

Abstract
In previous studies we provided evidence that the γ isoform of protein kinase C (PKCγ) is an important contributor to the increased pain sensitivity that occurs after injury. Here we combined electrophysiological and behavioral approaches in wild-type and PKCγ-null mice to compare the hyperexcitability of wide dynamic range neurons in lamina V of the spinal cord dorsal horn with the behavioral hyperexcitability produced by the same injury [application of a C-fiber irritant, mustard oil (MO), to the hindpaw]. Wild-type and null mice did not differ in their response to mechanical or thermal stimuli before tissue injury, and the magnitude of the response to the MO stimuli was comparable. In wild-type mice, MO produced a dramatic and progressive enhancement of the response of lamina V neurons to innocuous mechanical and thermal stimuli. The time course of the neuronal hyperexcitability paralleled the time course of the MO-induced behavioral allodynia (nocifensive behavior in response to a previously innocuous mechanical stimulus). Neuronal hyperexcitability was also manifest in the PKCγ-null mice, but it lasted <30 min. By contrast, the behavioral allodynia produced by MO in the PKCγ-null mice, although reduced to approximately half that of the wild-type mice, persisted long after the lamina V hyperexcitability had subsided. Because the MO-induced behavioral allodynia was completely blocked by an NMDA receptor antagonist, we conclude that PKCγ mediates the transition from short- to long-term hyperexcitability of lamina V nociresponsive neurons but that the persistence of injury-induced pain must involve activity within multiple NMDA-dependent spinal cord circuits.
Keywords: mustard oil, protein kinase C, NMDA, γ isoform, persistent pain, spinal cord
There is considerable evidence that the excitability of spinal cord neurons is increased after tissue or nerve injury, resulting in enhanced transmission of nociceptive messages (for review, see Millan, 1999). In this condition, non-noxious stimuli not only produce pain (allodynia), but the allodynia can also persist even after the peripheral injury has resolved. Thus, alterations in the responsiveness of spinal cord neurons are critical for the development and maintenance of pathophysiological pain states. The mechanisms by which short-term enhancement in spinal cord activity is transformed into long-lasting increases in neuronal excitability and associated behavioral hypersensitivity are unclear. Spinal NMDA receptors clearly contribute to these phenomena (King et al., 1988;Sher and Mitchell, 1990; Ren et al., 1992; Woolf and Salter, 2000). However, because activation of NMDA receptors alone produces only transient increases in spinal cord neuronal excitability (Cumberbatch et al., 1994), it has been hypothesized that sustained hyperexcitability depends on NMDA receptor-activated changes in downstream second messenger systems (Tölle et al., 1996).
Despite the evidence of an important contribution of excitatory amino acid receptors and second messengers in the development of central sensitization, the functional consequences of activating different spinal “pain” circuits are not known. For example, there are at least five different subpopulations of spinal cord pain transmission neurons (Millan, 1999), all of which express the NMDA receptor (Tölle et al., 1995). These can be distinguished by their location (e.g., laminas I, II, or V), by the types of stimuli to which they respond, and by the pathways and targets via which they access the brain. Neurons within each of these subpopulations respond to noxious stimulation and can undergo sensitization, but the extent to which they are involved in allodynia associated with injury is not clear. Moreover, the possibility that the neurochemical basis for neural and behavioral sensitization differs among these distinct populations of neurons has not been studied.
Recently, we reported that mice with a targeted deletion of the gene that encodes the γ isoform of protein kinase C (PKCγ) show normal acute pain responses in the absence of tissue or nerve injury but exhibit a significant decrease in the behavioral allodynia that occurs days after the injury (Malmberg et al., 1997). Importantly, in the dorsal horn, PKCγ is restricted to a population of local circuit interneurons in lamina II that does not overlap with the projection neurons that transmit the pain message to the CNS (Tanaka and Saito, 1992; Malmberg et al., 1997). It follows that injury-induced alterations in sensory processing occur within a circuit that contains these PKCγ-containing interneurons and the dorsal horn output neurons.
Here, we used in vivo extracellular recordings to study the sensitization of a subpopulation of nociresponsive neurons, namely, those in lamina V of the dorsal horn, to an injury stimulus that produces an NMDA receptor-mediated allodynia. We report that deletion of PKCγ completely blocks long-term hyperexcitability and/or sensitization of these neurons but that this loss is associated with only a partial reduction of the behavioral allodynia. Thus, PKCγ is necessary for the sensitization of lamina V neurons, but hyperexcitability within multiple, neurochemically distinct, circuits in the spinal cord must contribute to the persistence of NMDA receptor-mediated postinjury pain states.
MATERIALS AND METHODS
Animals and surgical preparation. PKCγ-null mice were generated as described previously (Abeliovich et al., 1993a,b) and were obtained from The Jackson Laboratory (Bar Harbor, ME). Wild-type and homozygous PKCγ-null littermates were used for breeding. Mice (20–30 gm) were anesthetized with 10% urethane (1.5 gm/kg, i.p.) and administered dexamethasone (0.2 mg, s.c.) and atropine (0.3 mg, s.c.). Heart rate was monitored continuously, and supplemental doses of urethane were given intraperitoneally as required to maintain a heart rate of ∼9–10 Hz. A laminectomy was performed at vertebral levels T13–L1 corresponding to spinal segments L4–L5. The mouse was placed into a specialized head holder, and the vertebral segments on both sides of the laminectomy were clamped firmly. The dura was retracted, and a spinal pool was formed and filled with 37°C saline. Core temperature was monitored continuously and maintained close to 37°C with a circulating hot-water pad. Bath temperature was also measured periodically. Mice breathed spontaneously throughout the experiment. All experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco.
