

Abstract
DNA damage has been implicated as one important initiator of cell death in neuropathological conditions such as stroke. Accordingly, it is important to understand the signaling processes that control neuronal death induced by this stimulus. Previous evidence has shown that the death of embryonic cortical neurons treated with the DNA-damaging agent camptothecin is dependent on the tumor suppressor p53 and cyclin-dependent kinase (CDK) activity and that the inhibition of either pathway alone leads to enhanced and prolonged survival. We presently show that p53 and CDKs are activated independently on parallel pathways. An increase in p53 protein levels, nuclear localization, and DNA binding that result from DNA damage are not affected by the inhibition of CDK activity. Conversely, no decrease in retinoblastoma protein (pRb) phosphorylation was observed in p53-deficient neurons that were treated with camptothecin. However, either p53 deficiency or the inhibition of CDK activity alone inhibited Bax translocation, cytochrome c release, and caspase-3-like activation. Taken together, our results indicate that p53 and CDK are activated independently and then act in concert to control Bax-mediated apoptosis.
Keywords: apoptosis, p53, CDK, neuronal, cell cycle, Bax
Neuronal apoptosis is an important component of brain ontogeny (Oppenheim, 1991) and is important in the progression of neuropathological conditions such as stroke and neurodegenerative disease (Stefanis et al., 1997; Cotman, 1998; Dirnagl et al., 1999). Previous work by ourselves and others demonstrated that DNA damage activates the apoptotic process in neurons. For example, irradiation (Enokido et al., 1996b), cytosine arabinoside (AraC;Winkelman and Hines, 1983; Wallace and Johnson, 1989; Martin et al., 1990; Tomkins et al., 1994; Park et al., 1998b), cisplatin (Gill and Windebank, 1998), topoisomerase-II inhibitors (Nakajima et al., 1994;Tomkins et al., 1994), and the topoisomerase-I inhibitor camptothecin (Morris and Geller, 1996; Park et al., 1997b, 1998a, 2000) all induce apoptotic neuronal cell death. A number of these agents cause peripheral neuropathies and neurodegeneration (Winkelman and Hines, 1983; Baker et al., 1991; Vogel and Horoupian, 1993; Mansfield and Castillo, 1994). DNA damage also may participate in initiating cell death in neuropathological conditions such as stroke (Chen et al., 1997). Given these observations, it has become increasingly important to understand the downstream signaling events that control DNA damage-evoked neuronal cell death.
Several molecular events that mediate death in some neuronal apoptosis paradigms have been described. For example, it has been suggested previously that proteins that normally function to control cell cycle progression in actively dividing cells may play required roles in the death of terminally differentiated postmitotic neurons (Farinelli and Greene, 1996; Park et al., 1996, 1997a,b, 1998a, 2000; Gill and Windebank, 1998). In support of this, changes have been reported in levels of cyclin-dependent kinases (CDKs) as well as changes of their activating cyclin partners during trophic factor deprivation (Gao and Zelenka, 1995), cisplatin-induced injury (Gill and Windebank, 1998), stroke (Timsit et al., 1999; Osuga et al., 2000), and in brains of Alzheimer's disease patients (McShea et al., 1997; Vincent et al., 1997; Busser et al., 1998). Specific to DNA damage, CDK inhibition, by both pharmacological and molecular means, prevents the death of sympathetic and/or cortical neurons evoked by UV irradiation, AraC, and/or camptothecin (Park et al., 1997b, 1998a,b, 2000). Furthermore, studies that use camptothecin have demonstrated an increase in cyclin D1-associated kinase activity and protection by the expression of dominant-negative CDK4/6 (Park et al., 1998a). These studies indicate that CDK4/6 activity plays a required role in DNA damage-evoked neuronal apoptosis.
At least three other molecular events have been suggested to be required for the neuronal death that follows DNA damage. These include the tumor suppressor p53 (Enokido et al., 1996a; Xiang et al., 1998;Giovanni et al., 2000), the proapoptotic Bcl2-related Bax (Xiang et al., 1998; Keramaris et al., 2000), and the various death effector protease enzymes, caspases (Stefanis et al., 1999; Keramaris et al., 2000). The obligate nature of p53 in some neuronal death paradigms is evidenced by significant neuroprotection in p53-deficient neurons exposed to excitotoxic injury (Xiang et al., 1996), ischemia (Crumrine et al., 1994), and DNA damage (Johnson et al., 1999; Giovanni et al., 2000). Similarly, multiple groups reported that Bax-deficient neurons are resistant to death that is evoked by apoptotic initiators, including trophic factor deprivation (Deckwerth et al., 1998) and DNA damage (Xiang et al., 1998). Finally, the inhibition of caspases also has been shown to inhibit apoptotic neuronal death that is evoked by numerous different apoptotic initiators, including DNA damage (Stefanis et al., 1999; Keramaris et al., 2000).
Although the individual components that play an obligate role in the neuronal death that is induced by DNA damage are being delineated, the biochemical relationship between these molecular signaling events is less clear. Accordingly, the present study explores the relationship among the apoptotic control elements, CDKs, p53, Bax, cytochromec, and caspases in camptothecin-induced neuronal death.
MATERIALS AND METHODS
Materials. Flavopiridol {L86-8275, [(−)cis-5,7-dihydroxy-2-(2-chlorophenyl)- 8[4-(3-hydroxy-1-methyl)-piperidinyl]-4H-benzopyran-4-one]} was a generous gift from Dr. Peter J. Worland (National Cancer Institute, Bethesda, MD). Camptothecin was obtained from Sigma (St. Louis, MO). Boc-aspartyl (OMe)-fluoromethylketone (BAF) and zVAD-fmk were purchased from Enzyme Systems Products (Dublin, CA).
Knock-out mice. p53 and Bax-deficient (C57BL/6 background) neurons were obtained from embryos derived from heterozygous pairings. Genotyping of each individual embryo was performed by PCR as follows: (1) p53 was genotyped by using GTATCTGGAAGACAGGCAGAC (O-p53-1) and TGTACTTGTAGTGGATGGTGG (O-p53-2) primers to detect the wild-type allele (450 bp) and TATACTCAGAGCCGGCCT (O-p53-X7) and TTCCTCGTGCTTTACGGTATC (O-neo-2) primers to detect the targeted allele (533 bp). PCR conditions included 94°C for 5 min (1 cycle), 94°C for 1 min, 55°C for 1 min, 72°C for 1 min (30 cycles), and 72°C for 10 min. (2) Bax was genotyped by using GTTGACCAGAGTGGCGTAGG (BaxIN5R), CCGCTTCCATTGCTCAGCGG (NEOR), and GAGCTGATCAGAACCATCATG (BaxEX5F) primers. BaxIN5R and BaxEX5F primers were used to amplify a 304 bp fragment from the wild-type Bax allele. BaxIN5R and NEOR were used to amplify a 504 bp fragment from the targeted Bax allele. PCR conditions included 94°C for 5 min (1 cycle), 94°C for 1 min, 62°C for 1 min, 72°C for 1.5 min (30 cycles), and 72°C for 7 min.
Culture and survival of cortical neurons. Mouse cortical neurons were cultured from embryonic day 15 (E15) mice (CD1, Charles River, Wilmington, MA; or p53 or Bax heterozygous breedings) as described previously (Giovanni et al., 2000). For the knock-out studies each individual embryo was dissected and plated individually. The neurons were plated into 24-well dishes (∼200,000 cells/well) or 6-well dishes (1–3 million cells/well) coated with poly-d-lysine (100 μg/ml) in serum-free medium [N2/DMEM (1:1) supplemented with 6 mg/ml d-glucose, 100 μg/ml transferrin, 25 μg/ml insulin, 20 nm progesterone, 60 μm putrescine, 30 nm selenium]. At 1–2 d after initial plating the medium was exchanged with serum-free medium supplemented with camptothecin (10 μm), flavopiridol (1 μm), and/or BAF/zVAD (100 μm), as indicated in this text and figures. At appropriate times of culture under the conditions described in this text, the cells were lysed and the numbers of viable cells were evaluated. Briefly, cells were lysed in 200 μl of cell lysis buffer (0.1× PBS, pH 7.4, containing 0.5% Triton X-100, 2 mm MgCl2, and 0.5 gm/100 ml cetyldimethylethylammonium bromide), which disrupts cells but leaves the nuclei intact. Then 10 μl of sample from each culture was loaded onto a hemacytometer, and the number of healthy intact nuclei was evaluated by phase microscopy. Nuclei that displayed characteristics of blebbing, disruption of nuclear membrane, phase-bright apoptotic bodies, and chromatin margination were excluded. All experimental points are expressed as a percentage of cells plated on day 0. Alternatively, the cells were collected and analyzed for biochemical analyses as described below.
