of dye fluorescence, and ratio images were obtained. Levels of intracellular calcium concentration are expressed as ratio values (ranging from 0.4 to 0.8).
Drugs. Drugs were applied by dissolving them to the desired final concentration in the saline Ringer's solution and by switching the perfusion from control saline to drug-containing saline. In addition, slices were incubated in 30 μmbicuculline to avoid contamination of glutamatergic EPSPs by depolarizing GABA-mediated potentials. The pH of the Ringer's solution containing 3-NP or MMA was adjusted to 7.4 with NaOH. AMPA,d(−)-2-amino-5-phosphonopentanoic acid (APV), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), SCH 23390,l-sulpiride, and forskolin were from Tocris Cookson. Quinpirole, SKF 38393, and PD 98059 were from RBI (Natick, MA). BAPTA, bicuculline, nifedipine, 3-NP, NMDA, MMA, and tetrodotoxin (TTX) were from Sigma (St. Louis, MO), and UO126 was from Alexis. Intracellular application of 100 μm H89 for a period lasting at least 20 min after the beginning of the recordings did not significantly change either the amplitude or the time course of EPSPs, the membrane potential, or the input resistance of the neurons (p > 0.05 for all the measured parameters).
Preparation of 6-OHDA-denervated rats and D2 KO mice. To obtain unilateral nigrostriatal lesions, rats (anesthetized with 45 mg/kg body weight pentobarbitone, i.p.) were injected with 6-OHDA (8 μg/4 μl of saline containing 0.1% ascorbic acid) via a Hamilton syringe through a cannula inserted just rostral to the substantia nigra using stereotaxic coordinates (Paxinos and Watson, 1986). Twenty days later, the rats were tested with apomorphine (0.5 mg/kg, s.c.), and contralateral turns were recorded with automatic rotometers for 3 hr. Only those rats that consistently made at least 200 contralateral turns were used for the electrophysiological studies. Rats were anesthetized with diethyl ether, and brain dissection confirmed that the nigrostriatal pathway was lesioned. This was established by the observation of a >95% loss of DA neurons in the substantia nigra compacta, and the almost complete absence of DA terminals in the striatum. This was monitored by using a monoclonal antibody for tyrosine hydroxylase (Calabresi et al., 1993b; Centonze et al., 1999).
The generation of mice lacking D2 receptors has been reported previously (Baik et al., 1995). The intrinsic and synaptic membrane properties of neurons recorded from D2 lacking striatal slices were similar to those recorded from wild-type (WT) mice and control rats, as reported previously (Calabresi et al., 1997).
Biochemical experiments. Corticostriatal slices (270 μm) were prepared as described above. Slices were maintained in saline Ringer's solution and where appropriate preincubated for 15 min with 10 μm PD98059 in DMSO or 0.1% DMSO alone. Slices were stimulated with 100 μm 3-NP or 100 μm 3-NP plus 1 μml-sulpiride for 15 min, after which the striatum was rapidly homogenized using a microfuge tube pestle in 40 μl of extraction buffer (10 mm Tris-HCl, 1% Triton X-100, 50 mm NaCl, 50 mmNaF, 5 μm ZnCl2, 30 mm sodium pyrophosphate, 1 mm DTT, titrated to pH 7.05, with freshly added protease and phosphatase inhibitors) (Hipskind et al., 1994). Insoluble proteins were removed by centrifugation at 10,000 × g, 15 min, 4°C, and supernatant proteins were immediately denatured in 50 mm Tris-HCl, pH 6.8, 2% SDS, 2% glycerol, 1% 2-mercaptoethanol. Western blots containing 8 μg of extract protein per lane were immunodetected as described (Bowler et al., 1999) using antisera specific for the activated forms of mitogen-activated protein (MAP) kinase and ERKs 1 and 2 activated by phosphorylation on Thr 202 and Tyr 204 (Cell Signaling Technology), as well as control antisera directed against ERKs 1 and 2 (CST). The complexes were visualized with Renaissance ECL (NEN Life Science Products) and exposure to Kodak XAR-5 film after incubation with HRP-coupled secondary antibodies. The immunoblot results were confirmed by pull-down kinase assays using the same extracts and recombinant glutathioneS-transferase–Elk308–428.
RESULTS
Electrophysiology of 3-NP and MMA
In striatal spiny neurons, AMPA receptor-mediated EPSPs were elicited by electrical stimulation of adjacent cortex at a physiological concentration of external magnesium (1.2 mm) and in the presence of 50 μm APV, an NMDA receptor antagonist. Conversely, NMDA receptor-mediated EPSPs were evoked in the absence of external magnesium and in the presence of 10 μm CNQX, an AMPA receptor antagonist (Calabresi et al., 1996a). Interestingly, these NMDA-mediated potentials were greatly enhanced by a 20 min bath application of 100 μm 3-NP (n = 55; p < 0.001) or 300 μm MMA (n = 46;p < 0.001), whereas the AMPA-mediated EPSPs were unaffected (3-NP: n = 28, p > 0.05; MMA: n = 24, p > 0.05) (Fig.1a,b). The potentiation induced by 3-NP was dose dependent: in the presence of 10 μm 3-NP (n = 5), the NMDA-mediated EPSP was increased to 131 ± 12% of the control value, whereas in the presence of 30 and 300 μm3-NP (n = 4 for each concentration), this synaptic potential was 195 ± 24 and 246 ± 16%, respectively, of the control value (data not shown). The toxin-induced potentiation measured in spiny neurons persisted after the washout of both drugs (Fig.1a,b). In 10 striatal spiny neurons, the long-lasting recordings allowed us to follow the 3-NP-LTP for >60 min after toxin washout. In similar experimental conditions, we observed that the MMA-induced potentiation lasted for >60 min (n = 7). In contrast to the results obtained with spiny neurons, striatal large cholinergic interneurons (n = 7; p > 0.05 for both 3-NP and MMA) and cortical pyramidal neurons (n = 5, p > 0.05 for 3-NP; n = 3, p > 0.05 for MMA) showed no change of NMDA-mediated EPSP amplitude after SD inhibition (Fig.2). Similarly, AMPA-mediated EPSPs of both striatal cholinergic interneurons (n = 6,p > 0.05 for 3-NP; n = 5,p > 0.05 for MMA) and prefrontal and frontal cortical pyramidal cells (n = 3; p > 0.05 for both 3-NP and MMA) were unaffected by this treatment (Fig. 2).
Fig. 1.

Inhibition of mitochondrial complex II but not complex I activity induces LTP in striatal spiny neurons. In spiny neurons, 3-NP enhanced the amplitude of NMDA-mediated corticostriatal EPSPs (in 0 mm Mg plus CNQX), whereas AMPA-mediated potentials (in 1.2 mm Mg plus APV) were unaffected.Traces on the right are an average of four single EPSPs (a). The effect of 3-NP was mimicked by MMA (b). Conversely, rotenone failed to affect either component of excitatory synaptic transmission (c). Calibration in a also applies to b and c. In all the experiments, the resting membrane potential of the recorded cells (dotted lines) was constant and ranged between −84 and −86 mV.
