

Abstract
Cathepsin D (CD) deficiency has been shown to induce ceroid-lipofuscin storage in lysosomes of mouse CNS neuron (Koike et al., 2000). To understand the behavior of microglial cells corresponding to these neuronal changes, CD-deficient (CD−/−) mice, which die at approximately postnatal day (P) 25 by intestinal necrosis, were examined using morphological as well as biochemical approaches. Light and electron microscopic observations revealed that microglia showing large round cell bodies with few processes appeared in the cerebral cortex and thalamus after P16. At P24, microglia often encircled neurons that were occupied with autolysosomes, indicating increased phagocytic activity. These morphologically transformed microglia markedly expressed inducible nitric oxide synthase (iNOS), which was also detected in the intestine of the mice. To assess the role of microglial nitric oxide (NO) in neuropathological changes in CD−/− mice,l-NG-nitro-arginine methylester (l-NAME), a competitive NOS inhibitor, or S-methylisothiourea hemisulfate (SMT), an iNOS inhibitor, was administered intraperitoneally for 13 consecutive days. The total number of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive cells counted in the thalamus was found to be significantly decreased by chronic treatment of l-NAME or SMT, whereas neither the neuronal accumulation of ceroid–lipofuscin nor the microglial phagocytic activity was affected by these treatments. Moreover, the chronic treatment ofl-NAME or SMT completely suppressed hemorrhage-necrotic changes in the small intestine of CD−/− mice, resulting in normal growth of the body weight of the mice. These results suggest that NO production via iNOS activity in microglia and peripheral macrophages contributes to secondary tissue damages such as neuronal apoptosis and intestinal necrosis, respectively.
Keywords: cathepsin D-deficient mouse, microglia, nitric oxide, l-NG-nitro-arginine methylester, apoptotic neuron, intestinal atrophy
It has been shown that cathepsin D (CD; EC 3.4.23.5)-deficient (CD−/−) mice, which are born normally but die at postnatal day (P) 26 ± 1 with signs of massive intestinal necrosis, thromboembolia, and lymphopenia (Saftig et al., 1995), exhibit prominent neuropathological changes in CNS (Koike et al., 2000). The mice suffering from seizures and blindness demonstrate an accumulation of lysosomes with granular osmiophilic deposits and fingerprint profiles in CNS neurons, which are features typical of neuronal ceroid lipofuscinoses (NCLs) (Dawson and Cho, 2000). White Swedish Landrace sheep expressing an inactive form of the point-mutated CD molecule have been found to show a severe loss of neurons in the cortical area and hippocampus and accumulation of protein-like storage materials in CNS neurons (Tyynelä et al., 2000). Subunit c of the mitochondrial F1F0ATPase, a common storage material of NCL except for the infantile form of NCL, is markedly accumulated in both the CNS and peripheral cells of CD−/− mice (Koike et al., 2000). The loss of CD activity causes a novel type of lysosomal storage disease associated with massive neurodegeneration. The mechanism underlying neuronal damage and death, however, remains unknown.
Microglia are normally present as ramified cells that have small cell bodies with numerous branching processes. Once fully activated, ramified microglia are morphologically transformed into cells that are characterized by large cell bodies with few processes and phagocytic activity. As an intermediate form, microglia appear to have large cell bodies with several thicker processes. These activated microglia appear to be directly involved in propagation of neuropathological events such as Alzheimer's disease because the microglial activation leads to produce mediators of inflammation-mediated neurodegeneration including nitric oxide (NO) and tumor necrosis factor-α (TNF-α) (Meda et al., 1995; Brown et al., 1996; Yan et al., 1996; Barger and Harmon, 1997; Tan et al., 1999;Tanabe et al., 1999; Kim et al., 2000; Wada et al., 2000). The direct involvement of NO in microglia-induced neuronal death has also been investigated in in vitro (Boje and Arora, 1992; Chao et al., 1992) and in vivo (Takeuchi et al., 1998; Matsuoka et al., 1999) studies. However, little is known about the relationship between microglia-induced inflammation and neurodegeneration associated with CD deficiency.
The present study attempted to characterize microglia in CNS tissues of CD−/− mice and elucidate its involvement in neurodegenerative change. We found a prominent expression of inducible NO synthase (iNOS) in both microglia and peripheral macrophages. The chronic treatment ofl-NG-nitro-arginine methylester (l-NAME), a competitive NOS inhibitor, and S-methylisothiourea hemisulfate (SMT), an iNOS inhibitor, revealed that NO production via iNOS activities of microglia and peripheral macrophages was significantly but differentially associated with pathological changes in the CNS and intestine.
MATERIALS AND METHODS
Animals. Heterozygous (+/−) mice (Saftig et al., 1995) were transferred to the Institute of Experimental Animal Sciences, Kyushu University Faculty of Dentistry, and kept in conventional facilities. Selection of CD−/− mice from littermates obtained by heterozygous coupling was performed according to the method of Saftig et al. (1995) in which template genomic DNA that was isolated from tail biopsies was examined by CD exon 4-specific PCR with primers of MCD14 (5′-AGACTAACAGGCCTGTTCCC-3′) and MCD15 (5′-TCAGCTGTAGTTGCTC AC ATG-3′). Heterozygous mice that were used as control animals in the present study show no pathological phenotypes when examined by histological, immunocytochemical, and biochemical methods. The day of birth was designated as P1.