Western blot analyses. Cortical neurons were dissociated and cultured as described above. The neurons were washed twice in PBS and harvested in SDS-loading buffer. Protein (10 μg) was loaded onto SDS-polyacrylamide gels and transferred onto nitrocellulose membrane as described previously. Blots were probed with anti-p53 (PharMingen, San Diego, CA), anti-Bax (Santa Cruz, Santa Cruz, CA), anti-phospho-pRb (NEN, Boston, MA), or anti-actin (Sigma) antibodies as indicated.
Caspase activity. Cortical neurons were harvested at the indicated times for caspase activity. Briefly, the cells were washed two times in PBS and collected in caspase lysis buffer as described previously (Stefanis et al., 1999). Then the cells were incubated on ice for 20 min and sonicated briefly for 3 sec. Next the extract was centrifuged for 15 min at 12,000 rpm on an Eppendorf tabletop centrifuge. The supernatant was collected and assayed for protein by Bradford (Bio-Rad, Hercules, CA). In any single experiment an equal amount of protein (∼5 μg of protein) was incubated with DEVDamino-fluoro-coumarin as described previously. An increase in fluorescence was measured with a fluorometer (400 nm excitation, 505 nm emission) as described previously (Stefanis et al., 1996).
p53 electrophoretic mobility shift assay (EMSA). Cortical neurons (E16) were harvested at the times indicated in a lysis solution containing (in mm) 100 HEPES buffer, 5 MgCl2, 2.5 EDTA, and 0.5 PMSF plus 20% glycerol, 0.5 m KCl, 20 μm sodium orthovanadate, and 0.1% NP40 as described previously (Macleod et al., 1996). Cell lysate (10 μg) was incubated with binding buffer [(in mm) 250 KCl, 100 HEPES, and 5 DTT plus 0.5% Triton X-100, 5 mg/ml BSA, 50% glycerol (Macleod et al., 1996)] containing 0.1 μg/μl sonicated herring sperm, 1 μl of anti-p53 antibody (AB-1; Oncogene Research Products, San Diego, CA), and32P-radiolabeled p53 double-stranded probe (CCTGCCTTGCCTGGACTTGC; 60,000 cpm). p53 probes were prepared by annealing single-strand oligonucleotides (each 1 μg/μl) in 50 mm final NaCl at 95°C for 5 min. Annealed DNA (50 μl) was incubated for 30 min in a reaction containing 10× reaction buffer, 25 mm dNTP (C, T, G only),32P-dATP (Amersham, Arlington Heights, IL), and Klenow. The reaction was purified through a Sephadex G-25 spin column, and radioactivity levels were evaluated. A 100-fold excess (100 μg/μl) of unlabeled p53 oligonucleotide was used as one control for binding specificity. Each sample was resolved on a 5% polyacrylamide gel and visualized by autoradiography.
Immunofluorescence. Cortical neurons were dissociated and cultured, as described above. After various times of camptothecin treatment, the neurons were fixed with 3.7% paraformaldehyde for 30 min at 4°C. The cells were washed with PBS and incubated in PBS containing 10% serum and 0.4% Triton X-100. Then the neurons were incubated with rabbit anti-p53 (1:20; Santa Cruz SC-6243) or anti-Bax (1:500; Santa Cruz P19; Trevigen YTH6A7) antibodies in PBS containing 2% normal goat serum (NGS) and 0.4% Triton X-100 for 1 hr. After washing, rhodamine-linked (1:100; Molecular Probes, Eugene, OR) or CY-3-linked (1:300 Jackson Laboratories, Bar Harbor, ME) secondary antibody was added in PBS containing 2% NGS and 0.4% Triton X-100 for 20 min. Hoechst dye 33342 (Sigma) or Yo-Yo (1:1500; Molecular Probes) then was added to stain the nuclei. Cells were visualized under fluorescent/confocal microscopy. For Bax staining, similar results were obtained with both antibodies. The antibodies also were tested in Bax-deficient neuronal cultures. Although diffuse and sometimes dim granular staining was observed in Bax-deficient neurons that were untreated or treated with camptothecin, the bright punctate labeling observed in camptothecin-treated wild-type cells was not present. Similar results have been reported previously (Putcha et al., 1999) for Bax staining in sympathetic neurons.
RESULTS
Death commitment of cortical neurons exposed to DNA damage
We showed previously that the treatment of embryonic cortical neurons with the DNA topoisomerase-1 inhibitor camptothecin induces apoptotic death (Morris and Geller, 1996; Park et al., 1997b, 1998a,2000; Keramaris et al., 2000). To investigate more closely the mechanisms of death evoked by this DNA-damaging agent, we first determined the commitment point for death in this paradigm. Cultured cortical neurons were pulse treated with camptothecin for various times, followed by its removal. As shown in Figure1A, the commitment point for neuronal death, as defined by the time at which 50% neuronal death occurs by 24 hr in response to pulse treatment, was ∼4 hr. This indicated that biochemical events before this time point likely control the irreversible decision to undergo apoptosis. Accordingly, we next examined the pathway of upstream events that control this commitment phase.
Fig. 1.
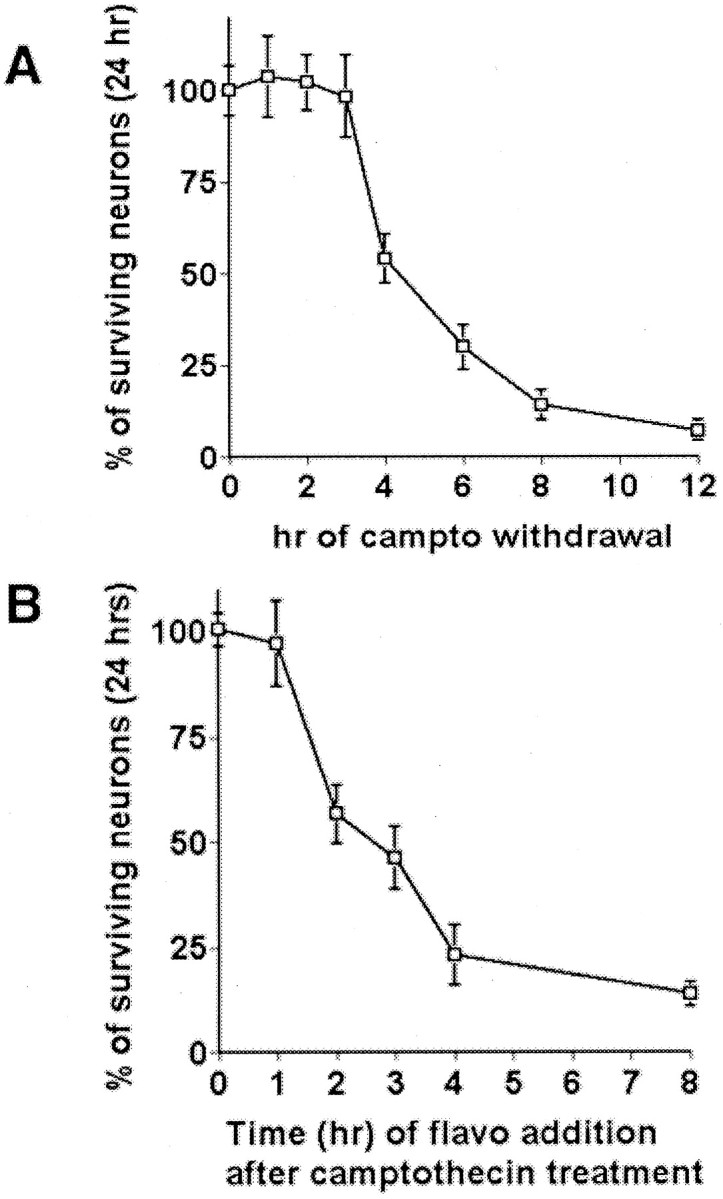
Determination of the commitment point for neuronal death by DNA damage and protection by CDK inhibitors. A, Determination of the commitment point for neuronal death by DNA damage. Cortical cultures were treated with camptothecin (campto; 10 μm) at 0 hr and removed at the indicated times; neuronal survival was determined at 24 hr after treatment. B, Determination of the commitment point for protection by CDK inhibition. The CDK inhibitor flavopiridol (1 μm) was added to cortical cultures treated with camptothecin at the indicated times; neuronal survival was determined at 24 hr after treatment. In each experiment the viability was assessed by nuclear counts. Eachpoint is the mean ± SEM of data from three cultures.
