

Abstract
Elevated levels of β-Amyloid (Aβ) are present in the brains of individuals with either the sporadic or familial form of Alzheimer's disease (AD), and the deposition of Aβ within the senile plaques that are a hallmark of AD is thought to be a primary cause of the cognitive dysfunction that occurs in AD. Recent evidence suggests that Aβ induces neuronal apoptosis in the brain and in primary neuronal cultures, and that this Aβ-induced neuronal death may be responsible in part for the cognitive decline found in AD patients. In this study we have characterized one mechanism by which Aβ induces neuronal death. We found that in cortical neurons exposed to Aβ, activated c-Jun N-terminal kinase (JNK) is required for the phosphorylation and activation of the c-Jun transcription factor, which in turn stimulates the transcription of several key target genes, including the death inducer Fas ligand. The binding of Fas ligand to its receptor Fas then induces a cascade of events that lead to caspase activation and ultimately cell death. By analyzing the effects of mutations in each of the components of the JNK–c-Jun–Fas ligand–Fas pathway, we demonstrate that this pathway plays a critical role in mediating Aβ-induced death of cultured neurons. These findings raise the possibility that the JNK pathway may also contribute to Aβ-dependent death in AD patients.
Keywords: Alzheimer's disease, β-amyloid, c-Jun N-terminal kinase, JNK, c-Jun, Fas, Fas ligand, apoptosis, neuronal cell death
Alzheimer's disease (AD) is a neurodegenerative disorder that is characterized by senile plaques, neurofibrillary tangles, and neuronal loss (Yankner, 1996; Selkoe, 1999). A common feature of AD is the accumulation of β-amyloid (Aβ), 39- to 43-amino acid peptides derived from the amyloid precursor protein (APP). Aβ peptides aggregate to form fibrillar deposits that are the principal component of senile plaques. The importance of Aβ in the pathogenesis of AD is suggested by several findings. Notably, mutations in APP or presenilin, two proteins that are implicated in familial forms of AD, lead to an increase in the amyloidogenic form of Aβ (Selkoe, 1999). In addition, fibrillar Aβ, but not soluble Aβ, is specifically toxic to cultured neuronsin vitro (Yankner et al., 1990; Yankner, 1996). The evidence that Aβ accumulation is a determining factor in AD makes it important to determine the mechanism by which Aβ induces neuronal cell death. Recent studies have shown that in AD brains and in cultures of neurons exposed to Aβ, the dying cells display the characteristics of apoptosis (Anderson et al., 1996; Estus et al., 1997; Stadelmann et al., 1999). Aβ-induced apoptosis has been suggested to involve oxidative stress and the perturbation of intracellular calcium homeostasis and to be protein synthesis-dependent (Takashima et al., 1993; Pike et al., 1996; Yankner, 1996; Imaizumi et al., 1999; Selkoe, 1999). However, the specific intracellular signaling pathways by which Aβ triggers cell death are not yet well defined.
Several features of the c-Jun N-terminal kinase (JNK) pathway suggested that this pathway might mediate Aβ-induced apoptosis. The JNK pathway is activated by oxidative stress, raising the possibility that Aβ might also activate the JNK cascade. In addition, activation of the JNK pathway triggers the induction of gene transcription. Thus the protein synthesis dependence of Aβ-induced apoptosis might reflect a requirement for JNK-dependent transcription.
Activated JNK phosphorylates and activates several transcription factors, c-Jun, activating transcription factor 2 (ATF2), and Elk-1 (Ip and Davis, 1998). Recent evidence suggests that JNK and its target c-Jun play an important role in triggering neuronal apoptosis (Xia et al., 1995; Yang et al., 1997; Herdegen et al., 1998; Luo et al., 1998; Behrens et al., 1999; Le-Niculescu et al., 1999). The inhibition of JNK or c-Jun activity in nerve growth factor (NGF)-deprived PC12 cells or sympathetic neurons inhibits apoptosis. Likewise, in JNK3−/− or c-Jun mutant mice, hippocampal neurons are highly resistant to kainic acid-induced cell death (Yang et al., 1997). These studies indicate that the JNK pathway mediates NGF withdrawal- and kainic acid-induced neuronal apoptosis. The precise mechanism by which activation of the JNK cascade leads to apoptosis is not known. However, it was demonstrated recently that Fas ligand transcription is activated on neurotrophic factor withdrawal by a JNK-dependent mechanism (Le-Niculescu et al., 1999). Once induced, Fas ligand contributes to cell death in an autocrine or paracrine manner by activating its receptor Fas, which in turn leads to activation of a caspase cascade and the demise of the cell.
In the present study, we show that Aβ induces the death of cortical neurons via the activation of c-Jun and the induction of the Fas ligand death cascade in a JNK-dependent manner. These findings raise the possibility that this signaling cascade may contribute to neuronal cell death in AD patients.
MATERIALS AND METHODS
Materials. Antibodies were obtained from the following sources: anti-phospho-specific JNK and anti-β-galactosidase (β-Gal) antibodies from Promega (Madison, WI); anti-phospho-specific c-Jun (Ser-73) and anti-JNK3 antibodies from Upstate Biotechnology (Lake Placid, NY); anti-JNK1 antibody from PharMingen (San Diego, CA); anti-c-Jun, anti-Fas, anti-caspase-8, and anti-Fas-associated death-domain-containing protein (FADD) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); anti-Fas ligand antibody from Transduction Laboratories (Lexington, KY); anti-TuJ1 antibody from Babco (Richmond, CA); and anti-Fas antibody SM1/23 from Alexis. The Aβ peptides Aβ25–35, Aβ35–25, and Aβ1–40 were obtained from Bachem (Torrance, CA). The Fas–Fc fusion protein was from R & D Systems (Minneapolis, MN), and the caspase-8 inhibitor IETD-fluoromethyl ketone (fmk) was from Calbiochem (La Jolla, CA). The caspase-8 assay kit was from Clontech (Cambridge, UK). Kainic acid, anisomycin, and potassium cyanide (KCN) were from Sigma (St. Louis, MO). The MTS reagent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) was obtained from Promega.
Plasmid constructs. Dominant negative (DN)-SEK-1, DN-c-Jun FlagΔ169, the JNK interacting protein (JIP) binding domain (JBD) of JIP-1, cytomegalovirus (CMV)-LacZ, and CMV-enhanced green fluorescent protein constructs used in this study have been described previously (Xia et al., 1995; Whitmarsh et al., 1998; Brunet et al., 1999). DN-FADD (Chinnaiyan et al., 1996) was kindly provided by Dr. V. M. Dixit (Genentech, San Francisco, CA).
Animals. Long–Evans rats were obtained from Charles River (Wilmington, MA) for primary neuronal cultures. The JNK3 wild-type and knock-out mice from strain C57BL/6 were described previously (Yang et al., 1997). gld and lpr mice from the C3H/HeJ background and mice of strain ddY were obtained from Japan SLC.
Cell culture conditions. Primary cortical and hippocampal neurons were cultured from embryonic day 17–18 rat fetuses or from embryonic day 16–17 mouse fetuses as described previously (Xia et al., 1996). The neurons were plated on glass coverslips coated with poly-d-lysine or polyethyleneimine in 24-well plates at a density of 200,000 cells per well for nontransfection experiments and 400,000 cells per well for transfection experiments. Neurons were cultured in DMEM containing 10% calf serum for 1 d, and the medium was changed to serum-free DMEM with B27 supplement (Life Technologies, Gaithersburg, MD). The experiments were performed after 4–6 d in culture.
Treatment of neurons with Aβ peptides. One millimolar stock solutions of Aβ25–35, Aβ35–25, and Aβ1–40 were prepared in sterile water and stored at −20°C. To prepare aggregated Aβ peptides, the same volume of PBS was added to the stock solution and incubated at 37°C for 3–5 d. The soluble form of Aβ1–40 was prepared by adding PBS without subsequent incubation. Preaggregated Aβ peptides were added to the cultures at 25 μm and incubated for 24–48 hr for the apoptosis assays.
