

Abstract
Gap junctions are conductive channels that connect the interiors of coupled cells. In the hippocampus, GABA-containing hippocampal interneurons are interconnected by gap junctions, which mediate electrical coupling and synchronous firing and thereby promote inhibitory transmission. The present study was undertaken to examine the hypothesis that the gap junctional proteins connexin 32 (Cx32; expressed by oligodendrocytes, interneurons, or both), Cx36 (expressed by interneurons), and Cx43 (expressed by astrocytes) play a role in defining cell-specific patterns of neuronal death in hippocampus after global ischemia in mice. Global ischemia did not significantly alter Cx32 and Cx36 mRNA expression and slightly increased Cx43 mRNA expression in the vulnerable CA1, as assessed by Northern blot analysis and in situ hybridization. Global ischemia induced a selective increase in Cx32 and Cx36 but not Cx43 protein abundance in CA1 before onset of neuronal death, as assessed by Western blot analysis. The increase in Cx32 and Cx36 expression was intense and specific to parvalbumin-positive inhibitory interneurons of CA1, as assessed by double immunofluorescence. Protein abundance was unchanged in CA3 and dentate gyrus. The finding of increase in connexin protein without increase in mRNA suggests regulation of Cx32 and Cx36 expression at the translational or post-translational level. Cx32(Y/−) null mice exhibited enhanced vulnerability to brief ischemic insults, consistent with a role for Cx32 gap junctions in neuronal survival. These findings suggest that Cx32 and Cx36 gap junctions may contribute to the survival and resistance of GABAergic interneurons, thereby defining cell-specific patterns of global ischemia-induced neuronal death.
Keywords: gap junctions, neural connexins, global ischemia, neuronal death, GABAergic interneurons, electrical transmission, Cx32 knock-out
Transient forebrain or global ischemia, observed in patients during cardiorespiratory arrest and cardiac surgery or induced experimentally in animals, induces selective, delayed neuronal death, particularly of pyramidal neurons of hippocampal CA1 (Kirino, 1982; Pulsinelli et al., 1982; Petito et al., 1987; Schmidt-Kastner and Freund, 1991). Influx of Ca2+ and Zn2+via Ca2+-permeable AMPA receptors (AMPARs) is implicated in the death of CA1 pyramidal neurons (Tanaka et al., 2000). GABA-containing interneurons of CA1 are resistant to ischemic damage and survive (Hsu and Buzsaki, 1993). The molecular mechanisms underlying the cell-specific pattern of global ischemia-induced neuronal death are not well understood. Recent evidence supports a role for interastrocytic gap junctions in the spread of secondary injury associated with focal ischemia (Lin et al., 1998). Astrocytic gap junctions remain functional in postischemic brain, and gap junction blockers limit secondary expansion of infarction. Gap junctions between dying glial cells can kill resistant neighboring glial cells via glial “fratricide” (bystander death) and thereby propagate and spatially amplify injury (Lin et al., 1998). On the other hand, gap junctional coupling of astrocytes attenuates neuronal death in models of oxidative stress (Blanc et al., 1998). The role of gap junctional coupling in propagation of neuronal injury in global ischemia, however, is as yet unclear.
Gap junctions are conductive channels that connect the interiors of coupled cells. Their large internal diameter (∼12 Å) allows the exchange of small ions and intracellular signaling molecules between neighboring cells. As a result, gap junctions synchronize activity of coupled cells and are thought to play an important role in intercellular signaling in brain development, morphogenesis, and pattern formation (Bennett et al., 1991; Bruzzone et al., 1996;Goodenough et al., 1996; Dermietzel and Spray, 1998). Gap junctions comprise connexins, integral membrane proteins encoded by a gene family of at least 17 structurally related members in mammals. Although connexins share sequence similarity and a common membrane topology, they assemble to form channels that differ in gating and permeability properties and in temporal and spatial patterns of expression. Three gap junctional proteins, connexin 32 (Cx32), Cx36, and Cx43, are expressed abundantly in mammalian brain but with differing cellular specificity. Whereas Cx43 is the most abundant connexin expressed by astrocytes (Giaume et al., 1991), Cx32 is expressed predominantly in oligodendrocytes (Dermietzel et al., 1989; Kunzelmann et al., 1997; Li et al., 1997) and interneurons (Venance et al., 2000); Cx36 protein expression is neuron-specific (Condorelli et al., 1998; Sohl et al., 1998; Rash et al., 2000).
In CA1, parvalbumin (PV)-positive interneurons form a vast dendrodendritic network extending many hundreds of micrometers and connected by anatomically identified electrical and mixed electrical–chemical synapses (Fukuda and Kosaka, 2000). This network of GABAergic interneurons is a candidate for the generator of synchronized oscillations in hippocampus. Electrophysiological (Galarreta and Hestrin, 1999; Gibson et al., 1999; Venance et al., 2000) and morphological (Fukuda and Kosaka, 2000) evidence indicates the presence of electrical coupling between GABAergic interneurons in the hippocampus and in visual cortex. Reverse transcription-PCR (RT-PCR) studies indicate that electrical coupling between interneurons in these regions is likely to be mediated by Cx36 (and possibly Cx32), which exhibits high expression in bipolar interneurons in layer 2/3 of visual cortex, interneurons and spiny stellate cells in layer 4 of barrel cortex, and basket cells in dentate gyrus (Venance et al., 2000). Electrical coupling between interneurons is thought to mediate synchronous firing and thereby to promote inhibitory transmission. These observations raise the possibility that Cx36 or Cx32 gap junctions, or both, might play a role in survival of hippocampal interneurons, death of pyramidal neurons after ischemia, or both.