Fig. 2.

Striatal cholinergic interneurons and cortical pyramidal neurons do not show 3-NP-LTP. In cholinergic interneurons, 3-NP did not enhance the amplitude of either NMDA- or AMPA-mediated corticostriatal EPSPs (a). Resting membrane potential = −60 mV. Similar lack of effects was observed in cortical pyramidal neurons (b). Resting membrane potential = −72 mV.
Glutamate NMDA receptors mediated the increase in EPSP amplitude, because APV fully suppressed the EPSPs after 3-NP-LTP induction in spiny neurons (n = 10) (Fig.3a). Prolonged application of 3-NP in the millimolar range depolarizes cultured neurons (Greene et al., 1998). In our experiments, the membrane potential of spiny neurons under control conditions (−85 ± 5 mV; n = 155) was unaffected by either 100 μm 3-NP (−86 ± 6 mV; n = 97; p > 0.05) or 300 μm MMA (−85 ± 5 mV; n = 29; p > 0.05). These toxins also did not affect the resting input resistance (control: 39 ± 8 MΩ, n= 95; 3-NP: 40 ± 9 MΩ, n = 55; MMA: 38 ± 8 MΩ, n = 25) (p > 0.05). Similarly, resting membrane potential (control: −60 ± 4 mV,n = 14; 3-NP: −61 ± 5 mV, n = 10; MMA: −58 ± 4 mV, n = 7; p > 0.05) and apparent input resistance (control: 158 ± 42 MΩ,n = 14; 3-NP: 163 ± 38 MΩ, n = 9; MMA: 153 ± 40 MΩ, n = 5; p> 0.05) of large cholinergic interneurons were not significantly altered by these toxins. 3-NP and MMA also failed to affect the resting membrane potential (control: −77 ± 4 mV, n = 14; 3-NP: −77 ± 3 mV, n = 10; MMA: −78 ± 5 mV, n = 7; p > 0.05), and input resistance (control: 88 ± 38 MΩ, n = 14; 3-NP: 89 ± 40 MΩ, n = 9; MMA: 87 ± 34 MΩ,n = 5; p > 0.05) of prefrontal and frontal cortical pyramidal neurons (Siniscalchi et al., 1997).
Fig. 3.
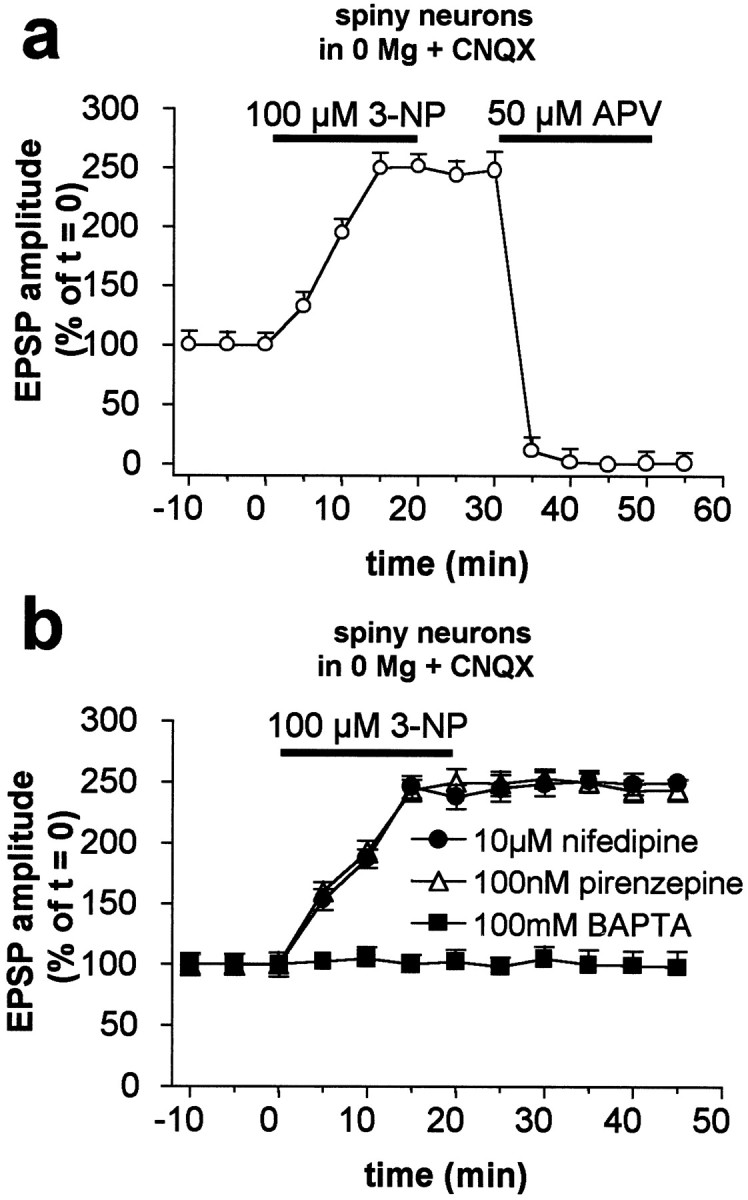
3-NP-LTP is caused by enhanced NMDA receptor-mediated synaptic transmission and requires intracellular calcium elevation. Cortically evoked EPSPs after the induction of 3-NP-LTP in spiny neurons were fully suppressed by the NMDA receptor antagonist APV (a). Intracellular injection of the calcium chelator BAPTA fully prevented 3-NP-LTP. Conversely, this form of synaptic plasticity was prevented neither by 10 μm nifedipine nor by 100 nm pirenzepine (b).
Rotenone does not induce LTP
To test whether the potentiation of the NMDA-mediated component of excitatory transmission recorded from spiny neurons was selectively induced by pharmacological inhibition of mitochondrial complex II activity or could also be achieved by the inhibition of other mitochondrial complexes, we incubated the slices in the presence of rotenone, a selective inhibitor of mitochondrial complex I (Betarbet et al., 2000; Luetjens et al., 2000). Bath application (20 min) of 1 μm rotenone failed to affect either the NMDA- or the AMPA-mediated striatal EPSPs recorded from spiny neurons (n = 8 for both experimental conditions) (Fig.1c). We also tested the effects of a higher concentration of this toxin: 3 μm rotenone produced a slow and irreversible membrane depolarization of striatal spiny neurons, which was not coupled, however, to a significant increase in NMDA-mediated EPSP amplitude (n = 5; data not shown).