Immunohistochemistry. Detailed immunohistochemical procedure has been described previously (Nakanishi et al., 1993, 1994, 1997). Briefly, specimens were obtained at P16, P20, and P24 from CD−/− mice and control littermates that were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and killed by intracardiac perfusion with isotonic saline, followed by a chilled fixative consisting of 4% paraformaldehyde in 0.2 m PBS, pH 7.4. After perfusion, the brain was removed, further fixed by immersion in the same fixative overnight at 4°C, and then immersed in 20% sucrose, pH 7.4, for 24 hr at 4°C. Floating parasagittal sections (30 μm thick) of the cerebral cortex and the thalamus were prepared by a cryostat and stained by F4/80 (100 μg/ml) (Serotec, Oxford, UK), anti-GFAP IgG (2.0 μg/ml) (Dako Japan, Kyoto, Japan) or anti-iNOS IgG (1.25 μg/ml) (Transduction Laboratories, Lexington, KY) with the avidin–biotin–peroxidase complex method as described previously. After PBS washes, sections were reacted with 0.025% 3′,3-diaminobenzidine/0.4% (NH4)2Ni(SO4)2/0.09% H2O2/0.1m Tris-buffered saline solution for 5–10 min. All sections were thoroughly rinsed with PBS, mounted, and coverslipped. As immunohistochemical control, the sections were incubated with nonimmune rabbit IgG or mouse IgG and followed by the same treatments described above.
For indirect fluorescent immunohistochemistry, floating parasagittal sections (30 μm thick) of the cerebral cortex and the thalamus were stained with F4/80 (200 μg/ml) and anti-GFAP IgG (8.2 μg/ml) for 90 min at 37°C. After a wash with PBS, F4/80 and anti-GFAP IgG-treated sections were stained with biotinylated anti-mouse and anti-rabbit IgG, respectively. After washes with PBS, sections were treated with 0.5% streptavidin-Alexa 488 (Molecular Probes, Eugene, OR) for 2 hr at room temperature. After wash with PBS, the sections were mounted in the anti-fading medium. For double staining, floating parasagittal sections (30 μm thick) of the cerebral cortex and the thalamus were stained with anti-iNOS IgG (2.5 μg/ml) for 90 min at 37°C. After a wash with PBS, the sections were treated with 0.5% anti-mouse IgG conjugated with fluorescein isothiocyanate (Amersham Pharmacia Biotech, Buckinghamshire, UK) for 2 hr at room temperature. To identify microglia in the iNOS-stained sections, we treated them further with biotinylated Griffonia Simplicifolia BS-I isolectin B4 (10 μg/ml) (GSA-I-B4; Sigma, St. Louis, MO) overnight at 4°C. After a wash with PBS, the sections were stained with 0.5% streptavidin-Cy3 (1:100) (Amersham Pharmacia Biotech) for 2 hr at room temperature. To identify astrocyte in the iNOS-stained sections, we treated them further with anti-GFAP IgG (2.0 μg/ml) (Dako Japan) overnight at 4°C. After a wash with PBS, the sections were stained with 0.5% streptavidin-Cy3 (Amersham Pharmacia Biotech) for 2 hr at room temperature. The sections were mounted in the anti-fading medium Vectashield (Vector Laboratories, Burlingame, CA).
Electron microscopy. CD-−/− mice at P18 and P25 were deeply anesthetized with sodium pentobarbital (25 mg/kg, i.p.) and fixed by cardiac perfusion with 2% paraformaldehyde/2% glutaraldehyde buffered with 0.1 m phosphate buffer, pH 7.2. After perfusion, the brain was removed and further immersed in the same fixative at 4°C overnight. After being washed thoroughly with the same buffer, containing 7.5% sucrose, samples were post-fixed with 1% OsO4 with the same buffer, containing 7.5% sucrose, at 4°C for 2 hr; the brain was block-stained with 2% aqueous solution of uranyl acetate for 1 hr. Then, the brain was dehydrated with a graded series of ethanol and embedded in Epon 812. Serial ultrathin sections were cut with the ultramicrotome, stained with lead citrate and uranyl acetate, and observed with a Hitachi H-7100 electron microscope as described previously (Koike et al., 2000).
Assay of NO2−accumulation. Specimens were obtained at P16, P20, and P24 from CD−/− mice and control littermates that were anesthetized with sodium pentobarbital (40 mg/kg, i.p.), and the brains and the small intestines were rapidly removed. The whole brain and the ileum were homogenized in 50 mm Tris-HCl, pH 7.4, containing 0.5 mm EGTA, 0.5 mm EDTA. After centrifugation at 50,000 × g for 30 min, the supernatant was used as the cytosolic fraction. The supernatant was transferred to 96-well dishes with Griess reagent (Griess Reagent Kit; Dojindo, Kumamoto, Japan) and incubated at room temperature for 15 min. The amount of NO2− in the cytosol was measured spectrophotometrically by using an ELISA plate reader (model 550; Bio-Rad, Richmond, CA) with the absorbance at 540 nm.
SDS-gel electrophoresis and immunoblotting. Detailed immunoblotting procedure has been described previously (Nakanishi et al., 1994; Amano et al., 1995). The whole brain and the ileum obtained from CD−/− mice and control littermates at P16, P20, and P24 were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and killed by intracardiac perfusion with isotonic saline. The soluble fractions obtained from the whole brain and the ileum homogenates were electrophoresed in SDS-polyacrylamide gels. For immunoblotting, proteins on SDS gels were transferred electrophoretically at 100 V for 12–15 hr from the gels to nitrocellulose membranes and then incubated at 4°C overnight under gentle agitation with the following primary antibodies: anti-iNOS IgG (0.1μg/ml) and anti-nNOS IgG (0.1 μg/ml). After washes, the membranes were incubated with 0.5% horseradish peroxidase (HRP)-labeled horse anti-mouse IgG (Amersham Pharmacia Biotech). Subsequently, membrane bound HRP-labeled antibodies were detected by the enhanced chemiluminescence detection system (ECL kit; Amersham Pharmacia Biotech) on x-ray film (X-Omat; Eastman Kodak, Rochester, NY) 30–60 sec after exposure. As a control, the primary antibody was replaced by nonimmune mouse IgG. The protein bands on x-ray film were scanned and densitometrically analyzed by a densitometer (Personal Scanning Imager PD110; Molecular Dynamics).