Whereas astrocytic gap junctions can propagate and spatially amplify injury (Lin et al., 1998), gap junctions between interneurons may promote inhibitory transmission and thereby may afford protection against neuronal injury. The present study was undertaken to examine the hypothesis that gap junctions play a role in the cell-specific patterns of global ischemia-induced neuronal death in CA1. We report here that global ischemia induces a selective upregulation of Cx36 (and Cx32) protein expression in parvalbumin-positive inhibitory interneurons in the vulnerable CA1 at times before the onset of neuronal death, consistent with a role in the survival of GABAergic interneurons. Moreover, transgenic Cx32-null mutant mice exhibit enhanced vulnerability to global ischemia-induced neuronal death.
MATERIALS AND METHODS
Global ischemia in mice. Animals were maintained in a temperature- and light-controlled environment with a 14 hr light/10 hr dark cycle and were treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Global ischemia was induced in adult male Cx32(Y/+) wild-type and Cx32(Y/−) knockout mice (a gift from K. Willecke, University of Bonn, Bonn, Germany) weighing 25–35 gm by temporary bilateral occlusion of the common carotid arteries (20 min) by a modification of the method of Oguro et al. (1999). Before surgery, animals were fasted overnight and anesthetized with halothane (2%), followed by halothane (0.5–1%) in a mixture of N2O:O2 (70:30) by means of a Vapomatic anesthetic vaporizer (CWE Inc., Ardmore, PA). Body temperature was maintained at 37°C with a rectal thermistor and heat lamp during the entire period of anesthesia. Control mice were sham-operated.
In situ hybridization.[35S]UTP- and [35S]CTP-labeled RNA probes directed against the Cx32, Cx36, and Cx43 cDNAs and [35S]UTP-labeled RNA probes directed against the AMPAR subunit Glu receptor 2 (GluR2) cDNA were transcribed by incubation of the corresponding cDNA (1 hr at 37°C) with T7 polymerase in the presence of labeled and unlabeled nucleotides using a Stratagene (La Jolla, CA) transcription kit. A radiolabeled probe was purified by phenol-chloroform extraction.
To examine patterns of ischemia-induced alterations in Cx32, Cx36, Cx43, and GluR2 mRNA expression in the hippocampus, experimental and control mice were anesthetized with chloroform and decapitated at 2, 8, 24, 48, and 72 hr after ischemia or 24 hr after sham operation. mRNA expression was assessed by in situ hybridization on coronal sections of mouse brain at the level of hippocampus by a modification of the methods of Pellegrini-Giampietro et al. (1992) and Gorter et al. (1997). In brief, brains were rapidly removed, frozen by immersion in 2-methylbutane at −35°C and stored at −70°C until sectioning. Coronal sections (18 μm) were cut on a cryotome and thaw-mounted onto slides. After fixation with 4% paraformaldehyde in 10 mm PBS containing 5 mmMgC12 (15 min at 4°C), sections were rinsed in PBS, dehydrated in graded ethanols, and stored in 95% ethanol (4°C) until use. For in situ hybridization, sections were acetylated, incubated with prehybridization solution (2 hr at 50°C), and hybridized by incubation with35S-labeled RNA probe (106 cpm/section, 1 ng/ml, overnight at 50°C). Sections were washed, treated with RNase A (20 μg/ml, 30 min at room temperature), and dehydrated in graded ethanols. Slides were apposed to Kodak XAR-5 film (Eastman Kodak, Rochester, NY) for 5 d.
For quantitation of mRNA expression, autoradiograms were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image analysis software. Films were scanned at 2000 dots/inch resolution, and images of each section (∼1 × 106pixels) were created. Mean OD values in regions of maximal labeling of individual hippocampal subfields were averaged for two sections per animal and film background-subtracted to give the mean OD for a single animal. OD values were expressed as grand means ± SD of individual means from a minimum of three mice per time point. OD values for samples of experimental animals were normalized to OD values for the corresponding brain regions of control animals on the same film to enable comparisons of sections apposed to different films. Statistical significance was assessed by means of Student's unpaired ttest.
Northern blot analysis. For Northern blot analysis of global ischemia-induced alterations in connexin mRNA expression in the hippocampus, mice were anesthetized with chloroform and killed by decapitation 24 hr after ischemia or sham operation. Hippocampi were rapidly dissected out and placed in ice-cold PBS, and total RNA was extracted by the thiocyanate-phenol-chloroform method (Chomczynski and Sacchi, 1987), as modified by Xie and Rothblum (1991). Samples of total RNA (20 μg) were loaded onto 1% agarose gels containing 0.5 mg/l ethidium bromide. Gels were capillary-blotted in 20× SSC onto nylon membranes (Hybond-N; Amersham Pharmacia Biotech, Buckinghamshire, UK) and fixed by UV light. For detection of Cx32, Cx36, and Cx43 mRNAs, membranes were prehybridized (30 min at 68°C) in QuickHyb solution (Stratagene) and hybridized (1 hr, 68°C) in prehybridization solution containing 32P-labeled Cx32, Cx36, or Cx43 cDNA (gifts from D. Paul, Harvard University, Cambridge MA). Membranes were washed twice in 2× SSC buffer containing 0.1% SDS at room temperature and once in 0.1× SSC buffer containing 1% SDS at 60°C before apposing to Kodak XAR-5 film for 12 hr (Cx32 and Cx43) or 48 hr (Cx36). Ethidium bromide-stained bands corresponding to 18 and 28 S ribosomal RNAs on each film were used to correct for variability in loading of samples.
