

Abstract
Benzodiazepines (BZDs) exert their effects in the CNS by binding to a modulatory site on GABAA receptors. Individual amino acids have been implicated in BZD recognition and modulation of the GABAA receptor, but the secondary structure of the amino acids contributing to the BZD binding site has not been elucidated. In this report we used the substituted cysteine accessibility method to understand the structural dynamics of a region of the GABAA receptor implicated in BZD binding, γ2Y72–γ2Y83. Each residue within this region was mutated to cysteine and expressed with wild-type α1 and β2 subunits inXenopus oocytes. Methanethiosulfonate (MTS) reagents were used to modify covalently the engineered cysteines, and the subsequent effects on BZD modulation of the receptor were monitored functionally by two-electrode voltage clamp. We identified an alternating pattern of accessibility to sulfhydryl modification, indicating that the region γ2T73–γ2T81 adopts a β-strand conformation. By monitoring the ability of BZD ligands to impede the covalent modification of accessible cysteines, we also identified two residues within this region, γ2A79 and γ2T81, that line the BZD binding site. Sulfhydryl modification of γ2A79C or γ2T81C allosterically shifts the GABA EC50 of the receptor, suggesting that certain MTS compounds may act as tethered agonists at the BZD binding site. Last, we present structural evidence that a portion of the BZD binding site undergoes a conformational change in response to GABA binding and channel gating (opening and desensitization). These data represent an important step in understanding allosteric communication in ligand-gated ion channels.
Keywords: benzodiazepine, binding site, allostery, ligand-gated ion channel, GABA, GABAA receptor, substituted cysteine accessibility method, Xenopus oocytes, secondary structure
Benzodiazepines (BZDs) are among the most commonly prescribed therapeutics in the treatment of panic disorder, sleeplessness, and epilepsy (Doble and Martin, 1996). BZDs exert their anxiolytic and hypnotic effects by binding to a unique site on the GABAA receptor, the main inhibitory ligand-gated ion channel (LGIC) in the CNS (Hevers and Lüddens, 1998). BZD ligands encompass a full spectrum of efficacy and can potentiate, inhibit, or have no effect on GABA currents, depending on the ligand that is bound. BZD agonists increase GABA-gated Cl− conductance by allosterically decreasing the GABA concentration needed to elicit half-maximal channel activity (EC50; Hevers and Lüddens, 1998), thus making them powerful modulators of inhibitory tone in the brain. Although several studies have made progress toward identifying amino acids on the GABAA receptor involved in BZD binding, a detailed structural map of the BZD binding pocket does not exist yet.
Both GABAA receptor α- and γ-subunits play critical roles in BZD binding and modulation of GABA-activated current (IGABA). It has been hypothesized that the BZD binding site is localized at the interface of these two subunits (for review, see Sigel and Buhr, 1997). To date, six residues in the γ2 subunit have been shown to affect ligand discrimination at the BZD site: γ2F77 (Buhr et al., 1997; Sigel et al., 1998), γ2A79 and γ2T81 (Kucken et al., 2000), γ2M130 (Buhr and Sigel, 1997; Wingrove et al., 1997), and γ2M57 and γ2Y58 (Buhr and Sigel, 1997; Kucken et al., 2000). Because these amino acids were identified by using chimeric and site-directed mutagenesis, none has been shown conclusively to line the BZD binding site itself.
The substituted cysteine accessibility method (SCAM) has been used previously to gain insight into the secondary structure of ion channels and ligand binding sites (for review, see Karlin and Akabas, 1998). In this study we used SCAM to examine the structure and dynamics of the γ2F77 region of the BZD binding site. We demonstrate that the polypeptide backbone surrounding γ2F77 is a β-strand, that γ2A79 and γ2T81 line the BZD binding pocket, and that the structure of the BZD binding site undergoes a conformational change during gating. Additionally, we provide evidence that modification of the BZD binding site by MTSEA-biotin or MTSEA-biotin-CAP, two sulfhydryl-specific reagents, allosterically shifts the sensitivity of the GABAA receptor for GABA. Our data provide a detailed molecular model of a portion of the BZD binding site and potentially describe the allosteric transitions that underlie BZD modulation of the GABAA receptor.
MATERIALS AND METHODS
Cysteine mutagenesis. Rat cDNAs encoding α1, β2, and γ2S GABAA receptor subunits were used for all molecular cloning and functional studies. γ2 Cysteine mutants were made by a modified form of recombinant PCR described previously (Kucken et al., 2000). Wild-type and mutant subunits were subcloned into pGH19 (Liman et al., 1992; Robertson et al., 1996) for expression in Xenopus laevis oocytes. All γ2 cysteine mutants were verified by double-stranded DNA sequencing and restriction enzyme analysis. The γ2 cysteine mutants are named with single letter amino acid code as follows: wild-type residue, residue number of the mature protein, mutant residue (e.g., A79C).
cRNA expression in Xenopus laevis oocytes. Capped cRNAs encoding individual γ2 cysteine mutants were transcribed in vitro from NheI-linearized cDNA template with the mMessage mMachine T7 kit (Ambion, Austin, TX). Oocytes were harvested from X. laevis and prepared for injection as described previously (Boileau et al., 1999). Briefly, oocytes were incubated in collagenase (0.25 mg/ml) in Ca2+-free ND96 [(in mm) 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.2] for 20 min at room temperature and defolliculated in osmotic shock solution [130 mmK2HPO4 and 1 mg/ml bovine serum albumin (BSA), pH 6.5] for 30 min at room temperature. Single oocytes were injected within 24 hr with 27 nl of cRNA (10–200 pg/nl per subunit) in the ratio 1:1:10 (α:β:γ; Boileau et al., 1998;Boileau and Czajkowski, 1999). Oocytes were stored for 2–14 d at 16°C in ND96 (as above, with 1.8 mmCaCl2) supplemented with 100 μg/ml gentamycin and 100 μg/ml BSA and were assayed functionally at least 2 d after cRNA injection.
Two-electrode voltage clamp. Oocytes were perfused continuously with ND96 (5 ml/min) while being held under two-electrode voltage clamp at −80 mV. The bath volume was ∼200 μl. Borosilicate electrodes used in recording (0.4–1.6 MΩ) were filled with 3m KCl. Electrophysiological data were acquired with a GeneClamp 500 (Axon Instruments, Foster City, CA) interfaced to a computer with an IT16 analog-to-digital device (Instrutech, Great Neck, NY). Dr. Sepinwall (Hoffman-La Roche, Nutley, NJ) generously supplied the BZDs used in this study. Working concentrations of flurazepam (FLZM) were made up in ND96 by diluting from a 10 mm stock made in water. Concentrations of Ro 15-1788 were made up in ND96 by diluting from a 10 mm stock made in DMSO. The final concentration of DMSO in solution was always <1% and did not affect GABAA receptor properties.
Methanethiosulfonate (MTS) reagents. Three derivatives of methanethiosulfonate (CH3SO2SCH2CH2X; MTS) were used to modify covalently the introduced cysteines: MTS ethylammonium (X = NH3+; MTSEA),N-biotinylaminoethyl MTS (X = NH-biotin; MTSEA-biotin), and N-biotinylaminoethyl CAP MTS (X = NHCO(CH2)5NH-biotin; MTSEA-biotin-CAP). MTSEA-biotin was used for initial accessibility studies. For rate determinations, MTSEA, MTSEA-biotin, and MTSEA-biotin-CAP were each used to modify accessible cysteines covalently. These reagents were chosen because they had the greatest effect on BZD potentiation of IGABAfor receptors containing γ2D75C, γ2A79C, and γ2T81C, respectively.
Concentration–response analysis. GABA concentration–responses were scaled to a low, nondesensitizing concentration of GABA (EC2-EC10) applied just before the test GABA concentration to correct for any slow drift inIGABA responsiveness over the course of the experiment. All concentration–response data were fit by the following equation:
where I is the current response,Imax is the maximal current response, [L] is the drug concentration, EC50is the drug concentration that evokes half-maximal current response, and n is the Hill coefficient. The FLZM potentiation ofIGABA was defined as:
where IGABA+FLZM is the current response in the presence of GABA and FLZM, andIGABA is the current evoked solely by GABA. FLZM potentiation was measured at low concentrations of GABA (EC2-EC10).
GABA concentration–response properties of γ2A79C- and γ2T81C-containing receptors also were measured after MTSEA-biotin and MTSEA-biotin-CAP modification. In these experiments the responses of α1β2γ2A79C or α1β2γ2T81C receptors to different concentrations of GABA were measured in the same oocyte before and after the application of 2 mm MTS reagent for 2 min. We also examined the ability of FLZM to shift the GABA EC50 of wild-type and γ2A79C-containing receptors by measuring GABA concentration–response curves in the presence of 1 μmFLZM. For both GABA and FLZM concentration–response curves, individual curve fits were obtained from single oocytes. Log EC50 values andnH values derived from the single curve fits were averaged and compared statistically by one-way ANOVA with Dunnett's post test for significance of differences. Data analysis and curve fitting were performed by using AxoGraph (Axon Instruments) and Prism software (GraphPad, San Diego, CA).