Immunoblotting and immunocytochemistry techniques. For immunoblotting, neurons treated with Aβ were washed with cold PBS, solubilized with SDS sample buffer, and then sonicated for 10 sec. The lysates were boiled for 5 min and centrifuged for 15 min. The supernatants were subjected to SDS-PAGE. Equal amounts of lysate protein were run on a 10% SDS polyacrylamide gel and transferred to nitrocellulose membranes. Nitrocellulose blots were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Triton X-100 (TBST) and then incubated with primary antibodies [anti-phospho-specific JNK, 1:5000; anti-JNK3, 1:1000; anti-JNK-1, 1:1000; anti-phospho-specific c-Jun (Ser-73), 1:5000; anti-c-Jun, 1:1000; anti-Fas ligand, 1:500; anti-Fas, 1:500; anti-caspase-8, 1:500; anti-FADD, 1:500; or anti-TuJ1, 1:1000] in TBST containing 3% BSA. Immunoreactivity was detected by sequential incubation of horseradish peroxidase-conjugated secondary antibody (1:15,000; Calbiochem) and ECL reagents (DuPont, Billerica, MA).
For β-Gal staining, cells were fixed in 4% paraformaldehyde and 2% sucrose, permeabilized with 0.1% Triton X-100, and blocked with 3% BSA. Cells were incubated with anti-β-Gal antibodies (1:300) and then incubated with the Cy3-conjugated secondary antibody (1:500, Jackson ImmunoResearch, West Grove, PA). For Fas ligand staining, cells were fixed in 4% paraformaldehyde and 2% sucrose and treated with cold MeOH. Cells were incubated with anti-Fas ligand antibody (1:500), biotin-conjugated secondary antibody (1:300; Calbiochem), and Cy3-labeled streptavidin (1:500; Jackson ImmunoResearch). Nuclei were stained with Hoechst 33258 (2.5 μg/ml; Sigma).
Transfection and apoptosis assay. Using the calcium phosphate transfection method (Xia et al., 1996; Brunet et al., 1999), cortical neurons were transfected with 2–4 μg of the construct of interest along with 0.3 μg of CMV-EGFP plasmid and 0.7 μg of CMV-LacZ plasmid. Two days after transfection, neurons were treated with Aβ25–35 for 24 hr, or 1 d after transfection, cells were treated with Aβ1–40 for 48 hr. The transfected neurons were fixed in 4% paraformaldehyde and 2% sucrose, and immunostaining for β-Gal was performed as described above. Nuclei were stained with Hoechst 33258. Apoptotic neurons among the β-Gal-positive neurons were counted in a blinded manner.
Cell death was determined under a fluorescence microscope after staining with Hoechst 33258 by counting the number of cells containing condensed or fragmented nuclei in two or three fields (100–200 cells) per well in a blinded manner. Data are expressed as percentage of apoptotic cells of the total cells counted. In some experiments, cell viability was assessed by measuring the ability of cellular dehydrogenase enzymes to reduce the MTS reagent leading to the formation of a color formazan dye. The data are expressed as percentage of loss of cell viability compared with the control condition.
Reverse transcriptase-coupled PCR analysis. Total RNA was isolated from cortical neurons using the RNAzole B reagent (Tel-Test). First-strand cDNA was synthesized by extension of oligo-dT primers with SuperScript II reverse transcriptase (Life Technologies) in a mixture containing 3 μg of total RNA according to the manufacturer's protocol. PCR amplifications were performed in a 25 μl reaction volume containing 0.2 mm dNTPs, 1.5 mmMgCl2, 1.8 U of ExpandTaq polymerase (Roche Molecular Biochemicals, Indianapolis, IN), and 0.4 μm of each primer. The PCR products were separated by 1% agarose gel electrophoresis and visualized by ethidium bromide staining. Primers used were as follows: rat Fas ligand sense, 5′-GCTCTGGTTGGAATGGGGTTAG-3′, and antisense, 5′-ATAGACCTTGTGGCTTAGGGGC-3′; rat Fas sense, 5′-CAAGGGACTGATAGCATCTTTGAGG-3′, and antisense, 5′-TCCAGATTCAGGGTCACAGGTTG-3′ (Le-Niculescu et al., 1999); rat S100 sense, 5′-GGATGTCTGAGCTGGAGAAG-3′, and antisense, 5′-ACTCCTGGAAGTCACACTCC-3′ (Freeman et al., 1994); mouse FasL sense, 5′-CAGCAGTGCCACTTCATCTTGG-3′, and antisense, 5′-TTCA-CTCCAGAGATCAGAGCGG-3′; mouse Fas sense, 5′-GAGAATTGCT-GAAGACATGACAATCC-3′, and antisense, 5′-GTAGTTTTCACTC-CAGACATTGTCC-3′ (Lee et al., 1999); and mouse β-actin sense, 5′-TGGATTCCTGTCGCATCCATGAAAG-3′, and antisense, 5′-CACTCC-AGAGATCAAAGCAGTTC-3′ (Le-Niculescu et al., 1999).
Caspase-8 activity assay. Fluorometric caspase-8 activity assays were performed according to the manufacturer's manual. In brief, cell lysates were prepared at 24 hr after Aβ25–35 or kainic acid treatment. The lysates were incubated with the fluorogenic substrate IETD-7-amino-4-trifluoromethyl coumarin at 37°C for 1 hr. Proteolytic cleavage of the substrate by caspase-8 emitted a fluorescent signal that was measured with a fluorometer (excitation, 400 nm; and emission, 505 nm).
RESULTS
Aβ activates the JNK pathway in cortical neurons
To study the mechanism of Aβ-induced cell death, we exposed primary cultures of rat cortical neurons to Aβ or control peptide for 24–48 hr and assessed in a blinded manner the effect on cortical neuron survival by visually monitoring nuclear condensation and neural process fragmentation. This approach is essentially identical to that used previously by Yankner et al. (1990) to model AD in vitro. As described previously, the fibrillar form of Aβ peptide (Aβ1–40), but not the soluble form of the peptide, effectively induced the death of cortical neurons within 48 hr (Fig.1A; Estus et al., 1997;Imaizumi et al., 1999). In addition, 10–50 μmconcentrations of a smaller Aβ peptide (Aβ25–35), but not a control peptide with the amino acid sequence inverted (Aβ35–25), were found to efficiently induce the death of cortical neurons within 24 hr (Fig. 1B,C). The Aβ25–35 peptide is a fragment of Aβ that has previously been shown to mimic the effects of the Aβ1–40 (Yankner et al., 1990; Imaizumi et al., 1999).
Fig. 1.