Effects of intracellular BAPTA and nifedipine on 3-NP-LTP
The integrity of mitochondrial function is crucial for cytosolic calcium homeostasis (Berridge, 1998). Thus, disruption of mitochondrial activity by SD inhibitors results in a disregulation of calcium buffering mechanisms (Murphy et al., 1999). To test whether increased intracellular calcium is critical for 3-NP-LTP, as has been found for physiological striatal synaptic plasticity (Calabresi et al., 1993a), we used recording electrodes filled with the calcium chelator BAPTA (100 mm). This treatment completely prevented 3-NP-LTP (n = 10; p > 0.05) (Fig.3b). To test the possible involvement of high-voltage activated L-type calcium channels, which have been shown to contribute to the potentiation of NMDA responses induced in spiny neurons by D1 receptor activation (Cepeda et al., 1998), we tested the ability of nifedipine (10 μm, 10 min bath application;n = 6) to influence 3-NP-LTP. As shown in Figure3b, this blocker failed to affect the amplitude and duration of this form of synaptic plasticity. Moreover, 10 μm nifedipine also failed to alter the amplitude and duration of control striatal EPSPs (n = 6; data not shown). Conversely, this concentration of nifedipine significantly reduced the duration of calcium-dependent plateau potentials recorded after blockade of potassium channels from striatal spiny neurons (Stefani et al., 1995). We also tested the possibility that acetylcholine, an endogenous neurotransmitter that increases intracellular calcium via M1 muscarinic receptors and favors striatal post-tetanic LTP (Calabresi et al., 1999), might also be involved in the formation of 3-NP-LTP. This possibility is unlikely, because 100 nm pirenzepine, an M1-like receptor antagonist, failed to affect 3-NP-LTP (n = 4; p > 0.05) (Fig. 3b).
3-NP-LTP is expressed only when the neurons are in the “up” state
Membrane potential of striatal spiny neurons recorded in intact animals oscillates between a hyperpolarized “down” state and a more depolarized “up” state, the latter being determined by converging excitatory synaptic inputs from the cortex (Stern et al., 1998). During the “up” state, the membrane potential of spiny neurons reaches a level (approximately −55 mV) capable of relieving the voltage-dependent magnesium blockade of the NMDA receptor channel. In the presence of a physiological concentration of magnesium (1.2 mm), membrane depolarization to −55 mV produced by intracellular injection of positive current reveals a component of synaptic potential that undergoes a long-term enhancement after SD inhibition (n = 10) (Fig.4a). In these experiments, 50 mm QX-314, a lidocaine derivative that blocks voltage-dependent sodium channels without affecting excitatory synaptic potentials (Calabresi et al., 1994), was used to block action potential discharge during current-induced membrane depolarization of the recorded neurons. APV (30 μm) significantly reduced the duration of EPSPs recorded at −55 mV and fully prevented the potentiation induced by 3-NP (n = 5) (Fig.4b). Conversely, the EPSPs recorded in the same neurons at resting membrane potential (down state, −85 mV) did not show 3-NP-induced potentiation and were insensitive to NMDA receptor antagonism. Thus, mitochondrial inhibition and prolonged cortical discharge, which lead to the generation of the up state of the neuron, may also cooperate to induce 3-NP-LTP in vivo.
Fig. 4.

3-NP-LTP is expressed when striatal neurons are in the up state. In the presence of physiological concentrations of external magnesium, 3-NP enhanced EPSPs recorded from neurons depolarized to −55 mV by intracellular injection of positive current (“up” state), but not EPSPs recorded at resting membrane potentials (−85 mV; “down” state) (a). Note that the data represented ina were obtained by shifting the membrane potential of each single neuron from the up to the down state and vice versa throughout the duration of 10 electrophysiological experiments. This experimental protocol was followed to test whether the induction of 3-NP-LTP on the up state EPSP also influenced the AMPA component of the EPSP as detected in the down state. The NMDA receptor antagonist APV fully prevented the formation of 3-NP-LTP observed in the up state (b). Traces in the bottom part represent examples of EPSPs recorded from two different spiny neurons in the absence (a) and presence (b) of APV at different membrane potential levels.
Effects of 3-NP and MMA on postsynaptic responses induced by application of AMPA and NMDA
We also investigated whether application of SD inhibitors could affect postsynaptic responses induced by application of AMPA and NMDA in spiny neurons and cholinergic interneurons. As shown previously (Calabresi et al., 1998), brief (20–60 sec) bath applications of these agonists induced inward currents in both of these neuronal subtypes. In the presence of 1 μm TTX, bath application of either 3-NP (30 μm; n = 9) or MMA (300 μm; n = 8) enhanced the inward currents induced by NMDA (p < 0.001) but not those caused by AMPA (p > 0.05) in spiny neurons (Fig.5a,b). Conversely, 3-NP (n = 6; p > 0.05) (Fig.5c) and MMA (n = 6; p> 0.05; data not shown) failed to alter agonist-induced inward currents recorded from striatal cholinergic interneurons.
Fig. 5.
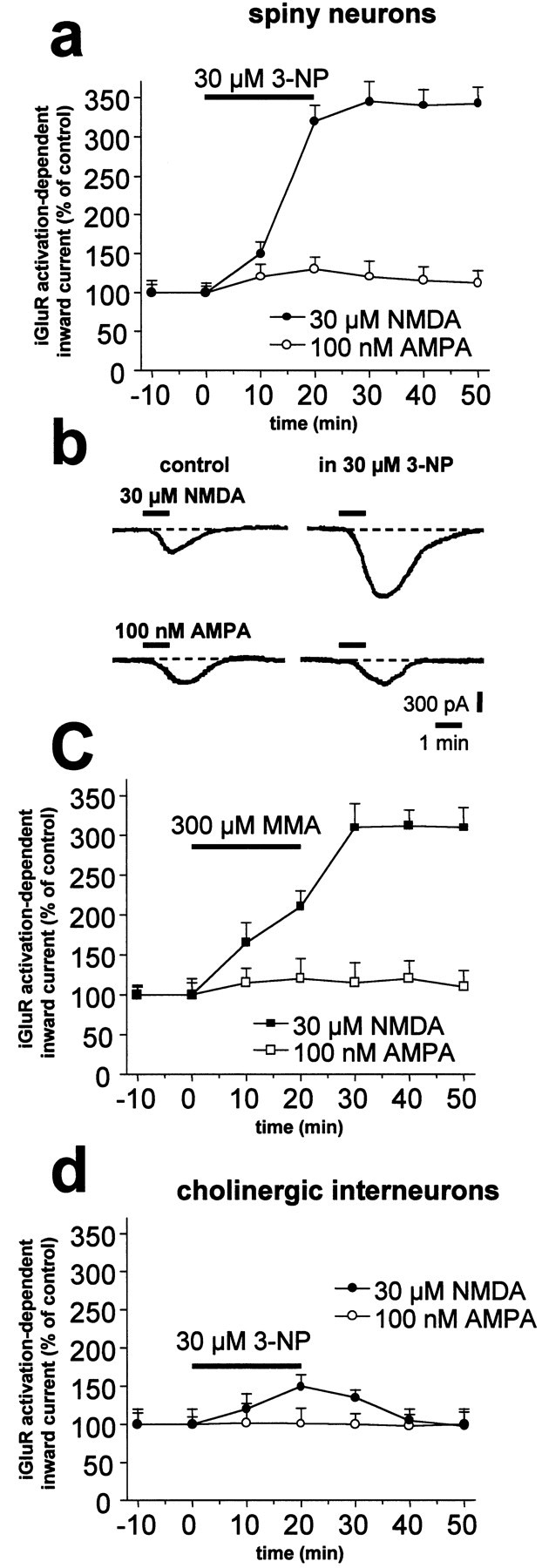
SD inhibitors enhance inward currents induced by bath application of NMDA in striatal spiny neurons but not in cholinergic interneurons. In spiny neurons, bath application of 3-NP significantly enhanced the inward current evoked by application of NMDA but not of AMPA (a). A single experiment obtained from a striatal spiny neuron voltage clamped at −80 mV is shown (b). The effect of 3-NP was mimicked by MMA (c). Application of 3-NP failed to affect NMDA- and AMPA-mediated currents in striatal cholinergic interneurons (d).