For quantitation of Cx32, Cx36, and Cx43 mRNA abundance in mouse hippocampus, Northern blots were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image analysis software. Mean OD values of bands were averaged for a minimum of two samples per animal and film background-subtracted to give the mean OD value for a single animal. OD values were expressed as grand means ± SD of individual means from a minimum of three mice. OD values for RNA samples were normalized to the OD value for 18 S ribosomal RNA on the same film to enable comparisons of OD values from blots apposed to different films. Statistical significance was assessed by means of Students's unpaired t test.
Western blot analysis. For quantitation of protein abundance in the hippocampal CA1, animals were anesthetized with chloroform and killed by decapitation at 8, 24, 48, and 72 hr after ischemia or 24 hr after sham operation. Hippocampi were quickly dissected out, and thick (1 mm) transverse slices were cut on a McIlwain tissue chopper starting at the dorsal end of the hippocampus. For biochemical analysis, the CA1 subfield was rapidly separated from the CA3 dentate gyrus by microdissection, placed in ice-cold PBS supplemented with the protease inhibitor phenylmethylsuflfonyl fluoride (PMSF, 1 mm; Sigma, St. Louis MO), and stored at −70°C until use. Tissue samples were homogenized by sonication in 200 μl of 1 mm NaHNO3 buffer, pH 6.8, containing PMSF, and lysed (overnight at 4°C) in Laemmli sample buffer (0.025m Tris-HCI, 5% glycerol, 1% SDS, 0.5% PBS, 0.1m dithiothreitol, 2.5 mm β-mercaptoethanol, 1 mm PMSF, and 0.5 mmNaHNO3 buffer, pH 6.8). Protein concentrations of samples were measured using the bicinochoninic acid protein assay kit (Pierce, Rockford, IL). Samples were diluted in Laemmli sample buffer to achieve the same final protein concentration, after which 10 μg samples were loaded onto 10% polyacrylamide minigels (Bio-Rad, Richmond, CA) and subjected to gel electrophoresis.
Protein bands were transferred to nitrocellulose membranes (Bio-Rad) in blotting buffer containing 0.192 m glycine and 20% methanol. Membranes were blocked with 25 mm Tris-buffered saline, pH 8.0, containing 0.1% Tween 20 and 4% skim milk (30 min at room temperature), and incubated with a primary antibody (1:1000, 1 hr at room temperature) followed by a secondary antibody (1 hr at room temperature). After reaction, membranes were treated with enhanced chemiluminescence reagents (Amersham Pharmacia Biotech) and apposed to Kodak XAR-5 film. Primary antibodies were as follows: (1) a mouse monoclonal antibody directed to a C-terminal epitope of Cx32 (anti-Cx32 antibody 7C7; Li et al., 1997); (2) a polyclonal antibody directed to the intracellular loop domain of skate Cx35, a structural homolog of Cx36 (O'Brien et al., 1996, 1998); (3) a rabbit polyclonal antibody directed to a segment within the cytoplasmic loop of Cx43 (Li et al., 1998; Zymed San Francisco, CA); and (4) a mouse monoclonal antibody directed to an N-terminal epitope of the AMPAR subunit GluR2 (Vissavajjhala et al., 1996; a gift from J. H. Morrison, Mt. Sinai Medical School, New York, NY). The antibody to Cx35 was used because a specific antibody to Cx36 was not yet available. Secondary antibodies were as follows: for Cx36 and Cx43, a horseradish peroxide (HRP)-conjugated anti-rabbit IgG; and for Cx32 and GluR2, an HRP-conjugated anti-mouse IgG (Vector Laboratories, Burlingame, CA).
To quantitate protein abundance, bands on Western blots were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 software. Band OD values for two samples per animal were averaged and film background-subtracted to give the mean OD value for a single animal. OD values were normalized to the corresponding control value and expressed as grand means ± SEM of individual means from a minimum of three animals. Statistical significance was assessed by means of Student's unpaired t test.
Histological analysis. Neuronal damage was assessed by histological examination of brain sections at the level of dorsal hippocampus from animals at 72 hr and 7 d after global ischemia or 72 hr after sham operation. Animals were placed under deep anesthesia and fixed by transcardiac perfusion with 4% paraformaldehyde in 10 mm PBS (150 ml, 20 min). Brains were removed and immersed in fixative (4°C, overnight). Coronal sections (30 μm) were cut with a vibratrome and stained with toluidine blue. Hippocampal injury was assessed quantitatively by the grading scale of Pulsinelli and Brierley (1979): 0, normal; 1, few neurons in CA1 damaged (<30%); 2, many neurons in CA1 damaged (30–70%); and 3, most neurons in CA1 damaged (>70%). Neuronal damage scores from a minimum of four microscopic sections per animal were statistically analyzed by means of Student's unpaired t test to determine significance and plotted as scatter graphs.