MTSEA-biotin modification. GABA and BZD current responses of oocytes expressing α1β2γ2or α1β2γ-mutant receptors were stabilized before exposure to MTS reagents (Toronto Research Biochemicals, Downsview, Ontario) by applying two to four pulses of each ligand over a 20 min period. Stability was defined as <3% variance of peak current responses to both GABA and FLZM. For all experiments, FLZM was used to measure the BZD potentiation ofIGABA before and after the MTS treatment. GABA concentrations ranged from EC2 to EC10, and FLZM concentrations were approximately EC80. Because γ2D75C- and γ2I76C-containing receptors exhibited a rightward shift in responsiveness to FLZM, these mutants were tested with 5 μm FLZM. The effects of covalent modification by MTSEA-biotin were tested as follows: after achieving current stability, IGABA andIGABA+FLZM were measured, followed by a 3 min wash; 2 mm MTSEA-biotin was bath-applied for 2 min, followed by a 5 min wash; thenIGABA andIGABA+FLZM were redetermined at the same concentrations that were used before MTSEA-biotin treatment. The covalent effect of MTSEA-biotin was taken as:
MTS rates of reaction. Rates of sulfhydryl-specific covalent modification of α1β2γ2D75C, α1β2γ2A79C, and α1β2γ2T81C receptors were determined by monitoring the effect of sequential subsaturating applications of MTS reagents on the potentiation ofIGABA by FLZM. Rates were determined as follows: after achieving current stability,IGABA andIGABA+FLZM were measured by applying 1 μm GABA and 1 μm GABA plus 1 μm FLZM, respectively (except in the case of receptors containing γ2D75C, when 5 μm FLZM was used); the oocyte was washed for 30 sec in ND96; the MTS reagent was applied by using a concentration and duration of application for which a robust effect could be observed but that did not result in a complete block of BZD potentiation; the oocyte was washed for 3 min in ND96; IGABAand IGABA+FLZM were redetermined, and the entire sequence was repeated. This protocol was continued until the reaction was complete (IGABA+FLZM no longer changed). Concentrations and durations of MTS application were as follows: γ2D75C, 200 μm MTS-EA, 10 sec; γ2A79C, 200 μmMTSEA-biotin, 5 sec; and γ2T81C, 20 μm MTSEA-biotin-CAP, 5 sec. The decrease in FLZM potentiation of IGABA was plotted versus cumulative time of MTS exposure and fit to the single-exponential decay equation:
where A is the initial response, k is the pseudo-first-order rate constant of the reaction, and t is the time in seconds (GraphPad). The derived pseudo-first-order rate constant was converted into a second-order rate constant (k2, m/sec) by dividing by the concentration of MTS reagent that was used (Pascual and Karlin, 1998). The effects of different drugs on the MTS reaction rates were assayed by the coapplication of GABA, FLZM, or Ro 15-1788 with the MTS reagent. Concentrations of drugs used in these experiments were as follows: γ2D75C, 5 μmFLZM, 1 μm Ro 15-1788, 100 μm GABA; γ2A79C, 5 μm FLZM, 5 μm Ro 15-1788, 100 μm GABA; γ2T81C, 5 μm FLZM, 1 μm Ro 15-1788, 100 μmGABA. With the exception of FLZM in experiments with γ2D75C-containing receptors, the concentrations of ligands reflect approximate EC95concentrations. In some cases, after treating the oocytes with MTS reagent in the presence of a BZD, we reexposed receptors to the same concentration of MTS reagent alone to demonstrate that a maximal decrease in FLZM potentiation of IGABAwas still obtainable.
Statistics. In all experiments the data were analyzed by one-way ANOVA, applying the Dunnett's post test for significance of differences between treatments (p < 0.05; GraphPad).
RESULTS
Expression and functional characterization of cysteine mutants
The 12 amino acids within the region γ2Y72–γ2Y83 were each mutated to cysteine (Fig. 1). This region of the γ2 subunit includes γ2F77, which has been shown previously to participate in BZD ligand discrimination and likely participates in the formation of the BZD binding site (Buhr et al., 1997; Sigel et al., 1998). To assess whether cysteine mutations affected GABAA receptor function and/or expression, we characterized the responsiveness of α1β2γ2mutant receptors to GABA and BZDs. Individual cysteine mutant γ2 subunits were coexpressed with wild-type α1 and β2 subunits inX. laevis oocytes, and GABA-elicited currents (IGABA) as well as FLZM potentiation of IGABA were measured with two-electrode voltage clamp.
Fig. 1.

Aligned partial sequences of the rat GABAA receptor γ1–3 and α1subunit isoforms. Numbering reflects alignment with the mature γ2 subunit. Residues in γ1 and γ3 that are identical to γ2 residues are represented by a dash. γ2F77, thecircled residue, has been implicated previously in BZD binding and modulation (Buhr et al., 1997; Sigel et al., 1998). Each amino acid in the region γ2Y72–γ2Y83 was mutated individually to cysteine and is denoted by a Cabove the corresponding wild-type γ2 residue.Underlined residues in α1 (Boileau et al., 1999) and γ2 represent amino acids that are accessible to sulfhydryl-specific modification after mutation to cysteine.
Cysteine substitution was well tolerated within the region γ2Y72–γ2Y83. The GABA EC50 values for eight cysteine mutants were not significantly different from wild-type values. For γ2Y72C-, γ2D75C-, and γ2F78C-containing receptors the GABA EC50 values were shifted less than fourfold (Table 1; Fig.2A). Cysteine substitutions had no effect on the calculated Hill coefficients for GABA-mediated activation of the receptor.
Table 1.
Summary of GABA and flurazepam concentration–response data from cysteine mutant and wild-type α1β2γ2 GABAAreceptors
| Receptor | GABA | Flurazepam | ||||||
|---|---|---|---|---|---|---|---|---|
| EC50(μm) | nH | n | EC50mut/EC50 αβγ | EC50(nm) | nH | n | EC50mut/EC50 αβγ | |
| αβγ | 18 ± 4.7 | 1.7 ± 0.3 | 3 | 1.0 | 250 ± 49 | 1.2 ± 0.1 | 3 | 1.0 |
| αβγY72C | 70 ± 121-160 | 1.3 ± 0.2 | 3 | 3.9 | 270 ± 64 | 1.3 ± 0.3 | 3 | 1.1 |
| αβγT73C | 20 ± 5.6 | 1.6 ± 0.3 | 3 | 1.1 | 160 ± 15 | 1.8 ± 0.3 | 3 | 0.6 |
| αβγI74C | 30 ± 4.5 | 1.2 ± 0.1 | 3 | 1.7 | 80 ± 12 | 2.9 ± 1.2 | 3 | 0.3 |
| αβγD75C | 39 ± 4.91-160 | 1.5 ± 0.3 | 3 | 2.2 | 4700 ± 13001-160 | 1.6 ± 0.6 | 3 | 18.8 |
| αβγI76C | 30 ± 13 | 1.4 ± 0.5 | 3 | 1.7 | 2600 ± 12001-160 | 2.4 ± 0.7 | 3 | 10.4 |
| αβγF77C | 24 ± 11 | 1.0 ± 0.3 | 3 | 1.3 | >10,000 | 3 | ||
| αβγF78C | 33 ± 5.8* | 1.3 ± 0.2 | 4 | 1.8 | 130 ± 35 | 1.8 ± 0.3 | 3 | 0.5 |
| αβγA79C | 30 ± 12 | 1.4 ± 0.3 | 9 | 1.7 | 310 ± 45 | 1.4 ± 0.3 | 3 | 1.2 |
| αβγQ80C | 9.0 ± 2.5 | 1.6 ± 0.4 | 3 | 0.5 | 260 ± 28 | 1.0 ± 0.1 | 3 | 1.0 |
| αβγT81C | 15 ± 1.6 | 1.5 ± 0.1 | 4 | 0.8 | 350 ± 170 | 1.5 ± 0.2 | 3 | 1.4 |
| αβγW82C | No expression | No expression | ||||||
| αβγY83C | 10 ± 1.9 | 1.6 ± 0.2 | 3 | 0.6 | 170 ± 17 | 1.1 ± 0.2 | 4 | 0.7 |
Data represent mean ± SD values. n, Number of independent experiments; nH, calculated Hill coefficient.
,
F1-160: Indicate values significantly different from wild-type receptors, withp < 0.05 and p < 0.01, respectively.
Fig. 2.