Electrophysiology and receptive field mapping. Fine-tip (<1.0 μm) tungsten microelectrodes (4–5 mΩ at 1 kHz; Frederick Haer & Co., Brunswick, ME) were used to record extracellular potentials, which were amplified and filtered using standard electrophysiological techniques. Unit activity was acquired, digitized, and discriminated by computer using Experimenter's Workbench (Datawave Technologies, Thornton, CO). Neurons were identified by continuously brushing the ipsilateral hindpaw with a sable-hair brush. After they were isolated, the neurons were characterized as multireceptive if they responded to brush, pressure, and noxious pinch. The receptive field was mapped with a blunt-tip 26 ga needle and marked with permanent marker. The audio threshold was set such that unit activity was detected aurally only when stimulation was within the defined receptive field. Only neurons with receptive fields located on the planter surface of the hindpaw were studied.
Experimental protocol. We measured paw thickness with a sensitive, spring-loaded caliper before and 2 hr after mustard oil (MO). Thermal stimuli were delivered via a 3 × 3 mm copper probe heated and cooled by a 9 W Peltier effect device that had a rate of rise of 2°C/sec. The probe was positioned firmly onto the center of the receptive fields and maintained at 35.5°C between the periods of stimulation. Mustard oil (3-isothiocynato-prop-1-ene; Sigma, St. Louis, MO), diluted to 10% with mineral oil, was painted onto the skin around the probe. Ten minutes after MO, thermal stimuli were presented for 10 sec at 5 min intervals, in an alternating manner to minimize thermal stimulus-induced sensitization. In separate experiments, we established baseline mechanical sensitivity by continuously brushing the receptive field (at ∼0.5 Hz) for 10 sec. After stable brush responses were recorded, MO was painted onto the skin of the receptive field (∼100 μl), and brush-evoked activity was again measured twice every 15 min (the two measurements separated by 1 min). Receptive fields were mapped every 30 min.
Behavior testing. Before testing, mice were habituated to the test environment for 60 min. We used von Frey hairs to test mechanical sensitivity (Malmberg and Basbaum, 1998) 15, 30, 60, and 120 min after MO (10%) was applied to the plantar surface. On the basis of preliminary experiments that characterized the 50% threshold, we initiated the testing paradigm with the 0.3 gm filament. The percentage increase in hypersensitivity was calculated by dividing the maximal hypersensitivity (post-MO threshold from the baseline threshold) by the maximal possible threshold reduction determined for each animal.d-2-Amino-5-phosphonovalerate (APV; 1.0 μg/5.0 μl) was administered by direct intrathecal injection in a volume of 5.0 μl, before application of MO. Within-group comparisons were made with Friedman's test, followed by Dunn's post hoc test; Mann–Whitney tests were used to make between-group comparisons.
Data analysis. When necessary (5 of 21 experiments), single units were discriminated off-line by a principal components method (Salganicoff et al., 1988), performed blind to genotype. This method distinguishes waveforms by simultaneously comparing the parameters that comprise the waveform (e.g., peak amplitude, peak time, peak width, valley amplitude, valley time, and valley width). Cluster analysis is used to separate single units, and an intercluster-weightedZ score is generated, with the distance matrix minimum set to 1.5. One second bins were used to generate peristimulus time histograms. Thermal stimulus-evoked activity was quantified by examining total spikes during and 15 sec after the stimulus (afterdischarges) and was analyzed by a Factorial ANOVA, followed by Fisher's PLSD tests. To minimize the potential confound that receptive field expansion could have on total brush-evoked spikes, we used peak brush-evoked activity as the measure of increased excitability to mechanical stimuli. Within-group comparisons were made with Friedman's test; Mann–Whitney tests were used for between-group comparisons.
Receptive field size was quantified from digitized diagrams of the mouse hindpaw using NIH Image software. The area of the receptive field was outlined, and the density of pixels within that area was measured, yielding a receptive field size as a percentage of the total size of the hindpaw. This analysis was repeated for each time point, and the changes in receptive field size after MO were expressed as a percentage of the pre-MO size. Receptive field expansion (within group) was analyzed by Friedman's test, followed by between-group comparisons with Mann–Whitney tests.
Histology. At the end of each experiment, we created a lesion (10 μA/10 sec) to mark the location of the recording site, and then the mice were perfused transcardially with 10 ml of 0.9% PBS, followed by 20 ml of 10% formalin. The lumbar segment of the spinal cord was removed and post-fixed in a 30% sucrose–formalin solution. Fifty micrometer sections were cut on a freezing microtome, mounted on slides, stained with cresyl violet, dehydrated, and coverslipped.
RESULTS
We recorded single-unit activity from 26 multireceptive neurons in the deeper part (lamina V) of the dorsal horn of the lumbar spinal cord (Fig. 1a). The neurons, which receive convergent input from large-diameter myelinated (A-fibers) and small-diameter unmyelinated (C-fibers) primary afferents, had excitatory peripheral receptive fields on the plantar surface of the ipsilateral hindpaw and exhibited very low levels of spontaneous activity (0–2 Hz). We studied these neurons because they respond to both noxious and innocuous stimulation, they encode thermal intensity, and they are readily made hyperexcitable by intense noxious input. We compared lamina V neuronal activity before, during, and after application of MO (10%; 60–100 μl) to the hindpaw. MO selectively activates C-fibers (Reeh et al., 1986), produces long-lasting changes in spinal cord excitability (Woolf and Wall, 1986), and evokes pain and allodynia in humans (Koltzenburg et al., 1994).
Fig. 1.

