

Abstract
Basic fibroblast growth factor (FGF2) has many roles in neuronal development and maintenance including effects on mitogenesis, survival, fate determination, differentiation, and migration. Using a conditionally immortalized rat hippocampal cell line, H19-7, and primary hippocampal cultures, we now demonstrate that FGF2 treatment differentially regulates members of the tumor necrosis factor (TNF) superfamily of death domain receptors and their ligands. H19-7 cells transferred from serum to defined (N2) medium undergo apoptosis by a Fas-dependent mechanism similar to primary neurons. In contrast, H19-7 cells treated with FGF undergo apoptosis by a Fas-independent mechanism. FGF suppresses the Fas death pathway but also induces apoptosis by activation of a TNFα death pathway in both H19-7 and hippocampal progenitor cells. Expression of the TNF receptor 1 (TNFR1) or TNFR2 in H19-7 cells is sufficient to sensitize the cells to TNFα, similar to the effects of FGF. Because TNFα can be either proapoptotic or antiapoptotic, these results provide an explanation for the divergent trophic effects of FGF2 treatment and the observation that multiple trophic inputs are required for the survival of specific neurons.
Keywords: TNFα, TNFR1, Fas, FasL, FGF, neuronal cell line, H19-7, apoptosis, hippocampal cells
In most regions of the brain, developing neurons can undergo programmed cell death, or apoptosis, during maturation (Oppenheim, 1991). In the nervous system, mechanisms of apoptosis differ depending on the initiating signal (Greenlund et al., 1995; Pan and Griep, 1995; Wood and Youle, 1995), and analysis of DNA fragmentation patterns suggests that a neuronal population can use different apoptotic mechanisms at different developmental stages (Wood et al., 1993). Although numerous conditions elicit neuronal apoptosis, the molecular pathways that execute the process are only partially defined in most cases. Moreover, factors can be proapoptotic, antiapoptotic, or neutral depending on the developmental status of the cell and the environment (Kuan et al., 1999). Understanding the mechanism by which specific factors such as basic fibroblast-derived growth factor (FGF2) regulate apoptosis is important for understanding their physiological roles.
In vivo, FGF2 is essential for normal neurogenesis in the brain and spinal cord. In mice lacking FGF2, there are neuronal deficits in the cerebral cortex and spinal cord, and phenotypically anomalous neurons in the hippocampus (Dono et al., 1998). In vitro, FGF2 has been characterized as a mitogen for neuronal progenitors (Palmer et al., 1995; Okabe et al., 1996), a differentiation factor for hippocampal neurons (Vicario-Abejón et al., 1995), a neuronal survival factor (Walicke and Baird, 1988; Abe et al., 1990), and a potential reprogrammer of neural stem cell fate (Palmer et al., 1999). Thus, for many neural progenitors, FGF2 may be essential to specify or direct a neuronal fate as well as functioning to regulate cell death.
Although some progress has been made in describing the apoptotic responses of mature neurons to injury or removal of neurotrophic agents, there is currently no widely accepted model for neuronal progenitor death or for understanding how trophic factors or stimuli rescue developing neurons. We have generated a cell line from embryonic day 17 (E17) rat hippocampus, termed H19-7, that has been conditionally immortalized with a temperature-sensitive SV40 large T antigen and has a number of properties characteristic of in vivo neuronal differentiation and apoptosis during development (Eves et al., 1992,1994). H19-7 cells undergo two types of apoptotic death dependent on the culture conditions. In serum-free defined medium, H19-7 cells undergo limited apoptosis that is probably p53 dependent and is largely rescued by Bcl2 or BclxL or Akt activation (Eves et al., 1996). After FGF2 treatment, H19-7 cells undergo apoptosis that is p53 independent but also rescued by Bcl2 or BclxL expression or Akt activation (Eves et al., 1996, 1998). In this study, we examine the regulation of these apoptotic pathways by FGF2.
The results presented here indicate that FGF2 causes a switch in death receptor pathways from Fas ligand-mediated death to tumor necrosis factor-α (TNFα)-mediated death in H19-7 cells. Furthermore, the upregulation of the TNFα-mediated death pathway by FGF2 promotes apoptosis in primary hippocampal cells as well. Because TNFα can elicit either proapoptotic or survival signals dependent on the cellular environment, these results provide a basis for understanding the variable success of FGF as a trophic factor for primary neurons.
MATERIALS AND METHODS
Cells and culture. H19-7 cells were conditionally immortalized from E17 rat hippocampus with a temperature-sensitive SV40 large T antigen (Eves et al., 1992). In culture, these cells proliferate at the permissive temperature 33°C. After FGF2 treatment at the nonpermissive temperature (39°C), H19-7 cells express a number of neuronal markers including neurite outgrowth, expression of neurofilament proteins (Eves et al., 1992, 1994), and activation of sodium channels (D. Hanck, E. M. Eves, and M. R. Rosner, unpublished observations). H19-7 cells are grown and differentiated on tissue culture plastic coated with 15 μg/ml poly-l-lysine. Cells proliferate at 33°C in DMEM containing 10% fetal bovine serum. For differentiation, cells are transferred to 39°C in DMEM with N2 supplements (N2) (Bottenstein, 1985) and treated with 10 ng/ml FGF2.
Hippocampi were dissected from E16.5 (plug date is E0.5) Sprague Dawley rats and triturated to produce a single-cell suspension in 124 mm NaCl, 5.37 mm KCl, 1 mmNaH2PO4, 14.5 mmd-glucose, 25 mm HEPES, pH 7.4, 27 μm phenol red, 1.2 mmMgSO4, and 3 mg/ml bovine serum albumin (BSA) (Novelli et al., 1988). Cell viability was determined by trypan blue exclusion, and 5 × 10−5 viable cells were plated per well in 12-well tissue culture dishes that had been coated with 15 μg/ml polyornithine and 1 μg/ml fibronectin. The cells were cultured in DMEM/F12 medium with supplements as described previously (Johe et al., 1996) and 10 ng/ml FGF2 for 4 d. Then fresh FGF2-free medium was added, and the cultures were further treated as described in the text.
Antibodies and ligands. Neutralizing antibodies for TNFα were goat anti-rat TNFα (R & D Systems, Minneapolis, MN) and a hamster anti-mouse TNFα monoclonal (clone TN3-19.12; PharMingen, San Diego, CA). TNFα on immunoblots was detected with rabbit anti-rat TNFα (Biosource, Camarillo, CA). Fas (M-20), Fas ligand (FasL; N-20), and TNF receptor 1 (TNFR1; E-20) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); the TNFR2 antibody was purchased from Research Diagnostics Inc. (Flanders, NJ). Recombinant mouse TNFα was from R & D Systems. FasL and its enhancer and the Fas:Fc FasL decoy and its enhancer were purchased from Alexis (San Diego, CA).
DNA constructs and transfections. T7-tagged human Fas-associated protein with death domain (FADD)-like apoptotic molecule (FLAME) [inhibitor of FADD-like interleukin converting enzyme (I-FLICE), MORT-1-associated CED-3 homolog-related inducer of toxicity (MRIT), caspase-like apoptosis regulatory protein (CLARP), Caspase 8 related protein (CASPAR), and FLICE-inhibitory protein (FLIP)] (Han et al., 1997; Hu et al., 1997; Inohara et al., 1997; Irmler et al., 1997; Shu et al., 1997; Srinivasula et al., 1997) and dominant-negative Caspase 9 cDNA mammalian expression vectors have been described previously (Li et al., 1997; Srinivasula et al., 1997). The mouse TNFR1 (in pBabe Bleo) and TNFR2 (in pBabe puro) expression vectors were kindly provided by Dr. Gökhan S. Hotamisligil (Harvard School of Public Health, Boston, MA) (Xu et al., 1999).
Double cesium chloride-purified DNA was transfected into cells using TransIT-LT1 (PanVera, Madison, WI) and following the protocol provided by the manufacturer. Subconfluent cultures of H19-7 cells in OPTI-MEM medium (Life Technologies, Gaithersburg, MD) were transfected with 10 μg of total DNA per 100 mm dish. The day after transfection the cells were split to multiple wells. On the second day after transfection the cells were shifted to 39°C in N2 ± 10 ng/ml FGF2. The day of this shift is designated day 0. Further treatments are described in the text and figure legends. For cotransfections with a green fluorescent protein (GFP) vector, the input ratio of experimental vector to GFP vector was always 4:1 by mass.
Viability and apoptotic cell counting. For cell survival determinations, cells from triplicate wells were harvested by trypsinization and counted. Survival of transfected cells in GFP cotransfections was determined by counting at least 500 cells in duplicate wells (or 300 in triplicate wells) on the day that the cultures were transferred to N2 ± FGF2 and 39°C and on the indicated subsequent days. Survival was calculated as green cells per total cells on day x divided by green cells per total cells on day 0. In all experiments survival was determined on at least 3 different days. Apoptotic cells were scored as nuclei with apoptotic morphology per total nuclei after the addition of 1 μg/ml Höechst 33342 (Molecular Probes, Eugene, OR) to cultures. Höechst 33342 was chosen because it permeates viable H19-7 cells making fixation unnecessary and thus avoiding the loss of apoptotic cells that occurs during fixation procedures. Unless otherwise noted, error bars on graphs represent SDs.
Statistical analysis. Experiments were done at least three times unless otherwise noted. The Wilcoxon Mann–Whitney test was used to determine statistical significance across multiple experiments. Thep values in the figure legends represent the confidence level with which the null hypothesis is rejected. For p> 0.05, the experimental results were not considered significantly different from controls.