p53 and cyclin-dependent kinases are activated independently and before death commitment
As previously reported, neurons that are deficient for p53 (Johnson et al., 1999; Giovanni et al., 2000) or are treated with the CDK inhibitor flavopiridol (Park et al., 1997b) are resistant to DNA damage-evoked death, indicating that the p53 and CDK signals play a required role in death in this paradigm. Our present evidence indicates that these signals are early temporal events that are activated before death commitment. As determined by Western blot analyses, total cellular p53 protein levels increase starting ∼2–4 hr after camptothecin exposure (Fig.2A,B). Importantly, the ability of p53 to bind consensus p53 DNA sequence as one marker of p53 activity also begins to increase at this time (Fig. 2C). By EMSA a p53-specific band increases starting ∼2 hr after DNA damage (Fig. 2C). This band could be competed by excess unlabeled oligonucleotide (Fig. 2D). In addition, this p53 band was not detectable in lysates prepared from camptothecin-treated p53-deficient neurons. Both controls indicate the p53-specific nature of this band. p53 binding was not observed in untreated control neurons.
Fig. 2.
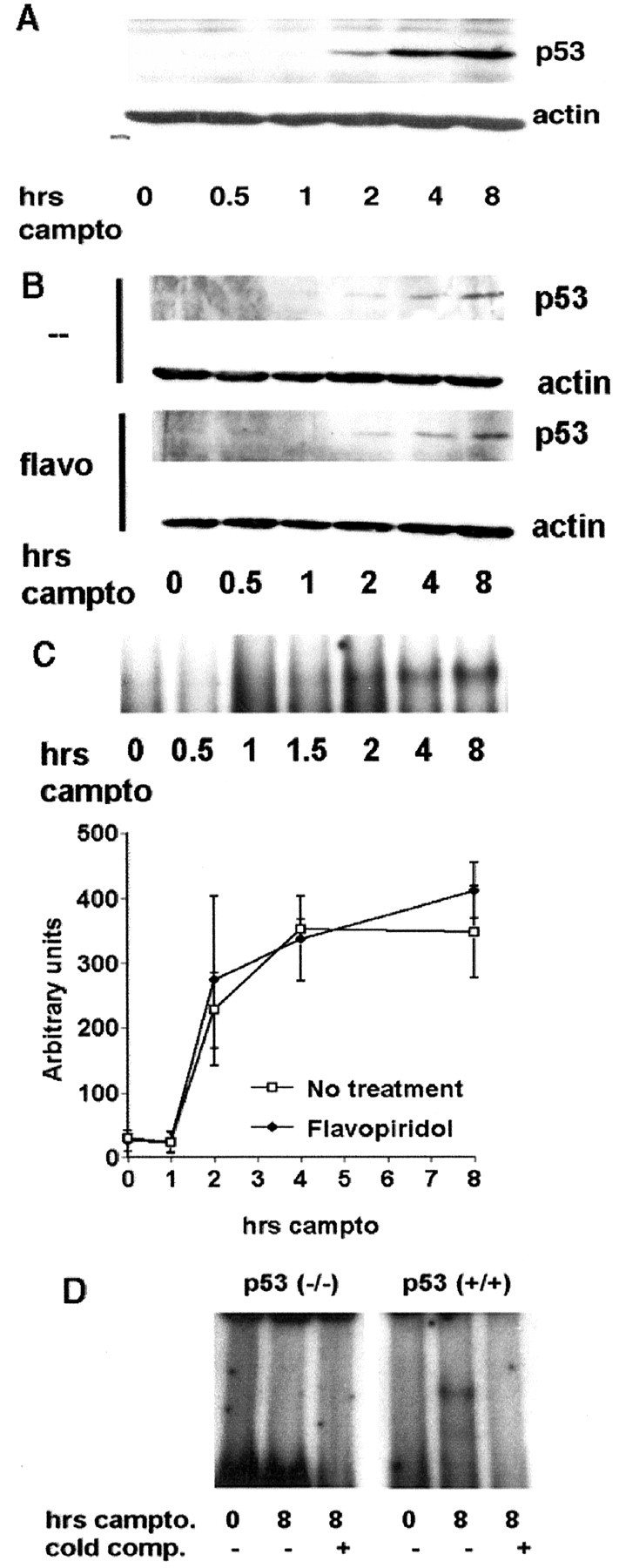
Induction of p53 expression and p53-specific DNA binding activity is not affected by cotreatment with the CDK inhibitor flavopiridol. Total cell lysates were prepared from wild-type cortical cultures treated with camptothecin (campto; 10 μm) alone or cotreated with flavopiridol (1 μm) and analyzed for p53 expression by Western blot analyses (A, B) or DNA binding by EMSA (C), as indicated in Materials and Methods. Densitometric analysis from three experiments (mean ± SEM) with and without flavopiridol cotreatment is shown. D, So that the specific nature of the EMSA band could be ensured, cell extracts from wild-type or p53-deficient neurons were untreated or treated with camptothecin (10 μm) for 8 hr and analyzed for p53 binding to a labeled p53 oligonucleotide probe by EMSA.cold comp, The addition of unlabeled probe as a competitor for radiolabeled probe. The band labeled p53 is the only band present in the camptothecin-treated wild-type or heterozygous extracts and is missing in the untreated and p53-deficient extracts.
Similar to early p53 induction, activation of CDKs, as determined by phosphorylation of retinoblastoma protein (pRb) on a CDK4/6 phosphorylation consensus site, occurs 1 hr after camptothecin treatment (Park et al., 2000). Consistent with this observation, flavopiridol must be added by 1 hr after the initiation of camptothecin treatment to rescue neurons fully from DNA damage (Fig.1B).
We next determined whether p53 and CDK were activated along the same pathway or whether they acted independently. In this regard, we first examined whether inhibition of the CDK pathway would prevent the activation of p53. Interestingly, cotreatment with the CDK inhibitor flavopiridol did not block the increase in total cellular p53 protein levels after camptothecin treatment (Fig. 2B). Because CDKs are known to regulate p53 function (Wang and Prives, 1995), we performed EMSA to determine the consensus DNA binding activity of p53 in neurons after DNA damage. However, no effect on DNA binding activity was observed with flavopiridol cotreatment (Fig.2C). Finally, to ensure that CDK activity was not affecting nuclear localization of p53, we examined p53 by immunofluorescence. Treatment of cortical cultures with camptothecin (5 hr) alone resulted in a profound increase in p53 staining in the nucleus (Fig.3). Flavopiridol cotreatment did not inhibit this nuclear accumulation of p53 (Fig. 3). In these experiments scattered cells with condensed nuclei were present in both control and experimental wells as a result of background death from the dissection and plating procedure. These cells were negative for p53 staining. Taken together, the above data indicate that CDK activity is not required for the DNA damage-inducible p53 response in neurons.
Fig. 3.
