

Abstract
Deleterious processes of extracellular proteolysis may contribute to the progression of tissue damage after acute brain injury. We recently showed that matrix metalloproteinase-9 (MMP-9) knock-out mice were protected against ischemic and traumatic brain injury. In this study, we examined the mechanisms involved by focusing on relevant MMP-9 substrates in blood–brain barrier, matrix, and white matter. MMP-9 knock-out and wild-type mice were subjected to transient focal ischemia. MMP-9 levels increased after ischemia in wild-type brain, with expression primarily present in vascular endothelium. Western blots showed that the blood–brain barrier-associated protein and MMP-9 substrate zonae occludens-1 was degraded after ischemia, but this was reduced in knock-out mice. There were no detectable changes in another blood–brain barrier-associated protein, occludin. Correspondingly, blood–brain barrier disruption assessed via Evans Blue leakage was significantly attenuated in MMP-9 knock-out mice compared with wild types. In white matter, ischemic degradation of the MMP-9 substrate myelin basic protein was significantly reduced in knock-out mice compared with wild types, whereas there was no degradation of other myelin proteins that are not MMP substrates (proteolipid protein and DM20). There were no detectable changes in the ubiquitous structural protein actin or the extracellular matrix protein laminin. Finally, 24 hr lesion volumes were significantly reduced in knock-out mice compared with wild types. These data demonstrate that the protective effects of MMP-9 gene knock-out after transient focal ischemia may be mediated by reduced proteolytic degradation of critical blood–brain barrier and white matter components.
Keywords: stroke, extracellular proteolysis, blood–brain barrier, myelin, neuroprotection, mouse
The matrix metalloproteinase (MMP) family consists of at least 23 endopeptidases with common motifs comprising propeptide and zinc-binding catalytic regions (Nagase and Woessner, 1999). Collectively, MMPs are extracellular proteases that are capable of modifying almost all components of the extracellular matrix. Physiological roles for MMPs include embyrological remodeling, angiogenesis, ovulation, and wound healing (Nagase and Woessner, 1999). Because MMPs can degrade many matrix components, enzyme activities are strictly regulated via gene transcription, proenzyme activation, and dynamic inhibition by tissue inhibitors of metalloproteinases (TIMPs) (Fini et al., 1998; Westermarck and Kahari, 1999).
Uncontrolled expression of MMPs can result in tissue injury and inflammation (Lukashev and Werb, 1998). In the CNS, MMPs have been implicated in neurodegenerative disorders such as multiple sclerosis and Alzheimer's disease (Yong et al., 1998; Hartung and Kieseier, 2000). Evidence is also accumulating to suggest that MMPs can be involved in acute brain injury. MMPs are upregulated after ischemia (Rosenberg et al., 1996; Mun-Bryce and Rosenberg, 1998b; Gasche et al., 1999; Heo et al., 1999), hemorrhage (Rosenberg and Navratil, 1997), and trauma (Morita-Fujimura et al., 1999; Ferguson and Muir, 2000; Vecil et al., 2000; Wang et al., 2000), and pharmacologic inhibition of MMPs can reduce tissue damage and edema (Romanic et al., 1998; Rosenberg et al., 1998). Recently, we showed that knock-out (KO) mice deficient in MMP-9 expression were protected against cerebral ischemia (Asahi et al., 2000b) and traumatic brain injury (Wang et al., 2000). These data implicate a pathophysiologic role for MMP-9, but the precise mechanisms involved remain to be fully elucidated. Previous studies have primarily focused on MMP-mediated degradation of collagen and laminin in the cerebrovascular basal lamina with subsequent perturbations in vessel integrity and edema. However, MMPs have broad substrate specificities (Imper and Van Wart, 1998), so additional targets may be involved.
In this study, we investigated the hypothesis that deleterious effects of MMP-9 in cerebral ischemia are mediated via proteolytic degradation of critical extracellular substrates in the brain. Although a broad range of substrates may be involved, we chose to focus on known MMP-9 substrates in the blood–brain barrier (BBB), white matter, and extracellular matrix because damage to these three compartments would play central roles in the pathophysiology of cerebral ischemia. For the BBB, we examined zonae occludens-1 (ZO-1), a protein that is associated with the functional expression of tight junctions in cerebral endothelium (Denker and Nigam, 1998; Harkness et al., 2000; Kniesel and Wolburg, 2000). For white matter, we focused on myelin basic protein (MBP) (Campagnoni, 1988; Chandler et al., 1995). For extracellular matrix, we examined laminin because it is widely expressed in brain and may mediate cell–matrix interactions necessary for neuronal survival (Tsirka et al., 1997a). Degradation of these substrates was examined after transient focal ischemia, and outcomes in MMP-9 knock-out mice were compared with wild-type (WT) littermates. To assess the specificity of our findings, non-MMP-9 substrates were also examined.
MATERIALS AND METHODS
Animal model. All experiments were performed following an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. In experiments to document the ischemic upregulation of MMP-9, normal male CD-1 mice were used. For all other experiments, male MMP-9 knock-out mice and their corresponding wild-type littermates were used. The MMP-9 knock-outs were bred from a CD-1 background (Vu et al., 1998). The major phenotypic alteration detected was a perturbed pattern of skeletal growth plate development during the first 3 weeks of life, that eventually remodeled, resulting in an axial skeleton of normal appearance (Vu et al., 1998).
The standard intraluminal middle cerebral artery occlusion method was used (Asahi et al., 2000b). Briefly, mice were anesthetized with 1% halothane in 30% oxygen and 70% nitrous oxide using a face mask. The right femoral artery was cannulated to record blood pressure and to obtain arterial blood samples. Core rectal temperature was maintained at 37.5°C with a thermostat-controlled heating pad. After a midline skin incision, the right external carotid artery was exposed, and its branches were electrocoagulated. A 7.0 nylon monofilament coated with silicon was introduced into the right internal carotid artery through the external carotid artery to occlude the origin of the middle cerebral artery. After 120 min of arterial occlusion, blood flow was restored by withdrawal of the nylon suture. Sham control animals were subjected to similar operations to expose the carotid arteries without occlusion of the middle cerebral artery. All animals were assessed with laser doppler flowmetry to confirm adequate induction of focal ischemia and successful reperfusion, as well as for purposes of comparisons of regional perfusion between experimental groups. Perfusion reductions during ischemia were assessed as a percentage of pre-ischemic baselines. To decrease anesthesia times, laser doppler flow probes were removed and then re-attached for the reperfusion phase. This results in baselines that can no longer be compared with pre-ischemic levels. Therefore, reperfusion profiles were assessed as fold-increase versus perfusion levels during ischemia just before unocclusion.