Cell extracts and immunoblotting. Cells were rinsed with cold PBS and extracted on the culture plates with radio immunoprecipitation assay buffer (Morrison et al., 1993) or with 2× Laemmli sample buffer (Laemmli, 1970). The latter was necessary for detection of Fas, FasL, and TNFα. After PAGE the samples were blotted to nitrocellulose, blocked for 2 hr at room temperature with 5% BSA (Jackson ImmunoResearch, West Grove, PA) in TBS with Tween (TBST; 10 mm Tris, pH 7.4, 150 mmNaCl, and 0.1% Tween 20), and incubated with primary antibodies overnight in TBST with 0.5% BSA at 4°C on a rotator. After washing three times with TBST, the blots were exposed to appropriate secondary antibodies conjugated with horseradish peroxidase (Sigma, St. Louis, MO) in TBST with 0.5% BSA and then to a chemiluminescence reagent (NEN, Boston, MA). In some experiments in which lysate protein concentration was not measurable (i.e., in sample buffer), equal protein loading on gels was verified by reprobing with an antibody to α-tubulin.
Caspase assays. Caspase (DEVD cleavage) activity in H19-7 cells was assayed using an ApoAlert Caspase Fluorescent Assay Kit (Clontech, Palo Alto, CA). Seventy percent confluent plates were treated with N2 or N2 + FGF2 at 39° for the indicated times. Cells were trypsinized, and 5 × 10−5 cells per sample were washed with PBS and pelleted. The cells were stored at −80°C until the assays were performed.
RNA isolation. mRNA was purified from H19-7 cells or adult rat tissues using the Poly (A)Pure mRNA Isolation Kit (Ambion, Austin, TX) and following the protocol provided by the manufacturer.
Reverse transcription-PCR. Reverse transcription (RT)-PCRs were performed by using either the Titan One Tube RT-PCR Kit [Boehringer Mannheim, Mannheim, Germany; FasL, TNFR2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] or the Advantage One-Step RT-PCR Kit (Clontech; TNFα and TNFR1) and following the manufacturers' protocols. For each reaction, 0.06 μg of mRNA template and 1 μm each of specific forward and reverse primers were used. For all reactions, the annealing temperature was 60°C. Specific primers were designed using MacVector 6.5 software (Oxford Molecular Group, Madison, WI) on the basis of the following rat cDNA sequences in GenBank, except where noted: TNFα forward (5′-CCTC AGCC TCTT CTCA TTCC-3′) and reverse (5′-CTCC GTGA TGTC TAAG TACT TGG-3′), TNFR1 forward (5′-TCTC AGTT GCAA GACA TGTC G-3′) and reverse (5′-TTGT GCCA GTTA CTAG GACC G-3′), TNFR2 forward (5′-CGTT CTCT GACA CCAC ATCA TCC-3′) and reverse (5′-GCTG CTGT TCAA GGCC TATT GC-3′), GAPDH forward (5′-GACA AGAT GGTG AAGG TCGG-3′) and reverse (5′-CATG GACT GTGG TCAT GAGC-3′), and FasL (Raoul et al., 1999).
Southern blotting. Ten microliters of each RT-PCR were electrophoresed per lane in a 1.2% agarose gel. The gels were stained with ethidium bromide to visualize the bands. The amplified DNA was then transferred to NytranN Nylon Transfer Membranes using the Turboblotter Rapid Downward Transfer System (Schleicher & Schuell, Dassel, Germany) and covalently cross-linked to the membrane by baking at 80°C for 2 hr. Membranes were prehybridized at 65°C for 1 hr in Rapid-hyb buffer (Amersham Pharmacia Biotech, Buckinghamshire, England). Then 1.5–2.5 × 10−6 cpm/ml nick-translated 32P-labeled specific probes were added to the buffers, and the membranes were hybridized for 2 hr at 65°C. The blots were washed once at 55°C for 30 min in 2× SSC with 0.1% SDS and three times at 55°C for 30 min in 0.2% SSC with 0.1% SDS. Specific probes were generated by using the following restriction digest fragments of mouse cDNAs as templates in nick translation reactions: TNFα, a 577 bpSpeI–EcoRI fragment (534–1110; American Type Culture Collection, Rockville, MD); TNFRI, a 759 bpNaeI–BamHI fragment (186–944); and TNFR2, a 882 bp BglII–SacI fragment (162–1043) (gifts from Dr. Gökhan S. Hotamisligil, Harvard School of Public Health). FasL (807 bp; 30–836) and GAPDH (538 bp; 25–562) probes were reverse transcribed and amplified from H19-7 mRNA, TA cloned into pCR2.1 (Invitrogen, Carlsbad, CA), and sequenced to confirm identity.
RESULTS
Caspase activity in H19-7 apoptosis
Previous studies of H19-7 cells suggest that there are two independent death pathways that regulate cell viability dependent on the cellular environment (Eves et al., 1996, 1998). To determine whether the time courses of activation of the two pathways differ at the level of the effector caspase, we assayed for caspase activity by DEVD cleavage. The major caspase activity responsible for DEVD cleavage in apoptotic cells is Caspase 3, which is downstream of the TNFα family of death receptors (for review, see Earnshaw et al., 1999), but other effector caspases such as Caspase 7 can also cleave this substrate (Faleiro et al., 1997). In H19-7 cells cultured either in defined medium (N2) or N2+ FGF2, caspase activity increased with time. However, caspase activity in cells cultured in N2 alone reached a maximum by 12 hr, whereas addition of FGF2 to the cells delayed both the onset and peak of caspase activation by 6–12 hr (Fig.1). These data indicate that FGF2 protects the cells from the rapid N2-induced caspase activation but causes a later robust apoptotic response.
Fig. 1.

Caspase activity in H19-7 cells. H19-7 cells were switched from serum-containing medium to N2 (N2) or N2 + 10 ng/ml FGF2 (FGF2) and cultured at 39°C. At the indicated times the cells were harvested and assayed for DEVD cleavage activity as described in Materials and Methods. These data are representative of three independent experiments in which the activity in N2 versus N2 + FGF2 was significantly different at 6 hr (p = 0.01), 12 hr (p = 0.01), and 24 hr (p = 0.05).
Activation of the effector Caspase 3 by death domain receptors occurs either directly via Caspase 8 or indirectly via activation of Caspase 8 and Bid followed by Caspase 9 (Zou et al., 1997; Li et al., 1998; Luo et al., 1998). It is likely that Caspase 9 plays a role in H19-7 cell death because antiapoptotic members of the Bcl2 family suppress the activation of Caspase 9 (Antonsson and Martinou, 2000; Kuan et al., 2000) and ectopically expressed Bcl2 or BclxLsuppresses death in H19-7 cells with or without FGF2 addition (Eves et al., 1996). To confirm that Caspase 9 mediates apoptosis in H19-7 cells, a dominant-negative Caspase 9 (dnCasp9) construct was transiently transfected into H19-7 cells (Li et al., 1997). As shown in Figure 2, expression of the dnCasp9 resulted in extended viability both for cells cultured in N2 as well as for cells treated with FGF2. The effect of dnCasp9 is believed to be specific for Caspase 9 because dnCasp9 acts at the level of Apaf-1 and no other caspases have been shown to interact with Apaf-1. These results are consistent with mechanisms involving both Caspase 9 and Caspase 8.
Fig. 2.

Ectopic dnCasp9 enhanced H19-7 cell survival. H19-7 cells were cotransfected with a dnCasp9 or a control vector and GFP. Transfected cell survival in N2 and FGF2-treated cultures was determined as described in Materials and Methods. Survival on day 5 is shown here. These data are representative of two independent experiments in which the dnCasp9 exhibited better survival than did wild type at day 5 in both N2 (p = 0.001) and N2 + FGF2 (p = 0.01).
To test whether Caspase 8 mediates apoptosis in H19-7 cells, we expressed the naturally occurring inhibitor of Caspase 8, FLAME (I-FLICE, MRIT, CLARP, CASPAR, and FLIP) (Han et al., 1997; Hu et al., 1997; Inohara et al., 1997; Irmler et al., 1997; Shu et al., 1997;Srinivasula et al., 1997), in H19-7 cells. Initially, FLAME was introduced into cells by transient transfection, and the viability of the FLAME-transfected cells in N2 or FGF2-containing medium was determined over time. However, the FLAME-transfected cells reproducibly died more rapidly than did controls shortly after transfection and were lost more slowly than were controls at later times (data not shown). Those data suggested that high levels of FLAME might be proapoptotic and that the lower levels were antiapoptotic. To avoid variability in FLAME expression, we selected a population of H19-7 cells stably expressing ectopic FLAME. A low level of T7-tagged FLAME was detectable on immunoblots of lysates from these cells (data not shown). As shown in Figure 3, FLAME extended viability for both N2 and FGF2-treated cultures. Thus Caspase 8, Caspase 9, and Caspase 3 appear to be components of the apoptotic pathways in both untreated and FGF-treated H19-7 cells.
Fig. 3.