Immunocytochemical methods were as described by Opitz et al. (2000)using the same antibodies as for the Western blot analysis for Cx32 and Cx36 and a parvalbumin antibody from Chemicon (Temecula, CA).
Generation of Cx32 mutant mice. The generation and initial characterization of Cx32-null [Cx32(−/−) female and Cx32(Y/−)] mice has been described (Nelles et al., 1996). Animals were generated from our colony at the Albert Einstein College of Medicine from two breeding pairs of Cx32-null mice obtained from K. Willecke (Nelles et al., 1996). Genotypes were established by PCR analysis of genomic DNA isolated from tail clips (Anzini et al., 1997) and confirmed by Southern blot analysis (see Fig. 7A).
Fig. 7.
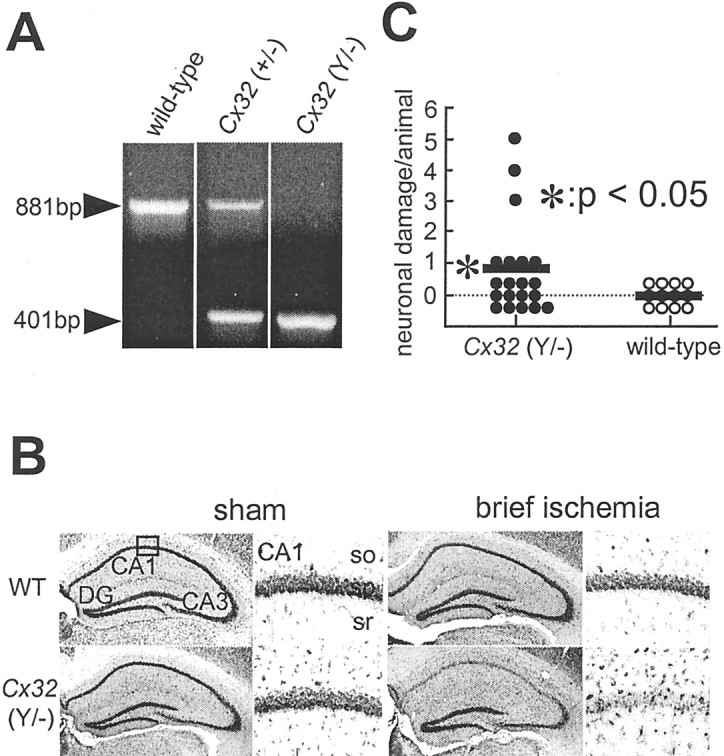
Cx32(Y/−) null mice exhibit enhanced vulnerability to global ischemia-induced neurodegeneration.A, Southern blot analysis of mice cDNAs. RT-PCR was performed on DNA samples obtained from tail biopsies of Cx32 wild-type and deficient mice and subjected to Southern blot hybridization.First lane, Wild-type; second lane, heterozygous female Cx32(+/−); third lane, homozygous Cx32(Y/−) null mice. The 881-bp amplicon indicates the presence of the wild-type Cx32 gene; the 441-bp amplicon indicates the presence of the Cx32 defective allele. B, Cx32(Y/−) null mice exhibit enhanced vulnerability to global ischemia-induced neurodegeneration. Toluidine blue staining of coronal brain sections shows the dorsal hippocampus of wild-type and Cx32(Y/−) null mice. Animals were killed 7 d after sublethal (brief, 10 min) global ischemia or sham operation. At 7 d after brief ischemia, wild-type (WT) mice exhibited no detectable neuronal loss; Cx32 null mice exhibited a range of neuronal loss. Scale bar:first and third columns, 100 μm;second and fourth columns, 50 μm.DG, Dentate gyrus; so, stratum oriens;sr, stratum radiatum. C, Quantitation of ischemia-induced damage. Neuronal damage scores are shown for Cx32(Y/−) null mice (left) and Cx32(Y/+) wild-type mice (right). Neuronal damage was assessed as follows: 0, no neuronal damage; 1, minor neuronal damage (<30% neurons are dead); 2, moderate neuronal damage (30–70% of neurons are dead); 3, severe (>70% neurons are dead); scores from right and left sides were summed for each animal's total score. *p < 0.01 for Cx32(Y/−) null mice versus Cx32(Y/+) wild-type mice.
RESULTS
Global ischemia induces delayed, selective neurodegeneration of mouse hippocampal CA1
To examine ischemia-induced alterations in connexin expression in hippocampus, male adult Cx32(Y/+) wild-type mice were subjected to ischemia by transient (20 min) bilateral occlusion of the common carotid arteries or to sham operation. Mice offer an advantage compared with rats in that they exhibit global ischemia in response to the relatively simple two-vessel occlusion surgical model and enable comparisons between animals with null mutations in a single gene of interest and their wild-type littermates (although strain differences in vulnerability to ischemic damage can complicate results; Schauwecker and Steward, 1997). To assess neuronal loss after induction of global ischemia in mice, brain sections of experimental and control animals were subjected to histological analysis. At 72 hr after ischemia, most animals exhibited no detectable cell loss in the hippocampal CA1 (Fig.1B,E) compared with control animals (Fig. 1A,D). Approximately 17% of animals subjected to global ischemia exhibited minor cell loss, and ∼17% of animals exhibited moderate cell loss in CA1 (data not shown). In contrast, nearly all animals at 7 d after ischemia exhibited marked loss in the pyramidal cell layer of CA1 (and slight to moderate loss in the pyramidal cell layer of CA3; Fig.1C,F). The few surviving neurons in CA1 of ischemic animals exhibited pyknotic nuclei, indicative of early neurodegeneration. In most animals, the hippocampal CA3 exhibited at most slight cell loss, and the dentate gyrus exhibited no cell loss at all times examined. These patterns of neurodegeneration observed in mice are similar to, but considerably more variable than, those observed in rats and gerbils (Kirino et al., 1992; Gorter et al., 1997).