Latex–carbon black perfusions of cerebrovascular angioarchitecture. Macroscopic examination of the KOs showed that the major cerebral arteries including the circle of Willis appeared normal, compared with WT littermates and normal CD-1 mice as described previously (Asahi et al., 2000b). Nevertheless, it is important to exclude the possibility that differences in ischemic outcome are not caused by differences in cerebrovascular structure between knock-out and wild-type mice. Therefore, an established vascular casting method (Coyle and Jokalainen, 1982; Maeda et al., 1999) was used to quantitatively assess the boundary zone between the middle and anterior cerebral arteries in these strains of mice. Briefly, mice were anesthetized with halothane as described above, and papaverine hydrochloride (40 mg/kg, i.v.) was injected to induce maximal vasodilation. Latex solution (Vultex, catalog number 563; Chicago Latex Production) was mixed with a small amount of carbon black ink (Bokusai, Fueki, Japan), warmed to 38°C, then used to perfuse the brain. After formalin fixation, the brains were extracted and digitally imaged. Morphometric analysis was performed using NIH Image software using the established technique published by Maeda et al. (1999). First, the total number of anastomoses per hemisphere was counted. Next, the boundary zone between the middle and anterior cerebral arteries on the dorsal brain surface was determined by identifying points of anastomoses, defined as the narrowest part of the vessel or halfway between the nearest branching points of the middle and anterior cerebral arteries. These points were connected into a line, and the distance from midline was measured at 2, 4, and 6 mm from the frontal pole. Measurements (n = 6 per group) were obtained for MMP-9 knock-outs, wild-type littermates, and commercially obtained CD-1 mice.
Measurement of neurological deficits. Mice were tested and scored for neurological deficits as follows: 0, no detectable neurological deficit; 1, failure to extend left forepaw fully; 2, spontaneous turning to the left; 3, spontaneous circling to left; 4, inability to move or retain normal upright posture. Assessments were made at 2 and 24 hr after ischemic onset.
Measurement of infarct volume. Mice were killed 24 hr after induction of focal ischemia. Eight coronal sections (1 mm thick) per brain were prepared and stained with 2,3,5-triphenyltetrazolium chloride (Sigma, St. Louis, MO). Infarct volume was quantified with a standard computer-assisted image analysis technique (n = 7 wild types; n= 8 knock-outs).
Antibodies and reagents. Rabbit polyclonal antibody against murine MMP-9 was a kind gift from Robert Senior (Washington University, St. Louis, MO). This antibody is able to recognize both latent and activated forms of MMP-9 (Betsuyaku et al., 2000). Rat monoclonal antibody against murine myelin proteolipid protein (PLP) (AA3) was a kind gift from Steve Pfeiffer (University of Connecticut, Farmington, CT). Rabbit polyclonal antibody against MBP was purchased from Chemicon (Temecula, CA). Rabbit affinity-isolated antigen-specific antibodies against actin and laminin were purchased from Sigma. Purified proteins of murine MMP-9, human MMP-2, and human MBP were purchased from Chemicon.
Preparation of tissue extracts. At 8, 16, and 24 hr after the onset of ischemic insult, mice (n = 5 per time point) were deeply anesthetized with halothane and then transcardially perfused with ice-cold PBS, pH 7.4. The brains were removed quickly and divided into ipsilateral and contralateral hemispheres. Ischemic tissue and matching tissue from the contralateral hemisphere was dissected, frozen immediately in liquid nitrogen, and stored at −80°C. Brain tissue extracts were prepared as previously described (Asahi et al., 2000b). Briefly, brain samples were homogenized in lysis buffer including protease inhibitors on ice. After centrifugation, supernatant was collected, and total protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA).
Western blot analysis. To investigate protein expression patterns in control and ischemic brains, equal amounts (30 μg) of total protein extracts were prepared. After mixing with 2× sample buffer, each sample was separated by Tris-glycine SDS-PAGE in native condition for MMP-9, myelin basic protein, and PLP, and in reducing condition for actin, laminin, occludin, and ZO-1. After separation, proteins were transferred to polyvinylidene difluoride membranes. All blots were blocked with 10% nonfat dry milk in PBS, pH 7.4, containing 0.1% Tween 20 (PBS-T) at 4°C overnight. Then, the filters were incubated with the primary antibodies diluted in blocking buffer for 1 hr at room temperature. The dilution rates of the primary antibodies were 1:4000, 1:500, 1:1000, 1:2000, 1:10,000, 1:250, and 1:250 for MMP-9, actin, laminin, myelin basic protein, myelin PLP, occludin, and ZO-1, respectively. After washing with PBS-T, the membrane was incubated with peroxidase-conjugated secondary antibody (anti-rabbit IgG made in donkey for MMP-9, actin, laminin, myelin basic protein, occludin, and ZO-1; anti-rat IgG made in goat for myelin PLP) (Amersham Pharmacia Biotech, Piscataway, NJ) at room temperature for 1 hr. Finally, antigen was detected by using the standard chemical luminescence method (ECL; Amersham Pharmacia Biotech).
SDS-PAGE zymogram. Similarly prepared protein samples (as in Western blot analysis) were loaded and separated by 10% Tris-glycine gel with 0.1% gelatin as substrate. After separation by electrophoresis, the gel was renatured and then incubated with developing buffer at 37°C for 24 hr as previously described (Asahi et al., 2000b). After developing, the gel was stained with 0.5% Coomassie Blue R-250 for 30 min and then destained appropriately.
Immunohistochemistry. To assess the spatial distribution of MMP-9 after transient focal ischemia, mice were transcardially perfused with ice-cold PBS, pH 7.4, followed with ice-cold 4% paraformaldehyde in PBS, pH 7.4, at 16 and 24 hr after the induction of ischemia (n = 3 wild types; n = 3 knock-outs). Sham-operated control mice (n = 3) were killed at 24 hr. The brains were removed, immersed with 4% paraformaldehyde in PBS overnight at 4°C, and cryoprotected in 30% sucrose in PBS at 4°C. Frozen coronal sections (30 μm thick) were prepared using a microtome. After quenching endogenous peroxidase in 0.3% H2O2 in PBS and blocking with 5% normal goat serum, sections were incubated overnight at 4°C with the MMP-9 rabbit polyclonal antibody (1:400). The sections were washed with PBS, incubated with biotinylated anti-rabbit IgG secondary antibody (Vector Laboratories, Burlingame, CA) at 1:200 dilution for 1 hr and followed by 1 hr of incubation with an avidin–biotin complex (Vector Laboratories). Peroxidase was visualized by incubation with diaminobenzidine substrate (Vector Laboratories). Negative control sections received identical treatment except for the primary antibody.
Quantitative evaluation of Evans blue extravasation.Vascular permeability was quantitatively evaluated using fluorescent detection of extravasated Evans blue dye (Uyama et al., 1988). Briefly, 2% Evans blue in PBS was infused (4 ml/kg, i.v.) as a BBB permeability tracer at the onset of reperfusion after 2 hr of transient focal ischemia (n = 7 wild types; n = 7 knock-outs). At 18–20 hr, mice were deeply anesthetized with halothane and transcardially perfused with ice-cold PBS to remove the intravascular dye. The brains were removed and divided into ipsilateral ischemic hemispheres and contralateral nonischemic hemispheres. The ipsilateral ischemic hemispheres were frozen immediately in liquid nitrogen and stored at −80°C before further analysis. Brain samples were homogenized in 1 ml of 50% trichloroacetic acid and centrifuged (10,000 rpm, 20 min). The supernatant was diluted fourfold with ethanol. A fluorescent plate reader (620 nm excitation, 680 nm emission) was used to quantify dye concentrations. Calculations were based on external standards (50–1000 ng/ml) dissolved in the same solvent (1:3; 50% trichloroacetic acid:ethanol). The amount of extravasated Evans blue was quantified as nanograms per ischemic hemisphere.