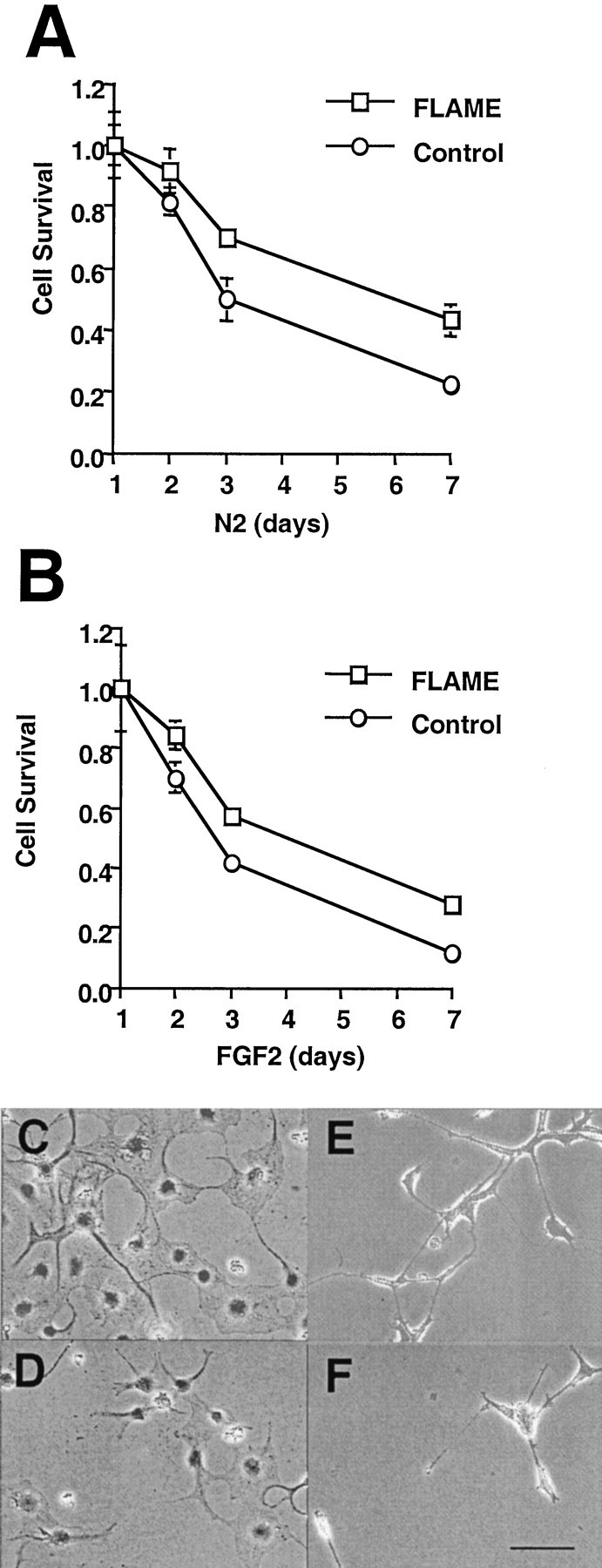
FLAME-enhanced H19-7 survival in N2 and FGF2-treated cultures. The survival of a population of H19-7 cells stably expressing ectopic FLAME was compared with that of cells stably expressing a control vector in both N2 (A, C, D) and FGF2-treated (B, E, F) conditions. A, B, Data that are representative of two independent experiments in which FLAME cells exhibited better survival than did control cells on day 2 in FGF2 (p = 0.005), on day 3 in both N2 (p = 0.05) and FGF2 (p = 0.005), and on day 7 in both N2 (p = 0.001) and FGF2 (p = 0.001). Representative fields were photographed (400×) on day 7. C, FLAME cells in N2.D, Control cells in N2. E, FGF2-treated FLAME cells. F, FGF2-treated control cells. Scale bar, 20 μm.
FasL, TNFα, and TNFR expression at the mRNA level
Caspase 8 is downstream of several death domain receptors including those for FasL and for TNFα. Expression of Fas, FasL, TNFα, and the TNF receptors (TNFR1 and TNFR2) has been demonstrated in the CNS and specifically in neurons during development (Sipe et al., 1996, 1998; Cheema et al., 1999). We have shown previously that Fas mRNA is induced in H19-7 cells after the switch from serum-containing proliferation medium to N2 medium (Gomes et al., 1999). Endogenous FasL has been shown to mediate apoptosis in NGF-deprived pheochromocytoma 12 (PC12) cells, in cerebellar granule neurons in low KCl (Le-Niculescu et al., 1999), and in trophic factor-deprived spinal motoneurons (Raoul et al., 1999). To determine whether the initial elements of the FasL or TNFα pathways are expressed in H19-7 cells, we analyzed the cell lysates by RT-PCR followed by Southern blotting of the cDNA products with specific probes for FasL, TNFα, TNFR1, and TNFR2. As shown in Figure4, mRNAs for all of these factors were expressed in H19-7 cells, and only TNFα appears to be regulated by FGF2 treatment. Because RT-PCR is qualitative rather than quantitative, these results indicate that mRNAs for the TNFα and FasL death pathways are present in the cells but do not preclude differential regulation of translation to protein or of function.
Fig. 4.

Southern blots of RT-PCRs. RT-PCRs were performed using purified mRNA from adult rat brain or thymus (lanes 1, 2) or H19-7 cells (lanes 3–7). H19-7 cells were serum starved in N2 medium at 39°C for 24 hr (lane 3) and then treated with 10 ng/ml FGF2 for 3 hr (lane 4), 6 hr (lane 5), 12 hr (lane 6), or 24 hr (lane 7). The identities of the amplified bands were verified by Southern blotting with the probes described in Materials and Methods. Br, Brain; Th, thymus.
Fas- and FasL-induced apoptosis in H19-7 cells in N2
When H19-7 cells in N2 were exposed to 10 ng/ml FasL, most of the cells died within 24 hr (Fig.5A), and all cells died within 2 d (data not shown). These results indicate that the cells express an active FasL-mediated death pathway. Correspondingly, the addition of a FasL decoy protein, Fas:Fc (Le-Niculescu et al., 1999;Raoul et al., 1999), significantly reduced the fraction of apoptotic H19-7 cells cultured in N2 (Fig. 5B), indicating that endogenous FasL is causing apoptosis. This conclusion is further supported by the observation that Fas protein increases in cells after the change from serum to defined N2 medium (Fig. 5C). We have also detected an increase in Fas ligand in cells cultured in N2 medium but not in FGF-treated cells (Fig. 5D). These results are consistent with the observations of others (Le-Niculescu et al., 1999) that trophic factor deprivation induces an upregulation of the Fas apoptotic pathway.
Fig. 5.
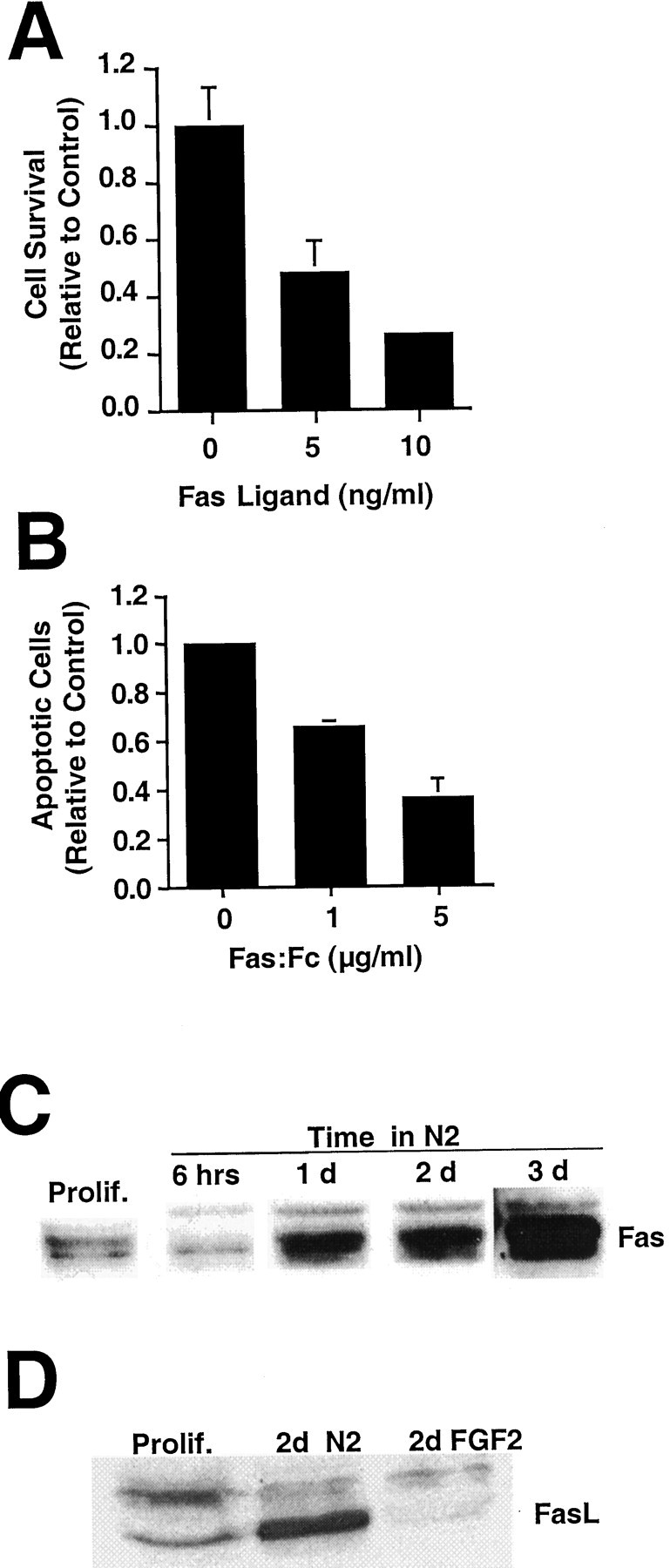
FasL kills H19-7 cells in N2 medium.A, H19-7 cells were transferred from proliferation conditions to 39°C in N2 medium and treated 2 d later with FasL (5 or 10 ng/ml) + 1 μg/ml enhancer. Duplicate cultures were counted 1 d later. These data are from one of three experiments. For 10 ng/ml FasL, p = 0.001. B, The Fas:Fc construct (1 or 5 μg/ml) and its enhancer (1 μg/ml) were added 1 d after the switch from proliferation conditions to N2, and apoptotic cells were counted 1 d later. These representative data are from one of three experiments for 5 μg/ml Fas:Fc (p = 0.005) and one of two experiments for 1 μg/ml Fas:Fc (p = 0.01). InA and B, the controls are untreated cells in N2. C, Proliferating H19-7 cells or cells cultured in N2 medium for the indicated times were harvested in 2× sample buffer, electrophoresed, blotted, and probed for Fas (45 kDa). D, Two days after the switch to N2 or N2 + FGF2 conditions, H19-7 cells were harvested in 2× sample buffer, blotted, and probed for FasL (37 kDa). Prolif., Proliferating cells.