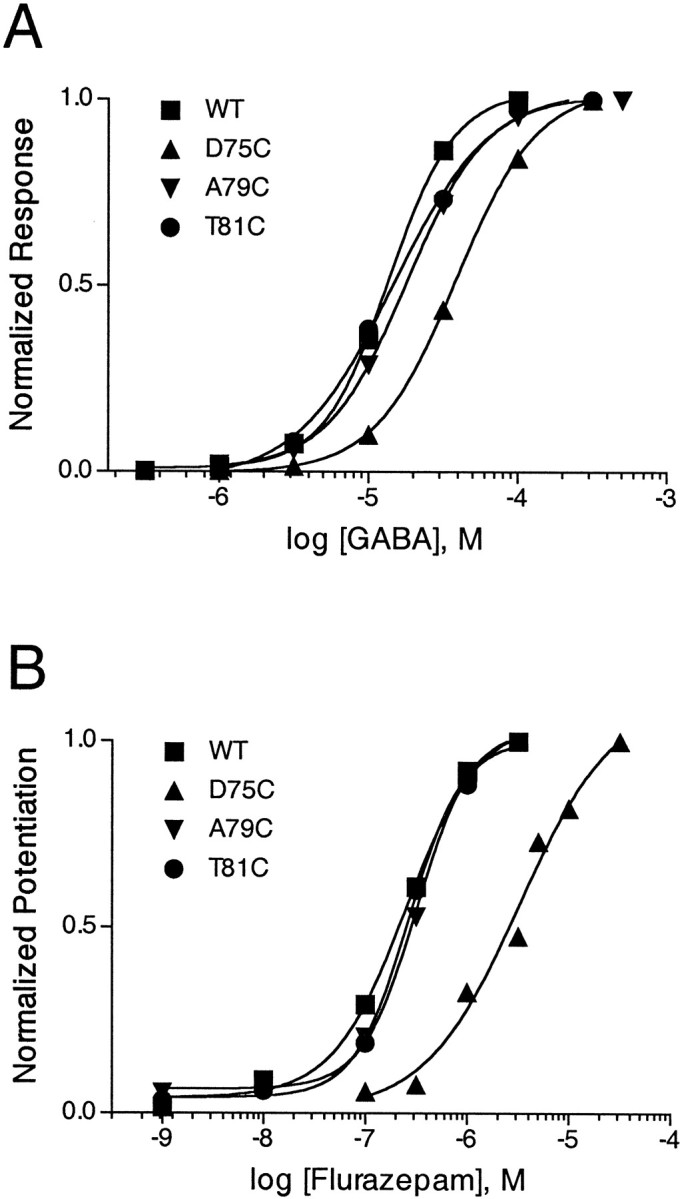
GABA (A) and BZD (B) concentration–response curves of wild-type α1β2γ2 GABAAreceptors and three representative mutant receptors: α1β2γ2D75C, α1β2γ2A79C, and α1β2γ2T81C. Oocytes expressing α1, β2, and γ2 or γ-mutant subunits were treated with increasing concentrations of GABA or flurazepam (FLZM) while current responses were recorded by using two-electrode voltage clamp. A, Responses to GABA are normalized to IGABA Max. B, FLZM potentiation ofIGABA was measured with 1 μmGABA. For each mutant, FLZM potentiation is normalized to maximal potentiation. Data were fit by nonlinear regression, as described in Materials and Methods. Experiments were performed at least three times with similar results. EC50 values obtained from the curve fits are reported in Table 1.
Because the presence of a γ2 subunit confers BZD sensitivity to GABAA receptors (Hevers and Lüddens, 1998), detectable potentiation ofIGABA in the presence of FLZM was taken to indicate functional expression of a mutant γ2 subunit. Cysteine substitution of eight mutants in the γ2 subunit did not disrupt sensitivity to FLZM. For γ2D75C- and γ2I76C-containing receptors, FLZM EC50 values were increased 19- and 10-fold, respectively (Table 1; Fig. 2B). FLZM-associated Hill coefficients were not noticeably different from wild-type values, except for γ2I74C- and γ2I76C-containing receptors, which displayed Hill numbers of 2.9 ± 1.2 and 2.4 ± 0.7, respectively. An increased Hill coefficient may be an indication of mutational gain of cooperativity (Colquhoun, 1998). However, because Hill coefficients are based on a scale of whole numbers, these numbers may not be different from wild-type values.
FLZM did not potentiate IGABA in γ2F77C- and γ2W82C-containing receptors. To determine whether these mutant subunits specifically disrupted FLZM potentiation or impaired receptor assembly, we assessed the Zn2+ sensitivities of α1β2γ2F77C and α1β2γ2W82C receptors. GABA receptors composed of α1β2 subunits are more sensitive to Zn2+ blockade than α1β2γ2receptors; thus Zn2+ sensitivity ofIGABA can be used to assess γ-subunit expression (Draguhn et al., 1990; Gingrich and Burkat, 1998). ZnCl2 (10 μm), when coapplied with 10 μm GABA, reducesIGABA by 80 ± 7% in α1β2 receptors but only by 22 ± 4% in α1β2γ2receptors (n = 3; Fig.3). For α1β2γ2W82C and α1β2γ2F77C receptors, ZnCl2 reducedIGABA by 80 ± 14% and 30 ± 3%, respectively (n = 3; Fig. 3). Because the Zn2+ block ofIGABA in α1β2γ2W82C receptors is indistinguishable from α1β2 receptors, it is likely that cysteine substitution at this residue is detrimental to assembly and/or cell surface expression of the γ2W82C subunit.
Fig. 3.

Zn2+ sensitivity of α1β2, α1β2γ2, and α1β2γ2 mutant GABAA receptors. GABA-activated current traces were recorded from oocytes expressing α1β2, α1β2γ2, α1β2γ2F77C, or α1β2γ2W82C receptors.Bars represent 10–20 sec applications of 10 μm GABA in the presence or absence of 10 μmZnCl2. Data reflect triplicate determinations.
In contrast, the small amount of Zn2+block observed for α1β2γ2F77C receptors indicates that cysteine substitution at γ2F77 does not impair γ-subunit assembly and/or surface expression; thus the inability of FLZM to potentiateIGABA is likely attributable to a direct effect of the mutation on BZD binding. FLZM was unable to potentiate IGABA in α1β2γ2F77C receptors even at high concentrations (>10 μm), suggesting that this mutation severely disrupts the BZD potentiation ofIGABA. Several structurally diverse BZD agonists also were applied to oocytes expressing α1β2γ2F77C receptors, including zolpidem and Cl 218–872, to identify a BZD for which this mutation did not disrupt recognition. None of the BZDs that were tested had an effect on IGABA, suggesting that cysteine substitution at γ2F77 disrupts BZD binding site architecture. In addition, α1β2γ2F77C receptors were expressed in human embryonic kidney (HEK) 293 cells, and the specific binding of [3H]flunitrazepam and [3H]Ro 15-1788 was measured. No specific binding was detected (data not shown). Taken together, these results suggest that cysteine substitution at γ2F77 disrupts BZD binding and supports previous evidence that this residue is crucial for BZD recognition (Buhr et al., 1997; Sigel et al., 1998).
Reaction of substituted cysteines with MTSEA-biotin
SCAM has been used previously to generate novel information about the secondary structure and conformational dynamics of the GABAA receptor agonist binding site (Boileau et al., 1999; Wagner and Czajkowski, 2001) and constituent ion channel (Xu and Akabas, 1996; Williams and Akabas, 1999, 2000). In this method, consecutive amino acids are mutated one at a time to cysteine, expressed heterologously in vitro, and treated with sulfhydryl-specific reagents. Accessibility is defined by observing whether changes in receptor function occur after treatment. A major assumption of SCAM is that the mutation of a candidate amino acid to cysteine does not disrupt the orientation or accessibility of the native side chain radically. Given our evidence that GABA and FLZM EC50 values for eight cysteine mutants have not been altered radically by mutation (see Table 1), it is likely that the positions of these introduced cysteine side chains reflect wild-type orientations. Although γ2D75C- and γ2I76C-containing receptors display decreased sensitivity to FLZM, GABA EC50 values for these cysteine mutants are unchanged (see Table 1), suggesting that mutation at these positions does not disrupt the native structure of the receptor protein fundamentally.
We measured FLZM modulation of IGABAin X. laevis oocytes expressing wild-type α1β2γ2or α1β2γ2-mutant GABAA receptors before and after treatment with 2 mm MTSEA-biotin for 2 min. Exposure of wild-type GABAA receptors to MTSEA-biotin had no significant effect on IGABA or on the FLZM potentiation of IGABA (Figs.4B, 6C). Therefore, if effects on FLZM potentiation were observed in cysteine mutant receptors after treatment with MTSEA-biotin, we interpreted this result as evidence that covalent modification occurred at the introduced cysteine. MTSEA-biotin treatment of receptors containing γ2Y72C, γ2I74C, γ2I76C, γ2F78C, γ2Q80C, or γ2Y83C had no effects on the FLZM potentiation ofIGABA (Fig. 4). Thus either these introduced cysteines were not accessible to MTSEA-biotin modification, or their modification by MTSEA-biotin had no observable effect on FLZM potentiation.