Fig. 1.
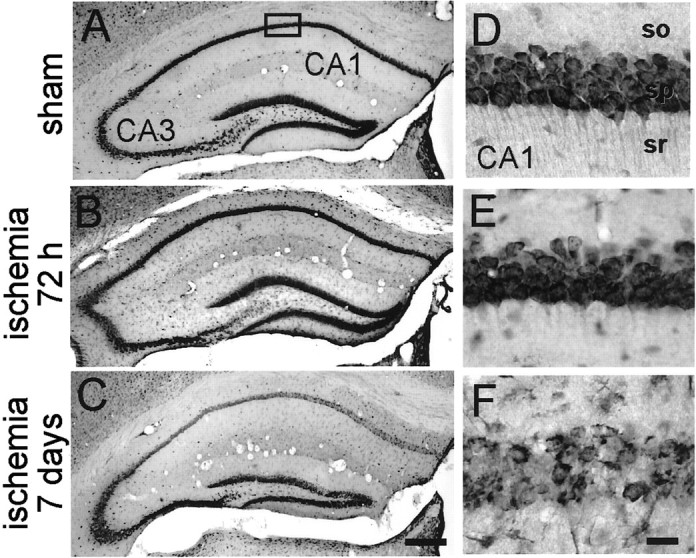
Histology at 72 hr after sham operation and 72 hr and 7 d after global ischemia in mice Toluidine blue labeling of coronal brain sections at the level of the dorsal hippocampus from control mice (n = 3; A, D) and experimental mice at 72 hr after ischemia (n = 3;B, E) revealed no detectable neuronal damage. At 7 d after ischemia (n = 3), cell loss was observed, primarily in CA1 (C, F). Brief (20 min) global ischemia was induced by bilateral occlusion of the common carotid arteries as described in Materials and Methods. Scale bars, 50 μm.so, Stratum oriens; sr, stratum radiatum.
Patterns of ischemia-induced alterations in Cx32, Cx36, and Cx43 mRNA expression
To examine patterns of connexin mRNA expression after global ischemia, in situ hybridization was performed on sections of brain from wild-type [Cx32(Y/+)] mice subjected to global ischemia at 2, 8, 24, 48, and 72 hr or to sham operation at 24 hr. In control animals, Cx32, Cx36, and Cx43 exhibited cell-specific patterns of expression throughout the hippocampus and neocortex generally in accord with patterns observed for rat brain (Srinivas et al., 1999; Condorelli et al., 2000; Rash et al., 2000). A notable exception was the essentially uniform expression of Cx36 throughout the pyramidal cell layer of mouse CA1 and CA3. Changes in connexin expression were assessed quantitatively by image analysis of autoradiographic film densities. Cx43 mRNA expression was increased in the pyramidal cell layer of CA1, evident at 48 and 72 hr after ischemia; Cx32 and Cx36 were unchanged in CA1 at all times examined (p< 0.01 for Cx43; n = 5 for ischemic mice at each time point; n = 5 for control mice for each connexin examined; Fig. 2A–C). The increase in Cx43 expression was first evident at 48 hr after ischemia and was slightly greater at 72 hr after ischemia. Expression of mRNAs encoding all three connexins was unchanged in dentate gyrus and in other brain regions examined (e.g., parietal cortex, area 1), except for Cx36, which showed a significant increase in dentate gyrus at 48 hr (p < 0.01) and 72 hr (p < 0.05). As a positive control, we also examined expression of mRNA encoding the AMPAR subunit GluR2, an mRNA known to undergo cell-specific downregulation in CA1 after global ischemia in rats (Pellegrini-Giampietro et al., 1992) and gerbils (Gorter et al., 1997). As expected, GluR2 mRNA expression was significantly and selectively downregulated in the CA1 after global ischemia; GluR2 mRNA expression was not detectably changed in either dentate gyrus (Fig. 2D) or CA3 (data not shown). GluR1 mRNA expression in CA1 was slightly decreased at 72 hr, presumably because of the onset of cell death. (Fig.2E).
Fig. 2.