H19-7 cells in N2 are insensitive to TNFα
Because a TNFα signaling cascade also leads to death in many cells, we tested TNFα for its effect on H19-7 cell viability and apoptosis in N2 cultures. Doses of up to 25 ng/ml TNFα had no significant effect on cell survival or the fraction of apoptotic cells (Fig. 6A,B). To determine whether this insensitivity was caused by saturating levels of endogenous TNFα, two TNFα-neutralizing antibody preparations were added to H19-7 cells in N2. Survival was not increased (Fig.6C), indicating that endogenous TNFα was not mediating apoptosis.
Fig. 6.
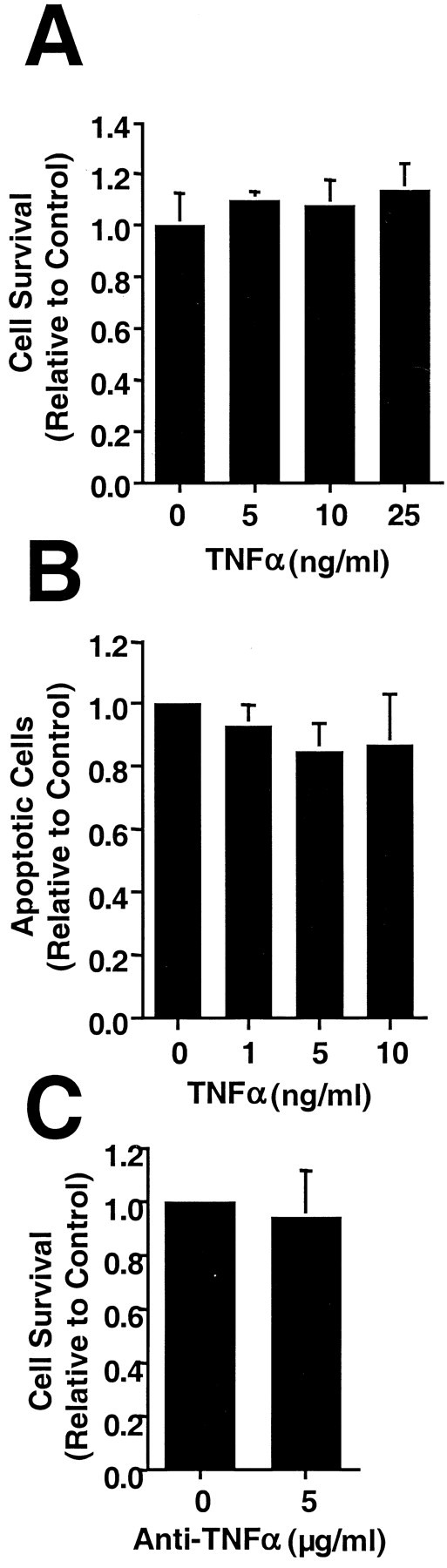
H19-7 cells in N2 medium are insensitive to TNFα. A, H19-7 cells were transferred from proliferation conditions to 39°C in N2 medium and treated 2 d later with the indicated concentrations of TNFα. Triplicate cultures were counted 1 d later. These representative data are from one of three experiments. B, Höechst 33342 was added to duplicate cultures 1 d after the addition of TNFα, and apoptotic cells were counted. These representative data are from one of three experiments. C, Antibodies to TNFα were added to H19-7 cells 1 d after the switch to N2. These data are representative of three experiments. The control in each panel is untreated cells in N2.
Ectopic TNFR expression induces TNFα sensitivity in N2
The observed insensitivity of the H19-7 cells in N2 to TNFα could be caused by the absence of functional TNFα receptors. Although mRNAs for both TNFα receptors (TNFR1 and TNFR2) were detected by RT-PCR (see Fig. 4), it is possible that functional receptors are not made. However, when immunoblotted with anti-TNFR1 receptor antibody, endogenous TNFR1 was detectable in whole-cell lysates, and the level increased with time in N2 (Fig.7A). In contrast, TNFR2 was detectable only after immunoprecipitation, and the levels did not appear to change with time in N2 (data not shown). To test whether the insensitivity to TNFα could be ascribed to limiting levels of a TNFα receptor, we transiently transfected constructs encoding either TNFR1 or TNFR2 (Xu et al., 1999) along with a GFP vector into H19-7 cells and compared the viabilities of TNFR-transfected cells with those of control vector-transfected cells in N2 and 10 ng/ml TNFα. As shown in Figure 7B, both TNF receptors conferred some sensitivity to TNFα. To confirm these transient expression data, we selected populations of H19-7 cells stably expressing TNFR1 or TNFR2. Both populations exhibited increased apoptosis when treated with 10 ng/ml TNFα in N2 (Fig. 7C). The finding that ectopic expression of either TNF receptor resulted in acquired sensitivity to TNFα in N2 implied that the level of one or both receptors in wild-type cells in N2 was insufficient to transmit an apoptotic signal. Furthermore, the requirement for exogenous TNFα to trigger apoptosis even in cells overexpressing a TNFR indicates that both the ligand and the receptor for the TNFα pathway were limiting in H19-7 cells cultured in N2 medium.
Fig. 7.
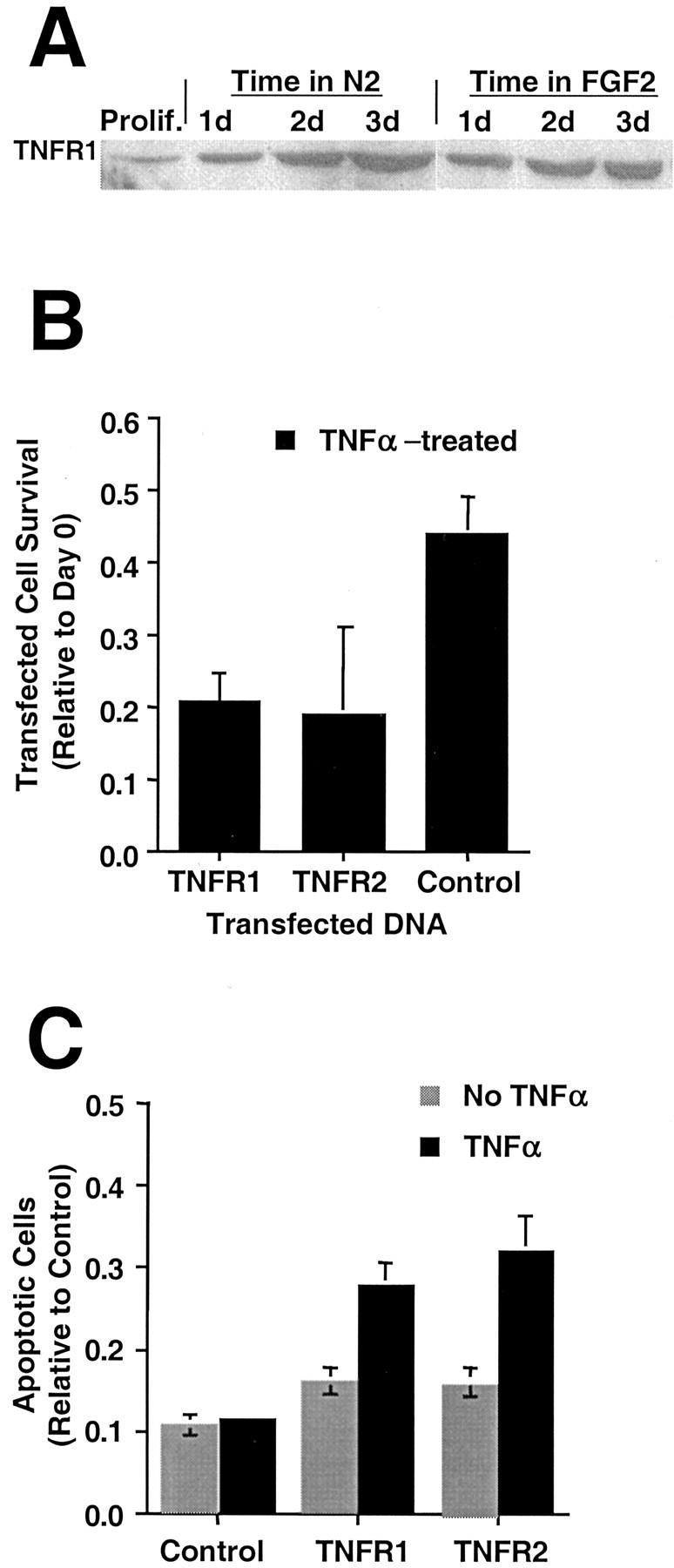
Ectopic expression of a TNFα receptor sensitizes H19-7 cells in N2 to TNFα. A, H19-7 cells were cultured as indicated and harvested in 2× buffer at the indicated times. The samples were electrophoresed and blotted as described in Materials and Methods and then probed for TNFR1 (55 kDa).B, TNFR1 or TNFR2 expression vector was transiently expressed in H19-7 cells along with a GFP vector. TNFα (10 ng/ml) was added to triplicate cultures 1 d after the switch to N2 medium, and the survival of transfected cells was determined 1 d later. These data are representative of three experiments for TNFR1 (p = 0.001) and four experiments for TNFR2 (p = 0.001). C, Populations of H19-7 cells stably expressing ectopic TNFR1 or TNFR2 or neither were shifted to N2 medium at 39°C, and some cultures were treated with TNFα (10 ng/ml) 1 d later. Höechst 33342 was added to the cultures, and triplicate counts of apoptotic cells were done 1 d after the addition of TNFα. These data are from one of two experiments for TNFR1 (with TNFα added, p = 0.025) and one of three experiments for TNFR2 (with TNFα added,p = 0.01).