Fig. 4.
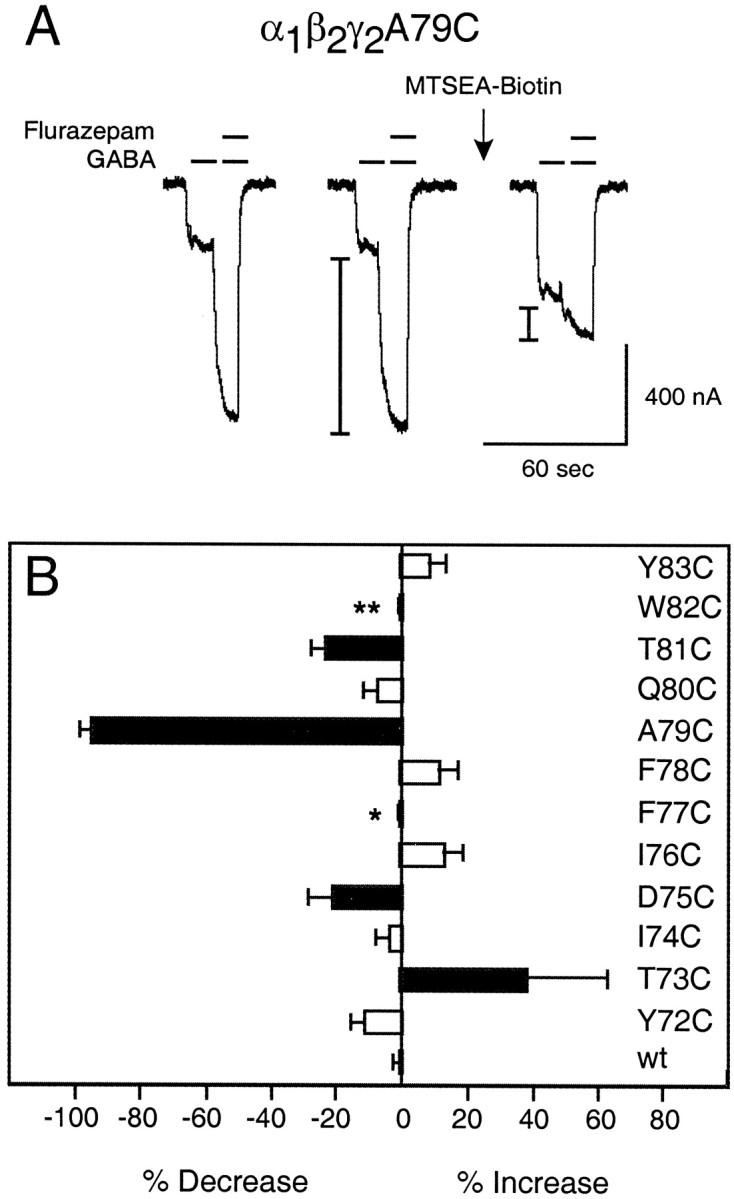
MTSEA-biotin effects on the γ2Y72C–γ2Y83C region. A, Representative current traces from α1β2γ2A79C receptors showing FLZM modulation of IGABA before and after a 2 min application of 2 mm MTSEA-biotin.I-bars denote potentiation ofIGABA measured during an application of 1 μm FLZM in the presence of 1 μm GABA. Note the decrease in FLZM potentiation and the increase inIGABA after MTSEA-biotin modification (arrow). B, Changes in FLZM potentiation after MTSEA-biotin modification of αβγ (wild-type;wt) and mutant receptors. The percentage of change in FLZM potentiation after modification is defined as [((FLZM PotentiationAfter/FLZM PotentiationBefore) − 1)·100]. A negative value represents a decrease in FLZM potentiation after MTSEA-biotin reaction, and a positive value represents an increase in FLZM potentiation after MTSEA-biotin reaction. Black barsindicate mutants in which the change in potentiation was significantly different (p < 0.01) from wtreceptor calculated by a one-way ANOVA with a Dunnett's post test. Data represent mean ± SD from at least three independent experiments. *No detectable BZD potentiation ofIGABA; **no detectable γ2 subunit expression.
Fig. 6.
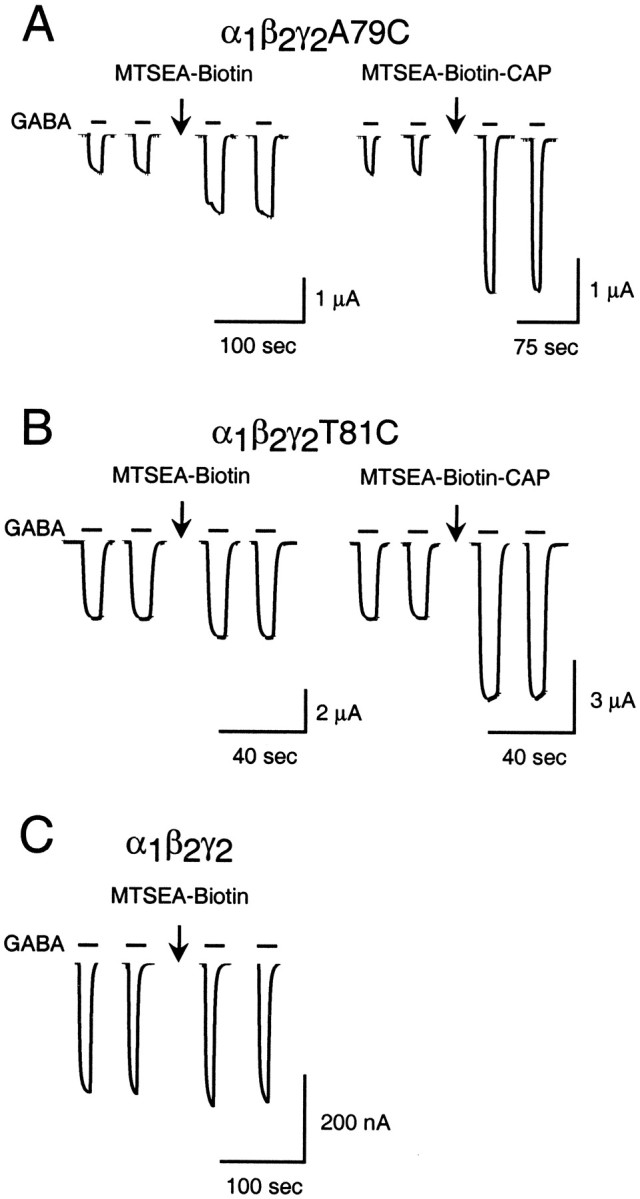
MTS modification of α1β2γ2A79C and α1β2γ2T81C receptors increases IGABA. Traces represent the effect of 2 min applications (arrows) of 2 mmMTSEA-biotin or 2 mm MTSEA-biotin-CAP on current evoked by 3 μm GABA in oocytes expressing receptors containing either γ2A79C (A) or γ2T81C (B) subunits. The application of 2 mmMTSEA-biotin to oocytes expressing α1β2γ2(C) or α1β2γ2T81C (B) receptors had no significant effect onIGABA.
In contrast, MTSEA-biotin treatment of receptors containing γ2T73C, γ2D75C, γ2A79C, and γ2T81C significantly altered the FLZM modulation ofIGABA (Fig. 4). After the application of MTSEA-biotin, the FLZM potentiation ofIGABA was increased by 38 ± 25% for γ2T73C-containing receptors, whereas potentiation was decreased by 22 ± 8%, 95 ± 2%, and 23 ± 4% for γ2D75C-, γ2A79C-, and γ2T81C-containing receptors, respectively. The alternating pattern of accessibility within the region bounded by γ2T73 and γ2T81 suggests that this domain of the BZD binding site forms a β-strand.
Identification of BZD binding site residues
We examined the extent to which both FLZM and Ro 15-1788 could slow the rate of reaction of MTS reagents with accessible cysteines to identify residues within γ2Y72–γ2Y83 that line the BZD binding pocket. Although Ro 15-1788 is a BZD antagonist that competitively blocks the binding of FLZM, it does not enhance or inhibit IGABA. Thus if the rate at which a MTS reagent reacts with an introduced cysteine is slowed by both FLZM and Ro 15-1788, then it is likely that both compounds are blocking the MTS reaction sterically and that the introduced cysteine is positioned in the BZD binding site.
MTS reaction rates were measured by examining the decrease in FLZM potentiation of IGABA after repeated exposure to MTSEA (α1β2γ2D75C), MTSEA-biotin (α1β2γ2A79C), or MTSEA-biotin-CAP (α1β2γ2T81C). The decrease in FLZM potentiation of the receptor was plotted against the cumulative time of MTS exposure, and the data were fit with a single-exponential decay curve. Second-order rate constants (k2) for the MTS reaction with the introduced cysteines were calculated from curve fits (Fig.5; see Materials and Methods).
Fig. 5.