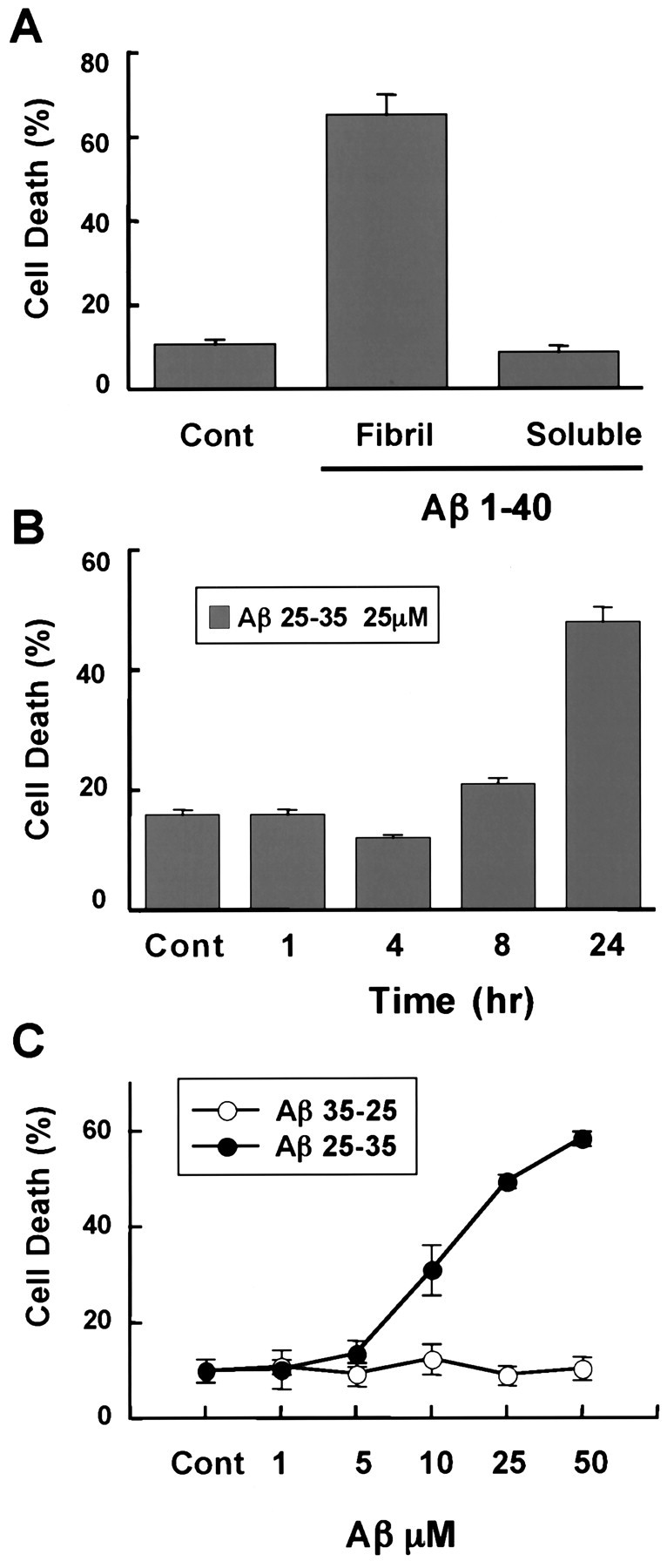
Aβ25–35 and Aβ1–40 promote neuronal cell death. A, Neurotoxicity of Aβ1–40. Rat primary cortical neurons were exposed to 50 μm fibrillar (Fibril) or soluble Aβ1–40 for 72 hr. Cells were fixed, nuclei were stained with Hoechst 33258, and apoptotic neurons were scored in a blinded manner as those cells that displayed condensed or fragmented nuclei. Experiments were performed at least three times, and data represent mean ± SEM of four wells from a representative experiment. B, Time course of Aβ25–35-induced neuronal apoptosis. Cultured neurons were treated with 25 μm Aβ25–35 for the indicated times, and apoptotic neurons were scored as described in A.C, Concentration-dependent neurotoxicity of Aβ25–35 and control peptide, Aβ35–25. Cortical neurons were exposed to Aβ25–35 or Aβ35-25 at various concentrations for 24 hr. Apoptotic neurons were scored as described in A.Cont, Control.
To test the hypothesis that Aβ-induced apoptosis of cortical neurons might involve the JNK signaling pathway, we asked whether the treatment of these neurons with Aβ leads to activation of components of the JNK pathway. JNK pathway activation was assessed by Western blotting using phosphorylation site-specific antibodies that recognize the phosphorylated and activated form of JNK or c-Jun (Fig.2). By Western blotting with the anti-phospho-JNK antibodies, it was possible to detect the two isoforms of JNK, 46 and 54 kDa. The anti-phospho-c-Jun antibodies specifically recognize c-Jun that is phosphorylated at Ser-73. The phosphorylation of c-Jun at Ser-73 has previously been shown to contribute to the induction of the transcriptional activity of c-Jun (Behrens et al., 1999). As shown in Figure 2A–D, exposure of cortical neurons to Aβ25–35 enhanced the phosphorylation of JNK and c-Jun in a time- and concentration-dependent manner. Western blotting with antibodies that recognize JNK1, JNK3, and c-Jun regardless of their state of phosphorylation indicated that the overall levels of these proteins was not affected by the addition of Aβ to cortical neurons. (Fig. 2A,C,D). The small change in the level of phosphorylated JNK was detected in multiple experiments and is statistically significant, as shown in Figure 2B. In addition, direct measurement of JNK activity revealed a consistent twofold increase in cells treated with Aβ25–35 (data not shown).
Fig. 2.

Aβ activates c-Jun and JNK in cortical neurons.A, Time course of Aβ-induced JNK and c-Jun phosphorylation. Rat cortical neurons were treated with 25 μm Aβ25–35 for the indicated number of hours. Whole-cell extracts were resolved by SDS-PAGE and immunoblotted with the antibodies directed against phospho-JNK or phospho-c-Jun (Ser-73). The total amount of JNK1, JNK3, and c-Jun protein was assessed using antibodies that recognize these proteins regardless of their phosphorylated state. B, Quantification of the phospho-JNK data (46 kDa isoform) from A. The densities of the bands were determined with a dual-wavelength Flying-spot scanner. *Statistical significance (p < 0.05) as assessed by the Wilcoxon test. C, Concentration-dependent activation of the JNK pathway by Aβ25–35. Cortical neurons were treated with Aβ25–35 at various concentrations for 8 hr. Western blotting analysis was done as described inA. D, Aβ1–40 activates the JNK pathway. Cortical neurons were incubated with 25 μmAβ1–40 or Aβ25–35 for the indicated times. Western blotting analysis was done as described in A. E, Aβ25–35 and fibril Aβ1–40 induce JNK phosphorylation in neurons. Cortical neurons were treated with PBS (lane 1), 25 μm Aβ25–35 (lane 2), 25 μm Aβ35–25 (lane 3), 25 μm fibrillar Aβ1–40 (lane 4), or 25 μm soluble Aβ1–40 (lane 5) for 8 hr. Western blotting analysis was performed using anti-phospho-specific c-Jun and anti-c-Jun as described in A.C, Control.
Given that the anti-phospho-JNK and anti-phospho-c-Jun antibodies recognize the activated forms of these proteins, these findings indicate that Aβ treatment of cortical neurons leads to the activation of c-Jun and raises the possibility that the activation of the JNK pathway may be relevant to Aβ-induced cell death in AD. Aβ25–35 peptide (25 μm) activation of JNK and c-Jun occurred within 4 hr of peptide addition, was sustained for at least 24 hr (Fig. 2A), and was found to precede the onset of apoptosis (Fig. 1B). This finding suggested that the activation of c-Jun might mediate Aβ induction of cortical neuron apoptosis. The possibility that the JNK–c-Jun pathway is critical for Aβ-induced neuronal apoptosis was supported by the observation that the concentrations of Aβ (25–50 μm) that induce JNK and c-Jun phosphorylation (Fig. 2C) are similar to the concentration of Aβ (10–50 μm) that are required to induce cell death (Fig. 1C). In addition, like Aβ25–35, exposure of cortical neurons to the fibrillar form of Aβ1–40, but not the soluble form, led to an induction of c-Jun phosphorylation (Fig. 2E). The control peptide Aβ35–25 had no effect on c-Jun phosphorylation (Fig.2E).