Fas and FasL apoptosis in FGF2-treated H19-7 cells
The previous results indicate that the apoptosis of undifferentiated H19-7 cells in N2 occurs, at least in part, via a Fas-mediated but not a TNFα-mediated death cascade. FGF2 addition causes H19-7 cell differentiation and subsequent apoptosis of the remaining cell population within a few days. To determine whether an apoptotic mechanism similar to that in N2 is responsible for cell death after FGF2 treatment, we initially analyzed the role of the Fas death pathway in this process. Previous results from our laboratory have shown expression of Fas mRNA in H19-7 cells (Gomes et al., 1999). Analysis of Fas protein levels by immunoblotting showed a transiently reduced level of protein in cells after temperature-shift, and this decrease occurred independent of the presence of FGF2 (Figs.8A, 5C). After 1 d, however, the levels of Fas protein in both N2 and FGF2-treated cells increased dramatically. Similarly, both FasL mRNA and FasL protein were detected in the cells. However, the level of FasL protein was greatly reduced in cells treated with FGF2 for 2 d relative to that in cells cultured in N2 alone (see Fig.5D). These results suggest that both Fas and Fas ligand are expressed in cells cultured in defined N2 medium but that Fas ligand expression is suppressed in FGF2-treated cells.
Fig. 8.
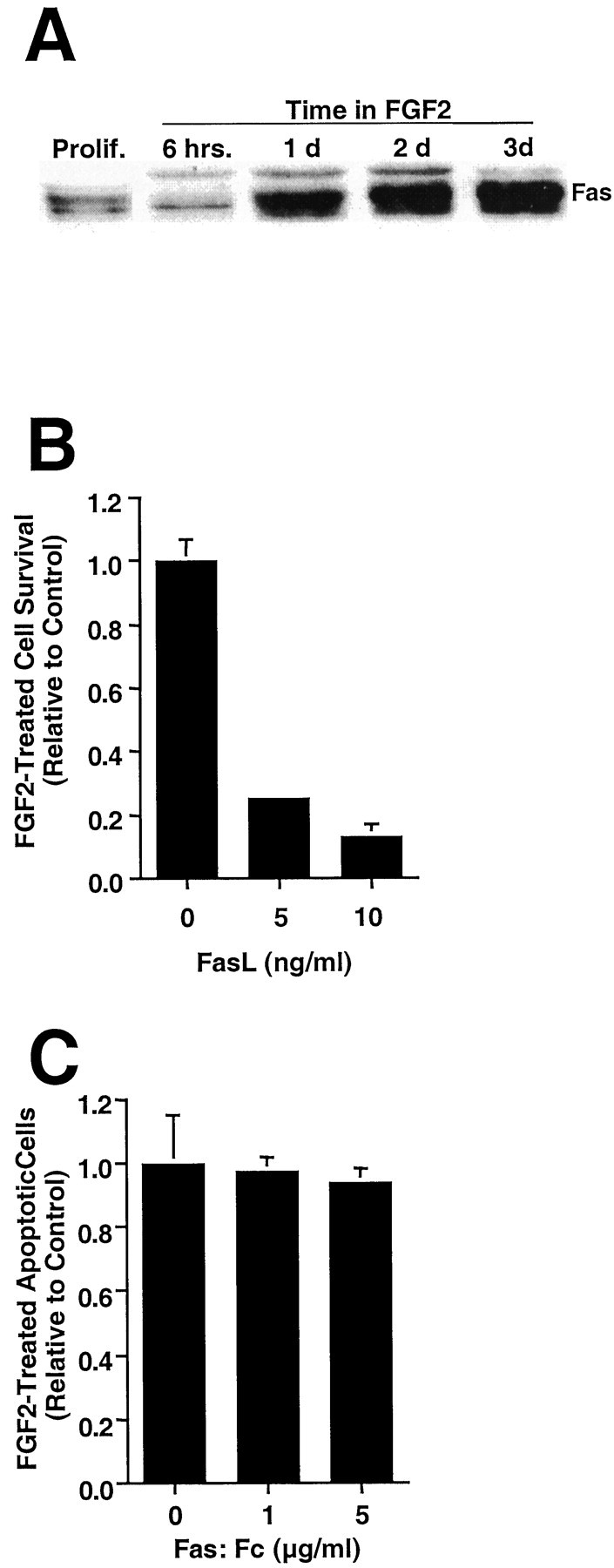
FGF2-treated H19-7 cells are killed by FasL.A, Proliferating H19-7 cells or cells cultured in N2 + FGF2 medium for the indicated times were harvested in 2× sample buffer, electrophoresed, blotted, and probed for Fas (45 kDa).B, H19-7 cells were transferred from proliferation conditions to 39°C in N2 medium + FGF2 and treated 2 d later with FasL (5 or 10 ng/ml) + 1 μg/ml enhancer. Triplicate cultures were counted 1 d later, and the data are normalized to the no-FasL control. These data are from one of three experiments. For 10 ng/ml FasL, p = 0.001. C, The Fas:Fc construct (1 or 5 μg/ml) and its enhancer (1 μg/ml) were added 1 d after the switch from proliferation conditions to N2, and apoptotic cells were counted 1 d later. The data are normalized to Fas:Fc-untreated cells. These data are representative of three independent experiments.
To determine whether the Fas death pathway can function after FGF2 treatment, the FGF2-treated cells were exposed to 5 or 10 ng/ml FasL. The cells died rapidly, similar to the cells in N2 medium alone (Fig. 8B). However, unlike the results in N2 cultures (Fig. 5B), administration of the FasL-binding decoy protein Fas:Fc did not reduce apoptosis, indicating that apoptosis was not being mediated by endogenously produced FasL (Fig. 8C). Taken together, the data indicate that FGF2 treatment inhibits Fas- and FasL-mediated apoptosis, despite the increase in Fas protein, via a reduction in FasL.
TNFα induces apoptosis in FGF2-treated H19-7 cells
Because a Fas and/or FasL pathway did not appear to be responsible for apoptosis in FGF2-treated H19-7 cells, we examined the TNFα pathway as a possible cause of cell death. RT-PCR analysis indicated that TNFα, TNFR1, and TNFR2 mRNAs were all expressed in FGF2-treated cells (see Fig. 4). FGF2-treated H19-7 cells were exposed to increasing concentrations of exogenous TNFα to test for a functional TNFα-responsive death pathway. Survival of H19-7 cells in FGF2 deceased after exposure to TNFα, and the effect on viability began to plateau at 10 ng/ml TNFα (Fig.9A; data not shown). Further analysis of TNFα treatment by an apoptotic assay showed significant effects at as low as 5 ng/ml (Fig. 9B). These results demonstrate the presence of a functional TNFα death pathway. Furthermore, addition of TNFα-neutralizing antibodies partially inhibited apoptosis in FGF2 cultures (Fig. 9C). These results, which are significantly different from those obtained for H19-7 cells in N2, suggest that endogenously produced TNFα was mediating a significant fraction of the cell death resulting from FGF2 treatment.
Fig. 9.
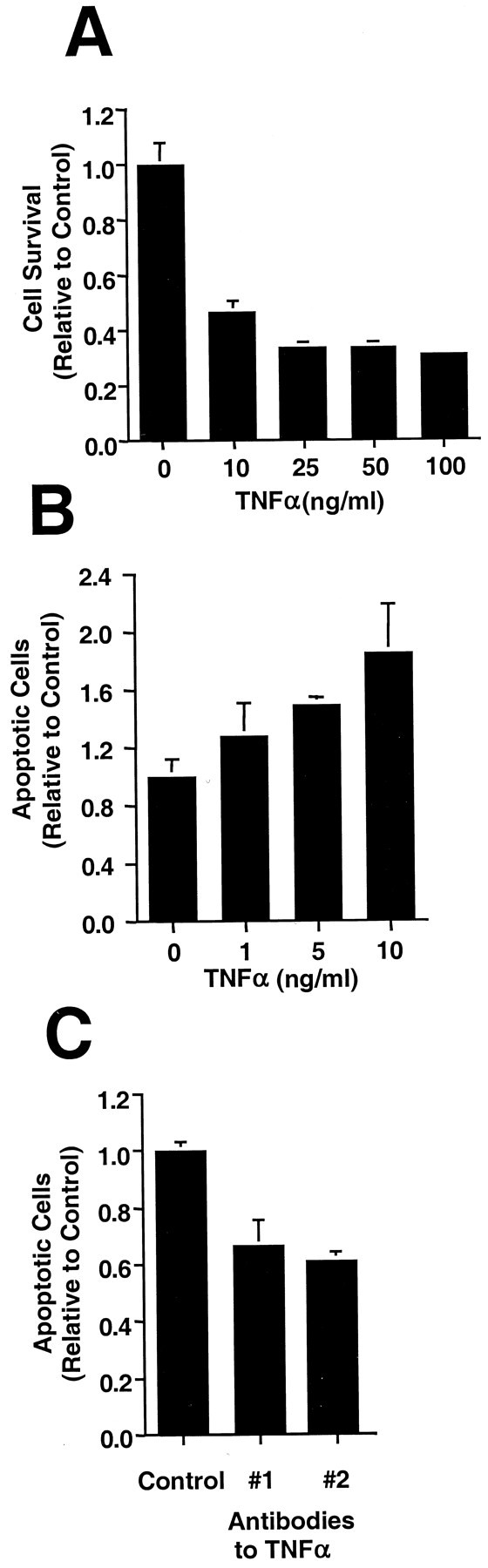
FGF2-treated H19-7 cells undergo apoptosis after TNFα treatment. H19-7 cells were shifted to N2 + FGF2 and treated with the indicated concentrations of TNFα. A, Cell survival was determined by counting 1 d after the addition of the TNFα. These data are from one of three experiments for all concentrations except 100 ng/ml (for 10, 25, or 50 ng/ml TNFα,p = 0.001). B, One day after TNFα treatment, apoptotic cells were detected with Höechst 33342. These data are representative of three experiments (for 5 or 10 ng/ml TNFα, p = 0.001). The control cells inA and B are treated with FGF2 but not TNFα. C, Control IgG, goat anti-rat TNFα (#1), or hamster anti-mouse TNFα (#2) at 5 μg/ml was added to the cultures at the time of the temperature and medium switch, and apoptotic cells were counted 2 d later. These data are from one of five experiments (p = 0.01).