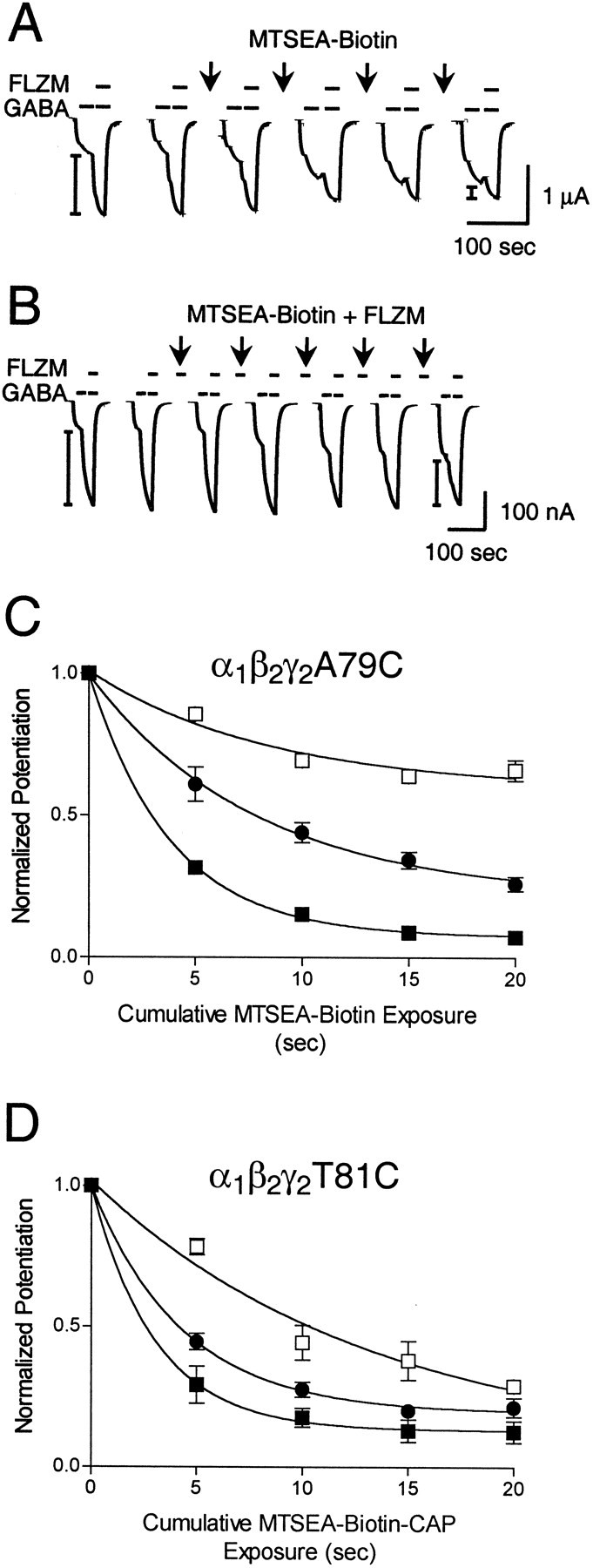
Rate of sulfhydryl modification of α1β2γ2A79C and α1β2γ2T81C receptors in the presence and absence of FLZM and Ro 15-1788. A, B, Representative GABA (1 μm) and GABA plus FLZM (1 μm each) current traces recorded from α1β2γ2A79C receptors.Arrows indicate 5 sec applications of 200 μm MTSEA-biotin alone (A) or 200 μm MTSEA-biotin plus 5 μm FLZM (B). FLZM potentiation ofIGABA was measured before and after each MTS treatment. I-bars on traces show BZD-potentiated current. C, Observed decreases in FLZM potentiation ofIGABA were plotted versus cumulative MTSEA-biotin exposure in α1β2γ2A79C receptors. Data obtained from individual experiments were normalized to the potentiation measured at t = 0 and fit to single-exponential decay curves (▪, MTS alone; ●, MTS + 5 μm FLZM; ■, MTS + 5 μm Ro 15-1788). Data points are mean ± SD from at least three independent experiments.D, Rate experiments were performed similarly for receptors containing γ2T81C, except that 5 sec applications of 20 μm MTSEA-biotin-CAP were used in place of MTSEA-biotin (▪, MTS alone; ●, MTS + 5 μm FLZM; ■, MTS + 5 μm Ro 15-1788). The calculated second-order rate constants for the MTS reaction are presented in Table 2.
Introduction of a cysteine at position γ2A79 created a free sulfhydryl that reacted with MTSEA-biotin. Both FLZM (∼EC94) and Ro 15-1788 (∼EC98) significantly slowed the rate of sulfhydryl modification of γ2A79C by MTSEA-biotin (p < 0.05; Table2; Fig. 5). α1β2γ2T81C receptors reacted robustly only with MTSEA-biotin-CAP. Modification of γ2T81C-containing receptors by MTSEA-biotin-CAP was slowed significantly by Ro 15-1788 (∼EC98;p < 0.05), but not by FLZM (∼EC93; Table 2; Fig. 5). The rate of modification of α1β2γ2D75C receptors by MTSEA was unchanged in the presence of FLZM (∼EC50) and Ro 15-1788 (∼EC98; Table 2). Taken together, these data indicate that γ2A79 and γ2T81, but not γ2D75, lie within the BZD binding site. In addition, Ro 15-1788, but not FLZM, protects γ2T81C from covalent modification, suggesting that γ2T81C may participate in forming an overlapping binding site subdomain for Ro 15-1788. We did not evaluate γ2T73 as a potential binding site candidate because sulfhydryl-specific derivitization of this residue resulted in an increase in BZD potentiation ofIGABA. This result suggests that γ2T73 is not within the BZD binding domain because it does not disrupt BZD recognition once it has been derivitized. It is possible that the increased BZD efficacy we have observed after modification of γ2T73 is attributable to conformational changes in the BZD site that correspondingly increase the sensitivity to FLZM.
Table 2.
Summary of second-order rate constants for reaction of MTS compounds with receptors containing γ2D75C, γ2A79C, or γ2T81C in the absence (control) or presence of BZD ligands
| Receptor | Reagent | Control | FLZM | Ro 15–1788 | GABA | ||||
|---|---|---|---|---|---|---|---|---|---|
| k2(m−1s−1) | n | k2(m−1s−1) | n | k2(m−1s−1) | n | k2(m−1s−1) | n | ||
| αβγD75C | MTSEA | 420 ± 200 | 3 | 530 ± 210 | 3 | 510 ± 250 | 3 | 580 ± 70 | 3 |
| αβγA79C | MTSEA-biotin | 1300 ± 200 | 5 | 670 ± 250* | 3 | 600 ± 1002-160 | 3 | 3700 ± 3002-160 | 3 |
| αβγT81C | MTSEA-biotin-CAP | 17,000 ± 4400 | 3 | 12,000 ± 1700 | 3 | 3700 ± 8002-160 | 3 | 22,000 ± 2300 | 3 |
Second-order rate constants (k2) were derived by dividing the fit pseudo-first-order rate constants by the concentration of MTS reagent used (see Materials and Methods). The concentration of MTS compounds used was as follows (in μm): MTSEA, 200; MTSEA-biotin, 200; MTSEA-biotin-CAP, 20. Data represent mean ± SD values.
,
F2-160: Indicate values significantly different from control (MTS alone), with p < 0.05 andp < 0.01, respectively.
Tethered MTSEA-biotin and MTSEA-biotin-CAP allosterically modulate GABA apparent affinity
After the reaction of MTSEA-biotin or MTSEA-biotin-CAP with α1β2γ2A79C receptors, we observed that IGABA was increased substantially (see Figs. 4A,6A). To gain insight into the chemical specificity of this effect, we examined whether other MTS reagents, including MTSEA, MTS-ethyltrimethylammonium (MTSET), MTS-ethylsulfonate (MTSES), and benzyl-MTS, also could modulateIGABA when tethered to γ2A79C. Although all of the MTS reagents that were tested reacted with γ2A79C, as evidenced by a decreased FLZM potentiation ofIGABA, only MTSET, MTSEA-biotin, and MTSEA-biotin-CAP increased IGABA (data not shown). MTSEA-biotin and MTSEA-biotin-CAP are the largest reagents that were tested, and MTSET is positively charged. The data suggest that large and/or positively charged compounds may be better suited to initiate allosteric changes in the receptor protein once they are attached covalently to the BZD binding site. Interestingly, robust increases in IGABA were observed after MTSEA-biotin-CAP, but not MTSEA-biotin, modification of γ2T81C-containing receptors (Fig.6B).