Procedure for l-NAME and SMT administration. CD−/− mice were divided into three subgroups and intraperitoneally treated with saline, l-NAME (10 mg/kg; Affinity BioReagents, Golden, CO), a competitive NOS inhibitor, and SMT (10 mg/kg; Research Biochemicals, Natick, MA), an iNOS inhibitor, twice a day for 13 consecutive days. The injections began on P12. The injection of saline to CD−/− mice was stopped at P23 because five of eight CD−/− mice were dead by P23, and the condition of the remaining animals was very serious at this stage. The wild-type littermates were also treated with saline for 13 consecutive days. After 12 hr of the final administration, mice were killed by intracardiac perfusion with isotonic saline, followed by 4% paraformaldehyde under pentobarbital anesthesia. After perfusion, the brain and the small intestine were removed, further fixed by immersion in the same fixative overnight at 4°C, and then immersed in 20% sucrose for 24 hr at 4°C. Floating parasagittal sections (30 μm thick) of the thalamus and coronal sections (30 μm thick) of the small intestine were prepared by a cryostat. Hematoxylin-eosin staining was performed according to standard procedures.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Thalamic sections prepared from CD−/− mice at P23, CD−/− mice at P24 treated withl-NAME, and age-matched wild-type control were treated with 2% H2O2 in PBS for 20 min at room temperature. After washes in PBS, sections were stained for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) using ApopTag kit (Intergen, Purchase, NY), following the protocol provided by the manufacturer.
Statistical analysis. Data are expressed as means ± SD. The statistical analyses of the data in Figures 3A, 6, and 9A were performed by Student's t test. The statistical analysis of the data in Figure 7 was performed by two-way ANOVA, followed by Scheffe's post hoc test for multiple comparison when F ratios reached significance.
Fig. 3.

Expression of iNOS in the brain of CD−/− mice.A, Accumulation of NO2−in the cytosolic fraction of the whole brain of CD−/− mice (−/−) and control littermates (+/+) at P20 and P24. *p < 0.05, **p < 0.01, when compared with control littermates (Student's t test). B, Alteration of iNOS and nNOS proteins in the soluble fraction of the whole brain of CD−/− mice (−/−) and control littermates (+/+) at P20 and P24. Arrowheads indicate the iNOS protein with molecular mass of 130 kDa and nNOS protein with molecular mass of 160 kDa.
Fig. 6.

The number of TUNEL-positive cells in the thalamus (TH) and the dentate gyrus (DG) of the hippocampus from control littermates (+/+), CD−/− mice (−/−), CD−/− mice treated withl-NAME or SMT. The mean total number of TUNEL-positive neurons in the thalamus of CD−/− mice was significantly decreased by chronic treatment of l-NAME or SMT without affecting that in the subgranular zone of the dentate gyrus. The TUNEL-positive cells were counted in semisequential sagittal sections of the thalamus and subgranular zone of the dentate gyrus. Each column and error bar represents the mean and SD, respectively. **p < 0.01, ***p < 0.001, when compared with the age-matched control littermates (Student'st test).
Fig. 9.

Expression of iNOS in the small intestine of CD−/− mice. A, Accumulation of NO2− in the cytosolic fraction of the small intestine of CD−/− mice (−/−) and control littermates (+/+) at P16 and P24. The number of experiments is shown in parentheses. **p < 0.01, when compared with control littermates (Student's t test). B, Expression of iNOS proteins in the soluble fraction of the small intestine of CD−/− mice (−/−) and control littermates (+/+) at P16 and P20. Arrowhead indicates the iNOS protein with molecular mass of 130 kDa. C, Immunostaining for iNOS in the small intestine of CD−/− mouse (−/−) and control littermate (+/+) at P24. Scale bar, 50 μm.
Fig. 7.

Effects of l-NAME and SMT on body weight of CD−/− mice. The mean body weight of CD−/− mice (−/−) that were treated with saline (n = 8),l-NAME (n = 5), and SMT (n = 5), and control littermates (+/+) (n = 5) was determined from P12 to P24.l-NAME was systematically administered twice a day from P12 to P24. Each symbol and error bar represents the mean and SD, respectively. ***p < 0.001, when compared with the age-matched CD−/− mice treated with saline (two-way ANOVA, followed by Scheffe's post hoc test for multiple comparison when F ratios reached significance).