Statistical analysis. Quantitative data were expressed as mean + SEM. Statistical comparisons were conducted using ANOVA followed by Tukey–Kramer tests for inter-group comparisons. Differences withp < 0.05 were considered statistically significant.
RESULTS
Cerebrovascular boundaries are similar in MMP-9 knock-out and wild-type mice
To exclude the possibility that differences in ischemic outcome may simply be caused by differences in cerebrovascular anatomy, an established technique involving latex and carbon black perfusion was used to delineate boundaries between the anterior and middle cerebral arteries (Fig. 1). Wild-type, MMP-9 knock-out, and commercially obtained CD-1 mice were compared. The use of the CD-1 strain was chosen because MMP-9 knock-outs were derived from this background. Quantitative analysis showed a similar vascular boundary and number of anastomoses in all three strains of mice (Table1).
Fig. 1.

Latex–carbon black perfusions of MMP-9 knock-out (KO), wild-type (WT), and CD-1 (CD) brains show a similar distribution of dorsal vessels and boundary zone for the anterior and middle cerebral arteries.
Table 1.
Vascular boundaries between anterior and middle cerebral arteries
| WT | KO | CD | ANOVA | |
|---|---|---|---|---|
| Distance of boundary from midline | ||||
| 2 mm from frontal pole | 2.0 ± 0.1 | 1.9 ± 0.3 | 1.9 ± 0.3 | F = 0.08; p = 0.92 |
| 4 mm from frontal pole | 2.2 ± 0.3 | 2.1 ± 0.3 | 2.3 ± 0.3 | F = 0.43; p = 0.66 |
| 6 mm from frontal pole | 2.5 ± 0.2 | 2.6 ± 0.2 | 2.7 ± 0.3 | F = 1.12; p = 0.35 |
| Number of anastomoses per hemisphere | 7.5 ± 1.8 | 9.0 ± 1.0 | 8.0 ± 1.8 | F = 1.72; p = 0.22 |
WT, Wild type; KO, MMP-9 knock-out; CD, commercial CD-1;n = 6 per group.
MMP-9 expression is upregulated after transient focal ischemia
In Western blot analysis, the 105 kDa latent form of MMP-9 was detected as early as 8 hr after start of the ischemic insult and expression continuously increased up to 24 hr (Fig.2A). MMP-9 was not detected in MMP-9 knock-out mice or sham-operated control brains at 24 hr after ischemia (Fig. 2A). Enzymatic activity of MMP-9 was also evaluated by zymogram using gelatin as a substrate. Similar profiles of elevated MMP-9 were documented from 8 to 24 hr after ischemia (Fig. 2B). Ischemic brains from MMP-9 knock-out mice showed no detectable MMP-9 activity at 24 hr (Fig.2B). Interestingly, a weak immunoreactive band on Western blots and subtle gelatinolytic activity on zymograms were detected in contralateral brain at 24 hr after ischemia. In both Western blot and zymogram analyses, no reliable detection of the activated form of MMP-9 was observed. However, in addition to MMP-9, faint bands of gelatinolytic activity were also visible at ∼72 kDa in ischemic brains from both wild-type and MMP-9 knock-out mice. These bands occur at the same level as a human MMP-2 standard, suggesting that low levels of induction of MMP-2 may occur in this model of focal ischemia.
Fig. 2.

A, Western blots of MMP-9 demonstrate the upregulation of protein expression after transient focal cerebral ischemia. MMP-9 expression was mainly limited to the l05 kDa zymogen. Extracts from sham-operated brain (S) did not show MMP-9 upregulation. Thelanes labeled 24h contra and 24h KO represent samples at 24 hr from contralateral brain and ischemic brain from MMP-9 knock-out mice, respectively. Murine MMP-9 was used as a standard. B, Gelatin zymograms demonstrate the upregulation of MMP-9 after transient focal cerebral ischemia. Mostly, the 105 kDa zymogen was detected, although these zymograms also showed some gelatinolytic activity at 97 kDa, corresponding with the cleaved–activated enzyme. Sham-operated brain (S) did not show MMP-9 upregulation. Thelanes labeled 24h contra and 24h KO represent samples at 24 hr from contralateral brain and ischemic brain from MMP-9 knock-out mice, respectively. Subtle gelatinolytic activity was also present close to the level for MMP-2. Murine MMP-9 and human MMP-2 were used as standards.
Immunohistochemistry suggested that there was very low level expression of MMP-9 in nonischemic sham-operated control brain (Fig.3A,B). After ischemia, an upregulation of MMP-9 was observed in cortex and striatum within the ischemic territory at 16 hr (Fig.3C,D); it became more pronounced by 24 hr (Fig.3E,F). MMP-9 expression was predominantly observed in endothelial cells of cerebral vasculature. However, MMP-9 immunoreactivity also appeared in a significant number of parenchymal cells. No immunoreactive cells were observed in brains from MMP-9 knock-out mice (Fig. 3G,H). No detection was observed in controls in which 24 hr ischemic brains were incubated without the primary antibody (Fig.3I,J).
Fig. 3.

Immunohistochemistry with anti-MMP-9. A, B, Sham-operated brains showed no MMP-9 expression.C–F, MMP-9 staining increased in wild-type mouse brain after transient focal ischemia. Diffuse MMP-9 immunoreactivity was observed within both the cortical and striatal areas of ischemic hemispheres. Immunoreactive MMP-9 mainly appeared in endothelial cells. In addition, MMP-9 expression was also detectable in parenchymal cells of ischemic regions. G, H, No immunoreactive cells were observed in ischemic brains of knock-out mice (KO).I, J, Negative controls incubated without primary antibody showed no staining. Scale bar, 50 μm.
Ischemic degradation of blood–brain barrier components is reduced in MMP-9 knock-out mice
The MMP substrate ZO-1 is a critical protein that is associated with the functional expression of tight junctions in the BBB. Western blot analysis detected a 220 kDa band for ZO-1, which was significantly degraded after ischemic injury (Fig.4A). Densitometric analysis showed that by 24 hr after ischemia, ZO-1 was decreased to ∼30% of baseline levels (Fig. 4A). Ischemic degradation of ZO-1 was significantly reduced in the MMP-9 knock-out mice compared with wild-type mice (Fig. 4A). Interestingly, however, no clear degradation of the other BBB-associated protein occludin was observed after ischemia, within the limits of our detection sensitivity (Fig. 4B).
Fig. 4.