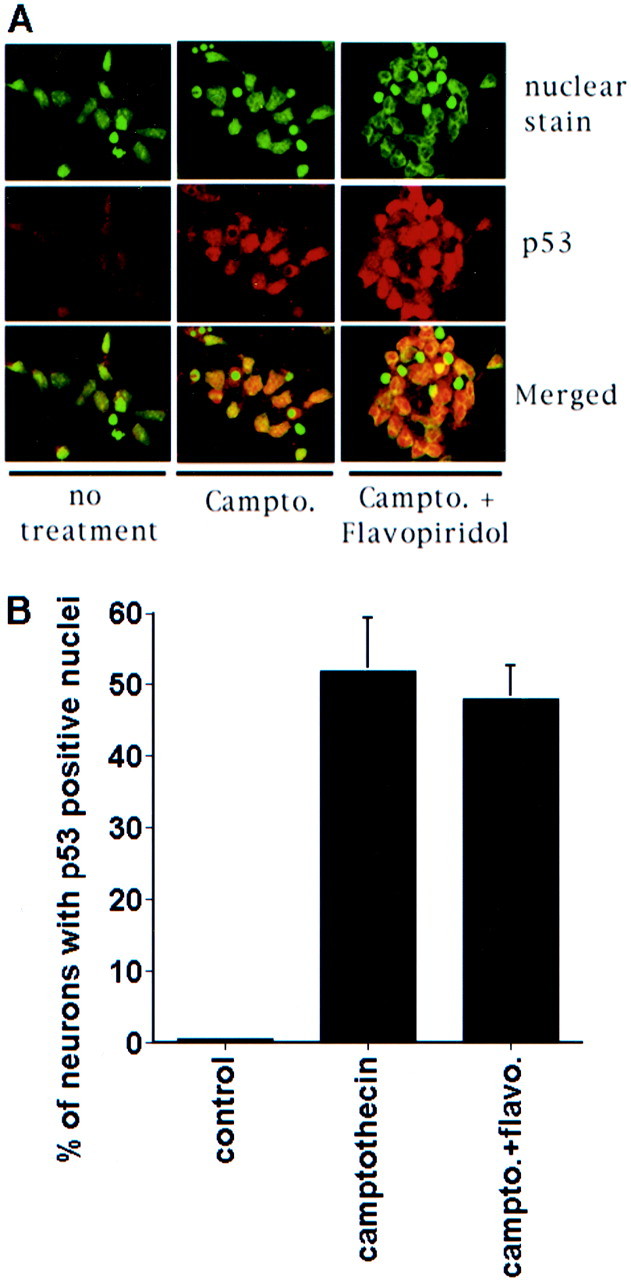
Nuclear localization of p53 is not inhibited by the inhibition of CDK. A, Immunofluorescence image of cultured cortical neurons stained with nuclear dye or anti-p53 antibody. Where indicated, the cultures were treated with camptothecin (campto; 10 μm) for 5 hr with and without flavopiridol (1 μm) treatment. B, Quantitation of neurons with positive nuclear staining for p53. Data are the mean ± SEM from three cultures.
To test whether p53 may act upstream of and activate the CDK pathway, we examined whether p53-deficient neurons display decreased CDK activity in response to camptothecin treatment. To determine this, we monitored the phosphorylation status of the CDK substrate pRb. We used a phosphospecific antibody that recognizes Ser795 in pRb, a specific kinase target of CDK4/6, to monitor the pRb phosphorylation after camptothecin treatment. After camptothecin treatment we observed rapid, inducible pRb phosphorylation in neurons within 1 hr (Fig.4). We have shown previously that total pRb levels do not change at these times after camptothecin treatment (Park et al., 2000). A comparison of neurons cultured from either p53-heterozygous or p53-nullizygous mice demonstrated no significant differences in the levels of phospho-pRb by Western analysis when normalized to actin levels (Fig. 4). Taken together, these data suggest that CDK activation and p53 induction mechanisms likely occur independently during the apoptosis of DNA-damaged neurons.
Fig. 4.
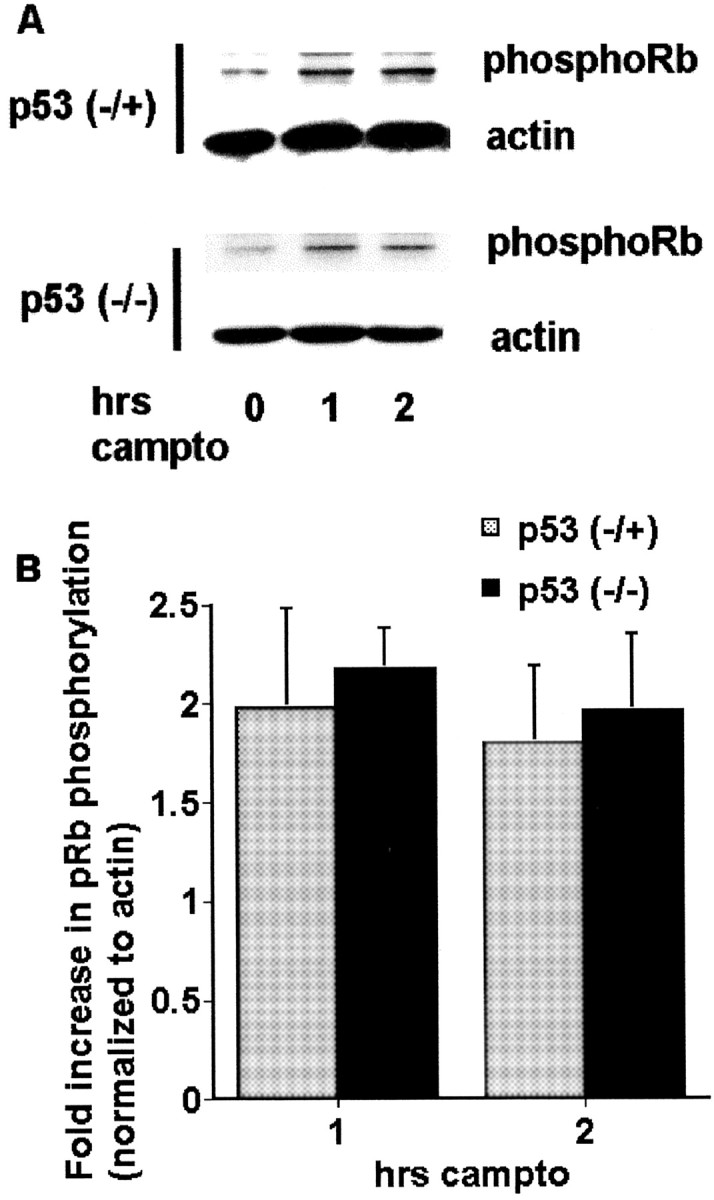
Phosphorylation of pRb is not inhibited by p53 deficiency during DNA damage-induced neuronal apoptosis.A, Western immunoblots (probed with anti-phospho-Rb Ser795 antibody) of whole-cell lysates of cortical neurons from p53-heterozygous or p53-nullizygous embryos after treatment with camptothecin (campto; 10 μm) for the indicated times. B, Densitometric analyses of the Western immunoblots. All data points were normalized to actin expression levels. Data are the mean ± SEM from three experiments and are expressed relative to the initial amount of phosphorylated pRb at time 0.
p53 and CDK-mediated pathways act cooperatively to mediate Bax activation, consequent cytochrome c release, and caspase activation
The observations described above indicate that the activation of p53 and CDKs occurs independently. We next determined whether putative downstream effectors of p53- or CDK-mediated pathways activate a common death element. Bax has been shown previously to be required for cytochrome c release, caspase activation, and apoptotic death after DNA damage. Accordingly, we examined whether the inhibition of either the p53 or CDK pathway was sufficient to affect Bax activity.
Previous results in several death paradigms have indicated that Bax expression can be induced by p53 activity. However, as shown in Figure5A, total cellular protein levels of Bax, as measured by Western blot analyses, do not change with camptothecin treatment. Instead, immunocytochemical analyses indicates that Bax translocates to mitochondria. As shown in Figure5B, Bax appears punctate after camptothecin treatment. Similar results were obtained for two Bax antibodies (see Materials and Methods). As would be expected if caspases act downstream of Bax, the general caspase inhibitor zVAD-fmk had no effect on Bax translocation (Fig. 5C). This punctate appearance is localized mitochondrially; 45% of neurons (n = 141) from cultures cotreated with camptothecin and BAF (100 μm) for 12 hr displayed colocalization of punctate Bax and mitotracker mitochondrial staining (data not shown). A significant percentage (35%), however, was positive for punctate Bax expression although negative for mitotracker staining (data not shown), presumably because of the ongoing death process and loss of membrane potential as previously reported after camptothecin treatment (Stefanis et al., 1999).