RESULTS
Morphological transformation of microglia in the CNS of CD−/− mice
To examine the morphological transformation of microglia during the course of neuropathological changes in CNS tissues of CD−/− mice, brain sections from the mice were immunostained with F4/80, a marker for microglia–macrophages. At P16 of CD−/− mice, a proliferation of F4/80-immunoreactive microglia was first observed especially in the cerebral cortex (Fig.1A) and the thalamus (Fig. 1B). Processes of microglia were usually attached to the cell bodies of large neurons in these brain regions. These cells were considered to be activated microglia because they had shortened and thicker processes. After P20, most microglial cells exhibiting expanded and round cell bodies with few thick processes appeared in the entire brain of CD−/− mice. F4/80 immunoreactivity formed an almost continuous rim in both the cortex (Fig.1C,E) and the thalamus (Fig.1D,F), suggesting that microglia phagocytosed cortical and thalamic neurons. The number of these phagocytic microglia that were characterized by their large round cell bodies with few thick processes was dramatically increased in an age-dependent manner; the mean incidence in the thalamus (mean ± SD cells per section; n = 5 or 6) was 31 ± 13 at P16, 67 ± 10 at P20, and 121 ± 13 at P24. By contrast, ramified microglia were only detected in these brain regions of control littermates even at P24 (Fig. 1G,H).
Fig. 1.
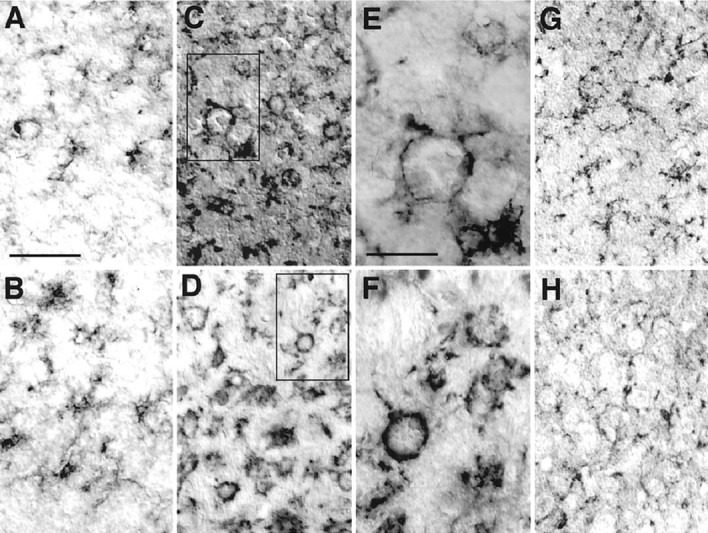
Accumulation of activated microglia stained with F4/80 in the cerebral cortex and the thalamus from CD−/− mice.A, C, E, G, the cortex;B, D, F, H, thalamus.A, B, CD−/− mouse at P16;C, D, CD−/− mouse at P24;E, F, enlargement of boxed areas in C and D, respectively;G, H, control littermate mouse at P24. Scale bars: A, 50 μm; E, 20 μm.
Phagocytosis of ceroid–lipofuscin-laden neurons by microglia
We have shown previously that neuronal cell bodies with autofluorescent bodies increased in number in various brain regions of CD−/− mice (Koike et al., 2000). In the brain sections prepared from CD−/− mice at P24, large autofluorescent bodies were evident in the cell bodies of neurons especially in the cerebral cortex and the thalamus of CD−/− mice. To further evaluate the possible phagocytosis of these ceroid–lipofuscin-laden neurons by microglia, we immunostained these sections by glial cell markers, F4/80 and anti-GFAP antibody. Under fluorescent microscope, F4/80-labeled microglia (green) were observed to uptake neurons laden with large autofluorescent bodies (orange) in the cortex (Fig.2A) and the thalamus (Fig. 2B). On the other hand, GFAP-labeled astrocytes showed no phagocytic activity in either the cortex (Fig.2B) or the thalamus (Fig. 2C). At the electron microscopic level, microglia often enclosed a large area of neuronal cell bodies that were characterized by the presence of a number of autophagosome–autolysosome-like structures containing part of the cytoplasm or heterogenously dense materials as reported previously (Koike et al., 2000) (Fig. 2E). Furthermore, microglia that completely surrounded pale neurons by their cytoplasmic processes were also often encountered, suggesting progressive microglial phagocytosis of neurons (Fig.2F). These observations clearly suggest that phagocytosis of ceroid–lipofuscin-laden neurons is responsible for morphological transformation of microglia in CNS of CD−/− mice.
Fig. 2.

Phagocytosis of neurons laden with ceroid–lipofuscin by microglia in the cortex and the thalamus of CD−/− mice. A–D, Immunostaining using F4/80 and anti-GFAP antibody in the cortex (A, C) and the thalamus (B, D) of CD−/− mouse at P24.A, B, Immunostaining for F4/80 (green) and autofluorescence of ceroid–lipofuscin (orange). C,D, Immunostaining for GFAP (green) and autofluorescence of ceroid–lipofuscin (orange). It was noted that microglia labeled by F4/80 extensively engulfed neurons that were laden with large autofluorescent bodies, whereas astrocytes labeled by anti-GFAP antibody showed no phagocytic activity. E, F, Electron micrographs of microglia attached and phagocytosed neurons laden with autophagosome–autolysosome-like bodies in the cortex (E) and the thalamus (F) of CD−/− mouse at P24. m, Microglia; n, neuron. Scale bars: A, 20 μm;E, 4.0 μm; F, 3.5 μm.