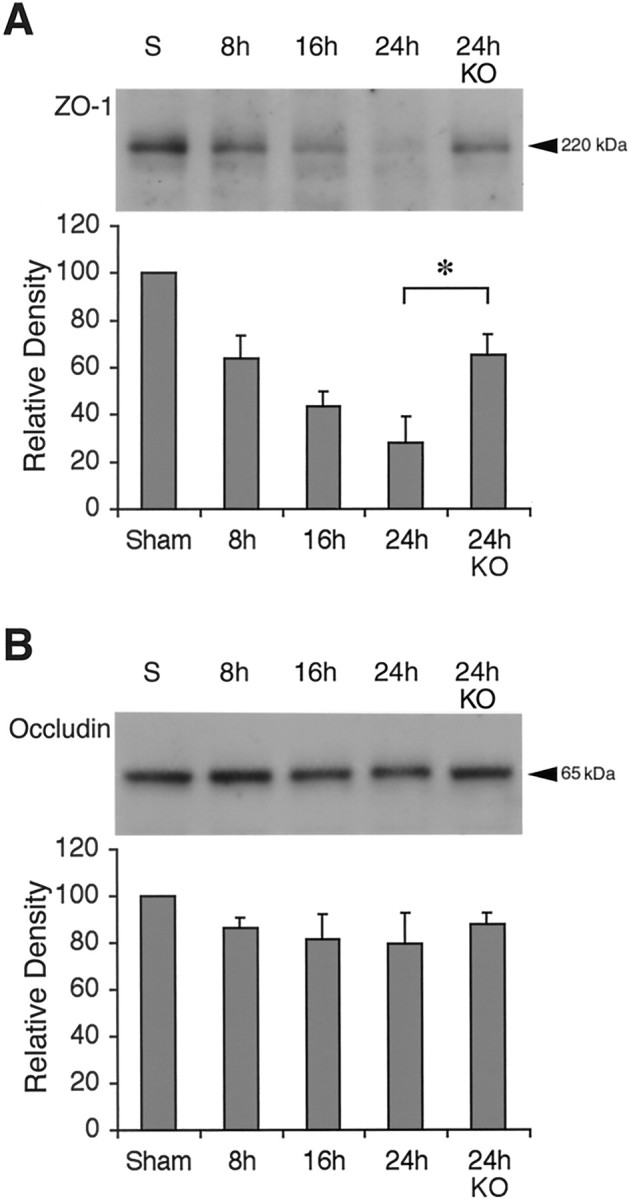
A, Western blots showed a time-dependent degradation of the BBB-associated protein ZO-1 after ischemic onset. Densitometric analysis demonstrated that ZO-1 degradation was significantly ameliorated in MMP-9 knock-out mice (KO). n = 5 animals per time point; *p < 0.05. B, Western blots showed no significant change in another BBB-associated protein, occludin.n = 5 animals per time point.
Blood–brain barrier disruption is attenuated in MMP-9 knock-out mice after transient focal ischemia
Fluorescence spectrophotometric analysis of Evans blue standards showed a linear correlation between fluorescence intensity and dye concentration within the 50–1000 ng/ml range (data not shown), thus allowing for a quantitative measurement of dye concentrations in the ischemic mouse brains. In all mice reperfused with Evans blue, leakage of the dye into brain parenchyma was observed at 18–20 hr after ischemia. The severity of blood–brain barrier disruption in the ischemic brains, expressed as Evans blue extravasation per ischemic hemisphere, was significantly reduced in the MMP-9 knock-out mice (1263 ± 529 ng/hemisphere; n = 7) compared with wild-type mice (3067 ± 621 ng/hemisphere; n = 7) (Fig.5A,B).
Fig. 5.
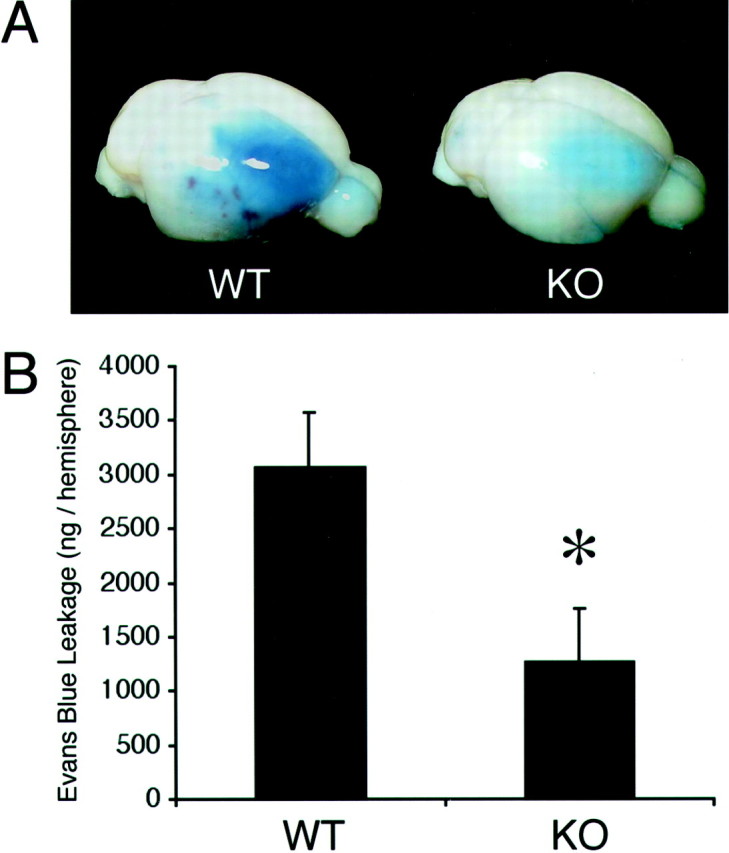
A, Representative brains showing Evans blue leakage in wild-type mice (WT) and reduced leakage in MMP-9 knock-outs (KO).B, Fluorescent quantitation of Evans blue showed that BBB leakage was significantly reduced in knock-out mice compared with wild-type mice. n = 7 per group; *p < 0.05.
Ischemic degradation of myelin basic protein is reduced in MMP-9 knock-out mice
The known MMP-9 substrate MBP was examined as a marker for white matter damage after transient focal ischemia. As expected, Western blots of MBP showed multiple bands corresponding to the four isoforms (21.5, 18.5, 17, and 14 kDa) (Fig.6A). After ischemic injury, MBP was degraded. Densitometric analysis showed that significant degradation occurred in 21.5, 18.5, and 14 kDa isoforms by 24 hr after ischemia (Fig. 6B). A degraded band recognized by the MBP antibody appeared at ∼10 kDa (Fig. 6A). The intensity of this band of degraded MBP significantly increased by 24 hr after transient focal ischemia (Fig.6C). Ischemic degradation of 18.5 and 14 kDa MBP and accumulation of the 10 kDa band of degraded MBP was significantly ameliorated in MMP-9 knock-out mice compared with wild types (Fig.6B,C). The other major myelin-associated proteins, PLP and DM20, were also examined. No detectable degradation in the 26 kDa band of PLP or the 20 kDa band of DM20 occurred after ischemic injury in wild-type or knock-out mice (Fig.7A,B).
Fig. 6.