Microfluorimetric measurements of intracellular calcium changes after 3-NP
In the presence of TTX (1 μm), under the control condition, bath application of NMDA (30 μm, 1 min) elicited a membrane depolarization coupled to a transient and reversible elevation in intracellular calcium (Fig.6). After pretreatment with 100 μm 3-NP (20 min), no significant change in resting membrane potential was observed, whereas the intracellular calcium level was moderately but significantly increased (n = 7; p < 0.05) (Fig. 6). In these conditions, NMDA evoked a marked membrane depolarization up to threefold higher than in controls that slowly returned to resting values. After the onset of this depolarization, intracellular calcium concentrations increased steadily, reaching a peak at the washout of NMDA (n = 7; p < 0.001). After a rapid decrease from this peak, however, the intracellular calcium level remained elevated and generally returned to basal levels only 10–15 min after the wash of the agonist (Fig. 6).
Fig. 6.

3-NP enhances basal and NMDA-induced intracellular calcium concentration in striatal spiny neurons. The histogram shows the mean percentage of increase ± SEM of intracellular calcium (expressed as ratio values) in response to NMDA, 3-NP alone, and NMDA in the presence of 3-NP (a). Thetraces in the top panel show the membrane depolarization induced by NMDA (30 μm) in the control condition (left) and after 20 min of 3-NP treatment (right). The bottom panel shows the simultaneous measurements of intracellular calcium levels in the same neuron (b). Resting membrane potential = −84 mV. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's ttest for paired observations).
Role of endogenous dopamine
The pathologic alterations found in HD, as well as those induced by inhibitors of SD, are selective for the striatum (Ferrante et al., 1985). The reason for this regional specificity is unknown. A unique feature of the striatum, compared with other brain areas, is the massive dopaminergic innervation arising from the substantia nigra (Graybiel, 1995). Endogenous DA and energy metabolism impairment caused by mitochondrial alteration might cooperatively produce the neuronal damage induced by 3-NP in the striatum by favoring 3-NP-LTP induction. Accordingly, we found that 3-NP-LTP was absent in slices obtained from rats in which the striatum was DA-depleted by homolateral nigral injection of 6-OHDA (n = 15). Conversely, slices obtained from the contralateral intact side expressed 3-NP-LTP (Fig.7a) (n = 11). In DA-denervated slices, 3-NP-LTP was restored by previous application (15 min) of 3 μm quinpirole (n= 8), a D2-like DA receptor agonist, but not by 10 μm SKF 38393 (n = 9), a D1-like DA receptor agonist (Fig. 7b). These results suggested that activation of D2- but not D1-like DA receptors is required for the induction of 3-NP-LTP. It is worth noting that DA denervation did not affect the resting membrane potential (−86 ± 5 mV;n = 12) and the input resistance (38 ± 8 MΩ;n = 12) of striatal spiny neurons.
Fig. 7.

Endogenous DA controls the formation of 3-NP-LTP through the activation of D2 DA receptors. 3-NP-LTP was abolished in slices obtained from DA-depleted striata, whereas it was normally expressed in tissue obtained from intact contralateral striata (a). Quinpirole, a D2-like DA receptor agonist, but not SKF 38393, a D1-like DA receptor agonist, restored 3-NP-LTP in DA-denervated neurons (b). 3-NP-LTP was blocked by l-sulpiride, a D2-like DA receptor antagonist, but not by SCH 23390, a D1-like DA receptor antagonist (c). 3-NP-LTP was also prevented by forskolin, an adenylyl cyclase activator, and by SKF 38393, a D1-like DA receptor agonist (d). Intracellular application of the PKA inhibitor H89 partially prevented the inhibitory effects ofl-sulpiride and forskolin on 3-NP-LTP formation (e). 3-NP-LTP was absent in striatal neurons recorded from mice lacking D2 receptors but not in WT animals. Intracellular injection of H89 partially restored 3-NP-LTP in mice lacking D2 (f). Note that in this latter graph data obtained from WT mice were significantly different from both D2 knock-out (KO) mice (p < 0.01) and D2 KO mice plus H89 (p < 0.001). Moreover, a statistical significance (p < 0.001) was detected between D2 KO mice and D2 KO mice plus intracellular H89.
To further evaluate the potential roles of D1- and D2-like receptors in 3-NP-LTP induction, slices prepared from control rats were incubated in 1 μml-sulpiride (n = 27;p > 0.05), a D2-like DA receptor antagonist, or 10 μm SCH 23390 (n = 25;p < 0.001), a D1-like DA receptor antagonist.l-sulpiride, but not SCH 23390, fully prevented 3-NP-LTP (Fig. 7c). Notably,l-sulpiride was unable to block 3-NP-LTP when applied after its induction (n = 8; p< 0.001), demonstrating that striatal DA is required for the induction but not for the maintenance of this phenomenon (data not shown). In the presence of endogenous DA, the blockade of D2-like DA receptors results in an increased activity of adenylyl cyclase. A similar effect is achieved by the stimulation of D1-like receptors by exogenous agonists (Jaber et al., 1996; Vallone et al., 2000). Accordingly, the inhibitory effect of l-sulpiride on 3-NP-LTP formation was mimicked by incubation of the slices in 30 μmforskolin (n = 14), an activator of adenylyl cyclase, or 10 μm SKF 38393 (n = 12) (Fig. 7d), indicating that high cAMP levels prevent induction of 3-NP-LTP and that the stimulation of D1-like receptors provides a negative modulation of the D2-like receptor-mediated 3-NP-LTP formation. Neither forskolin (n = 3) nor SKF 38393 (n = 4) affected 3-NP-LTP when applied after its induction (data not shown). Because the available pharmacological tools do not distinguish between the subtypes composing the D2-like DA receptor family (D2, D3, and D4), we used mice lacking D2 DA receptors (Baik et al., 1995). Corticostriatal slices prepared from these mice failed to show 3-NP-LTP (n = 6), whereas slices from WT animals showed 3-NP-LTP (n = 7) (Fig.7f), thereby confirming an essential role for D2 receptors in this process. When protein kinase A (PKA) activity was blocked with H89 by previous intracellular (100 μm; n = 5) (Fig. 7e) or bath application (10 μm; n = 5) (data not shown), l-sulpiride, forskolin (n = 8) (Fig. 7e), or D2 receptor disruption (n = 5) (Fig. 7f) only partially prevented 3-NP-induced 3-NP-LTP. Thus 3-NP-LTP induction apparently requires the D2-receptor-mediated inhibition of PKA at the postsynaptic site.