Characterization of wide dynamic range neurons in the mouse spinal cord. a, Histological confirmation of recording sites in the deep dorsal horn.Left, An example of a Nissl-stained transverse hemisection of mouse spinal cord illustrating the recording site in lamina V of the lumbar segment that receives input from the hindpaw (asterisk). Right, Schematic representation of the recording sites that were identified by a lesion (filled dots). Sites were effectively reconstructed in 15 of 21 mice. In the cases in which lesions were not identified, the depth of the sites was consistent with those in other mice (mean depth from surface of spinal cord = 542 ± 21 μm). b–d, Acute responses of nociceptive spinal cord neurons to mechanical (b), thermal (c), and chemical (d) stimuli. There was no difference in evoked activity between the wild-type (filled squares or bars) and PKCγ knock-out (open squares orbars) mice in response to brush (b), heat (c), or mustard oil (d) application.
In agreement with the finding that acute pain processing is intact after PKCγ deletion (Malmberg et al., 1997), we found no significant difference in the responses of lamina V neurons in wild-type and null mice before the MO stimulus. Neither the magnitude of the response to an innocuous mechanical stimulus (Fig. 1b) nor the temperature dependence of the firing in response to stimuli of 41–49°C (Fig. 1c) differed in wild-type or PKCγ-null mice. Furthermore, neither the magnitude (4–6 Hz) nor the duration (2–3 min) of the MO-induced activity of these neurons differed in the wild-type and PKCγ-null mice (Fig. 1d).
By contrast, using three different measures of hyperexcitability, namely, spontaneous activity, stimulus-evoked discharge, and poststimulus afterdischarge, we found a profound hyperexcitability of lamina V neurons after MO. The duration of the hyperexcitability, however, differed greatly in wild-type and PKCγ-null mice. Short-term hyperexcitability was present in both groups of mice, but only in wild-type mice did this persist. Figure 2illustrates the pre-MO and post-MO responses to a noxious thermal stimulus (45°C). Before the sensitizing application of MO, lamina V neurons in wild-type and PKCγ-null mice exhibited comparable spontaneous activity, stimulus-evoked responses, and afterdischarges. Ten minutes after MO, we recorded a significant increase in stimulus-evoked activity in both wild-type (Fig. 2c) and null (Fig. 2d) mice. By 55 min after MO, the 45°C stimulus-evoked responses and poststimulus afterdischarges were still enhanced compared with preinjury levels in the wild type (Fig.2e), but the sensitization was completely lost in the null mice (Fig. 2f). During the 2 hr post-MO period, spontaneous activity in the wild-type mice was also significantly greater (162 ± 18%) than that observed in the PKCγ-null mice (−10 ± 15%; p < 0.05, post hoc).
Fig. 2.

Mean peristimulus time histograms illustrate short- and long-term sensitization of nociresponsive spinal cord neurons to a 45°C stimulus (demarcated by arrowheads).a, b, Before the sensitizing application of MO, there is a comparable response to a 45°C stimulus in wild-type (+/+;a) and PKCγ-null (−/−; b) mice. Afterdischarges, i.e., neuronal activity in the 15 sec period after the thermal stimulus, were comparable (47.8 ± 23.4 and 30.6 ± 15.0 spikes/15 sec in +/+ and −/−, respectively). c, d, Ten minutes after MO, peak evoked firing was significantly enhanced in both +/+ and −/− mice. Spontaneous activity and poststimulus discharges were also enhanced, compared with baseline, but only in +/+ mice. e, f, At 55 min after MO, the neuronal responses evoked by the 45°C stimulus and the afterdischarges were still greatly elevated in +/+ mice (e), but sensitization was lost in −/− mice (f) (n = 6 cells/group).
This differential long-term neuronal hyperexcitability was manifest in response to innocuous as well as noxious temperatures. Before MO, the neurons in both groups of mice encoded thermal stimulus intensity (Fig.3a), and in both groups of mice we observed a rapid and pronounced sensitization to both non-noxious and noxious stimuli soon after the MO was applied (p < 0.01 in both groups). Compared with baseline, the post-MO response to an innocuous stimulus (41°C) increased by 10- and 6-fold in the wild-type and PKCγ-null mice, respectively (Fig. 3a). The increases to noxious stimuli were as follows: to 45°C, 175 versus 225% (Figs.2c,d, 3a), and to 49°C, 75 versus 50% (Fig.3a) in the wild-type and PKCγ-null mice, respectively. The magnitude of the increased discharges did not differ statistically in the two groups of mice (Fig. 3b). However, 70–80 min after MO, we observed a striking difference in the stimulus-evoked responses, i.e., in the magnitude of the neuronal hyperexcitability (p < 0.05). The marked sensitization was maintained in the wild-type mice (Fig. 3c), but the neural responses to all thermal stimuli returned to pre-MO levels in the PKCγ-null mice (Fig. 3c,d). Finally, we found that the magnitude and duration of the afterdischarges also increased over time and in a temperature-dependent manner, but only in the wild-type mice (p < 0.001, wild type vs null). Importantly, the differences in neuronal hyperexcitability between the wild-type and null mice occurred even though the magnitude of the MO-induced inflammation (paw thickness) did not differ in the two groups of mice (46 and 41%, respectively). These data suggest that the changes in excitability of lamina V neurons result from the sensitization of spinal cord circuits in which PKCγ is expressed.
Fig. 3.