Accumulation of NO2− and expression of iNOS in microglia
Microglia have been suggested to be directly involved in propagation of neuropathological events via production of NO (Boje and Arora, 1992; Chao et al., 1992; Meda et al., 1995; Brown et al., 1996;Yan et al., 1996; Barger and Harmon, 1997; Takeuchi et al., 1998;Matsuoka et al., 1999; Tan et al., 1999; Kim et al., 2000; Wada et al., 2000). For this reason, we measured the accumulation of NO2−, a major metabolite of NO, in the cytosolic fraction of the brain of CD−/− mice. At P20, concentrations of NO2−measured in the cytosolic fractions of the whole brain in CD−/− mice were significantly higher than those from wild-type littermate mice and further increased at P24. At P24, the level of NO2− that was measured from CD−/− mice was approximately threefold higher than that from the wild-type littermate mice (Fig.3A). Additionally, the expression level of iNOS in the cytosolic fraction of the brain of CD−/− mice was examined by Western blotting. A single band with a molecular mass of ∼130 kDa indicating iNOS immunoreactivity was observed in the soluble fraction of the brain from CD−/− mice at P20 (Fig. 3B). At P24, the level of iNOS was increased by approximately twofold. In the soluble fraction of the wild-type littermate mice, no band corresponding to iNOS was detectable. Moreover, a single 160 kDa band representing nNOS was observed in the soluble fraction of both CD−/− mice and control littermates at P20. The level of nNOS showed no age-related change (Fig.3B).
Immunohistochemical localization of iNOS
To clarify the localization of iNOS in the brain of CD−/− mice, we conducted immunohistochemical staining using anti-iNOS antibody. Immunoreactivity for iNOS was observed especially in the cerebral cortex and the thalamus from CD−/− mice after P16. Positive staining for iNOS in these regions at P16 was localized in activated microglia-like cells characterized by their shortened and thicker processes. At P24, phagocytic microglia-like cells that accumulated in both the cerebral cortex (Fig.4C) and the thalamus (Fig.4D) were intensely stained by anti-iNOS antibody. The number of iNOS-positive microglia in the thalamus of CD−/− mice (mean ± SD cells per section; n = 4 or 5) was 158 ± 26 at P16, 184 ± 16 at P20, and 216 ± 22 at P24. On the other hand, no immunoreactivity for iNOS was detected either in the cerebral cortex (Fig. 4A) or the thalamus (Fig. 4B) of the wild-type littermate mice. To identify the iNOS-positive cells, iNOS-stained sections of the thalamus were treated further with glial cell markers. At P24, the iNOS-positive cells (Fig. 4E) corresponded well with GSA-I-B4-positive microglia (Fig. 4G), whereas iNOS (Fig.4F) was not expressed in astrocytes identified by anti-GFAP antibody (Fig. 4H). These observations clearly indicate that iNOS is exclusively expressed in activated microglia.
Fig. 4.

Immunostaining for iNOS in the brain.A–D, Immunostaining using anti-iNOS antibody in the cerebral cortex (A, C) and the thalamus (B, D) of the CD−/− mice and control littermates at P24. A, B, control; C,D, CD−/−. E–H, Double immunostaining for iNOS (E) and GSA-I-B4 (G) in the same thalamus section of CD−/− mouse at P24. Double immunostaining for iNOS (F) and GFAP (H) in the same thalamus section of CD−/− mouse at P24. Scale bars: A, 50 μm;E, 20 μm.
Effects of l-NAME and SMT on neurodegeneration in CNS of CD−/− mice
Although NO is known to work as a biologic messenger molecule regulating immune function and blood vessel dilation and serving as a neurotransmitter in both CNS and peripheral nervous system, a sustained production and high levels of NO have cytotoxic properties (Lowenstein and Snyder, 1992; Schmidt and Walter, 1994). We next analyzed for neuronal death in the described brain regions of CD−/− mice because iNOS expressed in microglia is thought to be the isoform that produces the large quantities of NO (Nathan and Xie, 1994). In the CNS of the wild-type littermate mice, TUNEL-positive cells were found to be densely distributed in the dentate gyrus of the hippocampus, the olfactory bulb, and the subependyma in which apoptosis plays the opposite role to neurogenesis in the regulation of cell numbers. Besides these three brain regions, TUNEL-positive cells were found in CD−/− mice particularly in the thalamus, where phagocytic iNOS-positive microglia accumulated most densely. At P16, only a few TUNEL-positive cells (ranging from two to five cells per section of the thalamus) were observed in CD−/− mice. At P24, TUNEL-positive cells were dramatically increased (24 ± 7 cells per section of the thalamus; n = 9) and mainly distributed in the thalamus (Figs. 5B, 6). The majority of TUNEL-positive cells in the thalamus were considered to be neurons because of their relatively large cell size and location. In control littermates, TUNEL-positive cells were hardly detectable in the thalamus, even at P24 (Fig. 5A).
Fig. 5.

TUNEL-stained sections in the thalamus and the dentate gyrus. A, The thalamus of control littermates at P24; B, thalamus of CD−/− mice at P24;C, thalamus of CD−/− mice treated withl-NAME at P24; D, dentate gyrus of CD−/− mice treated with l-NAME at P24. Scale bar, 100 μm.
To assess the role of microglial NO in neuropathological changes in CD−/− mice, the competitive NOS inhibitor l-NAME (10 mg/kg, n = 6) or the iNOS inhibitor SMT (10 mg/kg,n = 4) was administered intraperitoneally to CD−/− mice at P12 and twice a day thereafter for 13 consecutive days. The chronic treatment of l-NAME or SMT partially but significantly reduced the total number of TUNEL-positive cells in the thalamus of CD−/− mice (Figs. 5C, 6) without affecting the total number of TUNEL-positive neurons in the dentate gyrus of the hippocampus (Figs. 5D, 6). By contrast, chronic treatment ofl-NAME or SMT affected neither the number of neurons laden with ceroid–lipofuscin nor the number of microglia that phagocytosed these damaged storage neurons (control group, 24 ± 7 cells per square millimeter;l-NAME-treated group, 20 ± 3 cells per square millimeter). It was also noted that nuclei of damaged storage neurons that were phagocytosed by microglia were not stained with TUNEL (data not shown).