We hypothesized that the increases inIGABA were attributable to changes in the GABA EC50 values of α1β2γ2A79C and α1β2γ2T81C receptors after MTS modification. To test this hypothesis, we measured complete GABA concentration–response curves in single oocytes expressing α1β2γ2A79C receptors before and after the application of 2 mm MTSEA-biotin (Fig.7A) or 2 mm MTSEA-biotin-CAP (Fig. 7B). Covalent modification of γ2A79C-containing receptors by MTSEA-biotin resulted in a significant ∼1.6-fold increase in GABA EC50 (p< 0.05; Table 3). Likewise, MTSEA-biotin-CAP modification of γ2A79C-containing receptors resulted in a ∼2.6-fold increase in GABA EC50(p < 0.01; Table 3). The GABA EC50 shifts that were measured after the covalent modification of γ2A79C were similar to the shift in GABA EC50 observed in the presence of FLZM. Coapplications of 1 μm FLZM during a GABA concentration–response protocol resulted in a significant ∼3.8-fold increase in the EC50 of α1β2γ2A79C receptors for GABA (p < 0.01; Fig.7C) and a ∼3.2-fold shift in wild-type receptors (Table3). Taken together, our data suggest that the addition of a MTS reagent to the BZD binding site may initiate structural changes in the receptor that mimic perturbation by an agonist. This effect can be explained most simply by a model in which MTSEA-biotin and MTSEA-biotin-CAP mechanistically act like BZD partial agonists when tethered to γ2A79C.
Fig. 7.
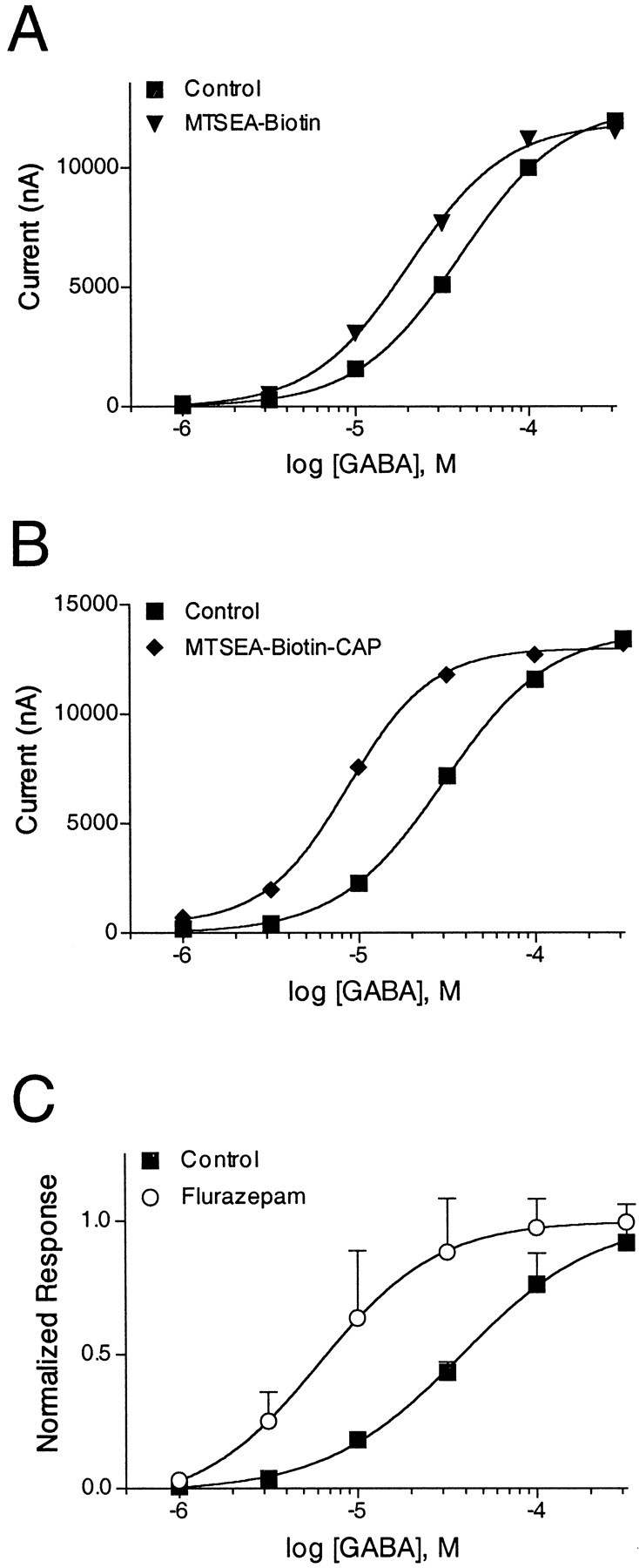
MTSEA-biotin and MTSEA-biotin-CAP shift GABA EC50 when linked covalently to γ2A79C.A, B, GABA concentration–response curves obtained from single oocytes expressing α1β2γ2A79C receptors before (▪) and after (▾) reaction with 2 mm MTSEA-biotin (A) or before (▪) and after (♦) reaction with 2 mm MTSEA-biotin-CAP (B). The experiments were repeated two additional times with similar results.C, GABA concentration–response curves obtained from α1β2γ2A79C receptors in the absence (▪) and presence (○) of 1 μm FLZM. Data were fit by nonlinear regression, as described in Materials and Methods. Data represent mean ± SEM from three independent experiments. EC50 values obtained from the curve fits are reported in Table 3.
Table 3.
GABA EC50 values of α1β2γ2 and α1β2γ2A79C receptors before and after treatment with 2 mm MTSEA-biotin, 2 mm MTSEA-biotin-CAP, or 1 μm FLZM
| Receptor | GABA EC50 (μm) before | n | Treatment | GABA EC50(μm) after | n | EC50before/EC50 after |
|---|---|---|---|---|---|---|
| αβγ | 18 ± 4.73-a | 3 | FLZM | 5.5 ± 0.93-160 | 3 | 3.3 |
| αβγA79C | 30 ± 123-a | 9 | FLZM | 7.8 ± 5.63-160 | 3 | 3.8 |
| αβγA79C | 43 ± 6.73-b | 3 | MTSEA-biotin | 28 ± 7.43-b,3-150 | 3 | 1.5 |
| αβγA79C | 29 ± 1.73-c | 3 | MTSEA-biotin-CAP | 11 ± 3.33-c,3-160 | 3 | 2.6 |
Data represent mean ± SD values.
F3-150: ,
F3-160: Indicate values significantly different from GABA before (control), with p < 0.05 and p < 0.01, respectively.
EC50 values taken from Table 1.
EC50 values from single oocyte experiments before and after MTSEA-biotin application.
EC50values from single oocyte experiments before and after MTSEA-biotin-CAP application.
Conformational changes detected within the BZD binding site
According to allosteric theory, modulators bind to a site on the receptor protein that is distinct from the agonist binding site and exert their effects by initiating an allosteric transition in the protein that indirectly modifies the conformation of the agonist binding site (Changeux and Edelstein, 1998). Both radioligand binding and electrophysiological studies of the GABAAreceptor have demonstrated functional interactions between the GABA and BZD binding sites (Skerritt and Johnston, 1983; Boileau and Czajkowski, 1999). Structural evidence, however, for GABA binding site–BZD binding site communication is scarce. To detect directly whether structural changes of the BZD binding site occur during GABA binding and activation of the receptor, we examined whether GABA (100 μm; approximately EC70-EC86) altered the rates of reaction of MTS reagents with α1β2γ2D75C, α1β2γ2A79C, and α1β2γ2T81C receptors. GABA significantly increased the rate of MTS modification of γ2A79C-containing, but not γ2D75C- or γ2T81C-containing, receptors (Fig.8; see Table 2). The ability of GABA to increase the accessibility of γ2A79C to sulfhydryl modification demonstrates that a domain of the BZD binding site undergoes an allosteric structural rearrangement during GABA binding and channel gating.
Fig. 8.