Role of MAP kinase ERK
D2 receptors and intracellular calcium interact to regulate the mitogen-activated protein kinase ERK in slices (Yan et al., 1999). All of these factors also participate in the generation of 3-NP-LTP. In addition, like 3-NP-LTP induction, ERK activity is controlled by glutamate acting through NMDA but not AMPA receptors (Kurino et al., 1995). ERK is an important intermediate in physiological synaptic plasticity (Kornhauser and Greenberg, 1997; Roberson et al., 1999) and excitotoxicity (Xia et al., 1996). We therefore evaluated the possibility that ERK might be involved in mediating the effect of D2 agonists in 3-NP-LTP. Indeed, treatment of corticostriatal slices with 3-NP induced ERK phosphorylation, a measure of its activation (Fig.8a). This was fully abolished by preincubation with 10 μm PD 98059, an inhibitor of the ERK-activating kinase MEK. Using the same conditions, this pharmacological agent also prevented the induction of 3-NP-LTP (n = 15; p > 0.05) (Fig.8b), thereby implicating the ERK cascade in this phenomenon. Accordingly, similar results were obtained by incubating the slices in the presence of 30 μm of UO126 (n = 5; data not shown), another specific inhibitor of MEK. Furthermore, we found that doses ofl-sulpiride able to block 3-NP-LTP (1 μm) significantly reduced the phosphorylation of ERK produced by 3-NP treatment (Fig. 8a).
Fig. 8.

3-NP-LTP induction is blocked by the ERK cascade inhibitor PD98059. Corticostriatal slices were treated with 100 μm 3-NP, with 3-NP plus MEK inhibitor PD 98059 (PD; 10 μm), or 3-NP together with 1 μml-sulpiride as indicated in Materials and Methods. After protein extraction, ERK activation was analyzed by immunoblotting with antibodies specific for phosphorylated, activated ERKs 1 and 2. Equal levels of ERK were confirmed by immunoblotting with anti-ERK1/2 (a). Inhibition of MAP kinase activity by 10 μm PD 98059 prevented 3-NP-induced 3-NP-LTP (b).
DISCUSSION
General findings
The present study provides two major novel findings. First, inhibition of mitochondrial complex II but not mitochondrial complex I activity results in a selective long-term enhancement of NMDA-mediated synaptic transmission in striatal spiny neurons. Second, endogenous DA is required for this form of synaptic plasticity via the activation of D2 DA receptors.
Interestingly, the non-NMDA-mediated component of glutamatergic EPSPs is not affected by SD inhibitors. This observation indicates that 3-NP-LTP is expressed at the postsynaptic site. In fact, a presynaptic site of expression of this phenomenon (i.e., an increased glutamate release after SD inhibition) would be detected as an increase of both NMDA and non-NMDA components of striatal EPSPs. Accordingly, we found that in spiny neurons, but not in cholinergic interneurons, both SD inhibitors selectively enhanced inward currents induced by application of NMDA, whereas responses induced by AMPA were unchanged. Because these experiments were performed in the presence of TTX, we assume that the facilitatory effect of NMDA responses caused by SD inhibitors is exerted at a postsynaptic site of action. However, a definitive demonstration that 3-NP does not influence excitatory transmission by enhancing glutamate release will require the analysis of spontaneous miniature potentials.
We have also evaluated the sensitivity to 3-NP of frontal and prefrontal pyramidal cortical neurons. These neurons have been shown to be sensitive to mitochondrial activity inhibition (Ohgoh et al., 2000), but at least in our experimental conditions, they did not show significant electrophysiological changes after application of 3-NP. It is possible that higher concentrations, or longer periods of exposure, or both, are required to affect the electrophysiological parameters in these neuronal subtypes.
Moreover, we have also tested whether the inhibition of mitochondrial complex I by rotenone, which has been reported to interfere with dopamine cell survival (Betarbet et al., 2000), could also mimic the 3-NP-LTP in striatal spiny neurons. We found that concentrations of rotenone able to inhibit mitochondrial complex I activity in vitro (Luetjens et al., 2000) do not induce potentiation of NMDA-mediated responses in striatal spiny neurons. This lack of effect of rotenone suggests that the enhancement of NMDA-mediated neurotransmission in the striatum is a pathophysiological response selectively triggered by inhibition of SD activity.
Role of intracellular calcium and glutamate receptors
An augmentation of postsynaptic intracellular calcium is a critical requirement for 3-NP-LTP, as demonstrated by the fact that blockade of this form of synaptic plasticity was achieved by buffering intracellular calcium. This observation is in agreement with the view that intact mitochondrial activity is of crucial importance for the maintenance of intracellular calcium homeostasis (Greene and Greenamyre, 1996; Murphy et al., 1999). Moreover, the capability of intracellular BAPTA to block 3-NP-LTP further supports the concept that, at least in the induction phase, 3-NP-LTP requires postsynaptic changes in striatal spiny neurons.
In the present study, by using combined electrophysiological recordings and microfluorimetric measurements of intracellular calcium from the same single striatal spiny neuron, we obtained two major results. First, although 3-NP did not alter somatic membrane potential of spiny neurons, it significantly enhanced basal intracellular calcium levels. Second, a dramatic and long-lasting enhancement of intracellular calcium levels was triggered by NMDA application in the presence of 3-NP. This large, lasting increase in intracellular calcium levels after combined inhibition of mitochondrial complex II and activation of NMDA receptors might initiate the cascade of metabolic events leading to selective neuronal death in the striatum.
It has recently been reported that transgenic mice expressing an HD mutation are resistant to quinolinic acid-induced striatal excitotoxicity (Hansson et al., 1999). This observation is in contrast to the excitotoxic hypothesis of HD pathology that our data support. Interestingly, in agreement with our findings, Levine et al. (1999)recently found an enhanced sensitivity to NMDA receptor activation in two animal models of HD.