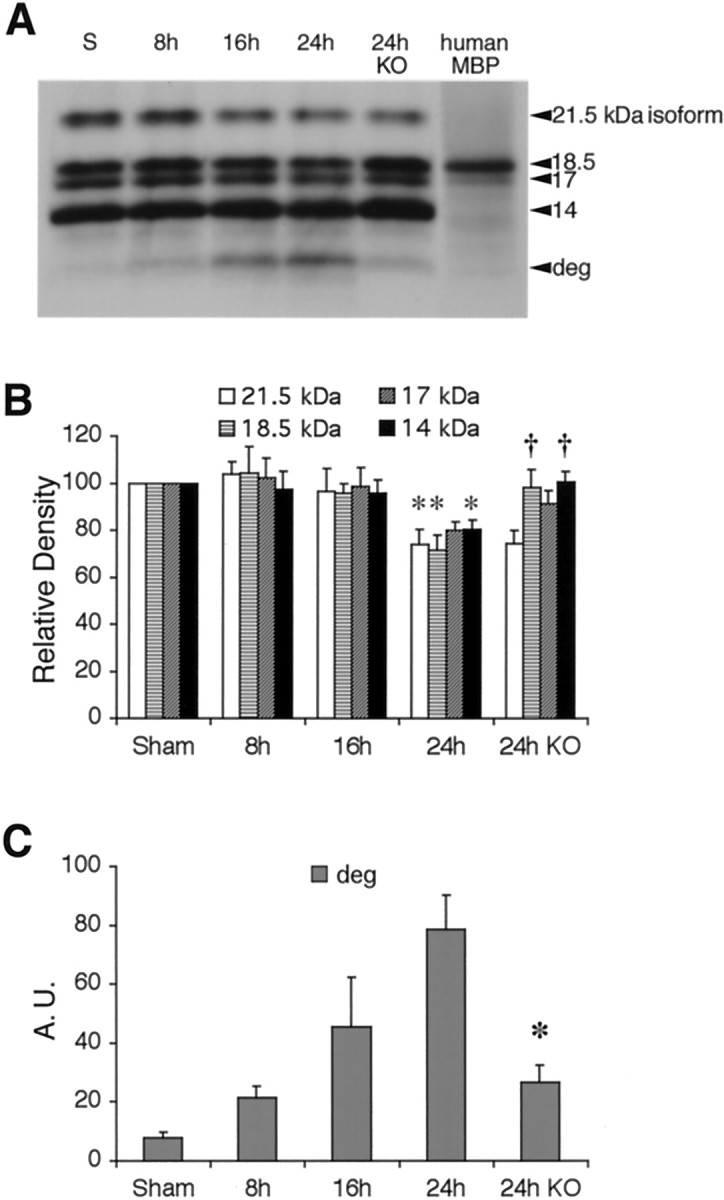
A, Western blots of MBP detected all four isoforms (21.5, 18.5, 17, and 14 kDa). After ischemia, a degraded band (deg) appeared at ∼10 kDa.B, Quantitative densitometric analysis showed that the 21.5, 18.5, and 14 kDa MBP bands were significantly degraded compared with sham controls by 24 hr after ischemia; *p < 0.05. In MMP-9 knock-out mice (KO), the band intensities for 18.5 and 14 kDa MBP were significantly higher compared with wild-type mice at 24 hr after ischemia; †p < 0.05; n = 5 per time point. C, Densitometric analysis showed that degraded MBP (deg) increased over time after ischemic onset in wild-type mice. This accumulation of degraded MBP was significantly reduced in MMP-9 knock-out mice (KO).n = 5 per time point; *p < 0.05. A.U., Arbitrary units.
Fig. 7.

A, Western blots of PLP and DM20 showed no detectable degradation after ischemia. B, Densitometric quantitation demonstrated that there were statistically significant effects in wild-type or MMP-9 knock-out mice (KO). n = 5 per time point.
To assess the relative specificity of these findings, we also examined laminin as a representative extracellular matrix protein and actin as a representative and ubiquitous intracellular protein. Western blots in our model system detected a major band for laminin at 220 kDa and actin at 42 kDa (Fig.8A,B). After transient focal ischemia, neither laminin nor actin showed significant signs of being degraded, within the limits of our detection sensitivity (Fig. 8A,B).
Fig. 8.
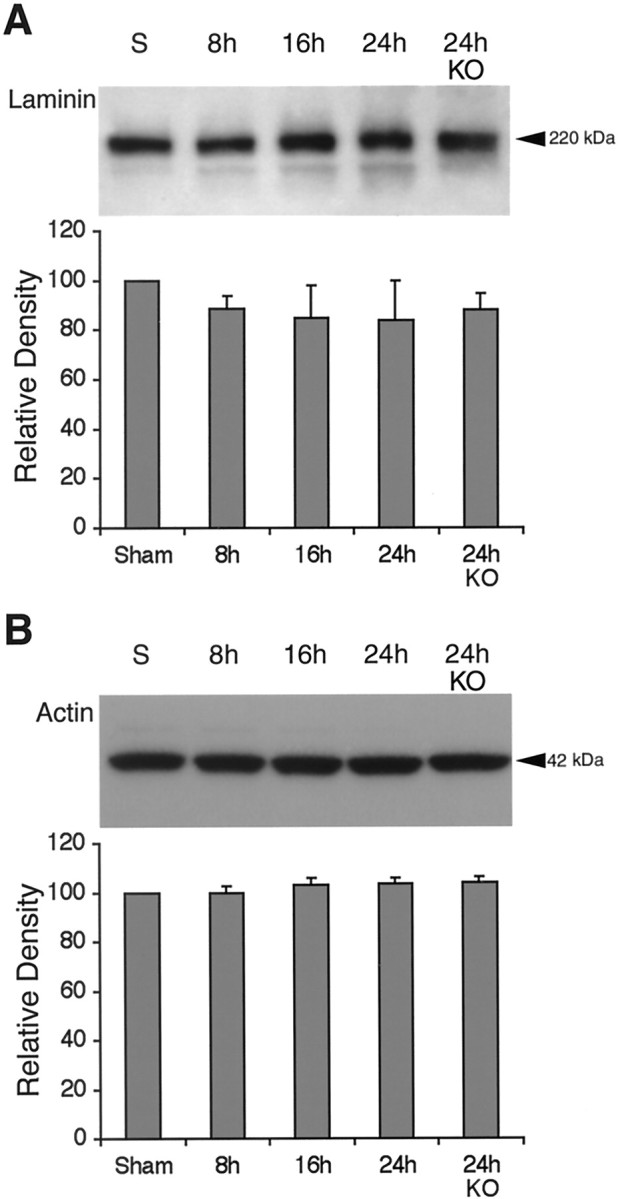
A, Western blots of the extracellular matrix component laminin showed no detectable degradation after ischemia in wild-type or MMP-9 knock-out mice (KO). n = 5 per time point.B, Western blots of the ubiquitous intracellular protein actin showed no detectable degradation after ischemia in wild-type or MMP-9 knock-out mice. n = 5 per time point.
MMP-9 gene knock-out reduces lesion volumes after transient focal ischemia
There were no differences in physiological parameters between MMP-9 knock-out mice and wild-type littermates (data not shown). Laser doppler flowmetry confirmed that consistent and similar levels of ischemia were achieved in wild-type and MMP-9 knock-out mice (9.9 ± 1.8% in wild types and 9.0 ± 0.8% in knock-outs, expressed as a percentage of pre-ischemic baselines). Similar levels of flow recovery were also observed after the onset of reperfusion (14.1 ± 1.2 in wild types and 13.7 ± 2.9 in knock-outs; expressed as fold-increase versus perfusion levels during ischemia).
Ischemic lesion volumes at 24 hr were significantly reduced in MMP-9 knock-out mice (54.3 ± 9.7 mm3;n = 8) compared with wild-type mice (86.4 ± 10.7 mm3; n = 7) (Fig.9A,B). Mean neurological deficits were slightly improved in the knock-out mice (2.3 ± 0.3; n = 8) compared with wild-type mice (2.9 ± 0.3; n = 7), but this difference did not reach statistical significance (p = 0.18).
Fig. 9.