The JNK pathway is involved in Aβ-induced apoptosis
To determine whether the activation of the JNK pathway is required for Aβ-induced apoptosis, we introduced into cortical neurons various dominant interfering forms of components of the JNK signaling pathway (DN-SEK-1 and the JBD of JIP-1) and assessed their effect on Aβ-induced apoptosis. DN-SEK-1 is a catalytically inactive form of SEK-1, the protein kinase that phosphorylates and activates the JNKs. When DN-SEK-1 is expressed at high levels in cells, it has been shown to block JNK activation (Xia et al., 1995; Luo et al., 1998). JIP-1 is a scaffolding protein that is thought to bind and localize components of the JNK signaling pathway to specific subcellular regions (Whitmarsh et al., 1998; Yasuda et al., 1999). A fragment of JIP-1 termed the JBD has been shown to bind to JNKs and to inhibit their ability to phosphorylate substrates such as c-Jun (Dickens et al., 1997). The JBD of JIP-1 thus functions as an inhibitor of the JNK signaling pathway (Dickens et al., 1997). We have previously shown that the expression of DN-SEK-1 or the JBD of JIP-1 in NGF-differentiated PC-12 cells effectively inhibits NGF withdrawal-induced apoptosis (Xia et al., 1995; Dickens et al., 1997). Therefore, we tested the ability of these JNK signaling pathway inhibitors to block Aβ-induced cortical neuron apoptosis.
We transfected cortical neurons with plasmids encoding DN-SEK-1 or the JBD of JIP-1, treated the cells with 25 μm Aβ25–35 for 24 hr or Aβ1–40 for 48 hr, and performed a blind analysis of cell survival and apoptosis in the transfected neurons. Of the Aβ25–35- or Aβ1–40-treated cortical neurons that were transfected with an empty vector, approximately half of the neurons underwent apoptosis (Fig. 3A–D). By contrast, in neurons transfected with DN-SEK-1 or the JBD of JIP-1, the number of neurons undergoing Aβ25–35- or Aβ1–40-induced apoptosis was significantly decreased (Fig. 3A–C). The inhibitory effects of the various dominant interfering forms of JNK signaling pathway components were found to be specific to Aβ, inasmuch as these JNK pathway inhibitors had little effect on the basal level of apoptosis detected in cortical neurons exposed to the vehicle control. Taken together, these findings suggest that Aβ induction of the SEK1-JNK signaling cascade is required for Aβ-induced cortical neuron apoptosis.
Fig. 3.

Dominant negative mutants of SEK-1 and c-Jun and the JBD of JIP-1 prevent Aβ-induced apoptosis in cortical neurons. Cortical neurons were cotransfected with 4 μg of an empty vector, DN-SEK-1, the JBD of JIP-1, or DN-c-Jun FlagΔ169 and 0.7 μg of CMV-LacZ and 0.3 μg of CMV-EGFP. Two or 1 d after the transfection, the neurons were treated with 25 μmAβ25–35 for 24 hr or 25 μm Aβ1–40 for 48 hr, respectively. The cells were fixed and immunostained with anti-β-Gal antibody. Nuclei were stained with Hoechst 33258. Transfected cells were identified as those that express green fluorescent protein. From the transfected cells, the apoptotic neurons were identified as those displaying condensed and fragmented nuclei. Data represent mean ± SEM of four wells from a typical experiment. *p < 0.05, significantly different from vector (ANOVA and Dunnett's test). Each experiment was repeated two or three times. A, Effect of DN-SEK-1 on Aβ25–35 toxicity. B, Effect of DN-SEK-1 on Aβ1–40 toxicity. C, Effect of the JBD of JIP-1 on Aβ25–35 toxicity. D, Effect of DN-c-Jun FlagΔ169 on Aβ25–35 toxicity.
c-Jun transcriptional activity is required for Aβ induction of apoptosis
A primary function of the JNK signaling pathway is the phosphorylation of key transcriptional regulators such as c-Jun, Elk1, and ATF2 at sites that lead to their activation. One of the best characterized targets of JNK is the c-Jun proto-oncogene. The exposure of cells to stress or the withdrawal of neurotrophic factors such as nerve growth factor leads to the phosphorylation of c-Jun at Ser-63 and Ser-73 within the c-Jun transactivation domain. The phosphorylation of c-Jun at these two sites potently induces the ability of c-Jun to activate transcription. As shown in Figure 2, Aβ promotes c-Jun phosphorylation at Ser-73, suggesting that Aβ treatment may activate c-Jun. To determine whether Aβ-induced phosphorylation of c-Jun is critical for Aβ induction of apoptosis, we blocked c-Jun function in cortical neurons and assessed the effect on cortical neuron death. DN-c-Jun FlagΔ169 is a form of c-Jun that lacks the c-Jun transactivation domain but is still capable of forming dimers and binding to the c-Jun DNA-binding element. When overexpressed in cells, DN-c-Jun FlagΔ169 effectively competes with endogenous c-Jun for binding to the c-Jun DNA regulatory element and thereby inhibits c-Jun-dependent transcription (Ham et al., 1995; Xia et al., 1995). We found that the introduction of DN-c-Jun FlagΔ169 into cortical neurons led to a significant decrease in the number of neurons undergoing Aβ-dependent cell death (Fig. 3D). These findings strongly suggest that Aβ induces cell death by a c-Jun-dependent mechanism that requires the activation of particular c-Jun target genes.
β-amyloid-induced death is attenuated in cortical neurons from JNK3 knock-out mice
To further investigate the involvement of the JNK signaling pathway for Aβ-induced apoptosis, we tested the ability of Aβ to induce the death of cortical neurons derived from JNK3 knock-out mice that bear a mutation leading to the loss of JNK3 expression. We first verified by Western blotting that neurons prepared from the JNK3 knock-out mice did not express JNK3 (Fig.4). We then found that Aβ25–35 is slightly less effective at inducing c-Jun phosphorylation at Ser-73 in the JNK3−/− neurons compared with wild-type neurons, consistent with the absence of Aβ25–35 induction of JNK3 activity in the JNK3−/− neurons (Fig. 4). Although the level of phosphorylation of c-Jun was reproducibly reduced in the JNK3−/−neurons, Aβ25–35 was still capable of inducing some phosphorylation of c-Jun, suggesting that the continued presence of JNK1, JNK2, and perhaps other kinases in JNK3−/− neurons are capable of phosphorylating c-Jun. Aβ25–35 treatment of cortical neurons induced the phosphorylation and activation of the JNK3 activator SEK1 in both wild-type and JNK3−/− neurons, indicating that, like the wild-type neurons, JNK3−/− neurons are still capable of responding to Aβ25–35 (data not shown).
Fig. 4.

Effect of Aβ on the expression levels of JNK1 and JNK3 and on the phosphorylation of c-Jun in cortical neurons from wild-type and JNK3−/− mice. Whole-cell extracts from wild-type and JNK3−/− cortical neurons treated with or without 25 μm Aβ25–35 (8 hr) were prepared, and equal amounts of proteins were resolved by SDS-PAGE. The levels of JNK1, JNK3, phosphorylated c-Jun, and total c-Jun were examined by immunoblotting. WT, Wild type;KO, knock-out.
When primary cortical neurons obtained from wild-type or JNK3−/− mice were treated with 25 μm Aβ25–35 for 24 hr, we found that Aβ25–35 was less effective at inducing apoptosis in the JNK3−/− neurons than in wild-type neurons. Approximately 55% of the cortical neurons from wild-type mice treated with Aβ were apoptotic, whereas only 35% of Aβ-treated JNK3-deficient neurons were undergoing apoptosis (Fig.5A). Similar findings were obtained when hippocampal neurons from wild-type or JNK3−/− mice were exposed to Aβ25–35 and when wild type and JNK3−/− cortical neurons were exposed to Aβ1–40 (Fig. 5B,C). By contrast, the number of neurons undergoing apoptosis was similar when wild-type or JNK3−/− neurons were treated with vehicle control, indicating that the decrease in apoptosis detected in JNK3−/− neurons specifically reflects a decrease in Aβ-mediated cell death.