Another novel finding of the present study is that SD inhibitors generate 3-NP-LTP only when the spiny neurons are in the up state. This experimental evidence could have profound pathophysiological implications, because it suggests that any event leading to continuous membrane depolarization, such as repetitive cortical activation, ischemia, or additional mitochondrial impairment, might facilitate 3-NP-LTP induction via the generation of a vicious cycle of continual reinforcement.
Our understanding of the genetics of HD has advanced substantially in recent years (Reddy et al., 1999; Yamamoto et al., 2000). The discovery of the link between an expanded polyglutamine repeat in the IT15 gene and the synthesis of an altered huntingtin protein represented a key step toward the definition of the molecular mechanisms underlying the pathogenesis of this disorder. Huntingtin is diffusely expressed in the brain and inhibits glyceraldehyde-3-phosphate dehydrogenase, a key enzyme in energy metabolism (Burke et al., 1996). Accordingly, brains of HD patients show decreased striatal glucose metabolism, abnormalities in mitochondrial electron transport chain, and increased concentrations of lactate (Gu et al., 1996; Browne et al., 1997). Nevertheless, it remained unclear how the inhibition of mitochondrial activity that affects the whole brain leads to the regional and cell type-specific vulnerability in HD. Our experiments provide novel insights into this process.
Is endogenous dopamine involved in striatal neurodegeneration and 3-NP-LTP?
Oxidative stress impairs DA uptake (Berman et al., 1996), which can account for increased levels of DA in the cerebrospinal fluid of HD patients (Garrett and Soares-da-Silva, 1992). Thus, elevated concentrations of endogenous DA, combined with mitochondrial impairment, might lead to increased sensitivity to excitotoxicity, resulting in preferential cell death in the striatum. It is also possible that DA metabolites, rather than DA itself, might contribute to cell death in animal models of HD. DA uptake is reduced in striatal synaptosomes exposed to mitochondrial poisons, and striatal DA levels are increased in rodents during infusion of the SD inhibitor malonate. Furthermore, animals treated with 3-NP for 5 d showed elevated levels of DOPAC but not DA, suggesting increased turnover of DA by monoamino-oxidases (for review, see Jakel and Maragos, 2000).
Our data suggest that the striatum is selectively vulnerable in HD because this structure is the main recipient of fibers releasing DA, a neurotransmitter that is critical for the formation of 3-NP-LTP. In support of this possibility, DA antagonists and DA denervation limit striatal damage after chronic administration of 3-NP and MMA (Maragos et al., 1998; Reynolds et al., 1998; Jakel and Maragos, 2000).
We found that D1 and D2 classes of DA receptors, which act in opposition to each other, are involved in the generation and modulation of 3-NP-LTP. Several experimental findings support this hypothesis. First, 3-NP-LTP was absent in 6-OHDA-treated rats and was restored by the administration of a D2 but not a D1 DA receptor agonist. Second, blockade of D2 but not of D1 DA receptors prevented the expression of 3-NP-LTP. Interestingly, 3-NP-LTP could also be inhibited by forskolin, an activator of adenylyl cyclase, or by the D1 receptor agonist SKF 38393, suggesting that inactivation of PKA is a crucial requirement for the generation of this form of synaptic plasticity. Accordingly, intracellular and bath application of H89, a PKA inhibitor, reverses the inhibition of 3-NP-LTP induced by l-sulpiride, SKF 38393, and forskolin. Third, 3-NP-LTP was absent in mice lacking D2 DA receptors, confirming that D2 rather than other D2-like DA receptor subfamilies (D3 and D4) are involved in this form of plasticity.
The exact mechanism by which D2 receptor activation produces 3-NP-LTP is still an open question. In Figure 9, we propose a possible model to explain the interaction between intracellular calcium, D2 DA receptors, PKA, and ERK (see below) in the generation of the 3-NP-LTP. It is possible that D2 receptors may favor release of calcium from intracellular stores (Vallar et al., 1990;Nemethy et al., 1998). Accordingly, it has recently been shown that activation of D2 DA receptors is able, via a phospholipase C-dependent mechanism, to mobilize intracellular calcium stores also in striatal spiny neurons (Hernandez-Lopez et al., 2000). This observation might well explain previous findings showing that activation of D2 receptors increases striatal calcineurin and ERK activity via calcium-dependent mechanisms (Nishi et al., 1997, Yan et al., 1999). Interestingly, the activation of this novel phospholipase C-dependent intracellular pathway by D2 receptors leads to a reduction of L-type calcium currents (Hernandez-Lopez et al., 2000). These data agree with our observation that D2- and calcium-dependent 3-NP-LTP are independent of L-type calcium channel activation. The D2-mediated mobilization of intracellular calcium might further amplify the augmented calcium levels induced by 3-NP-induced impairment of mitochondrial activity and NMDA receptor activation. Moreover, in this scenario, D2 receptor activation blocks PKA activity and thereby might suppress PKA-dependent inhibition of Raf-1, leading to activation of the MAP/ERK cascade. It should be stressed, however, that the interaction between PKA and the ERK pathway is complex and remains controversial (for review, seeSweatt, 2001). Thus, it is possible that an alternative pathway, not examined in this study, might account for ERK activation in 3-NP-LTP. Nevertheless, the intracellular cascade underlying 3-NP-LTP seems to be specific for DA via D2 receptor activation. In fact, the blockade of M1-like muscarinic receptors, which also increases intracellular calcium and favors striatal post-tetanic LTP (Calabresi et al., 1999), fails to alter 3-NP-LTP.
Fig. 9.

Hypothetical model to account for the receptor and post-receptor mechanisms underlying 3-NP-LTP in striatal spiny neurons. D2 and D1 receptors exert opposing actions on adenylyl cyclase (AC) activity. The D2 receptor-mediated reduction of cAMP levels leads to the inhibition of protein kinase A (PKA) activity. This suppresses PKA-dependent inhibition of Raf-1 and consequently activates the MEK/ERK cascade. MEK activity is also stimulated by increased intracellular calcium levels ([Ca2+]i). The increase in [Ca2+]i is secondary to NMDA receptor stimulation, impairment of mitochondrial buffering properties caused by SD inhibition, and D2 receptor-mediated stimulation of phospholipase C (PLC). The final result of this cascade of biochemical events is the ERK-dependent induction of nuclear events leading to protein synthesis and altered NMDA receptor-channel complex function.
Is 3-NP-LTP a synaptic phenomenon specific for neurons vulnerable in HD?