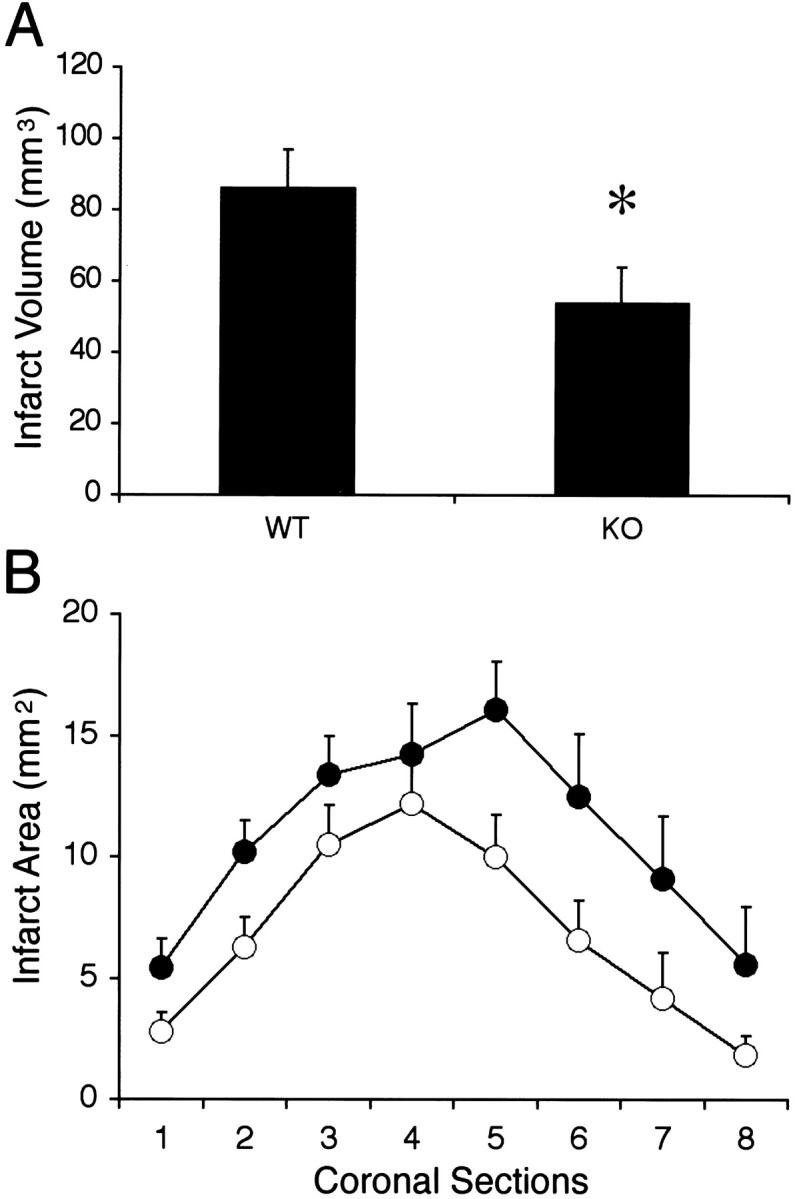
Twenty-four hour ischemic lesion volumes (A) and areas (B) showed that knock-out mice (KO; n = 8) had significantly reduced ischemic injury compared with wild-type mice (WT; n = 7). *p< 0.05.
DISCUSSION
Abnormal activation of intracellular proteolytic cascades contributes to the pathophysiology of cerebral ischemia, with extensive evidence documenting the deleterious effects of enzymes such as calpain (Bartus et al., 1995) and caspases (Schulz et al., 1999). Accumulating data now suggest that extracellular proteolytic cascades may also be involved (Turgeon and Houenou, 1997; Gingrich and Traynelis, 2000;Cuzner and Lo, 2001). In particular, the MMP family of zinc endopeptidases has been implicated (Mun-Bryce and Rosenberg, 1998b).
In the present study, we showed for the first time that critical components of the BBB and white matter were degraded after transient focal ischemia, and these effects were significantly ameliorated in MMP-9 knock-out mice compared with wild-type littermates. Of relevance was the fact that these components were MMP-9 substrates, whereas non-MMP-9 substrates were not affected. ZO-1, the BBB protein associated with endothelial tight junction formation, showed a time-dependent degradation after ischemic onset, and this was significantly attenuated in MMP-9 knock-out mice. Correspondingly, BBB disruption was reduced in the knock-outs. In white matter, three of the four MBP isoforms were degraded after ischemia, and this was also significantly attenuated in the MMP-9 knock-out mice. Concomitant with these alterations in substrate proteolysis, infarction volumes were reduced. Taken together, these data suggest that MMP-9 contributes to the pathophysiology of cerebral ischemia by degrading critical components in BBB and white matter compartments.
Previous investigations into the role of MMP-9 in ischemia have focused on the cerebrovascular basal lamina. Both collagen IV and laminin are known to be MMP-9 substrates, so it was expected that proteolysis of these basal lamina components would perturb BBB integrity and lead to vasogenic edema (Mun-Bryce and Rosenberg, 1998a) In fact, the involvement of MMP-9 in BBB dysfunction may not be restricted to ischemia alone, and may occur in a wide range of neuroinflammatory responses to bacterial meningitis (Kieseier et al., 1999), cold injury (Morita-Fujimura et al., 1999), and excitotoxicity (Zhang et al., 2000). However, BBB function is not solely determined by the basal lamina. Another major physiologic determinant is the tight junction present between cerebral endothelial cells. The functional integrity of these tight junctions is mediated via specialized proteins that include the zonae occludens family (ZO-1, ZO-2, ZO-3), occludin, cingulin, and claudin (Denker and Nigam, 1998; Kniesel and Wolburg, 2000). Among these proteins, ZO-1 is an MMP-9 substrate (Harkness et al., 2000). This is consistent with our data showing that ischemic degradation of ZO-1 was significantly reduced in the MMP-9 knock-out mice. ZO-1 is a specific molecular marker that corresponds to BBB maturation in developing brain (Nico et al., 1999). In this study, amelioration of ZO-1 proteolysis in the knock-outs was accompanied by an attenuation in BBB disruption. Interestingly, we were unable to detect degradation in another BBB protein, occludin. This difference may be related to the specificity of MMP-9-mediated proteolysis after ischemia. However, it is noted that occludin has also been identified as a substrate for unidentified metalloproteases (Wachtel et al., 1999). It is unclear why we did not detect significant degradation in occludin after ischemia and what might account for the differential sensitivity of the various BBB associated proteins.
In the present study, we could not detect a statistically significant degradation of laminin. Laminin is a substrate for several MMPs (Imper and Van Wart, 1998) and other proteases including plasmin (Chen and Strickland, 1997; Tsirka et al., 1997a). Previous studies have documented reductions in laminin immunostaining in brain after focal ischemia (Hamann et al., 1995) and excitotoxic injury (Chen and Strickland, 1997; Tsirka et al., 1997b). It is possible that Western blots were not sensitive enough to detect these changes. However, it is interesting to note that ZO-1 degradation was much more prominent in our model, with levels of proteolysis that were easily detected with Western blots. It is possible that the differential levels of degradation may be related to the relative contributions of ZO-1 versus laminin proteolysis in ischemic BBB disruptions.