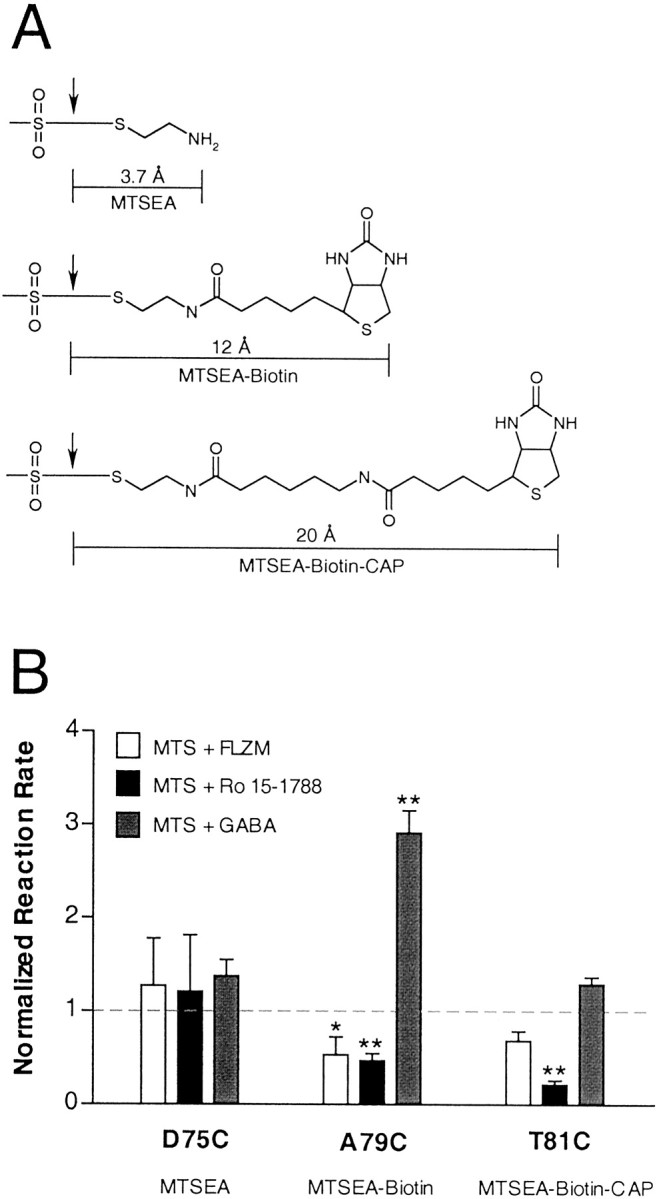
A, Structures and lengths (in angstroms) of the different MTS reagents used in our experiments. Lengths were measured after energy minimization (<0.5 kcal/Å) and represent only the portion of the MTS reagent that covalently modifies an introduced cysteine. Cleavage points of each MTS reagent are indicated by an arrow. B, Summary of the second-order rate constants calculated for MTS derivitization of γ2D75C-, γ2A79C-, and γ2T81C-containing receptors. Oocytes expressing mutant receptors were incubated in the presence of MTS alone (control),MTS + FLZM, MTS + Ro 15-1788, orMTS + GABA. MTS reagents used were as follows: γ2D75C, MTSEA; γ2A79C, MTSEA-biotin; γ2T81C, MTSEA-biotin-CAP. Second-order rate constants were calculated for each MTS reaction and were normalized to the rate measured in the absence of ligand (control). Displayed values are mean ± SD from at least three independent experiments. *,**Indicate values significantly different from control MTS values, with p < 0.05 and p < 0.01, respectively.
DISCUSSION
We used SCAM to examine the structure and dynamics of a region of the GABAA receptor implicated in BZD binding, γ2Y72–γ2Y83 (Buhr et al., 1997; Sigel et al., 1998). Our data indicate that this region is a β-strand. We directly demonstrate that two residues that had been implicated previously in BZD binding, γ2A79 and γ2T81 (Kucken et al., 2000), line the BZD binding site. We show that MTSEA-biotin and MTSEA-biotin-CAP have the ability to act as covalent agonists of the BZD binding site. Last, we demonstrate that a portion of the BZD binding site undergoes structural rearrangements during GABA binding and/or gating.
Identification of amino acids in the BZD binding site
Four residues within γ2Y72–γ2Y83 are accessible to MTSEA-biotin: γ2T73C, γ2D75C, γ2A79C, and γ2T81C. Of these four accessible residues, both γ2A79C and γ2T81C are protected from MTS modification by Ro 15-1788, whereas only γ2A79 is protected from MTS modification by FLZM. Although antagonists may induce conformational changes in the BZD binding site, it is unlikely that these binding-associated structural movements are similar to those induced by an agonist. Thus protection observed at an introduced cysteine in the presence of both an agonist and antagonist is good evidence that the cysteine lines the binding site. Therefore, we believe that γ2A79 is facing into the BZD binding pocket. Because only Ro 15-1788 is able to protect γ2T81C from sulfhydryl modification, we cannot conclude definitively that γ2T81 lines the BZD binding site by our criteria. However, other evidence also suggests that γ2T81 is facing into the binding site. In our study we demonstrate that MTSEA-biotin-CAP acts as a tethered agonist at this site. Moreover, we have shown previously via chimeric mutagenesis studies that both γ2A79 and γ2T81 are important determinants of BZD binding (Kucken et al., 2000). Although we could not evaluate the accessibility of γ2F77C, it has been well established in previous studies that this residue is a critical determinant of BZD binding (Buhr et al., 1997; Sigel et al., 1998). Thus our data support a model in which γ2F77, γ2A79, and γ2T81 line the BZD binding site.
Secondary structure of the γ2Y72–γ2Y83 region of the BZD binding site
Alternating residues within the region γ2T73–γ2T81 are accessible to MTSEA-biotin. These data are consistent with a model in which this region forms a β-strand. Because the accessibility of γ2F77C could not be tested, a strict pattern of alternating exposure has not been established absolutely. The residues accessible to MTSEA-biotin, with the exception of γ2A79, are hydrophilic amino acid residues. Because MTSEA-biotin is relatively impermeant (Chen et al., 1998) and MTS reagents react from 109 to 1010 times faster with ionized sulfhydryl groups than protonated sulfhydryls (Roberts et al., 1986) and ionization of a sulfhydryl is much more probable in an aqueous environment, the accessible residues likely are exposed at the water-accessible surface of the protein. The inaccessible residues are mostly hydrophobic residues and are likely to be buried within the protein. We must be cautious, however, in our interpretation of apparently unreactive residues, because we cannot rule out reactions that appear to have no functional consequences. Nevertheless, it is unlikely that the addition of a large biotin moiety would have no effect on BZD modulation of IGABA if γ2Y72C, γ2I74C, γ2I76C, γ2F78C, or γ2Q80C actually face into the BZD binding pocket. We were unable to test the accessibility of γ2W82C, because cysteine substitution at this residue impaired receptor assembly and/or expression. This tryptophan is highly conserved across many ligand-gated ion channel subunits and previously has been shown to regulate GABAAreceptor α1 subunit assembly (Srinivasan et al., 1999). Thus, it is reasonable to assume that γ2W82 is not solvent-accessible, because it is hydrophobic and likely participates in intraprotein contacts that are associated with subunit assembly.
Taken together, the results of this study strongly suggest that the polypeptide chain from γ2T73 to γ2T81 forms a β-strand and that a portion of this strand lines the BZD binding site. In agreement with our experimental results, this region is predicted by secondary structure modeling algorithms (Chou and Fasman, 1978) to adopt a β-strand conformation. Interestingly, an aligned region of the α1 subunit has been shown to form part of the GABA binding site and displays a similar secondary structure (Boileau et al., 1999).
Structural rearrangements in the BZD binding site
A central question in GABAA receptor pharmacology is how the binding of BZD ligands is transduced into allosteric modulation of the GABAA receptor. It is likely that functional coupling between the BZD and GABA binding sites is accompanied by structural rearrangements in the receptor protein that change the apparent affinity of both sites (Changeux and Edelstein, 1998; Colquhoun, 1998). We demonstrate that a residue that faces into the BZD binding pocket (γ2A79) experiences an increase in accessibility to MTSEA-biotin modification during GABA binding and channel gating (see Fig. 8). In the time course of our experiments GABA induces both channel opening and desensitization; thus we cannot distinguish which gating transition is responsible for the increase in accessibility. Nevertheless, our results are consistent with a model in which γ2A79 (or residues near γ2A79) move(s) during GABA-associated gating transitions. We hypothesize that GABA gating causes movement within the BZD binding site that makes it easier for MTS reagents or BZDs to approach physically and interact with the site. Alternatively, an increase in accessibility could reflect a change in the ionization state of the introduced cysteine. Regardless of the mechanism, these data provide direct physical evidence that confirms allosteric theory; structural rearrangements occur within the BZD binding site in response to GABA binding to its own distinct site on the receptor. A recent study also has detected movements within the third transmembrane domain of the GABAA receptor during allosteric modulation by BZDs (Williams and Akabas, 2000).
Theoretical model of the BZD binding site
We demonstrate that γ2T73, γ2D75, γ2A79, and γ2T81 line the accessible surface of a β-strand in the γ2 subunit of the GABAA receptor, with γ2A79 and γ2T81 in close proximity to the BZD ligand binding domain. We hypothesize that FLZM is topologically close to both γ2F77 and γ2A79 in the BZD binding site. Previous reports have speculated that the 5′-phenyl substituent of classical BZDs, such as FLZM, may participate in π–π stacking interactions with γ2F77 (Buhr et al., 1997; Sigel et al., 1998), whereas others have suggested that these interactions also may include α1H101 (Davies et al., 1998; McKernan et al., 1998). We hypothesize that FLZM is oriented such that its 5′-phenyl is in close contact with γ2F77 and that it occupies space within the binding site that is in close proximity to γ2A79. Although it is unlikely that FLZM chemically interacts with this alanine, the small size of the methyl group at this position may be important in creating an open volumetric space to accommodate BZD ligands of different sizes. Our data suggest that Ro 15-1788 binds near γ2A79, but with the additional contribution of γ2T81 to its binding site. Experiments that use a variety of chemically diverse MTS reagents that can modify γ2A79C and γ2T81C will be helpful in characterizing these structurally distinct binding subdomains further. Neither FLZM nor Ro 15-1788 appears to bind near γ2D75 or γ2T73. However, we propose that γ2D75 may be important in maintaining the architecture of the BZD site because cysteine substitution at this position reduces the FLZM sensitivity of the receptor.