Increasing TNFR1 or TNFα induces apoptosis in FGF2-treated H19-7 cells
To identify the mechanism by which FGF2 was promoting TNFα-mediated cell death, we determined whether the ligand, TNFα, and/or its receptors were rate limiting. First, H19-7 cells stably expressing TNFR1 or TNFR2 were treated with FGF2, and the extent of cell death was compared with that of the parent cells. As shown in Figure 10A, apoptosis of cells stably expressing TNFR1 increased ∼70% compared with that of wild-type cultures, whereas the increase in apoptosis of cells stably expressing TNFR2 was somewhat more variable and, over several experiments, not statistically significant. Neutralizing antibodies to TNFα were able to block most of the increased sensitivity for TNFR1-expressing cells, indicating that endogenously produced TNFα was primarily responsible for the increased apoptosis (data not shown). Thus, TNFR1 is an effective mediator of cell death in FGF2-treated H19-7 cells in response to endogenous TNFα, and the level of TNFR1 receptors is rate limiting. In contrast, ectopically expressed TNFR2 resulted in only a slight increase in apoptosis, suggesting that it is not the primary transducer of the endogenous TNFα apoptotic signal. Interestingly, after treatment with exogenous TNFα, the fractions of apoptotic cells in both TNF receptor-overexpressing and wild-type H19-7 cells increased to approximately the same level (Fig.10A). Thus, when the levels of TNFα are not rate limiting, the endogenous levels of TNF receptor are sufficient to mediate maximal death in FGF2-treated H19-7 cells. These results are significantly different from those for cells in N2 that need both exogenous TNFα and TNFR1 to promote TNFα-mediated cell death. Taken together, these results are consistent with the possibility that TNFα and TNFR1 are increased in FGF2-treated cells.
Fig. 10.
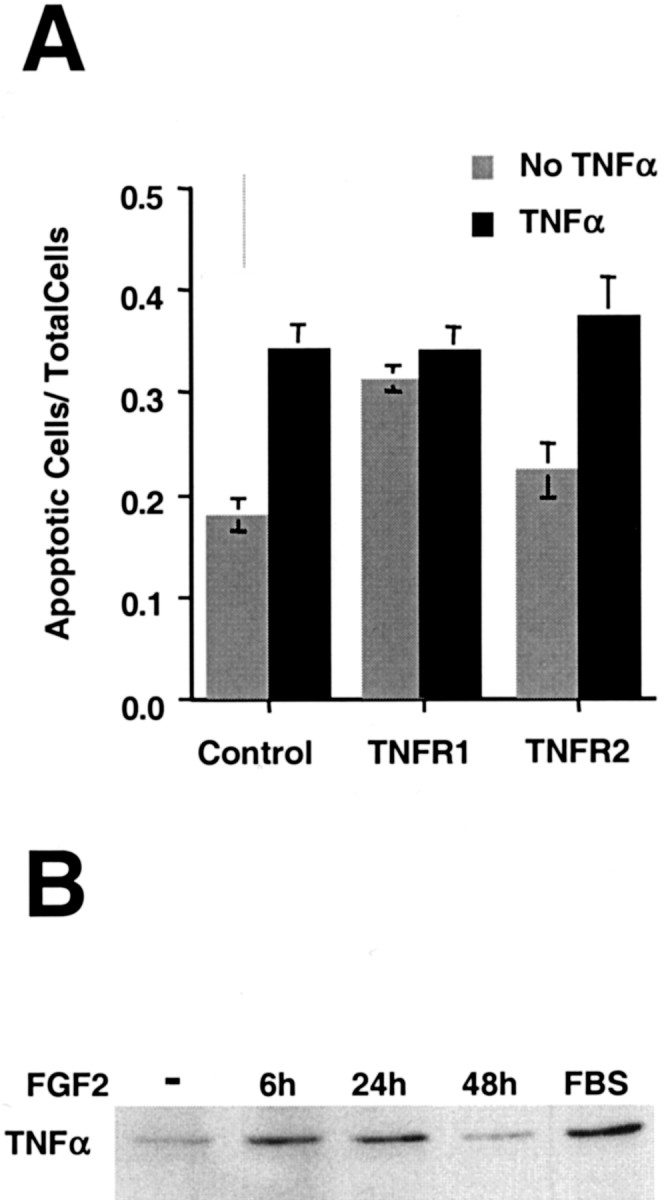
Overexpression of TNFR1 increases spontaneous apoptosis after FGF2 treatment. A, Populations of H19-7 cells expressing ectopic control vector, TNFR1, or TNFR2 were switched to N2 + FGF2. TNFα (10 ng/ml) was added to some cultures the following day. One day later apoptotic cells were detected with Höechst 33342 and counted. These data are representative of three experiments for TNFR1 (without TNFα, p = 0.01) and four experiments for TNFR2. B, H19-7 cells were treated as indicated, harvested in 2× sample buffer at the indicated times, electrophoresed, blotted, and probed for membrane-bound TNFα (30 kDa). FBS, Cells in medium containing 10% fetal bovine serum and no added FGF2.
Further support for this hypothesis comes from studies of the regulation of TNFα expression. Two lines of evidence indicate that FGF2 increases the rate of TNFα transcription. First, analysis of TNFα transcripts by PCR showed a distinct increase after addition of FGF2 to cells in N2 (see Fig. 4). Second, immunoblots showed that the amount of TNFα was elevated for at least 24 hr after the addition of FGF2 (Fig. 10B). The regulation of the TNFα receptors is less clear. RT-PCR did not show a dramatic increase in TNFR mRNA levels (see Fig. 4). Furthermore, immunoblotting analysis of TNFR1 protein revealed that, after shifting cells from serum to N2, the level of TNFR1 increased over time independent of FGF2 treatment (see Fig. 7A). Also, there were no evident differences in the levels of TNFR2 between N2 cells and FGF2-treated cells (data not shown). However, we cannot exclude the possibility that functional TNFR1 increased in response to FGF2, because the assays used do not measure receptor activity. These results indicate that TNFα protein is induced by FGF2 in H19-7 cells and suggest that the signaling pathway is additionally enabled, possibly by increasing functional TNFR1.
FGF2 induces TNFα-mediated apoptosis in primary hippocampal cells
TNFα has been reported to have either proapoptotic or antiapoptotic effects on primary neuronal cells dependent on the developmental stage and/or environment (Pan et al., 1997). Because H19-7 cells were derived from E17 rat hippocampal cells, we determined whether FGF2 also induces a TNFα-mediated apoptotic pathway in primary hippocampal cells. Rat E17 hippocampi were isolated, and the cells were expanded in culture as described in Materials and Methods. The cells were switched to defined medium lacking FGF2 for 24 hr, and then some cultures were treated with 10 ng/ml FGF2. Immunostaining has shown that ∼90% of the cells in these cultures are nestin positive, indicative of a neural progenitor state (Corbit et al., 2000). To determine whether endogenous TNFα was promoting apoptosis of these cells, TNFα-neutralizing or control antibodies were added to control and FGF2-treated cultures. As shown in Figure11A, the neutralizing antibodies reduced the fraction of apoptotic cells by up to 50% in FGF2-treated cultures but not in the control cultures. This reduction in apoptotic cells resulted in a comparable increase in cell survival (Fig. 11B). Thus, FGF2 induces a switch to a TNFα apoptotic pathway in primary neural progenitors as well as in H19-7 neuronal cells.
Fig. 11.
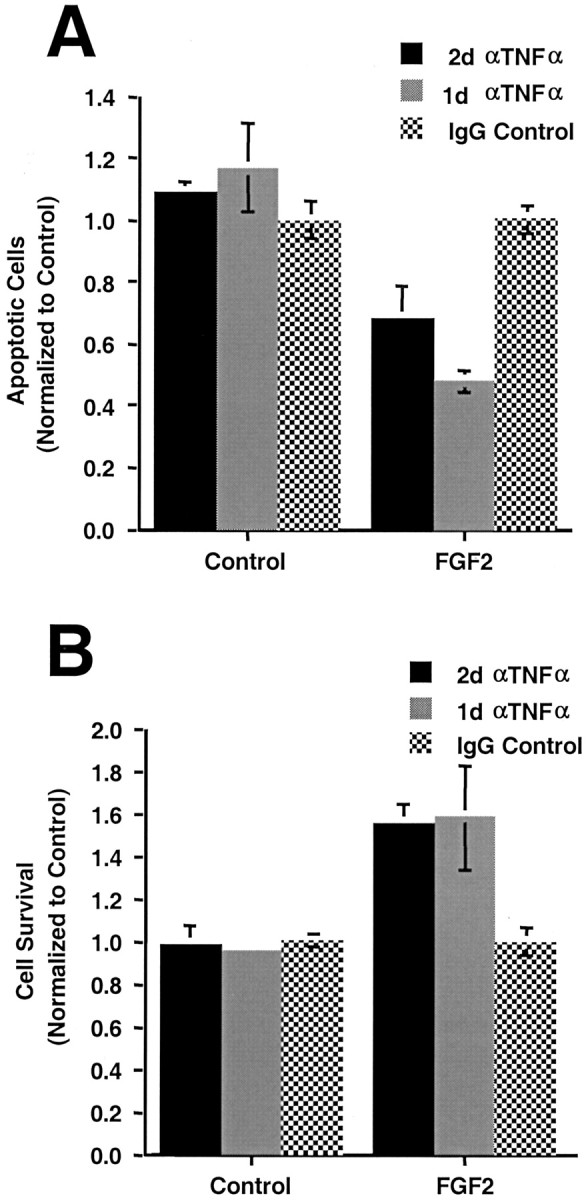
FGF2 induces TNFα-mediated apoptosis in primary hippocampal cells. Primary embryonic rat hippocampal cells were isolated and expanded as described in Materials and Methods. After 1 d in defined medium, some cultures were treated with FGF2 (10 ng/ml). Cultures were treated with antiserum to TNFα (αTNFα; 5 μg/ml) as indicated.A, Apoptotic cells were counted 2 d after the addition of FGF2 (for 2 d of antibody treatment,p = 0.005; for 1 d of antibody treatment,p = 0.001). B, Cell survival was determined for the same conditions (p = 0.001 for 1 d or 2 d of antibody treatment). These data are representative of three independent experiments.