The relationship between MMP and cerebrovascular integrity may extend beyond the development of vasogenic edema. It is conceivable that proteolytic weakening of the vessel walls may also increase risks of rupture and hemorrhage (Mun-Bryce and Rosenberg, 1998b). In a baboon model of focal ischemia, there was a positive correlation between local elevations in MMP-9 and the development of hemorrhagic foci (Heo et al., 1999), and the MMP inhibitor BB-94 reduced rates of hemorrhagic transformation after experimental embolic stroke (Lapchak et al., 2000). In our present mouse ischemia model, hemorrhagic transformation was not seen, most likely because we used a mechanical method of arterial occlusion. It is known that embolic occlusions with blood clots result in ischemic models more prone to hemorrhage (Asahi et al., 2000a). Nevertheless, it will be important for future studies to dissect MMP pathways in cerebral hemorrhage using embolic approaches in these knock-out preparations.
Experimental investigations into cerebral ischemia have primarily focused on damage to gray matter. Yet in clinical stroke, damage to white matter is extremely important (Petty and Wettstein, 1999). Here, we showed for the first time that three isoforms of MBP were degraded after cerebral ischemia, and this was significantly ameliorated in MMP-9 knock-outs. Of relevance was the fact that MBP is an MMP-9 substrate (Chandler et al., 1995). Proteolysis was specific to MBP, and no degradation was observed for other myelin proteins PLP and DM20. PLP and DM20 are alternatively spliced transmembrane proteins encoded by the same gene (Nave et al., 1987), and account for up to 50% of protein content in the myelin sheath. The lack of degradation in PLP and DM20 suggests that the MBP findings may be specific for MMP-9 proteolysis. MMP-9 participates in the proteolytic processing of immunogenic fragments of MBP in multiple sclerosis (Cuzner and Opdenakker, 1999; Hartung and Kieseier, 2000). In the context of cerebral ischemia, it is possible that similar inflammatory actions may occur. In general, it is difficult to quantify white matter injury and its contribution to total tissue infarction in cerebral ischemia, especially in rat or mouse models where volumes of the white matter component are low. Recently, immunohistochemical analysis has been developed as one possible approach (Valeriani et al., 2000). Our findings here also suggest that loss of MBP might be used as a marker to assess pathways of injury in white matter ischemia.
We had previously shown that MMP-9 knock-out mice were protected against permanent focal ischemia (Asahi et al., 2000b). Here, in addition to the attenuated proteolysis of critical BBB and white matter components, infarct volumes were also significantly reduced after transient focal ischemia. Important differences may exist between the pathophysiology of permanent versus transient cerebral ischemia with reperfusion. Therefore, the fact that MMP-9 gene knock-out was beneficial in both paradigms further supports the hypothesis that upregulation in MMP-9 may play a central role in the ischemic pathophysiology. The deleterious mechanisms of MMP-9 are likely to be broad-based because these knock-outs are also protected against traumatic brain injury (Wang et al., 2000).
There are a few caveats associated with the present study. First, MMP-9 has a wide range of substrates beyond those examined here. Here, we focused on representative substrates that would be critical targets relevant to the pathophysiology of cerebral ischemia. Other targets will have to be examined as well because ischemic injury is clearly multifactorial. Second, our immunohistochemistry data suggest that MMP-9 was primarily upregulated in vascular compartments, consistent with the fact that endothelial cells can secrete this protease (Herron et al., 1986). Hence, the MMP-9 enzyme would be ideally situated to degrade ZO-1. However, it is known that neurons, astrocytes, oligodendrocytes, and microglia can also secrete MMP (Gottschall and Deb, 1996; Oh et al., 1999; Rosenberg et al., 2001). We observed some cellular staining in the parenchyma, but additional colocalization studies are needed to assess specific cell types in our model, especially with regard to MMP-9 activity in white matter. A third caveat involves MBP degradation. Although this will be a useful marker for white matter injury, our data cannot determine whether loss of myelin integrity may have functional effects on the axon itself. Fourth, the present data support a deleterious role for MMP-9. However, these are acute experiments. MMPs are known to play important roles in remodeling and cell migration (Yong et al., 1998; Nagase and Woessner, 1999). Therefore, it is possible that MMPs may play beneficial roles during the chronic recovery from ischemic damage. Long-term studies with these knock-out mice would be useful to address this question. Finally, accumulating data suggest that proteolysis via the plasminogen axis may also contribute to the pathophysiology of cerebral ischemia (Wang et al., 1998; Ahn et al., 1999). MMPs are known to act in concert with the plasminogen axis (Cuzner and Opdenakker, 1999). Therefore, it will be important to carefully dissect the role for MMP-9 within the context of other proteases in the cascade.
In conclusion, the present study demonstrated that ischemic degradation of critical components in BBB and white matter were significantly ameliorated by MMP-9 gene knock-out. Knock-out mice were also protected against transient focal ischemia, further supporting our previous findings in permanent ischemia. These data suggest that MMP-9 may play a critical role in the broad pathophysiology of cerebral ischemia. Further characterization of the substrates and mechanisms of MMP actions may allow for the delineation of new therapeutic targets for stroke.
Footnotes
This work was funded in part by National Institutes of Health Grants R01-NS37074, R01-NS38731, R01-NS40529, R01-EY12651, and P50-NS10828. M.E.F. is a Stein Research to Prevent Blindness Professor. We thank Robert Senior and J. Michael Shipley for permission to use the MMP-9 knock-out mice and the anti-MMP-9 antibody. We thank Stephen Pfeiffer, Vijay Kuchroo, and Marjorie Lees for providing the anti-PLP antibody, Jean-Paul Vonsattel for helpful advice with the immunohistochemistry studies, Keiichiro Maeda and Konstatin Hossmann for assistance with the latex–carbon black perfusion technique, and Greg del Zoppo for critical discussions and his unstinting intellectual support.
Correspondence should be addressed to Dr. Eng H. Lo, Neuroprotection Research Laboratory, Departments of Neurology and Radiology, Harvard Medical School, Massachusetts General Hospital East 149-2322, Charlestown, MA 02129. E-mail:Lo@helix.mgh.harvard.edu.