Patterns of global ischemia-induced alterations in Cx32, Cx36, and Cx43 mRNA expression in mouse hippocampus, as assessed by in situ hybridization of coronal brain sections.A–C, In control animals, Cx32, Cx36, and Cx43 mRNA expression was prominent in the pyramidal cell layers of the hippocampal CA1 and CA3 and in the granule cell layer of dentate gyrus (DG). Global ischemia induced a marked increase in Cx43 (C) mRNA expression in CA1 (closed circles). Cx32 (A) and Cx36 (B) were unchanged at all times examined in CA1. Connexin mRNA expression was unchanged in the dentate gyrus except for Cx36 (open circles). D, Global ischemia also downregulated mRNA encoding the AMPAR subunit GluR2 in CA1.E, GluR1 mRNA expression was unchanged in all subfields as late as 48 hr; GluR1 was modestly reduced at 72 hr, presumably because of onset of cell loss. Data shown are mean densities ± SEM of autoradiographic films for in situ hybridization of Cx32, Cx36, and Cx43 mRNAs in the hippocampal CA1 at 2, 8, 24, 48, and 72 hr after the ischemic insult.
Global ischemia induces downregulation of Cx32 mRNA abundance in mouse hippocampus
To assess quantitatively the effects of global ischemia on Cx32, Cx36, and Cx43 mRNA expression, we performed Northern blot analysis on RNA samples isolated from the hippocampus of experimental and control Cx32(Y/+) wild-type mice. Animals were killed at 24 hr after ischemia or sham operation. RNA samples were subjected to gel electrophoresis and probed with highly specific32P-labeled RNA probes directed to the Cx32 (Paul, 1986), Cx36 (Condorelli et al., 1998), and Cx43 (Beyer et al., 1987) mRNAs. Global ischemia induced a marked reduction in Cx32 mRNA abundance at 24 hr; Cx36 and Cx43 mRNA abundance was unchanged (Fig. 3A). Quantitative analysis of band densities on Northern blots revealed a reduction to 46.7 ± 16.2% of the control value for Cx32 mRNA expression; Cx36 and Cx43 mRNA was not detectably altered (n = 3 for ischemic animals; n = 3 for control animals; for Cx32,p < 0.05 for experimental vs control animals; Fig.3B). In situ hybridization showed no reduction in Cx32 mRNA in CA1, CA3, or dentate gyrus (Fig. 2A). Most of the Cx32 mRNA in control brains presumably is in oligodendrocytes, which are diffusely distributed. This Cx32 mRNA and a decrease therein after ischemia might be difficult to detect by in situ hybridization. Thus, the Northern blot analysis and in situ hybridization results are not contradictory.
Fig. 3.

Global ischemia-induced downregulation of Cx32 mRNA expression in mouse hippocampus, as assessed by Northern blot analysis. A, Representative film autoradiograms of Northern blots probed with highly specific 35S-labeled RNA probes directed to the Cx32, Cx36, and Cx43 mRNAs. Samples of total RNA extracts isolated from control animals at 24 hr after sham operation (n = 3) and experimental animals at 24 hr after ischemia (n = 3) were subjected to Northern blot analysis as described in Materials and Methods. Global ischemia induced a significant downregulation in Cx32 mRNA. B, Quantitation of Northern blot analysis data. Means ± SEM of band densities are shown from three independent experiments involving samples from three experimental and three control animals. Band densities for samples from experimental animals were normalized to the corresponding band densities for samples from control animals.
Global ischemia selectively upregulates Cx32 and Cx36 protein abundance in CA1
To assess quantitatively effects of global ischemia on Cx32, Cx36, and Cx43 protein abundance in CA1, we performed quantitative Western blot analysis. CA1 was microdissected from brains of mice at 8, 24, 48, and 72 hr after ischemia or at 24 hr after sham operation. Samples of whole-cell protein were subjected to gel electrophoresis, and gels were probed with highly specific antibodies to the Cx32 (Paul, 1986; Li et al., 1997), Cx35 (skate homolog of mammalian Cx36; O'Brien et al., 1996; Condorelli et al., 1998), and Cx43 (Li et al., 1998) proteins. As a positive control, we also examined GluR2 protein abundance in CA1 with a GluR2 subunit-specific antibody (Vissavajjhala et al., 1996). Protein abundance was assessed by image analysis of band densities on Western blots. Global ischemia produced a marked increase in Cx32 and Cx36 protein abundance in CA1 at 24 hr (to 134 ± 10.7%;p < 0.01 for experimental vs control for Cx32; and to 138 ± 6.5%; p < 0.01 for experimental vs control for Cx36; n = 3 for control animals;n = 3 for ischemic animals at each time point; Fig.4A,B). Maximal increases in connexin protein were observed at 24 hr after ischemia, with some decline toward control levels by 48 and 72 hr. In contrast, Cx43 expression was essentially unchanged in CA1 after global ischemia (to 107 ± 4.5% of control at 24 hr; n = 3 for control animals; n = 3 for ischemic animals at each time point; Fig. 4C). As expected, GluR2 protein abundance in CA1 decreased after global ischemia to 66 ± 2.1% at 48 hr after ischemia and to 60 ± 2.3% at 72 hr after ischemia (n = 3 for control animals; n = 3 for ischemic animals at each time point; p < 0.01 for experimental vs control at each time point). Similar results were observed in gerbil hippocampus (Opitz et al., 2000). These findings indicate that global ischemia induces alterations in connexin protein expression, stability, or both in a subunit- and subfield-specific manner.
Fig. 4.