The issue of cellular specificity is particularly important in HD. We have shown that 3-NP-LTP is expressed in spiny striatal neurons but not in cholinergic interneurons. Although this represents only a partial step toward demonstrating true “striatal” specificity, we point out that in our experimental system, 3-NP failed to enhance excitatory transmission in frontal and prefrontal pyramidal cortical neurons.
It is well known that in HD the striatal enkephalinergic neurons of the “indirect” pathway are much more sensitive than are striatal substance P neurons of the “direct” pathway (Antonini et al., 1998;Mitchell et al., 1999). This corresponds to their expression of D2 and D1 receptors, respectively. In the current view of striatal organization, the D2 receptor is expressed only in a part of spiny neurons. Nevertheless, it is worth noting that striatal neurons projecting to the internal segment of the globus pallidus and substantia nigra also can send axon collaterals to the external segment of the globus pallidus, suggesting that direct and indirect pathways are not truly segregated (Kawaguchi et al., 1990). In addition, although D1 and D2 receptors have been reported to be largely segregated, single-cell RT-PCR experiments have also demonstrated a substantial coexpression of these receptors (Surmeier et al., 1996;Nicola et al., 2000). Further support of the hypothesis that D1 and D2 receptors are coexpressed in striatal spiny neurons arises from a recent anatomical and physiological study showing that both D1 and D2 receptors are coexpressed in cultured striatal spiny neurons (Aizman et al., 2000). Thus, it is not surprising that we did not observe a heterogeneous response of striatal spiny neurons to 3-NP. It is possible, in fact, that although low levels of D2 receptors are not detectable by morphological analysis, they could play a significant functional role.
Role of the MAP kinase ERK in 3-NP-LTP
Pathological events like energy metabolism impairment and excitotoxicity have been found to activate ERK in central neurons (Ghosh and Greenberg, 1995; Sugino et al., 2000). We found that 3-NP induced a significant activation of ERK, as measured by its phosphorylation, and that PD 98059 and UO126, two inhibitors of the ERK-activating kinase MEK, block 3-NP-LTP. One would expect that the ERK cascade exerts its effect selectively on late phases of synaptic plasticity by altering the pattern of gene expression. However, we found that the pharmacological inhibitors of the ERK cascade also blocked early stages of 3-NP-LTP, thereby suggesting a role early in this phenomenon. In agreement with this, PD 98059 completely prevents LTP in the dentate gyrus and LTD in the prefrontal cortex, indicating that the activation of the MAP kinase ERK cascade can play a role in the induction phases of different forms of synaptic plasticity in several brain areas (Coogan et al., 1999; Otani et al., 1999; Sweatt, 2001).
Conclusions
Future studies investigating the possible modulation of excitatory transmission in slices obtained from rats that have been chronically intoxicated with systemic 3-NP injection should be performed to compare the acute effects of this toxin with those produced by the long-term inhibition of SD activity.
Novel approaches in the therapy of HD have been proposed on the basis of mouse genetic models (Ona et al., 1999). Our data suggest that drugs interfering with the mechanisms underlying the induction of 3-NP-LTP might represent an additional therapeutic strategy to treat HD in its presymptomatic phase.
Footnotes
This work was supported by the following grants: BIOMED Grant BMH4-97-2215 to P.C. and R.A.H.; Cofin-Ministero dell'Universitáe della Ricerca Scientifica e Tecnologica (MURST) and Telethon (Grant E.729) to P.C. and G.B.; MURST–Consiglio Nazionale delle Ricerche (legge 95/95) to G.B.; and Grants MH-40899 and DA-10044 to P.G. We thank Dr. Karima Chergui for helpful discussion and critical reading of this manuscript. We also thank Massimo Tolu for technical assistance.
Correspondence should be addressed to Prof. Paolo Calabresi, Clinica Neurologica, Dipartimento di Neuroscienze, Università di Tor Vergata, Via di Tor Vergata 135, Rome 00133, Italy. E-mail:calabre@uniroma2.it.
REFERENCES
- 1.Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- 2.Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington's disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253–255. doi: 10.1002/ana.410430216. [DOI] [PubMed] [Google Scholar]
- 3.Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice like dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 4.Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem. 1996;67:593–600. doi: 10.1046/j.1471-4159.1996.67020593.x. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 6.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 7.Bowler WB, Dixon CJ, Halleux C, Maier R, Bilbe G, Fraser WD, Gallagher JA, Hipskind RA. Signaling in human osteoblasts by extracellular nucleotides. Their weak induction of the c-fos proto-oncogene via Ca2+ mobilization is strongly potentiated by a parathyroid hormone/cAMP-dependent protein kinase pathway independently of mitogen-activated protein kinase. J Biol Chem. 1999;274:14315–14324. doi: 10.1074/jbc.274.20.14315. [DOI] [PubMed] [Google Scholar]
- 8.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 9.Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ. Huntington and DRPLA proteins selectively interact with the enzyme GADPH. Nat Med. 1996;2:347–350. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Lithium treatment blocks long-term synaptic depression in the striatum. Neuron. 1993a;10:955–962. doi: 10.1016/0896-6273(93)90210-i. [DOI] [PubMed] [Google Scholar]
- 11.Calabresi P, Mercuri NB, Sancesario G, Bernardi G. Electrophysiology of dopamine denervated striatal neurons. Implications for Parkinson's disease. Brain. 1993b;116:433–452. [PubMed] [Google Scholar]
- 12.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Post-receptor mechanisms underlying striatal long-term depression. J Neurosci. 1994;14:4871–4881. doi: 10.1523/JNEUROSCI.14-08-04871.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calabresi P, Pisani A, Mercuri NB, Bernardi G. The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci. 1996a;19:19–24. doi: 10.1016/0166-2236(96)81862-5. [DOI] [PubMed] [Google Scholar]
- 14.Calabresi P, Siniscalchi A, Pisani A, Stefani A, Mercuri NB, Bernardi G. A field potential analysis on the effects of lamotrigine, GP 47779, and felbamate in neocortical slices. Neurology. 1996b;47:557–562. doi: 10.1212/wnl.47.2.557. [DOI] [PubMed] [Google Scholar]
- 15.Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci. 1997;17:4536–4544. doi: 10.1523/JNEUROSCI.17-12-04536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calabresi P, Centonze D, Pisani A, Sancesario G, Gubellini P, Marfia GA, Bernardi G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- 17.Calabresi P, Centonze D, Gubellini P, Bernardi G. Activation of M1-like muscarinic receptors is required for the induction of corticostriatal LTP. Neuropharmacology. 1999;38:323–326. doi: 10.1016/s0028-3908(98)00199-3. [DOI] [PubMed] [Google Scholar]
- 18.Calabresi P, Centonze D, Gubellini P, Marfia GA, Pisani A, Sancesario G, Bernardi G. Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog Neurobiol. 2000;61:231–265. doi: 10.1016/s0301-0082(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 19.Centonze D, Gubellini P, Picconi B, Calabresi P, Giacomini P, Bernardi G. Unilateral dopamine denervation blocks corticostriatal LTP. J Neurophysiol. 1999;82:3575–3579. doi: 10.1152/jn.1999.82.6.3575. [DOI] [PubMed] [Google Scholar]