Our data demonstrate that MTSEA-biotin and MTSEA-biotin-CAP, after the modification of γ2A79, are oriented in a manner such that they are able to modulate allosterically the EC50 of the GABA binding site for GABA. Interestingly, although MTSEA-biotin shifts the GABA EC50 for γ2A79C-containing receptors within the range expected for a BZD partial agonist, this reagent has little-to-no effect on the GABA EC50 of γ2T81C-containing receptors. In contrast, MTSEA-biotin-CAP modification of γ2A79C-containing receptors shifts the GABA EC50 within the range of a full agonist and partially shifts the GABA EC50 of γ2T81C-containing receptors. Because MTSEA-biotin-CAP is 8 Å longer than MTSEA-biotin (see Fig. 8), these data suggest that γ2T81 lies farther than γ2A79 from a domain of the BZD binding site that drives allosteric interaction with the GABA binding site.
We speculate that MTSEA-biotin and MTSEA-biotin-CAP bridge the BZD binding site and are capable of exerting their allosteric effects on the GABA binding site by inducing shifts in the distance of α1 and γ2 subunits relative to each other. This mechanism may represent one set of conformational changes that may be required to transduce the binding of BZDs into allosteric modulation of the GABA binding site. Further studies that use the cross-linking of α1 and γ2 residues to span the BZD binding site will be necessary to test this hypothesis. Our results confirm the long-held belief that structural changes in the GABAAreceptor protein underlie allosteric communication between the GABA and BZD binding sites.
Footnotes
This work was supported in part by National Institute of Mental Health Grant MH12966 (J.A.T.) and by National Institute of Neurological Disorders and Stroke Grant NS34727 (C.C.). C.C. is a recipient of the Burroughs Wellcome Fund New Investigator Award in the Basic Pharmacological Sciences. We thank Drs. David Wagner, Andrew Boileau, and J. Glen Newell, as well as Amy Kucken, for a careful reading of this manuscript and invaluable discussion, and Erin McCarthy and Jeff Malik for oocyte preparation.
Correspondence should be addressed to Dr. Cynthia Czajkowski, Department of Physiology, University of Wisconsin, Room 197 MSC, 1300 University Avenue, Madison, WI 53706. E-mail:czajkowski@physiology.wisc.edu.
REFERENCES
- 1.Boileau AJ, Czajkowski C. Identification of transduction elements for benzodiazepine modulation of the GABAA receptor: three residues are required for allosteric coupling. J Neurosci. 1999;19:10213–10220. doi: 10.1523/JNEUROSCI.19-23-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boileau AJ, Kucken AM, Evers AR, Czajkowski C. Molecular dissection of benzodiazepine binding and allosteric coupling using chimeric γ-aminobutyric acidA receptor subunits. Mol Pharmacol. 1998;53:295–303. doi: 10.1124/mol.53.2.295. [DOI] [PubMed] [Google Scholar]
- 3.Boileau AJ, Evers AR, Davis AF, Czajkowski C. Mapping the agonist binding site of the GABAA receptor: evidence for a β-strand. J Neurosci. 1999;19:4847–4854. doi: 10.1523/JNEUROSCI.19-12-04847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buhr A, Sigel E. A point mutation in the γ2 subunit of the γ-aminobutyric acid type A receptors results in altered benzodiazepine binding specificity. Proc Natl Acad Sci USA. 1997;94:8824–8829. doi: 10.1073/pnas.94.16.8824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhr A, Baur R, Sigel E. Subtle changes in residue 77 of the γ-subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J Biol Chem. 1997;272:11799–11804. doi: 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- 6.Changeux J-P, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 7.Chen JG, Liu-Chen S, Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. J Biol Chem. 1998;273:12675–12681. doi: 10.1074/jbc.273.20.12675. [DOI] [PubMed] [Google Scholar]
- 8.Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 9.Colquhoun D. Binding, gating, affinity, and efficacy: the interpretation of structure–activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies M, Bateson AN, Dunn SMJ. Structural requirements for ligand interactions at the benzodiazepine recognition site of the GABAA receptor. J Neurochem. 1998;70:2188–2194. doi: 10.1046/j.1471-4159.1998.70052188.x. [DOI] [PubMed] [Google Scholar]
- 11.Doble A, Martin IL. The GABAA/benzodiazepine receptor as a target for psychoactive drugs. Landes; Austin, TX: 1996. [Google Scholar]
- 12.Draguhn A, Verdorn TA, Ewert M, Seeburg PH, Sakmann B. Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+. Neuron. 1990;5:781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- 13.Gingrich KJ, Burkat PM. Zn2+ inhibition of recombinant GABAA receptors: an allosteric, state-dependent mechanism determined by the γ-subunit. J Physiol (Lond) 1998;506:609–625. doi: 10.1111/j.1469-7793.1998.609bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hevers W, Lüddens H. The diversity of GABAA receptors: pharmacological and electrophysiological properties of GABAA channel subtypes. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- 15.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 16.Kucken AM, Wagner DA, Ward PR, Teissére JA, Boileau AJ, Czajkowski C. Identification of benzodiazepine binding site residues in the γ2 subunit of the γ-aminobutyric acidA receptor. Mol Pharmacol. 2000;57:932–939. [PubMed] [Google Scholar]
- 17.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 18.McKernan RM, Farrar S, Collins I, Emms F, Asuni A, Quirk K, Broughton H. Photoaffinity labeling of the benzodiazepine binding site of α1β2γ2 γ-aminobutyric acidA receptors with flunitrazepam identifies a subset of ligands that interact directly with His102 of the α-subunit and predicts orientation of these within the benzodiazepine pharmacophore. Mol Pharmacol. 1998;54:33–43. doi: 10.1124/mol.54.1.33. [DOI] [PubMed] [Google Scholar]
- 19.Pascual JM, Karlin A. State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor: inferences from rates of reaction of thiosulfonates with substituted cysteines in the M2 segment of the α-subunit. J Gen Physiol. 1998;111:717–739. doi: 10.1085/jgp.111.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA. Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry. 1986;25:5595–5601. doi: 10.1021/bi00367a038. [DOI] [PubMed] [Google Scholar]
- 21.Robertson GA, Warmke JM, Ganetzky B. Potassium currents expressed from Drosophila and mouse eag cDNAs in Xenopus oocytes. Neuropharmacology. 1996;35:841–850. doi: 10.1016/0028-3908(96)00113-x. [DOI] [PubMed] [Google Scholar]
- 22.Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- 23.Sigel E, Schaerer MT, Buhr A, Baur R. The benzodiazepine binding pocket of recombinant α1β2γ2 γ-aminobutyric acidA receptors: relative orientation of ligands and amino acid side chains. Mol Pharmacol. 1998;54:1097–1105. doi: 10.1124/mol.54.6.1097. [DOI] [PubMed] [Google Scholar]
- 24.Skerritt JH, Johnston GA. Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur J Pharmacol. 1983;89:193–198. doi: 10.1016/0014-2999(83)90494-6. [DOI] [PubMed] [Google Scholar]
- 25.Srinivasan S, Nichols CJ, Lawless GM, Olsen RW, Tobin AJ. Two invariant tryptophans on the α1 subunit define domains necessary for GABAA receptor assembly. J Biol Chem. 1999;274:26633–26638. doi: 10.1074/jbc.274.38.26633. [DOI] [PubMed] [Google Scholar]
- 26.Wagner DA, Czajkowski C. Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci. 2001;21:67–74. doi: 10.1523/JNEUROSCI.21-01-00067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams DB, Akabas MH. γ-Aminobutyric acid increases the water accessibility of M3 membrane-spanning segment residues in γ-aminobutyric acid type A receptors. Biophys J. 1999;77:2563–2574. doi: 10.1016/s0006-3495(99)77091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams DB, Akabas MH. Benzodiazepines induce a conformational change in the region of the γ-aminobutyric acid type A receptor α1 subunit M3 membrane-spanning segment. Mol Pharmacol. 2000;58:1129–1136. doi: 10.1124/mol.58.5.1129. [DOI] [PubMed] [Google Scholar]
- 29.Wingrove PB, Thompson SA, Wafford KA, Whiting PJ. Key amino acids in the γ-subunit of the γ-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine binding site. Mol Pharmacol. 1997;52:874–881. doi: 10.1124/mol.52.5.874. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, Akabas MH. Identification of channel lining residues in the M2 membrane-spanning segment of the GABAA receptor α1 subunit. J Gen Physiol. 1996;107:195–205. doi: 10.1085/jgp.107.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]