Fig. 5.

JNK3-deficient neurons are resistant to Aβ-induced apoptosis. A, B, Cortical (A) and hippocampal (B) neurons were cultured from wild-type and JNK3−/−mice embryos and treated with 25 μm Aβ25–35 for 24 hr. Cells were fixed, and nuclei were stained with Hoechst 33258. Apoptotic neurons were scored in a blinded manner. Data represent mean ± SEM of four wells from a typical experiment. *p < 0.05, significantly different from wild type (ANOVA and Dunnett's test); n = 4. C, Wild-type and JNK3−/− cortical neurons were treated with 25 μm Aβ1–40 for 48 hr, and apoptotic neurons were counted as described in A. D, Wild-type and JNK3−/− cortical and hippocampal neurons were treated with 25 μm Aβ25–35 as described inA. Cell viability was assayed by metabolic integrity (MTS assay). Data represent mean ± SEM (n = 4). *p < 0.05, significantly different from wild type (Student's t test). KO, Knock-out; WT, wild type.
In addition to determining the percentage of wild-type or JNK3−/− neurons undergoing apoptosis by assessing the number of cells containing fragmented or condensed nuclei, we obtained a measure of cell viability within the neuronal cultures by measuring cellular bioreducing activity using the MTS reagent (see Materials and Methods). When the MTS assay was used to measure the viability of Aβ-treated cortical or hippocampal neurons from wild-type or JNK3−/− mice, significantly more viable neurons were detected in the cultures prepared from JNK3−/− mice compared with cultures derived from wild-type mice (Fig. 5D). This biochemical analysis of apoptosis confirms the findings obtained by visual quantification of apoptotic nuclei and supports the conclusion that Aβ promotes neuronal apoptosis by a JNK3-dependent mechanism.
Aβ leads to Fas ligand induction
Because the ability of Aβ to induce apoptosis is dependent on the ability of Aβ to activate JNK3- and c-Jun-dependent transcriptional events, we next sought to identify JNK3 and c-Jun targets that might mediate Aβ-induced apoptosis. Several potential JNK3 and c-Jun targets have recently been identified whose transcription might directly or indirectly be induced on Aβ activation of the JNK3–c-Jun signaling pathway. These potential targets include Fas ligand, its receptor Fas, the p53 tumor suppressor gene, and tumor necrosis factor-α (TNF-α).
Previous studies have shown that survival factor withdrawal leads to the induction of Fas ligand mRNA and protein in cerebellar granule neurons and PC12 cells (Le-Niculescu et al., 1999). In addition, inhibition of the interaction of Fas ligand with its receptor Fas leads to a reduction in apoptosis (Faris et al., 1998a; Ishiyama et al., 1998; Kasibhatla et al., 1998; Brunet et al., 1999; Le-Niculescu et al., 1999; Kolbus et al., 2000). Consistent with the idea that Fas ligand is a target of the JNK3–c-Jun signaling pathway, the Fas ligand gene has c-Jun binding sites within its promoter (Faris et al., 1998a,b; Kasibhatla et al., 1998).
To determine whether the Fas ligand gene is a target of Aβ-dependent c-Jun activation, we first asked whether the exposure of cortical neurons to Aβ affects the expression of Fas ligand. Treatment of cortical neurons with Aβ25–35 led to a small but reproducible increase in the level of the Fas ligand mRNA but not the S100 control mRNA, as assessed by reverse transcriptase-coupled PCR (RT-PCR; Fig.6A). The increase in Fas ligand transcription was observed at 8 or 16 hr after Aβ25–35 treatment, indicating that it occurred subsequent to JNK3 and c-Jun activation. The Aβ-induced increase in the Fas ligand mRNA level was accompanied by an increase in Fas ligand protein, as indicated by Western blotting and immunofluorescence microscopy using anti-Fas ligand antibodies (Fig. 6B,C). Quantification of these results indicated a statistically significant induction of the Fas Ligand protein in response to Aβ induction (Fig.6B). In addition, like Aβ, the glutamate receptor agonist kainic acid, which has previously been shown to induce JNK3-dependent neuronal apoptosis (Yang et al., 1997), was also found to induce Fas ligand mRNA and protein levels (Fig.6B,C).
Fig. 6.
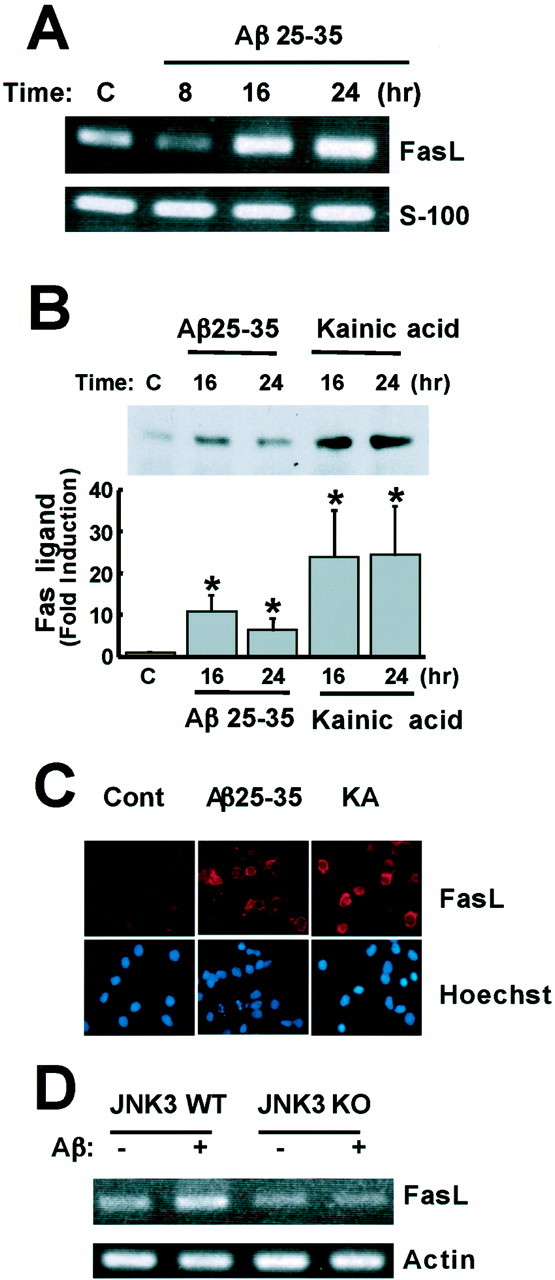
Aβ upregulates Fas ligand expression in cortical neurons. A, Aβ induction of Fas ligand (FasL) mRNA. Cortical neurons were treated with 25 μm Aβ25–35, and total RNA was extracted at the indicated times. Expression of Fas ligand and S100 mRNAs was examined by semiquantitative RT-PCR. B, Induction of Fas ligand protein level by Aβ and kainic acid. Cortical neurons were treated with 25 μm Aβ25–35 or 200 μm kainic acid, and total cell extracts were prepared at the indicated times. The level of Fas ligand was determined by immunoblotting using anti-Fas ligand antibodies and quantified using the Wilcoxon test (*p < 0.05). C, Immunostaining using anti-Fas ligand (FasL) antibody. Neurons were treated with Aβ25–35 or kainic acid (KA) for 16 hr and fixed. The cells were incubated with anti-Fas ligand antibody, followed by anti-mouse biotin-conjugated secondary antibody and then Cy3-labeled streptoavidin and Hoechst 33258. D, Cortical neurons derived from wild-type and JNK3−/− mice were treated with 25 μm Aβ25–35, and total RNA was extracted at 24 hr. Expression of Fas ligand and actin mRNAs was examined by semiquantitative RT-PCR. WT, Wild type;KO, knock-out.