Amelioration of the decrease in body weight and intestinal atrophy in CE−/− mice by chronic treatment of l-NAME or SMT
The decline of the body weight after P14 is one of the typical pathological features of CD deficiency. In CD−/− mice injected with saline, body weight started to decline at approximately P14 as described previously (Saftig et al., 1995) (Fig.7). The mean weight of the CD−/− mice at P22 was 3.7 ± 0.9 gm (n = 8), which is only 50% of that of wild-type littermates (7.5 ± 0.2 gm;n = 5). However, CD−/− mice that were treated withl-NAME or SMT did not show a significant decrease in their body weight (Fig. 7). Although CD−/− mice treated withl-NAME or SMT showed a tendency for decline of their body weight after P18, the difference of body weight between these treated CD−/− mice and their wild-type littermates did not reach the statistical significance throughout the experimental period. Furthermore, five of eight CD−/− mice treated with saline died by P23, whereas all CD−/− mice treated withl-NAME (n = 5) or SMT(n = 5) were alive at this stage. In some experiments, CD−/− mice continued to be treated with l-NAME to determine whether l-NAME could significantly prolong the survival of CD−/− mice. The mean life span ofl-NAME-treated CD−/− mice was 28.0 ± 0.8 d (n = 5), which is significantly longer than that of nontreated CD−/− mice (25.0 ± 0.6 d;n = 8; p < 0.001; Student'st test). Thus l-NAME significantly prolonged the survival of CD−/− mice but could not abrogate the death of CD−/− mice.
The small intestine of CD−/− mice at P24 was found to exhibit hemorrhage-necrotic appearance and atrophic changes of the ileal mucosa as described previously (Saftig et al., 1995) (Fig.8B,E), but no pathological changes were observed in these tissues of the wild-type littermates (Fig. 8A,D). Both the hemorrhage-necrotic appearance and the atrophic changes of the ileal mucosa were markedly ameliorated by chronic treatment ofl-NAME (Fig. 8C,F) or SMT (data not shown).
Fig. 8.

Effects of l-NAME on pathological changes in the small intestine of CD−/− mice.A–C, The small intestine from control littermate at P24 (A), CD−/− mouse at P24 (B), and CD−/− mouse at P24 (C) treated with l-NAME. The treatment of l-NAME markedly ameliorated hemorrhage-necrotic appearance of the small intestine from CD−/− mice. D–F, Longitudinal sections through ileum from control littermate at P24 (D), CD−/− mouse at P24 (E), and CD−/− mouse at P24 (F) treated with l-NAME. The treatment of l-NAME markedly ameliorated atrophic changes of the ileal mucosa of CD−/− mice. Scale bars: A, 5 mm; D, 100 μm.
Accumulation of NO2− and expression of iNOS in peripheral macrophages
We also measured the accumulation of NO2− in the cytosolic fraction of the small intestine of CD−/− mice. At P16, the cytosolic fractions of the small intestine from the wild-type littermate mice contained NO2− at a very low concentration and its level was not significantly changed even at P24. (Fig. 9A). On the other hand, the mean concentration of NO2− measured in the cytosolic fractions of the small intestine from CD−/− mice was increased after P16; the NO2− level was approximately eightfold higher in CD−/− mice at P24, compared with P16. By Western blotting, a single band with a molecular mass of ∼130 kDa representing iNOS was observed in the soluble fraction of the small intestine of CD−/− mice at P20, but not at P16 (Fig. 9B). In corresponding samples from littermate mice at P16 and P24, iNOS was not detectable. Indirect immunofluorescent staining with anti-iNOS antibody revealed that iNOS was expressed in macrophage-like cells in the small intestine of CD−/− mice at P24 (Fig. 9C). No staining was observed in the small intestine of control littermates.
DISCUSSION
In the CNS of CD−/− mice after P20, we observed a marked accumulation of morphologically transformed microglia exhibiting expanded and round cell bodies with few thick processes. They phagocytosed damaged storage neurons laden with ceroid–lipofuscin and expressed iNOS. Furthermore, TUNEL-positive cells appeared especially in the thalamus at the terminal stage of CD−/− mice. NO and the super oxide anion, which is generated in mitochondria, react rapidly to form a peroxinitrite anion (Beckman et al., 1990). This, in turn, generates highly toxic hydroxyl radicals and hydrogen peroxide. Although NO is synthesized from l-arginine by NOS, iNOS is thought to be the isoform that produces the large quantities of NO that can result in tissue damage or death (Nathan and Xie, 1994). In the present study, iNOS was found to be markedly expressed in activated microglia of CD−/− mice after P20. To directly address the possible involvement of NO in tissue damage and cell death in the CNS of CD−/− mice, we examined effects of l-NAME, a potent competitive NOS inhibitor, and SMT, an iNOS inhibitor. The chronic treatment ofl-NAME or SMT partially but significantly decreased the number of TUNEL-positive cells in the thalamus at the terminal stage of CD−/− mice without affecting the number of TUNEL-positive cells in the dentate gyrus of the hippocampus, in which apoptosis plays the opposite role to neurogenesis in the regulation of cell numbers. However, the accumulation of ceroid–lipofuscin in neurons and the phagocytosis of these damaged storage neurons by microglia were not changed by these treatments.