Summary of thermal stimulus-evoked activity of lamina V neurons 10–20 and 70–80 min after MO. a, c, Stimulus–response functions to 41, 45, and 49°C (presented every 5 min for 80 min) are shown for +/+ (filled squares) and −/− (open circles) mice. Thedashed lines in a indicate the preinjury stimulus–response profile. b, d, The integrated average of all temperatures is illustrated. The horizontal dashed lines denote the mean pre-MO evoked activity in +/+ (filled bars) and −/− (open bars) mice. At 10–20 min after MO, there was a significant increase in total stimulus-evoked activity at all temperatures. However, by 70–80 min, there is a significant difference in stimulus-evoked activity between +/+ and −/− mice, and neurons in the −/− mice no longer encode stimulus intensity. *p < 0.05.
To determine whether the loss of lamina V cell hyperexcitability to thermal stimuli in PKCγ-null mice is also manifest as a decreased response to mechanical stimuli [as occurs behaviorally days after nerve injury (Malmberg et al., 1997)], we next examined the brush-evoked activity of lamina V neurons after MO-induced sensitization. We monitored peak brush-evoked activity every 15 min for at least 2 hr after MO and found a marked difference between the groups of mice. Compared with baseline responses, which were equivalent in the two genotypes (Fig. 4a,b), peak brush-evoked activity was significantly enhanced 15 min (Fig.4c,d,g) and 30 min (Fig.5a) after the MO in both wild-type (p < 0.0001) and PKCγ-null mice (p < 0.0001). In fact, at these early time points there was no difference between the genotypes. By 75 min, however, the magnitude of sensitization to brush stimuli was significantly greater (p < 0.05) in wild-type mice compared with PKCγ-null mice (Fig. 5a). Although the enhanced neural response to an innocuous mechanical stimulus persisted for as long as the neurons were studied (up to 4 hr), in the PKCγ-null mice the neuronal sensitization disappeared completely within 2 hr (Fig. 4e–g, 5a).
Fig. 4.
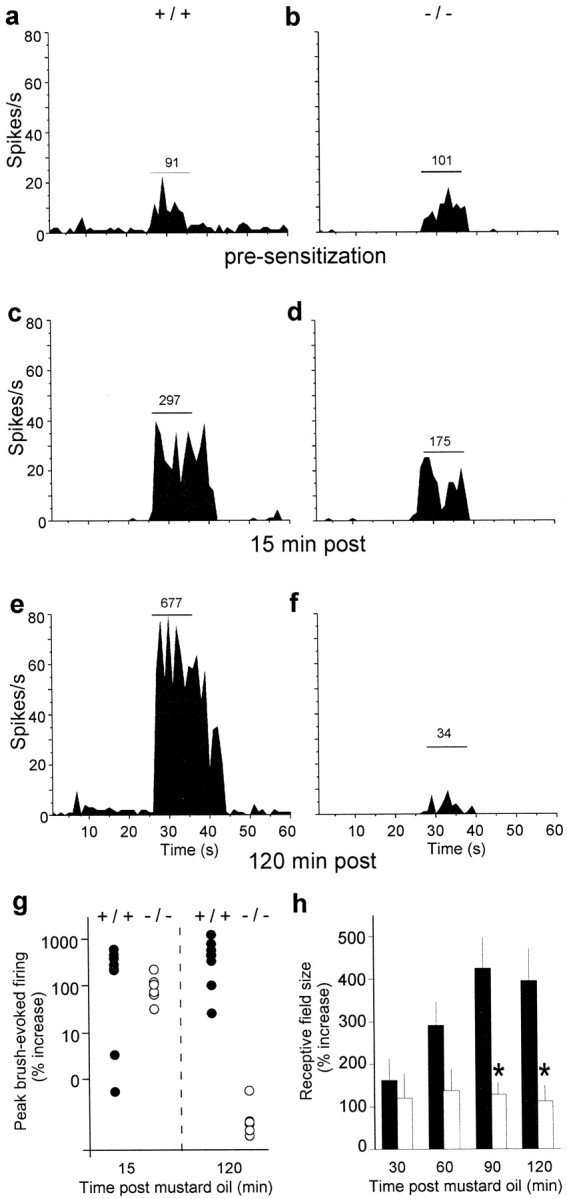
a–f, Representative peristimulus time histograms of the response to an innocuous mechanical stimulus (brush) before and after MO from a single +/+ cell and a single −/− cell are shown. The brush stimulus is denoted by the horizontal line; the total number of spikes per stimulus is shown above the line. a, b, Before MO application, the brush produced equivalent increases in neuronal activity in both +/+ (a) and −/− (b) mice.c, d, Fifteen minutes after MO, brush-evoked activity was enhanced in +/+ mice (c) and to a lesser extent in −/− mice (d). e, f, By 120 min, there is a dramatic sensitization in +/+ mice (e), but in −/− mice the sensitization is lost; the response to the brush returned to pre-MO levels (f). g, Summary of brush-evoked activity at 15 and 120 min after MO in +/+ (n = 8;filled circles) and −/− (n = 6;open circles) mice is shown, illustrating comparable degrees of sensitization at 15 min in the two groups of mice and a complete loss of sensitization 2 hr after MO in the −/− mice. Data are presented as a percentage of preinjury brush-evoked activity, and each cell is represented by a single circle.h, MO produced a significant increase in receptive field size in both groups of mice 30 min after its application (+/+, filled bars; −/−, open bars), but only in the +/+ mice was there a progressive increase in receptive field size over the course of the experiment. By 90 min, the magnitude of the expansion was significantly greater in +/+ than in −/− mice. Receptive fields in both groups were significantly greater than those in control (Friedman test); +/+ mice were different from −/− mice at 90 and 120 min (*p < 0.05, Mann–Whitney tests). Results are expressed as the mean ± SEM.
Fig. 5.