DISCUSSION
Apoptosis has been particularly difficult to study in neuronal systems because the reported roles of various growth factors in neuronal development and survival have been diverse and often apparently contradictory. Cytokines such as TNFα have been reported to be neuroprotective in some conditions and neurotoxic in others. Developing or mature neurons may lose sensitivity to some factors that they require early in development or that were toxic early in development. In vivo or in primary cell cultures, the effects of such factors are further confused by the presence of multiple cell types. In the present study, we have shown that FGF2 treatment can upregulate a TNFα-mediated death pathway in both a hippocampal progenitor cell line and primary hippocampal cells. Furthermore, FGF2 can downregulate the Fas death pathway by suppressing Fas ligand. These results demonstrate that FGF2 can induce a switch in death receptor pathways from the FasL-mediated cascade to a cascade mediated by TNFα. Because TNFα can act as a proapoptotic or antiapoptotic factor, these data provide a framework for understanding why FGF is not always neuroprotective and why multiple input signals may be required for neuronal survival.
Although FGF2 can function as a neurotrophic factor in certain conditions, it is insufficient to rescue H19-7 cells or cultured hippocampal neurons in serum-free medium (Baird, 1994). However, the survival of both primary E17 hippocampal neurons (Banker and Cowan, 1977) and differentiated H19-7 cells (E. M. Eves and M. R. Rosner, unpublished observations) can be prolonged in the presence of fresh glial-conditioned medium. Furthermore, like other conditionally immortalized neuronal cell lines (Shihabuddin et al., 1995; Whittemore et al., 1997), H19-7 cells are capable of migration and neural differentiation when grafted into the hippocampi of postnatal rats (U. Englund, R. Fricker, E. M. Eves, M. R. Rosner, and K. Wictorin, unpublished observations). Thus, the apoptotic death of H19-7 cells and primary hippocampal neurons after differentiation appears to be caused by lack of the appropriate environmental trophic stimuli.
Apoptosis of neuronal cells can be induced by many stimuli including trophic factor deprivation, hypoxia, death receptor ligation, and a variety of other insults. Most but not all neuronal apoptosis involves Caspase 3 (Troy et al., 1996; Pettmann and Henderson, 1998). Studies with knock-out mice and cells derived from them have shown that Caspase 3 and Caspase 9 are essential for normal brain development (Pettmann and Henderson, 1998; Earnshaw et al., 1999; Kuan et al., 2000), andin vivo, Caspase 3 activation in the brain requires functional Caspase 9 (Kuida et al., 1998). In agreement with these findings, we see robust activation of DEVDase activity as well as inhibition of apoptosis by Caspase 9 inhibitors in both untreated and FGF2-treated H19-7 cells. Receptors of the tumor necrosis factor receptor family such as Fas and TNFR activate Caspase 3 via two different pathways, both of which activate Caspase 8 initially. In H19-7 cells the inhibition of apoptosis by the Caspase 8 inhibitor FLAME as well as by Caspase 9 inhibitors (Bcl2, BclxL, and dnCasp9) indicates that both Caspase 8 and Caspase 9 are active in cells in N2 as well as in FGF2-treated cells. Because Caspase 8, Caspase 9, and a Caspase 3-like activity are involved in both conditions, the changes that occur with FGF2 treatment are likely to be upstream of caspase activation.
Only recently has Fas been shown to be expressed and active in the developing rat brain. Fas was mapped by RT-PCR and immunohistochemistry to the cerebrum, cerebellum, and hippocampus of the young mouse brain and in primary cultures of hippocampus and cerebrum (Park et al., 1998). In the developing rat cortex, Fas mRNA and protein were detected as well as receptor-interacting protein (RIP) in vivo, and Fas, FADD, RIP, and FLIP (FLAME) were detected in dissociated cortical neuroblasts in culture (Cheema et al., 1999). Interestingly, more extensive Fas expression was found in dissociated cells than was seen in situ, suggesting that the in vivocellular environment might be suppressing Fas expression. Karin and colleagues demonstrated that FasL is induced by potassium reduction in cultures of cerebellar granular neurons as well as by NGF withdrawal from differentiated PC12 cells and that death resulted from activation of the Fas death cascade (Le-Niculescu et al., 1999). Similarly, in embryonic spinal motoneurons deprived of trophic factors, programmed cell death can be inhibited by a Fas:Fc decoy receptor (Raoul et al., 1999). Moreover, Greenberg and colleagues have published data suggesting that phosphorylation of Forkhead by Akt suppresses FasL expression (Brunet et al., 1999). We have shown previously that ectopic expression of constitutively active Akt inhibits apoptosis in H19-7 cells (Eves et al., 1998). The results presented here showing that H19-7 cells in N2 (i.e., deprived of trophic factors) die via a Fas- and/or FasL-mediated pathway and that FGF2 can suppress this pathway are consistent with these observations. An alternate apoptotic pathway from Fas activates Jun N-terminal kinase via apoptosis signal-regulating kinase 1 and Daxx (Chang et al., 1998). This apoptotic pathway is not operative in H19-7 cells because ectopic expression of Daxx or dnDaxx failed to affect the timing or extent of apoptosis (data not shown).
Although Fas signaling can trigger multiple death cascades, TNFα signaling is even more complex (for review, see Natoli et al., 1998). TNF receptors are expressed in many regions of the brain, including the hippocampus (Pan et al., 1997; references therein). Interestingly, TNFα can act as a survival factor or a death-promoting factor in primary hippocampal neurons. Marinovich and colleagues noted that exposure of rat hippocampal neurons to trimethyltin induced apoptosis and that the level of apoptosis was increased in the presence of glial cells because of release of TNFα (Viviani et al., 1998). Thus, in this instance, chemically induced apoptosis was potentiated by a TNFα stimulus. Alternatively, TNFα can induce Bcl2 and Bclx expression via nuclear factor κB activation in primary hippocampal neurons (Tamatani et al., 1999). In that study, TNF protected neurons against hypoxia or nitric oxide-induced injury. Thus, TNFα can have dual roles in the brain depending on both the extracellular stimuli and the intracellular environment.
There is little known about the regulation of TNFα or its receptors by FGF. Studies of the TNFα promoter have used lipopolysaccharide (LPS) to stimulate expression in a variety of cell types. LPS activates several signaling pathways resulting in the activation of a number of transcription factors that bind to and activate the TNFα promoter (Tsai et al., 2000; Zhu et al., 2000). At least one of these factors, Elk-1, is activated by FGF2 treatment of H19-7 cells (Chung et al., 1998). Furthermore, protein kinase C induces transcription of the TNFα gene in primary rat astrocytes (Chung et al., 1992), and protein kinase C is activated by FGF in H19-7 cells and in cultured neuronal cells (Corbit et al., 1999, 2000). There are very few studies in the literature involving regulation of the TNFRs. TNFR1 is constitutively expressed in most cells. FGF has been shown to upregulate the mRNA for TNFR2 in articular cartilage (Alsalameh et al., 1999) and in C6 glioma cells (Huang et al., 1998). The interaction between membrane-associated TNFα and TNFR2 can potentiate cell death in the N1E-15 neuronal cell line (Sipe et al., 1998). In primary microvascular endothelial cells, both TNFR1 and TNFR2 are required for direct TNF-induced apoptosis (Horie et al., 1999). Taken together, these studies suggest that FGF can upregulate TNFα and the TNFR2 receptor and that TNFR2 as well as TNFR1 can promote apoptosis in neuronal cells. In H19-7 cells TNFR1 protein increases in N2 cultures independent of FGF2 treatment, and there is no detectable increase in TNFR2. However, ectopic expression of either receptor is sufficient to render cells in N2 medium sensitive to added TNFα, suggesting that the TNFα receptors are rate limiting.
The mechanism by which FGF2 induces the TNFα death pathway is not entirely clear. Our data show that FGF2 upregulates the membrane-bound form of TNFα. However, added TNFα is not sufficient to induce the death of cells in N2 medium. Similarly, overexpression of TNFR2 receptors is not sufficient to increase significantly the rate of cell death in either untreated or FGF2-treated cells without addition of exogenous TNFα. However, overexpression of TNFR1 is sufficient to saturate the TNFα death pathway in FGF2-treated cells, suggesting that death is mediated via the TNFR1 pathway. Although the total expression of TNFR1 protein does not appear to differ significantly between untreated (N2) and FGF2-treated cells, it is possible that FGF2 treatment does selectively increase the number of cell surface receptors. Alternatively, FGF2 may induce a downstream effector or suppress an inhibitor of the TNFα death pathway that acts at a rate-limiting step in the cascade. Inhibitor candidates include the silencer of death domain (Jiang et al., 1999) that can inhibit TNFR1 signaling possibly by competing for the TNFR1-associated death domain binding site. Depletion of cytosolic TNFR associated factor 2 has also been shown to enhance apoptosis in response to TNFα (Arch et al., 2000). Regardless of the precise target, it is clear that FGF treatment results in the upregulation of both TNFα and a rate-limiting component of the TNFR death pathway in immortalized and primary hippocampal cells.
Footnotes
This work was supported by the National Institutes of Health Grant NS33858 and by the Cornelius Crane Trust for Eczema Research (M.R.R.). We thank Suzana Gomes for expert technical assistance and Jane Booker for aid in preparing this manuscript.
Correspondence should be addressed to Dr. Marsha R. Rosner, Department of Neurobiology, Pharmacology, and Physiology, University of Chicago, 5841 South Maryland Avenue, MC6027 (Room N711), Chicago, IL 60637. E-mail: mrosner@midway.uchicago.edu.