Fig. 5.

DNA damage-induced Bax translocation is inhibited by either p53 deficiency or CDK inhibition. A, Bax protein levels do not change during the death of cortical neurons evoked by DNA damage. Shown are Western immunoblots of whole-cell lysates of cortical neurons after various periods of treatment with camptothecin (campto; 10 μm). Antibody specificity is demonstrated for Western analysis by using lysates from Bax-deficient neurons. Control for protein loading is indicated by actin expression. B, Immunofluorescence micrograph of Bax translocation in cortical neurons of Bax (−/−), p53 (−/+), or p53 (−/−) backgrounds. Where indicated, the cultures were treated with camptothecin (CA; 10 μm; 10 hr) alone or cotreated with the caspase inhibitor zVAD (100 μm) or the CDK inhibitor flavopiridol (FL; 1 μm). Punctate staining is indicative of Bax translocation from the cytosol and is quantitated in C. Data are the mean ± SEM from three cultures.
Interestingly, inhibition of either the CDK or p53 pathway individually is sufficient to inhibit Bax translocation when compared with untreated control cultures (Fig. 5C). Total Bax levels in neurons from p53-deficient and littermate control animals, as measured by Western blot analyses, were identical (data not shown). Consistent with these findings, p53-deficient neurons exposed to DNA damage did not show a loss of punctate immunocytochemical cytochrome c staining when compared with littermate controls (Fig.6). Inhibition of cytochrome crelease by flavopiridol has been reported previously (Stefanis et al., 1999). Conversely, p53 protein levels and DNA binding activity were not impaired in Bax-deficient neurons after camptothecin treatment when compared with littermate controls (Fig.7). Consistent with the notion of caspase activity as a more distal death effector, the induction of p53 protein and DNA binding also is still observed in neurons cotreated with general caspase inhibitors (Fig. 8). Taken together, these findings indicate that both p53 and CDK pathways act upstream of and are required to mediate Bax translocation, consequent cytochrome c release, and caspase activation.
Fig. 6.
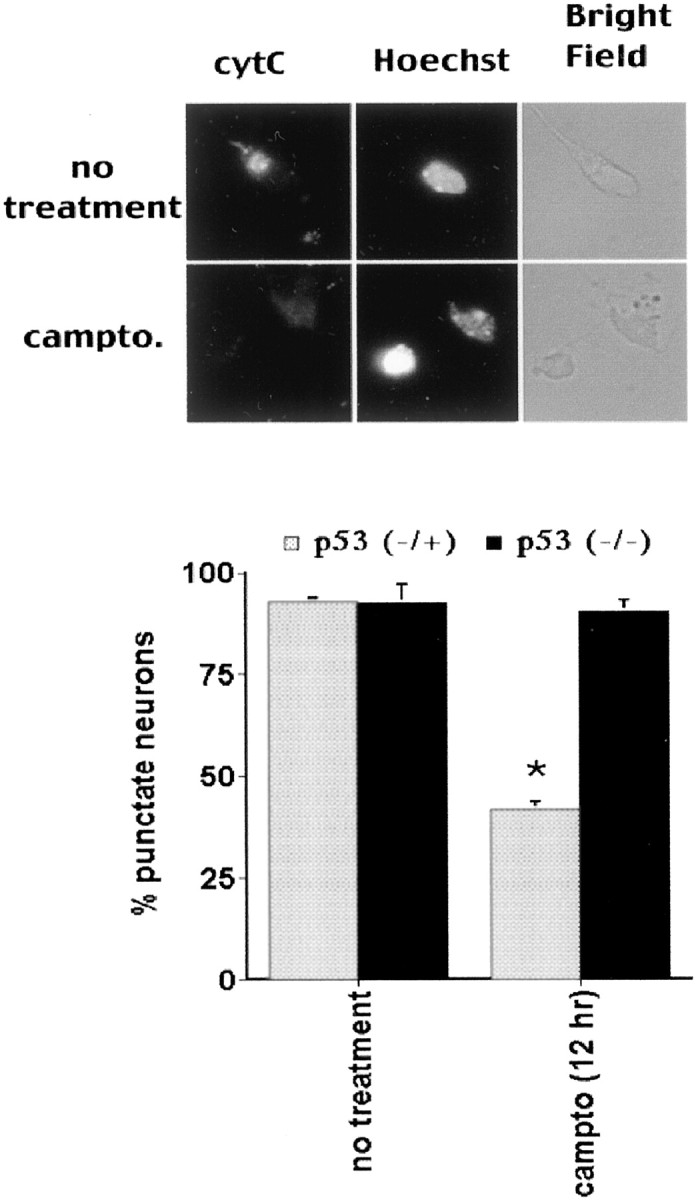
Cytochrome c (cytC) release is blocked by p53 deficiency during DNA damage-induced neuronal apoptosis. Cortical neurons from p53-heterozygous or p53-nullizygous embryos were cultured and treated with camptothecin (campto; 10 μm) for 12 hr. Quantitation of cytochromec release (bottom; as indicated by punctate-to-diffuse expression in top) is shown. Data are the mean ± SEM from three cultures. * indicates significance (p < 0.05, as derived from Student's ttest).
Fig. 7.
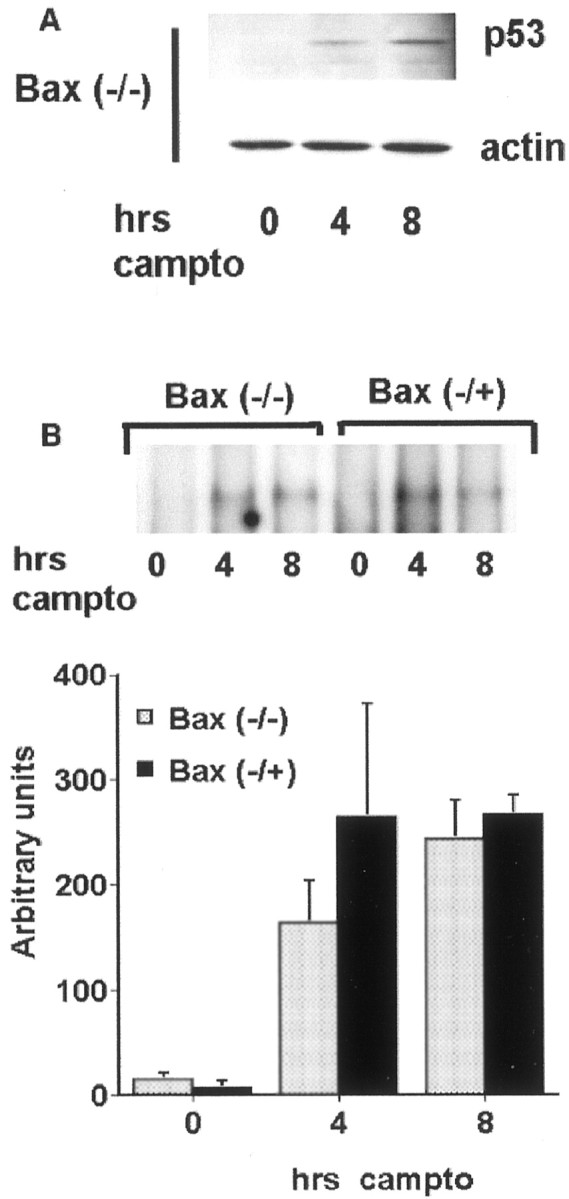
Bax deficiency does not inhibit p53 induction or DNA binding in neurons exposed to DNA damage. Bax-heterozygous or Bax-nullizygous neurons were treated with camptothecin (campto; 10 μm) for the indicated times and analyzed for p53 protein levels by Western blot analyses (A) and for DNA binding by EMSA (B). For B, representative radiographs and densitometric analysis from three experiments (mean ± SEM) are shown.