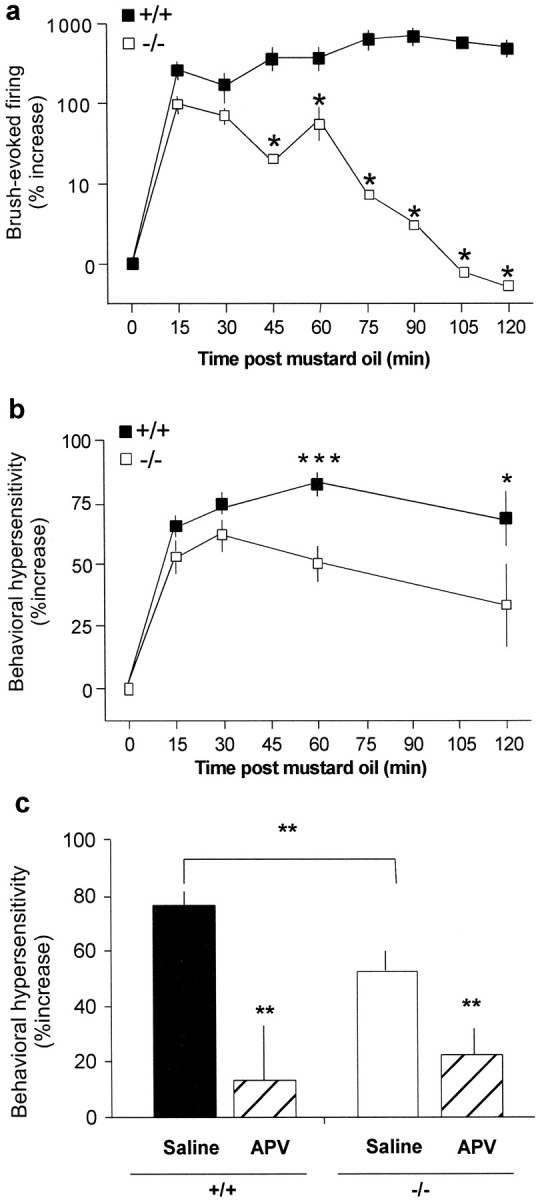
Time course of neuronal hyperexcitability and behavioral hypersensitivity to an innocuous mechanical stimulus after MO. a, At 15 min, brush-evoked neuronal activity was significantly enhanced in both +/+ and −/− mice; this increase did not differ statistically between the groups. By 60 and 120 min, innocuous mechanical stimulus-induced hyperexcitability was still present in +/+ mice but was greatly reduced (60 min) or absent (120 min) in −/− mice. b, The time course of the increased behavioral sensitivity (allodynia) paralleled the electrophysiological change. Mechanical withdrawal thresholds in the +/+ (filled squares; n = 15) and −/− (open squares; n = 16) mice did not differ before MO. After MO, the threshold for withdrawal significantly decreased in the ipsilateral paw in both +/+ and −/− mice (Friedman test, p < 0.001 each group), and there was no difference in the magnitude of sensitization at 15 or 30 min. However, behavioral allodynia was significantly attenuated in −/− mice compared with +/+ mice at both 60 and 120 min after MO (Mann–Whitney, *p < 0.05; ***p < 0.001). c, Mechanical thresholds in the +/+ and −/− mice pretreated with either saline (5.0 μl, i.t.) or APV (1.0 μg/5.0 μl; n = 8/genotype) are shown. APV prevented the development of MO-induced allodynia in both the +/+ and −/− mice. The 60 min time point is summarized (Mann–Whitney, **p < 0.01). Results are expressed as the mean ± SEM.
Another hallmark of sensitization in the spinal cord is an increased receptive field size of dorsal horn neurons after sustained C-fiber activation (Cook et al., 1987). We found that mechanosensitive receptive field size on the plantar surface of the hindpaw increased significantly in both wild-type and PKCγ-null mice (p < 0.01 for each genotype; Fig.4h), but with notable differences in the magnitude of the expansion over time. By 30 min (the earliest time point tested), receptive fields in wild-type and null mice increased by ∼100% (Fig.4h). Receptive field size stabilized at this level in PKCγ-null mice but continued to increase in the wild-type mice, such that by 2 hr after MO the receptive fields had increased by ∼400%. Because receptive fields of dorsal horn neurons are subject to central modulation (Zieglgansberger and Herz, 1971) and because receptive fields of nociceptive primary afferents exhibit little expansion after injury (Hylden et al., 1989), the progressive receptive field expansion observed in the wild-type but not the PKCγ-null mice is likely to be centrally mediated.
If the hyperexcitability of lamina V nociresponsive spinal cord neurons underlies a behavioral allodynia, then the transient sensitization in the PKCγ-null mice should be manifest as a short-lived MO-induced behavioral hypersensitivity. By contrast, the allodynia should persist in the wild-type mice. Because mechanical thresholds after MO have not been systematically examined, we applied MO to the hindpaws of wild-type and PKCγ-null mice and measured behavioral responses to innocuous mechanical stimuli over the next 2 hr. MO application produced an immediate licking response that was indistinguishable between the wild-type and null mice. In agreement with the electrophysiological finding of a profound and rapid hyperexcitability of lamina V neurons (Fig. 5a), within 15 min of the application of MO we observed a significant increase in behavioral sensitivity of the injured paw in both groups of mice (p < 0.001 for each genotype; Fig.5b). In the wild-type mice, the decreased mechanical threshold for evoking paw withdrawal peaked at 60 min and remained at this level for the duration of the experiment. By contrast, the peak allodynia in the PKCγ-null mice occurred at 30 min after MO and was significantly reduced compared with wild-type mice at 60 and 120 min. Importantly, pretreatment with the NMDA receptor antagonist APV completely blocked the behavioral hypersensitivity in both wild-type and PKCγ-null mice (Fig. 5c). APV did not alter paw withdrawal thresholds in the limb contralateral to the mustard oil injection, indicating that APV is, as expected, without effect on acute nociceptive thresholds. These results demonstrate that both the short- and long-term allodynia are NMDA receptor-mediated, including the component that persists during the period when the post-MO hyperexcitability of lamina V cells is completely lost in the PKCγ-null mice. Finally, because increased sensitivity to mechanical stimuli outside the original site of injury is an important component of central sensitization and persistent pain (Woolf, 1983; Koltzenburg et al., 1994), we also examined the behavioral thresholds contralateral to the MO application. At the 60 min time point, we observed a significant and persistent (at least 2 hr) allodynia contralateral to the injury in the wild-type mice (39 ± 8%;p < 0.01) but no change in mechanical sensitivity in the contralateral paw of the PKCγ-null mice (6.5 ± 8%;p > 0.05; data not shown). Thus, the lack of neuronal sensitization of the lamina V cell on the side of the injury in the PKCγ-null mice corresponded to an absence of allodynia contralateral to the injury.