New Zealand sheep that express an inactive form of the point-mutated CD molecule exhibit some neuropathological changes similar to CD−/− mice such as neuronal storage of autofluorescent granules and neuronal death (Tyynelä et al., 2000). Thus, it appears that both neuronal storage and neuronal death are directly attributable to the defective activity of CD. The activation of microglia and subsequent production of NO through iNOS are considered to be responsible for neuronal death of CD−/− mice, because microglial phagocytosis of storage neurons and iNOS expression were found to precede an appearance of TUNEL-positive cells in the thalamus. In both in vitro and in vivo studies, NO has been demonstrated to be a major causal factor for microglia-mediated neuronal death (Boje and Arora, 1992; Chao et al., 1992; Meda et al., 1995; Brown et al., 1996; Yan et al., 1996; Barger and Harmon, 1997;Takeuchi et al., 1998; Matsuoka et al., 1999; Kim et al., 2000).Takeuchi et al. (1998) reported that iNOS was induced in activated microglia surrounding necrotic lesions induced by an injection of ethanol. They also clearly indicated that iNOS can be induced in microglia to produce NO sufficient to cause neuronal death on acute brain injury without bacteria or molecules from their cell walls (lipopolysaccharides).
One possible mechanism underlying expression of iNOS high enough to produce NO causing neuronal death is binding and/or phagocytosis of damaged storage neurons by microglia. After engulfment of apoptotic cells, peripheral macrophages have been reported to increase the secretion of anti-inflammatory cytokines such as transforming growth factor-β (TGF-β) and decrease secretion of the proinflammatory cytokines such as TNF-α, interleukin (IL)-1 β, IL-8, and IL-12 (Voll et al., 1997; Fadok et al., 1998;Freire-de-Lima et al., 2000). TGF-β further depresses the expression of iNOS and the production of NO by shifting the arginine metabolic pathway to the ornithine synthetic one. On the other hand, phagocytosis of opsonized apoptotic cells via the complement receptor type 3 (CR3) has no significant effect on the secretion of pro- or anti-inflammatory cytokines (Fadok et al., 1998). However, CR3 contributes to the induction of iNOS and NO production in peripheral macrophages (Matsumoto et al., 1998). Furthermore, gangliosides that are particularly rich in neuronal cell membrane have been reported recently to activate microglia to produce proinflammatory mediators, including NO and TNF-α (Pyo et al., 1999). Our observations here showed that degenerating storage neurons in the thalamus exhibited morphological features distinct from apoptosis at both the light and electron microscopic levels. In our preliminary experiments, TNF-α was found to be increased in the thalamus of CD−/− mice after P20 (∼45 pg/mg protein at P24). More recently,Wada et al. (2000) have demonstrated that the inflammatory response initiated by activated microglia play a pivotal role in neuronal apoptosis of Sandhoff disease, an inherited glycolipid neuronal storage disease. Therefore, it is considered likely that microglia is activated to produce NO through iNOS by binding and/or phagocytosis of damaged storage neurons to initiate an intensive inflammatory response in the CNS leading to secondary neuronal damage evidenced by an appearance of TUNEL-positive cells. Although presumptive neuronal apoptosis has been investigated by TUNEL staining, it is now recognized that this assay can no longer define cell death as apoptosis (Grasl-Kraupp et al., 1995; Wang et al., 1999). We therefore referred to an appearance of TUNEL-positive neurons as neuronal damage rather than neuronal apoptosis. In summary, CD deficiency causes lysosomal storage in neurons, which stimulate and become internalized by microglia. Microglial NO induces neuronal damage in adjacent neurons and an inflammatory response in the CNS. The neuronal damage induced by NO can effectively be prevented by treatment withl-NAME or SMT.
In the course of our experiments, we have unexpectedly found that the chronic treatment of l-NAME or SMT almost completely suppressed the decrease in the body weight of CD−/− mice after P14. The closer histochemical analysis here revealed that this treatment markedly ameliorated a severe hemorrhage-necrotic appearance of the small intestine and atrophic changes of the ileal mucosa of CD−/− mice. Furthermore, the accumulation of NO2− and expression of iNOS were also found in the small intestine of CD−/− mice. These observations are consistent with the notion that NO has potential cytotoxic effects on the intestine (Boughton-Smith et al., 1993;Laszlo et al., 1994). Furthermore, l-NAME significantly prolonged the survival of CD−/− mice but could not abrogate the death of CD−/− mice. We speculate that most ofl-NAME-treated CD−/− mice died from seizure because they still showed signs of seizure without severe loss of body weight and intestinal necrosis. On the basis of these observations, NO production through iNOS expressed in peripheral macrophages may lead to a breakdown of mucosa in the small intestine of CD−/− mice. Although the mucosal damage is one of the most typical pathological features of CD−/− mice, no such pathological change in the small intestine was observed in White Swedish Landrace sheep (Tyynelä et al., 2000). Thus, the expression of iNOS and resultant mucosal damage is not directly attributable to the defective activity of CD. Further studies will be needed to clarify the factor responsible for the expression of iNOS in the intestine of CD−/− mice.
Footnotes
This work was supported by Grants-in-Aid on Priority Area from the Ministry of Education, Science, Sports and Culture, Japan.
Correspondence should be addressed to either Dr. Hiroshi Nakanishi, Laboratory of Oral Aging Science, Division of Oral Biological Sciences, Faculty of Dental Sciences, Kyushu University, Fukuoka 812-8582, Japan, E-mail: nakandeg@mbox.nc.kyushu-u.ac.jp., or Dr. Yasuo Uchiyama, Department of Cell Biology and Neuroscience, Osaka University Graduate School of Medicine, Suita, Osaka 565-0871, Japan, E-mail:uchiyama@anatl.med.osaka-u.ac.jp.