Fig. 8.
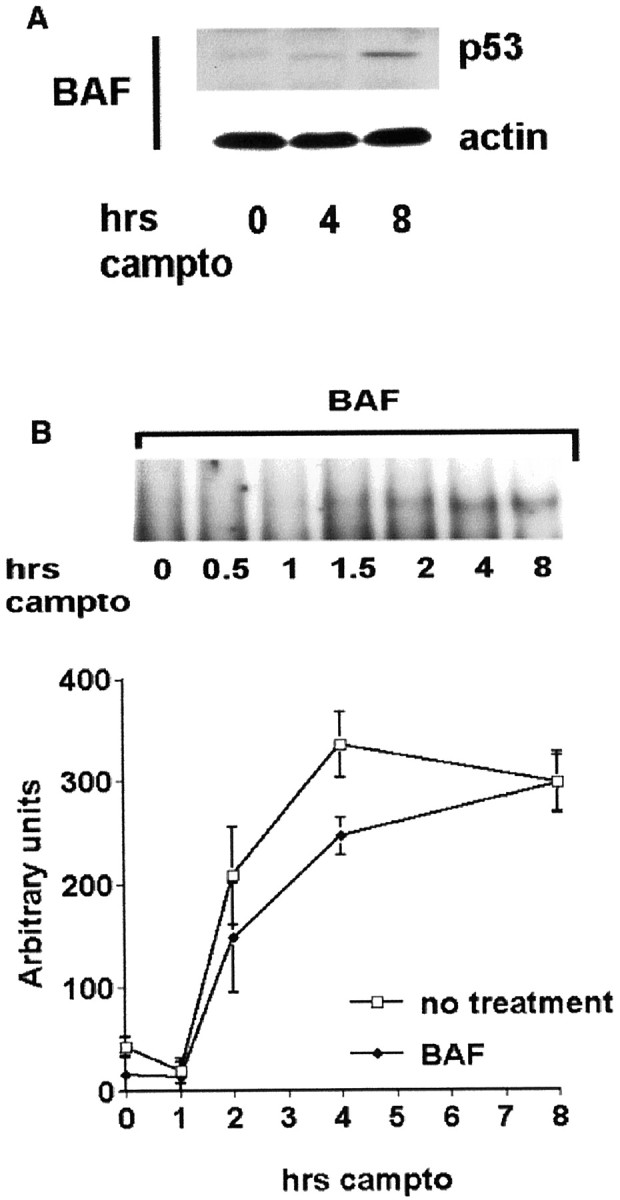
The general caspase inhibitor BAF does not inhibit p53 induction or DNA binding in neurons exposed to DNA damage. Cortical neurons were treated with camptothecin (campto; 10 μm) alone or cotreated with BAF (100 μm) for the indicated times and analyzed for p53 protein levels by Western blot analyses (A) and DNA binding by EMSA (B). For B, representative radiographs and densitometric analysis from three experiments (mean ± SEM) are shown.
p53, CDKs, Bax, and delayed death
p53-Deficient neurons are protected from DNA damage almost completely for 48 hr. However, survival is not complete at later time points (Fig. 9). Because p53 and CDKs appear to regulate both Bax translocation and caspase activity after short periods of camptothecin treatment, we explored the possibility that death in p53-deficient neurons after prolonged camptothecin exposure may result from the parallel proximal CDK pathway, with consequent delayed activation of caspase activity. Control cultures treated with camptothecin showed robust DEVD-AFC cleavage activity at 24 hr. However, no significant caspase activity was detected in p53-deficient cultures treated with camptothecin, even at 72 hr when delayed death occurs (Fig. 9B). Unfortunately, flavopiridol in and of itself is variably toxic at 72 hr, making analyses of delayed death difficult (Fig. 9A; data not shown). Death in these cultures at 72 hr, which may be a combination of delayed death and/or toxicity, also is not accompanied by caspase-3-like activity (Fig.9A). Finally, Bax-deficient neurons displayed some death at 72 hr (Fig. 9C). No caspase-3-like activity was detected in Bax-deficient neurons undergoing delayed death. Taken together, these results suggest that death in the presence of prolonged camptothecin treatment in the absence of p53 is caspase-independent. These results also suggest that the CDK and p53 pathways cooperate to mediate caspase activity and do not activate the conserved distal death effector pathway individually.
Fig. 9.
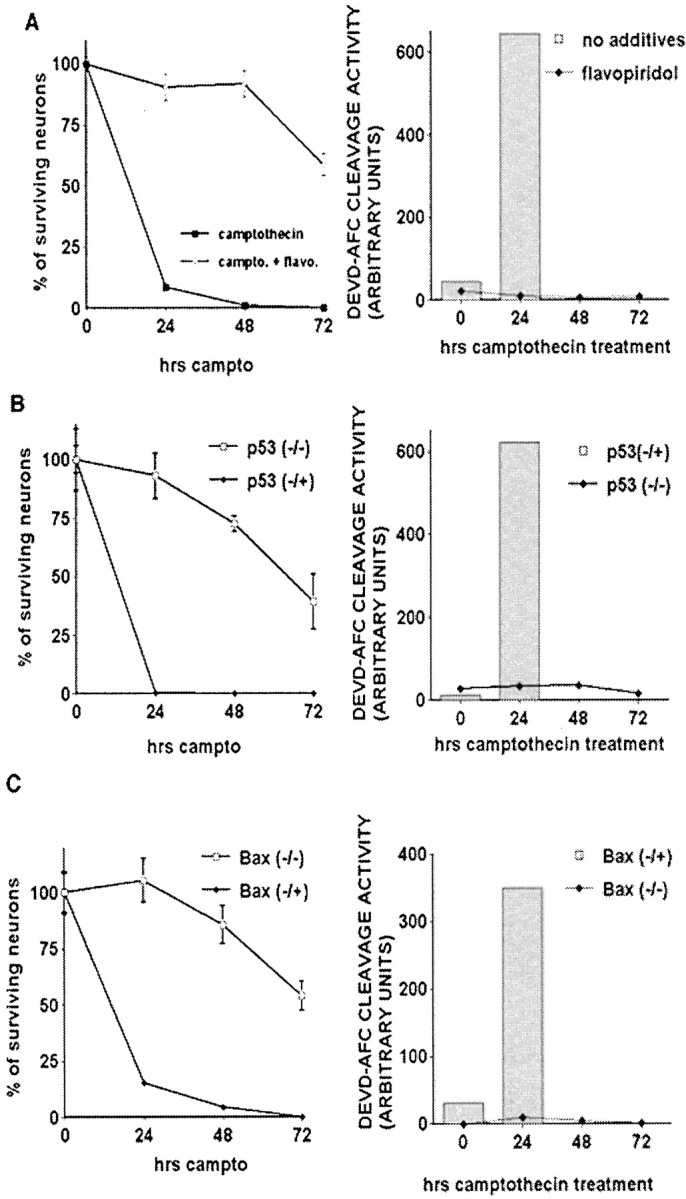
Delayed death in flavopiridol-treated, p53-deficient, or Bax-deficient mice is not accompanied by caspase-3-like activity. DNA damage-induced caspase activation in neurons is CDK-, p53-, and Bax-dependent. Cortical neurons were treated with camptothecin (campto; 10 μm) and assessed for survival (left column) or DEVD-AFC cleavage activity (right column). For survival experiments, eachpoint is the mean ± SEM of data from three cultures. Cleavage activity in camptothecin-treated wild-type or p53/Bax littermate cultures was assessed only at 0 and 24 hr because of low levels of total protein observed at later time points.A, Wild-type cortical neurons were cotreated with flavopiridol (1 μm). B, p53-Deficient neurons or littermate controls. C, Bax-deficient neurons or littermate controls.