REFERENCES
- 1.Abe K, Takayanagi M, Saito H. A comparison of neurotrophic effects of epidermal growth factor and basic fibroblast growth factor in primary cultured neurons from various regions of fetal rat brain. Jpn J Pharmacol. 1990;54:45–51. doi: 10.1254/jjp.54.45. [DOI] [PubMed] [Google Scholar]
- 2.Alsalameh S, Mattka B, Al-Ward R, Lorenz HM, Manger B, Pfizenmaier K, Grell M, Kalden JR. Preferential expression of tumor necrosis factor receptor 55 (TNF-R55) on human articular chondrocytes: selective transcriptional upregulation of TNF-R75 by proinflammatory cytokines interleukin 1beta, tumor necrosis factor-alpha, and basic fibroblast growth factor. J Rheumatol. 1999;26:645–653. [PubMed] [Google Scholar]
- 3.Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- 4.Arch RH, Gedrich RW, Thompson CB. Translocation of TRAF proteins regulates apoptotic threshold of cells. Biochem Biophys Res Commun. 2000;272:936–945. doi: 10.1006/bbrc.2000.2873. [DOI] [PubMed] [Google Scholar]
- 5.Baird A. Fibroblast growth factors: activities and significance of non-neurotrophin neurotrophic growth factors. Curr Opin Neurobiol. 1994;4:78–86. doi: 10.1016/0959-4388(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 6.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 7.Bottenstein JE. Growth and differentiation of neural cells in defined media. In: Bottenstein JE, Sato G, editors. Cell culture in the neurosciences. Plenum; New York: 1985. pp. 3–43. [Google Scholar]
- 8.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 9.Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 10.Cheema ZF, Wade SB, Sata M, Walsh K, Sohrabji F, Miranda RC. Fas/Apo [apoptosis]-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-kappaB. J Neurosci. 1999;19:1754–1770. doi: 10.1523/JNEUROSCI.19-05-01754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung IY, Kwon J, Benveniste EN. Role of protein kinase C activity in tumor necrosis factor-alpha gene expression. Involvement at the transcriptional level. J Immunol. 1992;149:3894–3902. [PubMed] [Google Scholar]
- 12.Chung KC, Gomes I, Wang D, Lau LF, Rosner MR. Raf and fibroblast growth factor phosphorylate Elk1 and activate the serum response element of the immediate early gene pip92 by mitogen-activated protein kinase-independent as well as -dependent signaling pathways. Mol Cell Biol. 1998;18:2272–2281. doi: 10.1128/mcb.18.4.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbit KC, Foster DA, Rosner MR. Protein kinase C delta mediates neurogenic but not mitogenic activation of mitogen-activated protein kinase in neuronal cells. Mol Cell Biol. 1999;19:4209–4218. doi: 10.1128/mcb.19.6.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbit KC, Soh JW, Yoshida K, Eves EM, Weinstein IB, Rosner MR. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol. 2000;20:5392–5403. doi: 10.1128/mcb.20.15.5392-5403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 17.Eves EM, Tucker MS, Roback JD, Downen M, Rosner MR, Wainer BH. Immortal rat hippocampal cell lines exhibit neuronal and glial lineages and neurotrophin gene expression. Proc Natl Acad Sci USA. 1992;89:4373–4377. doi: 10.1073/pnas.89.10.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eves EM, Kwon J, Downen M, Tucker MS, Wainer BH, Rosner MR. Conditional immortalization of neuronal cells from postmitotic cultures and adult CNS. Brain Res. 1994;656:396–404. doi: 10.1016/0006-8993(94)91484-2. [DOI] [PubMed] [Google Scholar]
- 19.Eves EM, Boise LH, Thompson CB, Wagner AJ, Hay N, Rosner MR. Apoptosis induced by differentiation or serum-deprivation in an immortalized central nervous system neuronal cell line. J Neurochem. 1996;67:1908–1920. doi: 10.1046/j.1471-4159.1996.67051908.x. [DOI] [PubMed] [Google Scholar]
- 20.Eves EM, Xiong W, Bellacosa A, Kennedy SG, Tsichlis PN, Rosner MR, Hay N. Akt, a target of PI 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol Cell Biol. 1998;18:2143–2152. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomes I, Xiong W, Miki T, Rosner MR. A proline- and glutamine-rich protein promotes apoptosis in neuronal cells. J Neurochem. 1999;73:612–622. doi: 10.1046/j.1471-4159.1999.0730612.x. [DOI] [PubMed] [Google Scholar]
- 23.Greenlund LJS, Korsmeyer SJ, Johnson EM., Jr Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 24.Han DK, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, Baskin DG, Schwartz SM, Hood L. MRIT, a novel death-effector domain-containing protein, interacts with caspases and Bcl-xL and initiates cell death. Proc Natl Acad Sci USA. 1997;94:11333–11338. doi: 10.1073/pnas.94.21.11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horie T, Dobashi K, Iizuka K, Yoshii A, Shimizu Y, Nakazawa T, Mori M. Interferon-gamma rescues TNF-alpha-induced apoptosis mediated by up-regulation of TNFR2 on EoL-1 cells. Exp Hematol. 1999;27:512–519. doi: 10.1016/s0301-472x(98)00058-7. [DOI] [PubMed] [Google Scholar]
- 26.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Lung HL, Leung KN, Tsang D. Selective induction of tumor necrosis factor receptor type II gene expression by tumor necrosis factor-alpha in C6 glioma cells. Life Sci. 1998;62:889–896. doi: 10.1016/s0024-3205(98)00006-x. [DOI] [PubMed] [Google Scholar]
- 28.Inohara N, Ding L, Chen S, Nunez G. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-xL. EMBO J. 1997;16:1686–1694. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 30. Jiang Y, Woronicz JD, Liu W, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science 283 1999. 543 546 [Erratum(1999)283:1852] [DOI] [PubMed] [Google Scholar]
- 31.Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996;10:3129–3140. doi: 10.1101/gad.10.24.3129. [DOI] [PubMed] [Google Scholar]
- 32.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 33.Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- 34.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 38.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 39.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 40.Morrison P, Takishima K, Rosner MR. Role of threonine residues in regulation of the epidermal growth factor receptor by protein kinase C and mitogen-activated protein kinase. J Biol Chem. 1993;268:15536–15543. [PubMed] [Google Scholar]
- 41.Natoli G, Costanzo A, Guido F, Moretti F, Levrero M. Apoptotic, non-apoptotic, and anti-apoptotic pathways of tumor necrosis factor signalling. Biochem Pharmacol. 1998;56:915–920. doi: 10.1016/s0006-2952(98)00154-3. [DOI] [PubMed] [Google Scholar]
- 42.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 43.Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M, McKay RD. Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro. Mech Dev. 1996;59:89–102. doi: 10.1016/0925-4773(96)00572-2. [DOI] [PubMed] [Google Scholar]
- 44.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 45.Palmer TD, Ray J, Gage FH. FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci. 1995;6:474–486. doi: 10.1006/mcne.1995.1035. [DOI] [PubMed] [Google Scholar]
- 46.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan H, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9:2157–2169. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 48.Pan W, Zadina JE, Harlan RE, Weber JT, Banks WA, Kastin AJ. Tumor necrosis factor-alpha: a neuromodulator in the CNS. Neurosci Biobehav Rev. 1997;21:603–613. doi: 10.1016/s0149-7634(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 49.Park C, Sakamaki K, Tachibana O, Yamashima T, Yamashita J, Yonehara S. Expression of fas antigen in the normal mouse brain. Biochem Biophys Res Commun. 1998;252:623–628. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- 50.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 51.Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–1062. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shihabuddin LS, Hertz JA, Holets VR, Whittemore SR. The adult CNS retains the potential to direct region-specific differentiation of a transplanted neuronal precursor cell line. J Neurosci. 1995;15:6666–6678. doi: 10.1523/JNEUROSCI.15-10-06666.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shu HB, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 54.Sipe KJ, Srisawasdi D, Dantzer R, Kelley KW, Weyhenmeyer JA. An endogenous 55 kDa TNF receptor mediates cell death in a neural cell line. Brain Res Mol Brain Res. 1996;38:222–232. doi: 10.1016/0169-328x(95)00310-o. [DOI] [PubMed] [Google Scholar]
- 55.Sipe KJ, Dantzer R, Kelley KW, Weyhenmeyer JA. Expression of the 75 kDa TNF receptor and its role in contact-mediated neuronal cell death. Brain Res Mol Brain Res. 1998;62:111–121. doi: 10.1016/s0169-328x(98)00221-6. [DOI] [PubMed] [Google Scholar]
- 56.Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, Armstrong RC, Alnemri ES. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 57.Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 58.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1beta in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol Cell Biol. 2000;20:6084–6094. doi: 10.1128/mcb.20.16.6084-6094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vicario-Abejón C, Johe KK, Hazel TG, Collazo D, McKay RDG. Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–114. doi: 10.1016/0896-6273(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 61.Viviani B, Corsini E, Galli CL, Marinovich M. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271–276. doi: 10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- 62.Walicke PA, Baird A. Neurotrophic effects of basic and acidic fibroblast growth factors are not mediated through glial cells. Dev Brain Res. 1988;40:71–79. doi: 10.1016/0165-3806(88)90009-0. [DOI] [PubMed] [Google Scholar]
- 63.Whittemore SR, Eaton MJ, Onifer SM. Gene therapy and the use of stem cells for central nervous system regeneration. Adv Neurol. 1997;72:113–119. [PubMed] [Google Scholar]
- 64.Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 66.Xu H, Sethi JK, Hotamisligil GS. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J Biol Chem. 1999;274:26287–26295. doi: 10.1074/jbc.274.37.26287. [DOI] [PubMed] [Google Scholar]
- 67.Zhu W, Downey JS, Gu J, Di Padova F, Gram H, Han J. Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J Immunol. 2000;164:6349–6358. doi: 10.4049/jimmunol.164.12.6349. [DOI] [PubMed] [Google Scholar]
- 68.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3 [see comments]. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]