- 20.Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- 21.Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 22.Ferrante RJ, Kowall NJ, Beal MF, Richardson EP, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;320:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 23.Garrett MC, Soares-da-Silva P. Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in Huntington's disease: evidence for an overactive dopaminergic brain transmission. J Neurochem. 1992;58:101–106. doi: 10.1111/j.1471-4159.1992.tb09283.x. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 25.Graybiel AM. Building action repertories: memory and learning function of the basal ganglia. Curr Opin Neurobiol. 1995;5:733–741. doi: 10.1016/0959-4388(95)80100-6. [DOI] [PubMed] [Google Scholar]
- 26.Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 27.Greene JG, Porter RH, Eller RV, Greenamyre JT. Inhibition of succinate dehydrogenase by malonic acid produces an “excitotoxic” lesion in rat striatum. J Neurochem. 1993;61:1151–1154. doi: 10.1111/j.1471-4159.1993.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 28.Greene JG, Sheu S-S, Gross RA, Greenamyre JT. 3-Nitropropionic acid exacerbates N-methyl-d-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience. 1998;84:503–510. doi: 10.1016/s0306-4522(97)00389-8. [DOI] [PubMed] [Google Scholar]
- 29.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 30.Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA. 1999;96:8727–8732. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez-Lopez S, Tkatch T, Perez-Garci E, Gallaraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hipskind RA, Baccarini M, Nordheim A. Transient activation of RAF-1, MEK, and ERK2 coincides kinetically with ternary complex factor phosphorylation and immediate-early gene promoter activity in vivo. Mol Cell Biol. 1994;14:6219–6231. doi: 10.1128/mcb.14.9.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodgson JG, Agopyan N, Gutekunst C-A, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li X-J, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model of Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 34.Jaber M, Robinson SW, Missale C, Caron MG. Dopamine receptors and brain function. Neuropharmacology. 1996;35:1503–1519. doi: 10.1016/s0028-3908(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 35.Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- 36.Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 1990;10:3421–3438. doi: 10.1523/JNEUROSCI.10-10-03421.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurons: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 38.Kornhauser JM, Greenberg ME. A kinase to remember: dual roles for MAP kinase in long-term memory. Neuron. 1997;18:839–842. doi: 10.1016/s0896-6273(00)80322-0. [DOI] [PubMed] [Google Scholar]
- 39.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 40.Levine MS, Klapstein GL, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet M-F. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- 41.Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitropropionic acid—exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- 42.Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maragos WF, Jakel RJ, Pang Z, Geddes JW. 6-Hydroxydopamine injections into the nigrostriatal pathway attenuate striatal malonate and 3-nitropropionic acid lesions. Exp Neurol. 1998;154:637–644. doi: 10.1006/exnr.1998.6918. [DOI] [PubMed] [Google Scholar]
- 44.McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF. Methylmalonate toxicity in primary neuronal cultures. Neuroscience. 1998;86:279–290. doi: 10.1016/s0306-4522(97)00594-0. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell IJ, Cooper AJ, Griffiths MR. The selective vulnerability of striatopallidal neurons. Prog Neurobiol. 1999;59:691–719. doi: 10.1016/s0301-0082(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 46.Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Nemethy Z, Horvath G, Makara GB, Acs Z, Barna I. Catecholaminergic control of intracellular free calcium and endorphin secretion of rat pituitary intermediate lobe cells. J Neuroendocrinol. 1998;10:85–91. [PubMed] [Google Scholar]
- 48.Nicola SM, Surmeier DJ, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 49.Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohgoh M, Shimizu H, Ogura H, Nishizawa Y. Astroglial trophic support and neuronal cell death: influence of cellular energy level on type of cell death induced by mitochondrial toxin in cultured rat cortical neurons. J Neurochem. 2000;75:925–933. doi: 10.1046/j.1471-4159.2000.0750925.x. [DOI] [PubMed] [Google Scholar]
- 51.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 52.Otani S, Auclair N, Desce J-M, Roisin M-P, Crépel F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington's disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 55.Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 1998;282:1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- 56.Reddy PH, Williams M, Tangle DA. Recent advances in understanding the pathogenesis of Huntington's disease. Trends Neurosci. 1999;22:248–255. doi: 10.1016/s0166-2236(99)01415-0. [DOI] [PubMed] [Google Scholar]
- 57.Reynolds DS, Carter RJ, Morton AJ. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siniscalchi A, Calabresi P, Mercuri NB, Bernardi G. Epileptiform discharge induced by 4-aminopyridine in magnesium-free medium in neocortical neurons: physiological and pharmacological characterization. Neuroscience. 1997;81:189–197. doi: 10.1016/s0306-4522(97)00178-4. [DOI] [PubMed] [Google Scholar]
- 60.Stefani A, Pisani A, De Murtas M, Mercuri NB, Marciani MG, Calabresi P. Action of GP 47779, the active metabolite of oxcarbazepine, on the corticostriatal system. II. Modulation of high-voltage-activated calcium currents. Epilepsia. 1995;336:997–1002. doi: 10.1111/j.1528-1157.1995.tb00958.x. [DOI] [PubMed] [Google Scholar]
- 61.Stern EA, Jaeger D, Wilson CJ. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature. 1998;394:475–478. doi: 10.1038/28848. [DOI] [PubMed] [Google Scholar]
- 62.Sugino T, Kazuhiko N, Yasushi T, Hattori I, Hashimoto N, Moriguchi T, Nishida E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20:4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 65.Vallar L, Muca C, Magni M, Albert P, Bunzow J, Meldolesi J, Civelli O. Differential coupling of dopamine D2 receptors expressed in different cell types. Stimulation of phosphatidylinositol 4,5-bisphosphate hydrolysis in LtK-fibroblasts, hyperpolarization, and cytosolic-free Ca2+ concentration decrease in GH4C1 cell. J Biol Chem. 1990;265:10320–10326. [PubMed] [Google Scholar]
- 66.Vallone D, Picetti P, Borrelli E. Structure and function of dopamine receptors. Neurosci Biobehav Rev. 2000;24:125–132. doi: 10.1016/s0149-7634(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 67.Wullner U, Young AB, Penney JB, Beal MF. 3-Nitropropionic acid toxicity in the striatum. J Neurochem. 1994;63:1772–1781. doi: 10.1046/j.1471-4159.1994.63051772.x. [DOI] [PubMed] [Google Scholar]
- 68.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 70.Yan Z, Feng J, Fienberg AA, Greengard P. D(2) dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci USA. 1999;96:11607–11612. doi: 10.1073/pnas.96.20.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]