DISCUSSION
DNA damage-induced neuronal apoptosis
DNA damage is a potent inducer of neuronal apoptosis. Although significant insight has been made regarding the requirement of individual death components after DNA damage, the way in which these signals are ordered remains less clear. Accordingly, we studied basic neuronal death signaling evoked by DNA damage, using the prototypical DNA topoisomerase-I inhibitor camptothecin (a neurotoxic, anticancer DNA-damaging agent; Morris and Geller, 1996). We and others have identified previously the required roles for various death-signaling pathways, including p53 (Johnson et al., 1999; Giovanni et al., 2000), CDKs (Park et al., 1997b, 1998a, 2000), Bax (Xiang et al., 1998;Keramaris et al., 2000), and caspases (Stefanis et al., 1999; Keramaris et al., 2000) in this paradigm. Presently, we studied the temporal and biochemical relationship between these signaling pathways.
CDKs and p53: Upstream death regulators are activated independently
Our previous evidence demonstrated that pharmacological CDK inhibitors (Park et al., 1997b) as well as the expression of DN Cdk4/6 (Park et al., 1998a) inhibit the death of cortical neurons exposed to camptothecin. In addition, we and others have shown that p53-deficient neurons are resistant to camptothecin-induced death (Johnson et al., 1999; Giovanni et al., 2000). Our present evidence indicates that these obligate death events are proximal signals that are initiated independently and temporally before death commitment, which occurs at ∼4 hr, as indicated by our camptothecin withdrawal experiment.
By delayed addition of the pharmacological CDK inhibitor flavopiridol for varying times, we also find that CDKs must be inhibited 2 hr after the initiation of camptothecin treatment. This time point also correlates well with the activation of camptothecin-induced, cyclin D1-associated kinase activity in neurons, an event that begins at 1 hr and peaks at 4 hr after treatment (Park et al., 1998a). Moreover, it is consistent with the CDK-dependent phosphorylation of pRb, which also occurs at 1 hr (Park et al., 2000), and strongly supports a role for CDK4/6 as a proximal death signal that is activated shortly after DNA damage.
Expression of the p53 tumor suppressor protein after DNA damage results in the transcriptional regulation of various genes for which the activity coordinates G1 arrest, DNA repair, and apoptosis (Jacks and Weinberg, 1996). Induction of cellular levels of p53 protein as well as DNA binding activity increases at ∼2 hr, a time point before death commitment. The observation that Bax deficiency or caspase inhibition had no effect on p53 activity also implicates p53 as a more proximal death effector.
The manner in which CDK and p53 are related biochemically is unclear. Numerous interactions between the CDK pathway and p53 have been described. In proliferating systems CDKs can regulate p53 activity by several mechanisms. p53 can be a direct downstream kinase substrate for CDKs, including cdc2/cyclin B and CDK2-cyclin A (Wang and Prives, 1995). In addition, p53 is regulated indirectly downstream of CDK4/6 via phosphorylation of pRb, consequent induction of p19ARF, and stabilization of p53 (Bates et al., 1998). However, our data indicate that CDK inhibition has no effect on p53 levels, DNA binding, or nuclear localization. This indicates that direct regulation of p53 by CDKs does not occur in neurons after DNA damage. In addition, it appears unlikely that the pRB/E2F/p19ARF pathway regulates the p53 signal in this death paradigm.
Conversely, support for p53 as an upstream regulator of CDKs comes from observations that p53 is a transcriptional activator of the CDK inhibitor p21WAF1/CIP1 (Dulic et al., 1994). However, in this case, p21 would inhibit CDK activity, and our data indicate that CDKs are activated after DNA damage. Moreover, our present data indicate that p53 deficiency has no effect on pRb phosphorylation. Finally, we have shown previously that p21 is not upregulated after camptothecin treatment (Park et al., 2000). Taken together, this evidence indicates that p53 and CDKs are activated independently and do not modulate their respective activities directly. However, we cannot rule out the possibility that other p53 members such as p73 (Pozniak et al., 2000; Stiewe and Putzer, 2000) may act upstream or downstream of CDKs.
CDKs and p53 cooperate to mediate Bax activation, cytochromec release, and caspase activation
The above results led us to examine whether p53 and CDKs activate completely separate pathways of death or whether they interact at a more distal point in the death program. We and others have shown that Bax activation is required for apoptotic death after DNA damage (Xiang et al., 1998; Keramaris et al., 2000). We presently show that this activation does not occur via the increase in Bax levels but by translocation to mitochondrial-like compartments. Interestingly, inhibition of either the p53 or CDK pathway alone is sufficient to block Bax activation. These results suggest functional cooperativity between p53- and CDK-mediated events in promoting Bax translocation. Consistent with this observation, p53 deficiency or CDK inhibition alone also inhibits cytochrome c release from the mitochondria and caspase activation.
The manner by which CDKs and p53 or its downstream effectors may cooperate to mediate Bax activity is unclear. We have shown that the activation of Cdk4/6 leads to pRb phosphorylation/inactivation after DNA damage and that expression of a mutant of pRb with multiple phosphorylation sites removed, including the CDK site, leads to neuroprotection from DNA damage (Park et al., 2000). One consequence of deregulated pRb activity may be the inappropriate activation of E2F members, the best-characterized target of pRb regulation. Indeed, E2Fs can induce death in both proliferating cell types (Qin et al., 1994;Hiebert et al., 1995) and neurons (Hou et al., 2000; O'Hare et al., 2000) when overexpressed; expression of DN DP-1, an obligate binding partner to E2F family members (Wu et al., 1996), is also protective in multiple neuronal death paradigms, including DNA damage (Giovanni et al., 1999; Park et al., 2000). Finally, E2F-1 deficiency can protect against numerous neuronal apoptotic initiators, including β-amyloid toxicity (Giovanni et al., 2000), low K+(O'Hare et al., 2000), stroke (MacManus et al., 1999), and staurosporine (Hou et al., 2000).
E2F members, however, are not the only factors that are regulated by pRb. At present, the list of pRb-interacting factors numbers >110 (Morris and Dyson, 2001). At least two pRb-interacting factors, nuclear factor-κB (NFκB; Tamami et al., 1996) and c-Jun kinase (JNK;Chauhan et al., 1999; Shim et al., 2000), have been described that also are modulated by p53. Interestingly, Vousden and colleagues (Ryan et al., 2000) reported that p53-mediated death requires NFκB activity. This is significant in light of the evidence that pRb also can repress NFκB transcriptional activity (Tamami et al., 1996), suggesting one connection between p53 and CDKs. In addition, JNK activity, a mediator of neuronal death in numerous circumstances (Xia et al., 1995; Yang et al., 1997; Eilers et al., 1998; Watson et al., 1998), is inhibited by pRb binding (Shim et al., 2000). p53 also can bind to (Buschmann et al., 2000) and mediate the activation of JNK (Mazzoni et al., 1999), suggesting an additional link between the CDK and p53 pathways.
It must be noted that the parallel/cooperative interaction between the p53 and CDK pathway is context-specific. For example, CDKs also mediate the activation of caspases in models of β-amyloid toxicity (Giovanni et al., 2000) and K+ deprivation, which do not appear to be p53-dependent (M. O'Hare, D. S. Park, unpublished results). Whether proximal death signals other than p53 cooperate with the CDK pathway in these instances remains to be determined.
Delayed death evoked by DNA damage is not dependent on the core conserved death program
Our present data indicate that, whereas p53 deficiency results in nearly full protection for 48 hr after camptothecin exposure, death occurs slowly thereafter. Similar death is observed in Bax-deficient neurons, and the activation of caspases does not accompany this delayed death. This evidence indicates that delayed death does not involve the core conserved apoptotic pathway. We have reported previously that caspase inhibition also results in delayed death, which is terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-negative (TUNEL-negative; Stefanis et al., 1999), suggesting a nonclassical apoptotic pathway of delayed death. Importantly, the observed absence of caspase activity during delayed death reinforces the cooperative nature of the downstream effectors of p53- and CDK-mediated pathways. In this regard, activation of the CDK pathway in the absence of p53 does not activate caspases, even during the delayed death process, suggesting that both pathways must be active for caspase activation to occur.