Global ischemia-induced changes in connexin subunit abundance in hippocampal CA1. Global ischemia induced an increase in Cx32 and Cx36 protein abundance in CA1 but did not detectably alter Cx43 protein abundance. Film autoradiograms (top panels) and band densities (bottom panels) of Western blots probed for Cx32 (A), Cx36 (B), Cx43 (C), and GluR2 (D) protein are shown. Samples of protein extracts from the CA1 of control animals at 24 hr after sham operation (n = 3) and experimental animals at 8, 24, 48, and 72 hr after ischemia (n = 3 for each treatment group at each time point) were subjected to Western blot analysis as described in Materials and Methods. Means ± SEM of band densities were from three independent experiments. Band densities for samples from experimental animals were normalized to those for control animals. Global ischemia induced a marked upregulation in Cx32 and Cx36 and a downregulation in GluR2 protein abundance in CA1 after ischemia. Cx43 protein abundance was unchanged at all times examined after global ischemia.
Global ischemia selectively increases Cx32 and Cx36 protein expression in parvalbumin-positive inhibitory interneurons in CA1
The results thus far indicate that Cx32 and Cx36 protein expression are enhanced in the hippocampal CA1 after global ischemia but do not distinguish between labeling of inhibitory interneurons and other cell types. To examine cell-specific changes in connexin expression in postischemic CA1, we performed double immunofluorescence of brain sections from control and experimental animals at times after ischemia. Consistent with the known organization of gap junctions in plaques or clusters of channels (>0.2 μm in diameter; Bukauskas et al., 2000), brain sections from control animals exhibited punctate immunolabeling of Cx32 (Fig. 5A,C) and Cx36 (Fig.6A,C) in the stratum radiatum and oriens of CA1, extending into the stratum pyramidale. Double immunofluorescence indicated that Cx32 and Cx36 immunoreactivity was found in the cell bodies of PV-positive neurons (Fig. 5, arrows inF, I, L, Fig.6J–L). Global ischemia markedly increased Cx32 (Fig. 5D,G) and Cx36 (Fig.6D,G) immunolabeling at 24 and 48 hr after ischemia. At 72 hr, Cx32 and Cx36 immunolabeling was somewhat reduced (Figs. 5J, 6J), although higher levels of Cx36 labeling were seen in PV-positive neurons. The structures forming puncta generally could not be identified in these preparations, but some appeared to be on PV-positive neurons (Fig.5D–I, Fig. 6G–I,arrow in I). These findings indicate that Cx32 and Cx36 are expressed by PV-positive inhibitory interneurons of hippocampal CA1 and that expression is enhanced after global ischemia. Immunolabeling of the three connexin proteins was unchanged in the hippocampal CA3 and dentate gyrus at all times examined (data not shown).
Fig. 5.

Global ischemia markedly increases Cx32 protein expression in parvalbumin-positive inhibitory interneurons in CA1. Cx32 (A, D, G, J), PV (B, E, H, K), and merged immunofluorescence images (C, F, I, L) show inhibitory interneurons within the CA1 pyramidal cell layer in brain sections from control animals (A–C) at 24 hr after sham operation and experimental animals at 24 hr (D–F), 48 hr (G–I) and 7 d (J–L) after global ischemia. Cx32 immunofluorescence in cell bodies exhibited a high coincidence with PV immunofluorescence (C, F, I, L) throughout the CA1 pyramidal cell layer. Cx32 immunofluorescence exhibited intense puncta along the dendrites of PV-positive inhibitory interneurons. Global ischemia markedly increased Cx32 immunofluorescence in parvalbumin-positive inhibitory interneurons in the CA1 pyramidal layer at 24 and 48 hr with some decline at 72 hr. Data are typical of 10 sections per animal from a minimum of three animals per time point and treatment group. Cx32 immunofluorescence is visualized in green, and PV immunofluorescence is visualized in red. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bar, 50 μm.
Fig. 6.

Global ischemia markedly increases Cx36 immunolabeling in CA1. Cx36 (A, D, G,J), PV (B, E, H,K), and merged immunofluorescence images (C,F, I, L) show Cx36 immunoreactivity and inhibitory interneurons in the CA1 pyramidal cell layer in brain sections from control animals (A–C) at 24 hr after sham operation and experimental animals at 24 (D–F), 48 (G–I), and 72 (J–L) hr after global ischemia. Global ischemia markedly increased punctate Cx36 immunolabeling in the pyramidal cell layer extending out into the stratum oriens and stratum radiatum at 24 and 48 hr. There was Cx36 labeling of PV-positive inhibitory neurons at 72 hr. Data are typical of a minimum of 10 sections per animal from three animals per time point and treatment group. Cx36 immunofluorescence is visualized in green, and PV immunofluorescence is visualized in red.so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bar, 50 μm.