DISCUSSION
The present results demonstrate that the sustained hyperexcitability of lamina V cells after MO-induced injury is dependent on the presence of PKCγ. Because the excitability of lamina V cells was transiently increased in the absence of PKCγ, it appears that PKCγ is required for the transition from short- to long-term hyperexcitability. We also show that hyperexcitability of lamina V cells is required for the full expression of the behavioral allodynia associated with MO-induced injury. Furthermore, because NMDA receptor antagonists completely blocked the MO-induced allodynia, it is likely that the PKCγ-containing nociceptive circuits are located downstream of the NMDA receptor-mediated sensitization. Finally, our results indicate that a component of injury-induced allodynia can be sustained by activity in pathways that do not derive from lamina V neurons and that the residual sensitization of these parallel nociceptive pathways is PKCγ independent.
These results confirm and extend previous findings. For example, Murase et al. (1999) demonstrated that input from deep dorsal horn neurons is required to evoke prolonged, C-fiber-induced, excitation in the substantia gelatinosa. Thus, dynamic interactions between interneurons in lamina II and neurons within lamina V are required for the expression of pain behavior. In agreement with this idea, our laboratory has provided evidence that a population of interneurons in the dorsal horn that express the γ isoform of PKC is critical to the development of the behavioral and anatomical changes that accompany nerve injury (Malmberg et al., 1997). Moreover, in the setting of persistent inflammation, PKCγ upregulates and translocates from the cytosol to the cell membrane of neurons in the superficial dorsal horn of the spinal cord over a time course that is consistent with behavioral allodynia (Martin et al., 1999).
There are several possible mechanisms via which the PKCγ-containing interneurons of lamina II could contribute to the altered responses of the lamina V neurons. Because most PKCγ-containing interneurons do not express markers of inhibitory interneurons, they are likely to be excitatory (Martin et al., 1999). These interneurons are then in a position to excite the lamina V cells, the dorsally directed dendrites of which often penetrate lamina II (Woolf and King, 1987; Light and Kavookjian, 1988). Moreover, because baseline neuronal and behavioral responses are comparable in the wild-type and null mice, the contribution of PKCγ must only develop in the setting of injury. Another possibility derives from the fact that many neurons in inner lamina II, where PKCγ is concentrated, respond to non-noxious stimulation (Bennett et al., 1980). Perhaps this non-nociceptive input is only transmitted to lamina V pain transmission neurons when PKCγ is activated in the setting of injury. On the other hand, PKCγ interneurons receive primary afferent input from a subset of C-fibers, distinguished by their ability to bind the lectin isolectin B4. These afferents express the vanilloid (VR1) receptor (Tominaga et al., 1998) and are, therefore, likely to be nociceptors. The importance of nociceptor input from this subpopulation of sensory neurons is supported by the finding that glial cell line-derived neurotrophic factor, the neurotrophin to which isolectin B4-positive neurons are preferentially sensitive (Snider and McMahon, 1998), reduces neuropathic pain (Boucher et al., 2000). Our results suggest that PKCγ may be essential for persistent behavioral hypersensitivity, in general, and not limited to pain that is neuropathic in origin (see Snider and McMahon, 1998).
These findings illustrate that multiple circuits can be rendered hyperexcitable after injury and that this sensitization must occur via biochemically distinct mechanisms, i.e., PKCγ dependent and independent. As noted above, there are multiple pathways that carry nociceptive information from the spinal cord to higher centers. Because there is evidence of an NMDA-mediated hyperexcitability of lamina I neurons (Ren et al., 1992), we suggest that ascending pathways that derive from lamina I neurons mediate the behavioral allodynia that persists when the sensitization of the lamina V cell is ablated. Indeed, lamina I nociresponsive neurons are readily sensitized by injury such that they can be activated by innocuous stimuli (Cook et al., 1987; Hylden et al., 1989). Moreover, selective ablation of a subset of lamina I neurons reduced pain behavior associated with persistent neuropathic or inflammatory conditions (Nichols et al., 1999). Because the dendrites of lamina I neurons arborize in the rostral caudal plane, only rarely penetrating into lamina II (Gobel, 1978; Beal, 1979), in contrast to the lamina V neuron, they probably do not come under the direct influence of the PKCγ-containing interneurons of inner lamina II. Independence of the different pain transmission circuits is also possible because the C-fiber input to lamina I and inner lamina II is distinct, involving peptidergic and nonpeptidergic afferents, respectively (Snider and McMahon, 1998). On the other hand, because nonselective blockers of PKC have been implicated in sensitization (Munro et al., 1994; Lin et al., 1996; Li et al., 2000), we suggest that the residual NMDA-mediated behavioral allodynia in the PKCγ-null mice involves other isoforms of PKC (Mao et al., 1993; Coderre, 1992, 1994; Tölle et al., 1996; Aley et al., 2000).