To determine whether Aβ induction of the Fas ligand mRNA level is dependent on Aβ activation of the JNK signaling pathway, we assessed whether Aβ was capable of inducing the level of Fas ligand mRNA expression in neurons from JNK3−/− mice. Cortical neurons obtained from wild-type and JNK3−/− neurons were left untreated or treated with Aβ. RNA was extracted from the cells, and the level of Fas ligand mRNA was determined by RT-PCR analysis (Fig.6D). Whereas a significant induction of Fas ligand mRNA was detected in Aβ25–35-treated wild-type neurons, no increase in the level of the Fas ligand mRNA was observed when neurons from JNK3−/− mice were exposed to Aβ25–35. These findings indicate that Aβ induction of the Fas ligand mRNA requires JNK3 and suggest that Aβ induces the expression of this gene by triggering the activation of JNK3, which in turn phosphorylates and activates c-Jun. Activated c-Jun may then bind to the promoter of the Fas ligand gene, leading to an increase in transcription.
Aβ-induced apoptosis is dependent on Fas ligand and Fas signaling
Given that binding of Fas ligand to Fas has previously been shown to induce apoptosis in lymphoid cells and in neuronal cells deprived of critical survival factors (Faris et al., 1998a,b; Kasibhatla et al., 1998; Brunet et al., 1999; Le-Niculescu et al., 1999), we next asked whether Aβ induction of cortical neuronal apoptosis is mediated by the Fas ligand–Fas signaling complex. Previous studies have shown that binding of Fas ligand to Fas triggers the formation of a death-inducing signaling complex (DISC), which consists of Fas ligand, Fas, the adapter protein FADD, and caspase-8. The interaction of caspase-8 with DISC leads to caspase-8 proteolysis and activation. This then triggers a cascade of caspase activation that promotes the demise of the cell by digestion of critical cellular constituents (Nagata, 1997).
To test whether the Fas ligand–Fas pathway plays a critical role in Aβ-induced toxicity, we used several approaches to interfere with Fas ligand–Fas function. The first approach was to block Fas signaling by incubating cortical neurons with a soluble Fas–Fc protein or an interfering anti-Fas antibody. Previous studies have shown that soluble Fas–Fc protein effectively competes with membrane-bound Fas for Fas ligand and thereby blocks the effects of Fas ligand (Faris et al., 1998a; Kasibhatla et al., 1998; Brunet et al., 1999; Le-Niculescu et al., 1999). Likewise, the interfering anti-Fas antibody (SM1/23) blocks the binding of Fas ligand to Fas (Ishiyama et al., 1998). We found that the incubation of cortical neurons with Fas–Fc or the neutralizing anti-Fas antibody, but not with control IgG, resulted in a significant reduction in Aβ25–35-induced cell death, as determined by both visualization of apoptotic cells and the MTS biochemical assay (Fig.7A–C). The interfering anti-Fas antibody also inhibited kainic acid-induced apoptosis (Fig.7D). The inhibitory effects of the anti-Fas antibody were specific, inasmuch as this antibody did not significantly block KCN-induced neuronal cell death (Fig. 7E). KCN-induced cell death has previously been shown to occur by necrosis rather than apoptosis (Shimizu et al., 1996) and presumably occurs by a Fas ligand-independent mechanism. Taken together, these experiments indicate that Aβ induces cortical neuron apoptosis via JNK3- and c-Jun-dependent activation of the Fas ligand–Fas signaling pathway.
Fig. 7.

Aβ induces apoptosis by a Fas ligand–Fas-dependent mechanism. A, B, Cortical neurons were pretreated with 15 μg/ml Fas–Fc or control IgG for 2 hr, and then 25 μm Aβ25–35 was added, and apoptosis was determined by scoring cells with condensed and fragmented nuclei (A) or using the MTS assay (B). *p < 0.05, significantly different from IgG (ANOVA and Dunnett's test or Student's t test). C–E, Cortical neurons were incubated with 2.5 μg/ml anti-Fas antibody or control IgG for 2 hr and then treated with 25 μm Aβ25–35 (C), 200 μm kainic acid (D), or 5 mm KCN (E). Cell death was assessed by the MTS assay. *p < 0.05, significantly different from IgG (Student's t test).
We further assessed the importance of the Fas ligand–Fas signaling complex for Aβ-induced cortical neuronal death by examining the efficacy with which Aβ induced the death of neurons isolated from mice that have mutations in either Fas ligand or Fas. gldand lpr mice carry mutations that lead to a loss of function of Fas ligand and Fas genes, respectively. We treated cortical neurons from wild-type, gld, and lpr mice with 25 μm Aβ25–35 or Aβ1–40 and scored viability of the neurons 24–72 hr after Aβ addition using the MTS assay. Whereas Aβ induced the death of a large percentage of the wild-type neurons (60%), Aβ was quite ineffective at inducing the death of neurons (15%) from gld and lpr mice (Fig.8A). By contrast, the viability of neurons from wild-type, gld, and lprmice was comparable when the neurons were grown in the absence of Aβ or treated with KCN, which induces necrotic cell death (data not shown). These findings provide further evidence that the Fas ligand–Fas signaling complex is a critical mediator of Aβ-induced neuronal death. We also examined whether the downstream signaling component of the Fas ligand–Fas signaling complex, caspase-8, is activated in response to Aβ. As shown in Figure 8B, Aβ or kainic acid treatment induced a twofold elevation in caspase-8 activity in rat and mouse neurons. The caspase-8 activity was completely inhibited in the presence of a specific caspase-8 inhibitor, Ζ-IETD-fmk.
Fig. 8.

The Fas ligand–Fas pathway is required for Aβ-induced apoptosis. A, Cortical neurons were prepared from wild-type, gld, and lprmice embryos and treated with 25 μm Aβ25–35 for 24 hr or 25 μm Aβ1–40 for 72 hr. Cell viability was assessed by the MTS assay. Data represent mean ± SEM (n = 5). *p < 0.05, significantly different from wild type (ANOVA and Dunnett's test).B, Neurons from rat and mouse were treated with 25 μm Aβ25–35 and the caspase-8 inhibitor, Ζ-IETD-fmk, or with 200 μm kainic acid. Caspase-8 activity was assessed by a fluorometric assay kit. Data represent mean ± SEM (n = 6 for rats; n = 4 for mice). C, Cortical neurons were cotransfected with 4 μg of an empty vector or DN-FADD and 0.7 μg of CMV-LacZ and 0.3 μg of CMV-EGFP. After 2 d, the cells were treated with 25 μm Aβ25–35 for 24 hr. The cells were fixed and immunostained with anti-β-Gal antibody. Nuclei were stained with Hoechst 33258. Transfected cells were identified as those that expressed green fluorescent protein, and of these cells, apoptotic neurons were scored as those that display condensed and fragmented nuclei. Data represent mean ± SEM of four wells from a typical experiment. D, Neurons were pretreated with DMSO or 100 μm caspase 8 inhibitor Ζ-IETD-fmk for 1 hr, and then 25 μm Aβ25–35 was added, and apoptosis was determined by the MTS assay. Data represent mean ± SEM of seven wells from a typical experiment. *p < 0.05, significantly different from vector or control (ANOVA and Dunnett's test or Student's t test).
We then determined whether downstream components of the Fas ligand–Fas signaling pathway are required for Aβ-induced apoptosis. A dominant interfering form of FADD (DN-FADD), which lacks 80 N-terminal amino acids of the death effector domain responsible for interaction with caspase-8, was introduced into neurons. Because DN-FADD is still able to associate with Fas, DN-FADD inhibits the interaction of Fas with endogenous FADD and blocks Fas signaling (Chinnaiyan et al., 1996). Caspase-8 activity was blocked by incubating cortical neurons with the selective caspase-8 inhibitor Ζ-IETD-fmk, before Aβ treatment. Consistent with the data by Ivins et al. (1999), the inhibition of FADD or caspase-8 function significantly reduced the number of neurons undergoing Aβ-induced apoptosis. (Fig.8C,D).