REFERENCES
- 1.Ahn MY, Zhang ZG, Tsang W, Chopp M. Endogenous plasminogen activator expression after embolic focal cerebral ischemia in mice. Brain Res. 1999;837:169–176. doi: 10.1016/s0006-8993(99)01645-5. [DOI] [PubMed] [Google Scholar]
- 2.Asahi M, Asahi K, Wang X, Lo EH. Reduction of tissue plasminogen activator induced hemorrhage and brain injury by free radical spin trapping after focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2000a;20:452–457. doi: 10.1097/00004647-200003000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role of matrix metalloproteinase 9 in focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000b;20:1681–1690. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Bartus RT, Elliott PJ, Hayward NJ, Dean RL, Harbeson S, Straub JA, Li Z, Powers JC. Calpain as a novel target for treating acute neurodegenerative disorders. Neurol Res. 1995;17:249–258. doi: 10.1080/01616412.1995.11740322. [DOI] [PubMed] [Google Scholar]
- 5.Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol. 2000;157:525–535. doi: 10.1016/S0002-9440(10)64563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campagnoni AT. Molecular biology of myelin proteins from the central nervous system. J Neurochem. 1988;51:1–14. doi: 10.1111/j.1471-4159.1988.tb04827.x. [DOI] [PubMed] [Google Scholar]
- 7.Chandler S, Coats R, Gearing A, Lury J, Wells G, Bone E. Matrix metalloproteinases degrade myelin basic protein. Neurosci Lett. 1995;201:223–226. doi: 10.1016/0304-3940(95)12173-0. [DOI] [PubMed] [Google Scholar]
- 8.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 9.Coyle P, Jokalainen PT. Dorsal cerebral arterial collaterals in the rat. Anat Rec. 1982;203:397–404. doi: 10.1002/ar.1092030309. [DOI] [PubMed] [Google Scholar]
- 10. Cuzner ML, Lo EH. Extracellular proteolysis and brain injury. Neuroprotection Lo EH, Marwah J. 2001. Prominent; Scottsdale, AZ, in press. [Google Scholar]
- 11.Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteinases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94:1–14. doi: 10.1016/s0165-5728(98)00241-0. [DOI] [PubMed] [Google Scholar]
- 12.Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol. 1998;274:F1–9. doi: 10.1152/ajprenal.1998.274.1.F1. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson TA, Muir D. MMP-2 and MMP-9 increase the neurite-promoting potential of Schwann cell basal laminae and are upregulated in degenerated nerve. Mol Cell Neurosci. 2000;16:157–167. doi: 10.1006/mcne.2000.0859. [DOI] [PubMed] [Google Scholar]
- 14.Fini ME, Cook JR, Mohan R, Brinckerhoff CE. Regulation of matrix metalloproteinase gene expression. In: Parks WC, Mecham RP, editors. Matrix metalloproteinases. Academic; New York: 1998. pp. 299–356. [Google Scholar]
- 15.Gasche Y, Fujimura Y, Copin J, Kawase M, Masengale J, Chan PH. Early appearance of activated MMP-9 after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Gingrich MB, Traynelis SF. Serine proteases and brain damage. Trends Neurosci. 2000;23:399–412. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- 17.Gottschall PE, Deb S. Regulation of matrix metalloproteinase expression in astrocytes, microgila and neurons. Neuroimmunomodulation. 1996;3:69–75. doi: 10.1159/000097229. [DOI] [PubMed] [Google Scholar]
- 18.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 19.Harkness KA, Adamson P, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain. 2000;123:698–709. doi: 10.1093/brain/123.4.698. [DOI] [PubMed] [Google Scholar]
- 20.Hartung HP, Kieseier BC. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol. 2000;107:140–147. doi: 10.1016/s0165-5728(00)00225-3. [DOI] [PubMed] [Google Scholar]
- 21.Heo JH, Lucero J, Abumiya T, Koizol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Herron GS, Banda MJ, Clark EJ, Gavrilovic J, Werb Z. Secretion of metalloproteinases by stimulated capillary endothelial cells: expression of collagenase and stromelysin activities is regulated by endogenous inhibitors. J Biol Chem. 1986;261:2814–2818. [PubMed] [Google Scholar]
- 23.Imper V, Van Wart HE. Substrate specificity and mechanisms of substrate recognition of the matrix metalloproteinases. In: Parks WC, Mecham RP, editors. Matrix metalloproteinases. Academic Press; New York: 1998. pp. 219–242. [Google Scholar]
- 24.Kieseier BC, Paul R, Koedel U, Seifert T, Clements JM, Gearing AJ, Pfister HW, Hartung HP. Differential expression of matrix metalloproteinases in bacterial meningitis. Brain. 1999;122:1579–1587. doi: 10.1093/brain/122.8.1579. [DOI] [PubMed] [Google Scholar]
- 25.Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- 27.Lukashev ME, Werb Z. ECM signalling: orchestrating cell behavior and misbehavior. Trends Cell Biol. 1998;8:437–441. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- 28.Maeda K, Hata R, Hossmann KA. Regional metabolic disturbances and cerebrovascular anatomy after permanent middle cerebral artery occlusion in C57Black/6 and SV129 mice. Neurobiol Dis. 1999;6:101–108. doi: 10.1006/nbdi.1998.0235. [DOI] [PubMed] [Google Scholar]
- 29.Morita-Fujimura Y, Fujimura M, Gasche Y, Copin J, Chan PH. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury induced brain trauma. J Cereb Blood Flow Metab. 1999;20:130–138. doi: 10.1097/00004647-200001000-00017. [DOI] [PubMed] [Google Scholar]
- 30.Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am J Physiol. 1998a;274:R1203–R1211. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- 31.Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998b;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Nagase H, Woessner JF. Matrix metalloproteinases: a minireview. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 33.Nave KA, Lai C, Bloom FE, Milner RJ. Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc Natl Acad Sci USA. 1987;84:5665–5669. doi: 10.1073/pnas.84.16.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nico B, Quondamatteo F, Herken R, Marzullo A, Corsi P, Bertossi M, Russo G, Ribatti D, Roncali L. Developmental expression of ZO-1 antigen in the mouse blood-brain barrier. Brain Res Dev Brain Res. 1999;114:161–169. doi: 10.1016/s0165-3806(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 35.Oh LYS, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petty MA, Wettstein JG. White matter ischaemia. Brain Res Brain Res Rev. 1999;31:58–64. doi: 10.1016/s0165-0173(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 37.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 38.Rosenberg GA, Navratil M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology. 1997;48:921–926. doi: 10.1212/wnl.48.4.921. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg GA, Cunningham LA, Wallace J, Aleander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- 42.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 43.Tsirka SE, Bugge TH, Degen JL, Strickland S. Neuronal death in the CNS demonstrates a non-fibrin substrate for plasmin. Proc Natl Acad Sci USA. 1997a;94:9779–9781. doi: 10.1073/pnas.94.18.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997b;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turgeon VL, Houenou LJ. The role of thrombin-like (serine) proteases in the development, plasticity, and pathology of the nervous system. Brain Res Brain Res Rev. 1997;25:85–95. doi: 10.1016/s0165-0173(97)00015-5. [DOI] [PubMed] [Google Scholar]
- 46.Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- 47.Valeriani V, Dewar D, McCulloch J. Quantitative assessment of ischemic pathology in axons, oligodendrocytes, and neurons: attenuation of damage after transient ischemia. J Cereb Blood Flow Metab. 2000;20:765–771. doi: 10.1097/00004647-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Vecil GG, Larsen PH, Corley SM, Herx LM, Besson A, Goodyer CG, Yong VW. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J Neurosci Res. 2000;61:212–224. doi: 10.1002/1097-4547(20000715)61:2<212::AID-JNR12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 49.Vu TH, Shipley JM, Bergers G, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9 is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wachtel M, Frei K, Ehler E, Fontana A, Winterhalter K, Gloor SM. Occludin proteolysis and increased permeability in endothelial cells through tyrosine phosphatase inhibition. J Cell Sci. 1999;112:4347–4356. doi: 10.1242/jcs.112.23.4347. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase 9 gene knockout on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7043. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang YF, Tsirka SE, Strickland S, Steig PE, Soriano SG, Lipton SA. TPA increases neuronal damage after focal cerebral ischemia in wild type and TPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 53.Westermarck J, Kahari W. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 54.Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and disease of the CNS. Trends Neurosci. 1998;21:75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- 55.Zhang JW, Deb S, Gottschall PE. Regional and age-related expression of gelatinases in brains of young and old rats after treatment with kainic acid. Neurosci Lett. 2000;295:9–12. doi: 10.1016/s0304-3940(00)01582-2. [DOI] [PubMed] [Google Scholar]