Our results demonstrate that the biochemical bases for the short- and long-term increases in lamina V neuronal excitability produced by injury are very different. Such temporally distinct forms of plasticity have been described in rat hippocampus (Ben-Ari et al., 1992;Abeliovich et al., 1993a; Malenka and Nicoll, 1993) and inAplysia sensory neurons (Fisher et al., 1997). We show that PKCγ is not required either for the acute injury-induced discharge or for the transient increase in the excitability of these neurons. Rather PKCγ is critical for converting short-term increases in spinal cord excitability into long-lasting hyperexcitability of lamina V neurons, some of which are likely to be projection neurons, and into a corresponding behavioral allodynia. However, a residual, although reduced, allodynia can occur in the absence of persistent lamina V hyperexcitability. We conclude that PKCγ is essential for the persistence of lamina V neuronal hyperexcitability but that integration of activity across PKCγ-dependent and -independent circuits is required for the full expression of injury-induced and NMDA receptor-mediated persistent pain. Although activity in lamina V neurons is sufficient to produce pain (Price and Mayer, 1975) and correlates with behavioral sensitization, separate pain-signaling pathways, not modulated by PKCγ, can maintain a hypersensitive behavioral state. This feature of the spinal cord may explain why anterolateral cordotomy for intractable pain, although initially successful, becomes ineffective with time (Cowie and Hitchcock, 1982).
Footnotes
This work was supported by National Institutes of Health (NIH) Grants DA 08377 and NS 14627 and by an RRP from Howard Hughes Medical Institute. W.J.M. was supported by a postdoctoral fellowship from the Merck/UNCF Science Initiative and by a training grant from the NIH. We thank H. Fields for useful discussions during experimental design, J. Trafton and G. Gurkoff for expert technical assistance, and A. Doupe, S. Lisberger, C. Boettiger, and D. Julius for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Allan I. Basbaum, Department of Anatomy, University of California, San Francisco, Box 0452, San Francisco, CA 94143-0452. E-mail: aib@phy.ucsf.edu.
W. J. Martin's present address: Merck Research Laboratories, 126 East Lincoln Avenue (RY80Y-145), Rahway, NJ 07065.
A. B. Malmberg's present address: NeurogesX, Inc., 969C Industrial Road, San Carlos, CA 94070.
REFERENCES
- 1.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell. 1993a;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- 2.Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993b;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- 3.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal JA. The ventral dendritic arbor of marginal (lamina I) neurons in the adult primate spinal cord. Neurosci Lett. 1979;14:201–206. doi: 10.1016/0304-3940(79)96148-2. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase C modulation of NMDA currents: an important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- 6.Bennett GJ, Abdelmoumene M, Hayashi H, Dubner R. Physiology and morphology of substantia gelatinosa neurons intracellularly stained with horseradish peroxidase. J Comp Neurol. 1980;194:809–827. doi: 10.1002/cne.901940407. [DOI] [PubMed] [Google Scholar]
- 7.Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290:124–127. doi: 10.1126/science.290.5489.124. [DOI] [PubMed] [Google Scholar]
- 8.Coderre TJ. Contribution of protein kinase C to central sensitization and persistent pain following tissue injury. Neurosci Lett. 1992;140:181–184. doi: 10.1016/0304-3940(92)90097-q. [DOI] [PubMed] [Google Scholar]
- 9.Coderre TJ. The role of excitatory amino acid receptors and intracellular messengers in persistent nociception after tissue injury in rats. Mol Neurobiol. 1994;7:229–246. doi: 10.1007/BF02769177. [DOI] [PubMed] [Google Scholar]
- 10.Cook AJ, Woolf CJ, Wall PD, McMahon SB. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature. 1987;325:151–153. doi: 10.1038/325151a0. [DOI] [PubMed] [Google Scholar]
- 11.Cowie RA, Hitchcock ER. The late results of antero-lateral cordotomy for pain relief. Acta Neurochir (Wien) 1982;64:39–50. doi: 10.1007/BF01405617. [DOI] [PubMed] [Google Scholar]
- 12.Cumberbatch MJ, Herrero JF, Headley PM. Exposure of rat spinal neurones to NMDA, AMPA and kainate produces only short-term enhancements of responses to noxious and non-noxious stimuli. Neurosci Lett. 1994;181:98–102. doi: 10.1016/0304-3940(94)90569-x. [DOI] [PubMed] [Google Scholar]
- 13.Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- 14.Gobel S. Golgi studies of the neurons in layer I and II of the dorsal horn of the medulla (trigeminal nucleus caudalis). J Comp Neurol. 1978;180:375–394. doi: 10.1002/cne.901800212. [DOI] [PubMed] [Google Scholar]
- 15.Hylden JLK, Nahin RL, Traub RJ, Dubner R. Expansion of receptive fields of spinal lamina I projection neurons in rats with unilateral adjuvant-induced inflammation: the contribution of dorsal horn mechanisms. Pain. 1989;37:229–243. doi: 10.1016/0304-3959(89)90135-8. [DOI] [PubMed] [Google Scholar]
- 16.King AE, Thompson SW, Urban I, Woolf CJ. An intracellular analysis of amino acid induced excitations of deep dorsal horn neurones in the rat spinal cord slice. Neurosci Lett. 1988;89:286–292. doi: 10.1016/0304-3940(88)90541-1. [DOI] [PubMed] [Google Scholar]