DISCUSSION
The accumulation of Aβ in the brains of AD patients has been implicated as a cause of the neuronal loss that occurs in Alzheimer's disease. However, the mechanisms by which Aβ induces neuronal death are not well understood. In this report we provide evidence that Aβ induces the activation of c-Jun in a JNK-dependent manner. JNK3 appears to promote apoptosis by phosphorylating and activating the transcription factor c-Jun. Our data show that Aβ induces a small but consistent phosphorylation of JNK and a relatively large increase in phosphorylation of c-Jun. The discrepancy between a small JNK activation and a robust c-Jun phosphorylation could reflect the fact that JNK activity is constitutively high in neurons compared with other cell types. These results further suggest that the induction of JNK alone is not sufficient for c-Jun phosphorylation and that there are other kinases contributing to the induction of c-Jun activity. However, it is clear that JNK activity is important for Aβ induction of neuronal death, inasmuch as the disruption of JNK3 function leads to a marked decrease in the percentage of neurons undergoing Aβ-induced neuronal apoptosis. In neurons from JNK3 knock-out mice, Aβ induction of c-Jun phosphorylation is partially blocked, again suggesting that kinases other than JNK may be involved in c-Jun phosphorylation. In addition, Aβ induction of neuronal apoptosis is inhibited when c-Jun function is blocked either by the expression of dominant interfering forms of c-Jun (this study) or by a targeted disruption of the c-Jun gene (Kihiko et al., 1999). Although a variety of JNK3–c-Jun targets have been identified, we find that Fas ligand specifically plays a role in Aβ-induced neuronal apoptosis. Aβ was found to induce Fas ligand expression in a JNK3-dependent manner. In addition, inhibition of Fas ligand and Fas function led to a decrease in Aβ-induced apoptosis. Taken together, these findings implicate the JNK–c-Jun–Fas ligand–Fas signaling cascade in Aβ-mediated death of cultured neurons.
Several recent observations also provide initial support for the possibility that the JNK3–c-Jun–Fas ligand–Fas pathway may mediate cell death in AD. First, in AD brains, JNK3 immunoreactivity is co-localized with ALZ-50 antigen, a marker for early neurofibrillary degeneration (Mohit et al., 1995), suggesting that JNK3-expressing neurons are highly vulnerable in AD brains. Second, JNK activation is detected in degenerating neurons in AD brains (Shoji et al., 2000; Zhu et al., 2001). Third, c-Jun is expressed at high levels specifically in apoptotic neurons that are detected in the AD brain (Anderson et al., 1994, 1996; Marcus et al., 1998). Finally, Fas expression is upregulated in the neurons of AD brains (de la Monte et al., 1997,1998; Seidl et al., 1999). Taken together, this correlative evidence suggests a role for the JNK–c-jun–Fas Ligand–Fas signaling cascade in AD, although additional experimentation will be required to confirm that this pathway mediates the neuronal cell death that occurs in AD.
A number of observations suggest that the JNK3–c-Jun–Fas ligand–Fas signaling pathway also mediates neuronal cell death that is initiated by apoptotic stimuli other than Aβ. The JNK pathway has been shown to play an important role in dopamine-induced apoptosis in a cellular model of Parkinson's disease (PD) (Luo et al., 1998). In addition, several recent reports suggest a significant role for Fas ligand and Fas in neurodegenerative disease. In an animal model of focal cerebral ischemia, Fas ligand and Fas are induced in neurons undergoing apoptosis, and the infarct volume after brain ischemia inlpr mice is significantly reduced compared with that in wild-type mice (Matsuyama et al., 1995; Herdegen et al., 1998;Martin-Villalba et al., 1999). Furthermore, in several neurodegenerative diseases such as PD and Down syndrome, the upregulation of Fas expression was detected specifically in neurons that were dying (de la Monte et al., 1997, 1998; Seidl et al., 1999). Thus it is possible that the JNK3–c-Jun–Fas ligand–Fas pathway may be a common mediator of cell death in a variety of neurodegenerative diseases.
With respect to AD, it is important to note that Aβ toxicity is not completely abolished in JNK3-deficient neurons or gld andlpr neurons. In addition, although c-Jun phosphorylation is reproducibly decreased in JNK3 knock-out cells, significant phosphorylation remains. This suggests that in addition to the JNK3–c-Jun–Fas ligand–Fas signaling pathway, there are other pathways that contribute to Aβ induction of apoptosis. It is likely that the other members of the JNK family (i.e., JNK1 and JNK2) play a role in Aβ-induced apoptosis. Consistent with this possibility, we have found that JNK1-deficient neurons are also resistant to the death-promoting effects of Aβ (data not shown). In addition to Fas ligand, there are likely to be other targets of c-Jun that contribute to its ability to mediate Aβ-induced apoptosis. One possible target is the death cytokine TNF-α, because the expression of TNF-α is known to be regulated by JNK and c-Jun (Ishizuka et al., 1997;Hoffmeyer et al., 1999). Finally, c-Jun is most likely just one of several JNK substrates that contribute to Aβ-induced apoptosis. The phosphorylation of Bcl-2 by JNK suppresses the prosurvival activity of Bcl-2 (Chang et al., 1997; Maundrell et al., 1997; Yamamoto et al., 1999). In addition, p53 and c-Myc, two potent inducers of apoptosis, are substrates of JNK. The phosphorylation of p53 and c-Myc by JNK leads to the induction of the proapoptotic function of these two proteins, consistent with the possibility that p53 and c-Myc might be mediators of the apoptotic effect of JNKs (Milne et al., 1995; Fuchs et al., 1998; Noguchi et al., 1999). It will be important to determine whether Aβ induction of JNK activity leads to Bcl-2, p53, and c-Myc phosphorylation and whether the phosphorylation of these proteins then contributes to Aβ-induced cell death.
It will also be of interest to establish the mechanism by which extracellular Aβ induces intracellular JNK activation. The fibrillar form of Aβ appears to bind to the surface of neurons through multiple receptors. Aβ has been shown to bind to a variety of proteins, including the Aβ precursor protein (Lorenzo et al., 2000), an endoplasmic reticulum amyloid β peptide binding protein/L-3-hydroxyacyl-coenzyme A dehydrogenase (Yan et al., 1997, 1999), and the receptor for advanced glycation end products, which mediates oxidative stress (Yan et al., 1996). Whether one or several of these Aβ-binding proteins mediates Aβ induction of JNK activation is not known.
A reasonable possibility is that one or several Aβ receptors promote apoptosis via a JNK-dependent pathway, whereas the other Aβ-binding proteins contribute to Aβ-mediated apoptosis through distinct signaling pathways. One such pathway could involve the action of calpain, p25, and cdk5 (Patrick et al., 1999; Lee et al., 2000). In a recent study, it was demonstrated in cultured neurons that Aβ induces the Ca2+-dependent activation of calpain I, which then cleaves p35, the regulatory subunit of cdk5, to p25, leading to constitutive cdk5 activation. Because p25 and cdk5 overexpression leads to the apoptosis of cultured neurons, it is likely that one way Aβ induces cell death is through the activation of p25 and cdk5. It remains to be determined whether there is cross-talk between JNKs and cdk5 that is important for Aβ induction of apoptosis. Further characterization of the multiple pathways that mediate the Aβ-dependent apoptosis of cultured neurons, and the importance of these pathways in AD, is likely to provide new avenues for treating this disease.
Footnotes
This research was supported by a grant to M.E.G. from Daiichi Pharmaceutical Co., Ltd. We thank Dr. Vishva M. Dixit for kindly providing plasmids of DN-FADD. We thank all members of the Greenberg laboratory for helpful discussions and support, particularly Dr. Anne Brunet.