In conclusion, select signal transduction pathways that occur in cortical neurons in response to DNA damage can be ordered into a simplified model, as represented in Figure10. DNA damage induces immediate early responses that activate at least two distinct signaling pathways involving (1) p53 induction and (2) CDK activation. We hypothesize that, whereas these two pathways are activated in a parallel manner, their downstream effectors cooperate to mediate the distal core death program (Bax translocation, cytochrome c release, and caspase activity). Because numerous other death/survival signals also are known to modulate neuronal apoptosis, it will be important to explore their requirements for death and place them temporally and biochemically in this growing signaling picture.
Fig. 10.

Proposed model for role of CDK, pRb, p53, Bax, and caspases in the death of cortical neurons evoked by DNA damage (see Discussion for summary).
Footnotes
This work was supported by the Heart and Stroke Foundation of Canada and the Medical Research Council of Canada (D.S.P.). D.S.P. is the recipient of a Glaxo- Wellcome Award in Stroke. L.S. is the recipient of a Burroughs Wellcome Award in Biomedical Sciences.
E.J.M. and E.K. contributed equally to this manuscript.
Correspondence should be addressed to Dr. David S. Park at the above address. E-mail: dpark@uottawa.ca.
REFERENCES
- 1.Baker WJ, Royer GL, Jr, Weiss RB. Cytarabine and neurologic toxicity [see comments]. J Clin Oncol. 1991;9:679–693. doi: 10.1200/JCO.1991.9.4.679. [DOI] [PubMed] [Google Scholar]
- 2.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14arf links the tumour suppressors Rb and p53 [letter]. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 3.Buschmann T, Adler V, Matusevich E, Fuchs SY, Ronai Z. p53 phosphorylation, association with murine double minute 2, c-Jun NH2-terminal kinase, p14ARF, p300/CBP during the cell cycle and after exposure to ultraviolet irradiation. Cancer Res. 2000;60:896–900. [PubMed] [Google Scholar]
- 4.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chauhan D, Hideshima T, Treon S, Teoh G, Raje N, Yoshihimito S, Tai YT, Li W, Fan J, DeCaprio J, Anderson KC. Functional interaction between retinoblastoma protein and stress-activated protein kinase in multiple myeloma cells. Cancer Res. 1999;59:1192–1195. [PubMed] [Google Scholar]
- 6.Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- 7.Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer's disease. Neurobiol Aging. 1998;19:S29–S32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 8.Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- 9.Deckwerth TL, Easton RM, Knudson CM, Korsmeyer SJ, Johnson EM., Jr Placement of the BCL2 family member BAX in the death pathway of sympathetic neurons activated by trophic factor deprivation. Exp Neurol. 1998;152:150–162. doi: 10.1006/exnr.1998.6846. [DOI] [PubMed] [Google Scholar]
- 10.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 11.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-Dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 12.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enokido Y, Araki T, Aizawa S, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996a;203:1–4. doi: 10.1016/0304-3940(95)12247-8. [DOI] [PubMed] [Google Scholar]
- 14.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996b;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 15.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao CY, Zelenka PS. Induction of cyclin B and H1 kinase activity in apoptotic PC12 cells. Exp Cell Res. 1995;219:612–618. doi: 10.1006/excr.1995.1271. [DOI] [PubMed] [Google Scholar]
- 17.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F/DP, in β-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- 19.Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack R, Park DS. E2F-1 mediates death of β-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase-3. J Biol Chem. 2000;275:11553–11560. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- 20.Hiebert SW, Packham G, Strom DK, Haffner R, Oren M, Zambetti G, Cleveland JL. E2F-1 × DP-1 induces p53 and overrides survival factors to trigger apoptosis. Mol Cell Biol. 1995;15:6864–6874. doi: 10.1128/mcb.15.12.6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou ST, Callaghan D, Fournier MC, Hill I, Kang L, Massie B, Morley P, Murray C, Rasquinha I, Slack R, MacManus JP. The transcription factor E2F-1 modulates apoptosis of neurons. J Neurochem. 2000;75:91–100. doi: 10.1046/j.1471-4159.2000.0750091.x. [DOI] [PubMed] [Google Scholar]
- 22.Jacks T, Weinberg RA. Cell-cycle control and its watchman [comment]. Nature. 1996;381:643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- 23.Johnson MD, Kinoshita Y, Xiang H, Ghatan S, Morrison RS. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci. 1999;19:2996–3006. doi: 10.1523/JNEUROSCI.19-08-02996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keramaris E, Stefanis L, Maclaurin J, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Involvement of caspase-3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 25.Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 26.MacManus JP, Koch CJ, Jian M, Walker T, Zurakowski B. Decreased brain infarct following focal ischemia in mice lacking the transcription factor E2F-1. NeuroReport. 1999;10:2711–2714. doi: 10.1097/00001756-199909090-00004. [DOI] [PubMed] [Google Scholar]
- 27.Mansfield SH, Castillo M. MR of cis-platinum-induced optic neuritis. AJNR Am J Neuroradiol. 1994;15:1178–1180. [PMC free article] [PubMed] [Google Scholar]
- 28.Martin DP, Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J Neurosci. 1990;10:184–193. doi: 10.1523/JNEUROSCI.10-01-00184.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci. 1999;19:9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 31.Morris EJ, Dyson NJ (2001) Retinoblastoma protein partners. Adv Cancer Res, in press. [DOI] [PubMed]
- 32.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakajima M, Kashiwagi K, Ohta J, Furukawa S, Hayashi K, Kawashima T, Hayashi Y. Etoposide induces programmed death in neurons cultured from the fetal rat central nervous system. Brain Res. 1994;641:350–352. doi: 10.1016/0006-8993(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 34.O'Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS. Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem. 2000;275:25358–25364. doi: 10.1074/jbc.M001725200. [DOI] [PubMed] [Google Scholar]
- 35.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 36.Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda J, Park DS. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA. 2000;97:10254–10259. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of postmitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 38.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997a;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997b;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998a;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998b;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 44.Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-κB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 47.Shim J, Park HS, Kim MJ, Park J, Park E, Cho SG, Eom SJ, Lee HW, Joe CO, Choi EJ. Rb protein down-regulates the stress-activated signals through inhibiting c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 2000;275:14107–14111. doi: 10.1074/jbc.275.19.14107. [DOI] [PubMed] [Google Scholar]
- 48.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 49.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stiewe T, Putzer BM. Role of the p53-homologue p73 in E2F-1-induced apoptosis. Nat Genet. 2000;26:464–469. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 51.Tamami M, Lindholm PF, Brady JN. The retinoblastoma gene product (Rb) induces binding of a conformationally inactive nuclear factor-κB. J Biol Chem. 1996;271:24551–24556. doi: 10.1074/jbc.271.40.24551. [DOI] [PubMed] [Google Scholar]
- 52.Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y, Khrestchatisky M. Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur J Neurosci. 1999;11:263–278. doi: 10.1046/j.1460-9568.1999.00434.x. [DOI] [PubMed] [Google Scholar]
- 53.Tomkins CE, Edwards SN, Tolkovsky AM. Apoptosis is induced in postmitotic rat sympathetic neurons by arabinosides and topoisomerase II inhibitors in the presence of NGF. J Cell Sci. 1994;107:1499–1507. doi: 10.1242/jcs.107.6.1499. [DOI] [PubMed] [Google Scholar]
- 54.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vogel H, Horoupian DS. Filamentous degeneration of neurons. A possible feature of cytosine arabinoside neurotoxicity. Cancer. 1993;71:1303–1308. doi: 10.1002/1097-0142(19930215)71:4<1303::aid-cncr2820710422>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 56.Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons: evidence that deoxycytidine may have a role in neuronal survival that is independent of DNA synthesis. J Neurosci. 1989;9:115–124. doi: 10.1523/JNEUROSCI.09-01-00115.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M, but not G1 cyclin-dependent kinases. Nature. 1995;376:88–91. doi: 10.1038/376088a0. [DOI] [PubMed] [Google Scholar]
- 58.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winkelman MD, Hines JD. Cerebellar degeneration caused by high-dose cytosine arabinoside: a clinicopathological study. Ann Neurol. 1983;14:520–527. doi: 10.1002/ana.410140505. [DOI] [PubMed] [Google Scholar]
- 60.Wu CL, Classon M, Dyson N, Harlow E. Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–3706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 62.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]