- 17.Koltzenburg M, Torebjörk HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- 18.Li KC, Zheng JH, Chen J. Involvement of spinal protein kinase C in induction and maintenance of both persistent spontaneous flinching reflex and contralateral heat hyperalgesia induced by subcutaneous bee venom in the conscious rat. Neurosci Lett. 2000;285:103–106. doi: 10.1016/s0304-3940(00)01039-9. [DOI] [PubMed] [Google Scholar]
- 19.Light AR, Kavookjian AM. Morphology and ultrastructure of physiologically identified substantia gelatinosa (lamina II) neurons with axons that terminate in deeper dorsal horn laminae (III-V). J Comp Neurol. 1988;267:172–189. doi: 10.1002/cne.902670203. [DOI] [PubMed] [Google Scholar]
- 20.Lin Q, Peng YB, Willis WD. Possible role of protein kinase C in the sensitization of primate spinothalamic tract neurons. J Neurosci. 1996;16:3026–3034. doi: 10.1523/JNEUROSCI.16-09-03026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 22.Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain. 1998;76:215–222. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- 23.Malmberg AB, Chen C, Tonegawa S, Basbaum AI. Preserved acute pain and reduced neuropathic pain in mice lacking PKCγ. Science. 1997;278:279–283. doi: 10.1126/science.278.5336.279. [DOI] [PubMed] [Google Scholar]
- 24.Mao J, Mayer DJ, Price DD. Patterns of increased brain activity indicative of pain in a rat model of peripheral mononeuropathy. J Neurosci. 1993;13:2689–2702. doi: 10.1523/JNEUROSCI.13-06-02689.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin WJ, Liu H, Wang H, Malmberg AB, Basbaum AI. Inflammation-induced upregulation of protein kinase Cγ immunoreactivity in rat spinal cord correlates with enhanced nociceptive processing. Neuroscience. 1999;88:1267–1274. doi: 10.1016/s0306-4522(98)00314-5. [DOI] [PubMed] [Google Scholar]
- 26.Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 27.Munro FE, Fleetwood-Walker SM, Mitchell R. Evidence for a role of protein kinase C in the sustained activation of rat dorsal horn neurons evoked by cutaneous mustard oil application. Neurosci Lett. 1994;170:199–202. doi: 10.1016/0304-3940(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 28.Murase K, Saka T, Asai T, Ikeda H. Functional circuitry for the induction of prolonged excitation in the rat spinal dorsal horn. Eur J Neurosci. 1999;11:3355–3358. doi: 10.1046/j.1460-9568.1999.00783.x. [DOI] [PubMed] [Google Scholar]
- 29.Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honoré P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- 30.Price DD, Mayer DJ. Neurophysiological characterization of the anterolateral quadrant neurons subserving pain in M. mulatta. Pain. 1975;1:59–72. doi: 10.1016/0304-3959(75)90005-6. [DOI] [PubMed] [Google Scholar]
- 31.Reeh PW, Kocher L, Jung S. Does neurogenic inflammation alter the sensitivity of unmyelinated nociceptors in the rat? Brain Res. 1986;384:42–50. doi: 10.1016/0006-8993(86)91217-5. [DOI] [PubMed] [Google Scholar]
- 32.Ren K, Hylden J, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50:331–344. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- 33.Salganicoff M, Sarna M, Sax L, Gerstein GL. Unsupervised waveform classification for multi-neuron recordings: a real-time, software-based system. I. Algorithms and implementation. J Neurosci Methods. 1988;25:181–187. doi: 10.1016/0165-0270(88)90132-x. [DOI] [PubMed] [Google Scholar]
- 34.Sher GD, Mitchell D. Intrathecal N-methyl-d-aspartate induces hyperexcitability in rat dorsal horn convergent neurones. Neurosci Lett. 1990;119:199–202. doi: 10.1016/0304-3940(90)90833-u. [DOI] [PubMed] [Google Scholar]
- 35.Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka C, Saito N. Localization of subspecies of protein kinase C in the mammalian central nervous system. Neurochem Int. 1992;21:499–512. doi: 10.1016/0197-0186(92)90081-2. [DOI] [PubMed] [Google Scholar]
- 37.Tölle TR, Berthele A, Laurie DJ, Seeburg PH, Zieglgansberger W. Cellular and subcellular distribution of NMDAR1 splice variant mRNA in the rat lumbar spinal cord. Eur J Neurosci. 1995;7:1235–1244. doi: 10.1111/j.1460-9568.1995.tb01114.x. [DOI] [PubMed] [Google Scholar]
- 38.Tölle TR, Berthele A, Schadrack J, Zieglgansberger W. Involvement of glutamatergic neurotransmission and protein kinase C in spinal plasticity and the development of chronic pain. Prog Brain Res. 1996;110:193–206. doi: 10.1016/s0079-6123(08)62575-3. [DOI] [PubMed] [Google Scholar]
- 39.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 40.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;308:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 41.Woolf CJ, King AE. Physiology and morphology of multireceptive neurons with C-afferent fiber inputs in the deep dorsal horn of the rat lumbar spinal cord. J Neurophysiol. 1987;58:460–479. doi: 10.1152/jn.1987.58.3.460. [DOI] [PubMed] [Google Scholar]
- 42.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 43.Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci. 1986;6:1433–1442. doi: 10.1523/JNEUROSCI.06-05-01433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zieglgansberger W, Herz A. Changes of cutaneous receptive fields of spino-cervical-tract neurones and other dorsal horn neurones by microelectrophoretically administered amino acids. Exp Brain Res. 1971;13:111–126. doi: 10.1007/BF00234080. [DOI] [PubMed] [Google Scholar]