REFERENCES
- 1.Amano T, Nakanishi H, Oka M, Yamamoto K. Increased expression of cathepsins E and D in reactive microglial cells associated with spongiform degeneration in the brain stem of senescence accelerated mouse. Exp Neurol. 1995;136:171–182. doi: 10.1006/exnr.1995.1094. [DOI] [PubMed] [Google Scholar]
- 2.Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implication for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boje LM, Arora PK. Microglia-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 5.Boughton-Smith NK, Evans SM, Laszlo F, Whittle BJR, Moncada S. The induction of nitric oxide synthase and intestinal vascular permeability by endotoxin in the rat. Br J Pharmacol. 1993;110:1189–1195. doi: 10.1111/j.1476-5381.1993.tb13940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 7.Chao CC, Hu S, Molitor TW, Shaakan EG, Peterson PK. Activated microglia mediate neuronal injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 8.Dawson G, Cho S. Batten's disease: clue to neuronal protein catabolism in lysosomes. J Neurosi Res. 2000;60:133–140. doi: 10.1002/(SICI)1097-4547(20000415)60:2<133::AID-JNR1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanism involving TGF-β, PGE2, and PFA. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freire-de-Lima CG, Nascimento DO, Soares MBP, Bozza PT, Castro-Faria-Neto HC, de Mello FG, DosReis GA, Lopes MF. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403:199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- 11.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Brush W, Schutle-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 12.Kim W-O, Mohney RP, Wilson B, Jeohn G-H, Liu B, Hong J-S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laszlo F, Whittle BJ, Moncada S. Time-dependent enhancement or inhibition of endotoxin-induced vascular injury in rat intestine by nitric oxide synthase inhibitors. Br J Pharmacol. 1994;111:1309–1315. doi: 10.1111/j.1476-5381.1994.tb14887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell. 1992;70:705–707. doi: 10.1016/0092-8674(92)90301-r. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto R, Aramaki Y, Arima H, Adachi Y, Ohno N, Yadomae T, Tsuchiya S. Contribution of CR3 to nitric oxide production from macrophages stimulated with high-dose of LPS. Biochem Biophys Res Commun. 1998;244:115–119. doi: 10.1006/bbrc.1998.8231. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka Y, Kitamura Y, Takahashi H, Tooyama I, Kimura H, Gebicke-Haerter PJ, Nomura Y, Taniguchi T. Interferon-γ plus lipopolysaccharide induction of delayed neuronal apoptosis in rat hippocampus. Neurochem Int. 1999;34:91–99. doi: 10.1016/s0197-0186(98)00053-9. [DOI] [PubMed] [Google Scholar]
- 18.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 19.Nakanishi H, Tsukuba T, Kondou T, Tanaka T, Yamamoto K. Transient forebrain ischemia induces increased expression and specific localization of cathepsins E and D in rat hippocampus and neostriatum. Exp Neurol. 1993;121:215–223. doi: 10.1006/exnr.1993.1088. [DOI] [PubMed] [Google Scholar]
- 20.Nakanishi H, Tominaga K, Amano T, Hirotsu I, Inoue T, Yamamoto K. Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp Neurol. 1994;126:119–128. doi: 10.1006/exnr.1994.1048. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi H, Amano T, Sastradipura DF, Yoshimine Y, Tsukuba T, Tanabe K, Hirotsu I, Ohono T, Yamamoto K. Increased expression of cathepsins E and D in neurons of the aged rat brain and their colocalization with lipofuscin and carboxy-terminal fragments of Alzheimer amyloid precursor protein. J Neurochem. 1997;68:739–749. doi: 10.1046/j.1471-4159.1997.68020739.x. [DOI] [PubMed] [Google Scholar]
- 22.Nathan C, Xie Q-W. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 23.Pyo H, Joe E, Jung S, Lee SH, Jou I. Gangliosides activate cultured rat brain microglia. J Biol Chem. 1999;274:34584–34589. doi: 10.1074/jbc.274.49.34584. [DOI] [PubMed] [Google Scholar]
- 24.Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Köster A, Hess B, Evers M, von Figra K, Peters C. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt HHHW, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 26.Takeuchi A, Isobe K-I, Miyaishi O, Sawada M, Fan Z-H, Nakashima I, Kiuchi K. Microglial NO induces delayed neuronal death following acute injury in the striatum. Eur J Neurosci. 1998;10:1613–1620. doi: 10.1046/j.1460-9568.1998.00168.x. [DOI] [PubMed] [Google Scholar]
- 27.Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after β-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 28.Tanabe K, Nakanishi H, Maeda H, Nishioku T, Hashimoto K, Liou S-Y, Akamine A, Yamamoto K. A predominant apoptotic death pathway of neuronal PC12 cells induced by activated microglia is displaced by an non-apoptotic death pathway following blockade of caspase-3-dependent cascade. J Biol Chem. 1999;274:15725–15731. doi: 10.1074/jbc.274.22.15725. [DOI] [PubMed] [Google Scholar]
- 29.Tyynelä J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M, Haltia M, Lobel P. A mutation in the ovine cathepsin D gene causes a cognitial lysosomal storage disease with profound neurodegeneration. EMBO J. 2000;19:2786–2792. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 31.Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H-D, Fukuda T, Suzuki T, Hashimoto K, Liou S-Y, Momoi T, Kosaka T, Yamamoto K, Nakanishi H. Differential effects of Bcl-2 overexpression on hippocampal CA1 neurons and dentate granule cells following hypoxic ischemia in adult mice. J Neurosci Res. 1999;57:1–12. doi: 10.1002/(SICI)1097-4547(19990701)57:1<1::AID-JNR1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 33.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]