Correspondence should be addressed to Michael E. Greenberg, Division of Neuroscience, Children's Hospital, and Department of Neurobiology, Harvard Medical School, Boston, MA 02115. E-mail:michael.greenberg@tch.harvard.edu.
Dr. Gotoh's present address: Institute of Molecular and Cell Bioscience, University of Tokyo, Tokyo 113-0032, Japan.
REFERENCES
- 1.Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease. Exp Neurol. 1994;125:286–295. doi: 10.1006/exnr.1994.1031. [DOI] [PubMed] [Google Scholar]
- 2.Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrens A, Sibilis M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 4.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phospholylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 5.Chang BS, Minn AJ, Muchmore SW, Fesik SW, Thompson CB. Identification of a novel regulatory domain in Bcl-xL and Bcl-2. EMBO J. 1997;16:968–977. doi: 10.1093/emboj/16.5.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 7.de la Monte SM, Sohn YK, Ganju N, Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci. 1997;152:73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- 8.de la Monte SM, Sohn YK, Ganju N, Wands JR. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest. 1998;78:401–411. [PubMed] [Google Scholar]
- 9.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 10.Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA, Nel A. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J Immunol. 1998a;160:134–144. [PubMed] [Google Scholar]
- 12.Faris M, Latinis KM, Kempiak SJ, Koretzky GA, Nel A. Stress-induced Fas ligand expression in T cells is mediated through a MEK kinase 1-regulated response element in the Fas ligand promoter. Mol Cell Biol. 1998b;18:5414–5424. doi: 10.1128/mcb.18.9.5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci USA. 1998;95:10541–10546. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 16.Herdegen T, Claret F-X, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmeyer A, Grosse-Wilde A, Flory E, Neufeld B, Kunz M, Rapp UR, Ludwig S. Different mitogen-activating protein kinase signaling pathways cooperate to regulate tumor necrosis factor α gene expression in T lymphocytes. J Biol Chem. 1999;274:4319–4327. doi: 10.1074/jbc.274.7.4319. [DOI] [PubMed] [Google Scholar]
- 18.Imaizumi K, Morihara T, Mori Y, Katayama T, Tsuda M, Furuyama T, Wanaka A, Takeda M, Tohyama M. The cell death-promoting gene DP-5, which interacts with BCL2 family, is induced during neuronal apoptosis following exposure to amyloid β protein. J Biol Chem. 1999;274:7975–7981. doi: 10.1074/jbc.274.12.7975. [DOI] [PubMed] [Google Scholar]
- 19.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)-from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 20.Ishiyama S, Hiroe M, Nishikawa T, Shimojo T, Abe S, Fujisaki H, Ito H, Yamakawa K, Kobayashi N, Kasajima T, Marumo F. The Fas/Fas ligand system is involved in the pathogenesis of autoimmune myocarditis in rats. J Immunol. 1998;161:4695–4701. [PubMed] [Google Scholar]
- 21.Ishizuka T, Terada N, Gerwins P, Hamelmann E, Oshiba A, Fanger GR, Johnson GL, Gelfand EW. Mast cell tumor necrosis factor a production is regulated by MEK kinases. Proc Natl Acad Sci USA. 1997;94:6358–6363. doi: 10.1073/pnas.94.12.6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivins KJ, Thornton PL, Rohn TT, Cotman CW. Neuronal apoptosis induced by beta-amyloid is mediated by caspase-8. Neurobiol Dis. 1999;6:440–449. doi: 10.1006/nbdi.1999.0268. [DOI] [PubMed] [Google Scholar]
- 23.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-κB and AP-1. Mol Cell. 1998;1:543–551. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- 24.Kihiko ME, Tucker HM, Rydel RE, Estus S. c-Jun contributes to amyloid β-induced neuronal apoptosis but is not necessary for amyloid β-induced c-jun induction. J Neurochem. 1999;73:2609–2612. doi: 10.1046/j.1471-4159.1999.0732609.x. [DOI] [PubMed] [Google Scholar]
- 25.Kolbus A, Herr I, Schreiber M, Debatin K-M, Wanger EF, Angel P. c-Jun-dependent CD95-L expression is a rate-limiting step in the induction of apoptosis by alkylating agents. Mol Cell Biol. 2000;20:575–582. doi: 10.1128/mcb.20.2.575-582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J, Richburg JH, Shipp EB, Meistrich ML, Boekelheide K. The Fas system, a regulator of testicular germ cell apoptosis, is differentially up-regulated in Sertoli cell versus germ cell injury of the testis. Endocrinology. 1999;140:852–858. doi: 10.1210/endo.140.2.6479. [DOI] [PubMed] [Google Scholar]
- 27.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 28.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret F-X, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci. 2000;3:460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 30.Luo Y, Umegaki H, Wang X, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- 31.Marcus DL, Strafaci JA, Miller DC, Masia S, Thomas CG, Rosman J, Hussain S, Freesman ML. Quantitative neuronal c-Fos and c-Jun expression in Alzheimer's disease. Neurobiol Aging. 1998;19:393–400. doi: 10.1016/s0197-4580(98)00077-3. [DOI] [PubMed] [Google Scholar]
- 32.Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin K-M. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuyama T, Hata R, Yamamoto Y, Tagaya M, Akita H, Uno H, Wanaka A, Furuyama J, Sugita M. Localization of the Fas antigen mRNA induced in the postischemic murine forebrain by in situ hybridization. Mol Brain Res. 1995;34:166–172. doi: 10.1016/0169-328x(95)00162-l. [DOI] [PubMed] [Google Scholar]
- 34.Maundrell K, Antonsson B, Magnenat E, Camp M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, Martinou J-C, Arkinstall S. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- 35.Milne DM, Campbell LE, Campbell DG, Meek DW. p53 is phosphorylated in vitro and in vivo by an ultraviolet radiation-induced protein kinase characteristic of the c-Jun kinase, JNK1. J Biol Chem. 1995;270:5511–5518. doi: 10.1074/jbc.270.10.5511. [DOI] [PubMed] [Google Scholar]
- 36.Mohit AA, Martin JH, Miller CA. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron. 1995;14:67–78. doi: 10.1016/0896-6273(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 37.Nagata S. Apoptosis by death factor. Cell. 1997;80:293–299. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 38.Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem. 1999;274:32580–32587. doi: 10.1074/jbc.274.46.32580. [DOI] [PubMed] [Google Scholar]
- 39.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 40.Pike C, Balazs R, Cotman CW. Attenuation of β-amyloid neurotoxicity in vitro by potassium-induced depolarization. J Neurochem. 1996;67:1774–1777. doi: 10.1046/j.1471-4159.1996.67041774.x. [DOI] [PubMed] [Google Scholar]
- 41.Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci Lett. 1999;260:9–12. doi: 10.1016/s0304-3940(98)00945-8. [DOI] [PubMed] [Google Scholar]
- 42.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, Tsujimoto Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–2050. [PubMed] [Google Scholar]
- 44.Shoji M, Iwakami N, Takeuchi S, Waragai M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular beta-amyloid accumulation. Mol Brain Res. 2000;85:221–233. doi: 10.1016/s0169-328x(00)00245-x. [DOI] [PubMed] [Google Scholar]
- 45.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brôck W, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proc Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 49.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;17:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 52.Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]
- 53.Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stern E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stern DM. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J Biol Chem. 1999;274:2145–2156. doi: 10.1074/jbc.274.4.2145. [DOI] [PubMed] [Google Scholar]
- 54.Yang DD, Kuan C-Y, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 55.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 56.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 57.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu X, Raina AK, Rottkamp CA, Aliev G, Peggy G, Boux H, Smith MA. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]