Cx32-null mice exhibit enhanced vulnerability to global ischemia-induced damage
A study involving immunogold indicates that within the brain the gap junctional protein Cx32 is expressed exclusively in oligodendrocytes (Rash et al., 2000). Single-cell RT-PCR studies indicate that Cx32 is expressed by interneurons in the neocortex, although less abundantly than Cx36 (Venance et al., 2000). To examine a possible role for Cx32 gap junctions in modulating vulnerability of neurons to insult and injury, we compared the degree of ischemia-induced neuronal death in the hippocampus of Cx32(Y/−) null mice with that of their wild-type littermates (Fig.7). Because Cx32 gap junctions formed between interneurons, within oligodendrocytes, or both, might be critical to inhibitory transmission in the hippocampus, and might thereby reduce vulnerability of neurons to ischemic insults, we reasoned that Cx32(Y/−) null mice would exhibit enhanced vulnerability. We therefore shortened the length of the ischemic episode from 20 to 10 min, an insult that is sublethal for CA1 neurons in wild-type mice. Brief (10 min) global ischemia induced a greater range of neuronal damage in Cx32(Y/−) null mice than in wild-type Cx32(Y/+) littermates (Fig. 7B,C). Seven of 20 Cx32(Y/−) null mice exhibited neurodegeneration after a 10 min ischemic episode, whereas 0 of 12 wild-type littermates exhibited detectable damage (Fig.7C). Quantification of the neuronal damage by the grading scale of Pulsinelli and Brierley (1979) revealed a range of damage in Cx32(Y/−) null mice (one animal scored 5; one animal scored 4; one animal scored 3; 4 animals scored 1; and 13 animals scored 0). The mean neuronal damage score of 20 animals was 0.9 ± 1.7 for Cx32(Y/−) null mice versus 0 for Cx32(Y/+) wild-type littermates (n = 20 for Cx32 null mice; n = 12 for wild-type mice; p < 0.05 for Cx32 null vs wild-type mice). These findings are consistent with a mechanism whereby Cx32 gap junctions between interneurons, within oligodendrocytes, or both serve to reduce neuronal vulnerability to ischemic insults.
DISCUSSION
The present study was undertaken to examine the role of connexins in global ischemia-induced neuronal death. We report here for the first time subtype- and cell-specific changes in connexin protein expression after global ischemia and at times preceding the onset of neuronal death. The new conclusions of our study are as follows: (1) global ischemia induces an upregulation in Cx43 mRNA expression in the vulnerable CA1 before the onset of neuronal death, but Cx32 and Cx36 are unchanged at all times examined in CA1; (2) global ischemia induces a selective upregulation of the gap junctional proteins Cx32 (expressed by resistant parvalbumin-positive inhibitory interneurons and oligodendrocytes) and Cx36 (expressed primarily by resistant parvalbumin-positive inhibitory interneurons) in the hippocampal CA1, but Cx43 protein is not detectably altered in any subfield or at any times examined; (3) ischemia-induced changes in Cx32 and Cx36 protein occur in the absence of detectable changes in the corresponding mRNAs, consistent with regulation of expression at the translational and post-translational levels rather than at the transcriptional level; and (4) Cx32-null mice exhibit enhanced vulnerability, consistent with a role for Cx32 gap junctions in neuroprotection against ischemia-induced cell death. Our finding that global ischemia induces upregulation of Cx32 and Cx36 protein expression, together with the observation that targeted deletion of Cx32 enhances vulnerability of CA1 to ischemia-induced damage, provides a basis for understanding a role for neuronal gap junctions in defining cell-specific patterns of neuronal injury after global ischemia. Because global ischemia targets pyramidal neurons of the hippocampal CA1 but spares GABAergic interneurons, these findings suggest the novel possibility that enhanced expression of Cx36 may play a critical role in protection and survival of CA1 interneurons after global ischemia.
Role of neuronal gap junctions in neuronal death
Morphological studies involving confocal and electron microscopic serial section analysis and three-dimensional reconstruction provide evidence that PV-positive GABAergic interneurons form a network in which distal dendrites of like neurons are interconnected by dendrodendritic gap junctions (Fukuda and Kosaka, 2000). Gap junctions linking the dendritic network are thought to facilitate synchronous oscillations generated in the interneuron network and thereby to reinforce inhibitory synaptic transmission (Galarreta and Hestrin, 1999; Gibson et al., 1999). Studies involving RT-PCR in combination with electrophysiology reveal expression of Cx32 and Cx36 in interneurons in the network and suggest that Cx32 and Cx36 gap junctions mediate synchronization of the network (Venance et al., 2000). These observations support a role for interneuronal gap junctions in the strengthening of inhibitory neurotransmission in the CA1. These observations, together with our finding that Cx32 and Cx36 protein expression is upregulated after ischemia, provide evidence for the novel hypothesis that interneuronal gap junctional communication promotes the resistance and survival of selected populations of interneurons to ischemia-induced damage. Because these interneurons also innervate pyramidal neurons in the hippocampal CA1, other possible consequences of upregulation of Cx36 include (1) reduced vulnerability of CA1 pyramidal neurons because of enhanced inhibitory input and reduced metabolic stress associated with activity or, on the other hand, (2) enhanced vulnerability of CA1 pyramidal neurons, which are kept hyperpolarized, because of increased Ca2+ influx through Ca2+-permeable AMPA receptors.
Further support for a role of Cx32 gap junctions in neuronal survival is provided by our finding that Cx32-null mice exhibit enhanced vulnerability to ischemia-induced neuronal damage. This finding is consistent with observations that Cx32-deficient mice display enhanced intrinsic excitability indicative of dysfunction of inhibitory synaptic transmission (Sutor et al., 2000). Electrophysiological recording from neocortical slices from Cx32-null mice indicates that ∼50% of pyramidal neurons exhibit enhanced intrinsic excitability and paroxysmal depolarizations resembling those induced by GABAA receptor antagonists. Moreover, late-depolarizing glutamatergic EPSPs are enhanced, indicative of reduced inhibitory input. Enhanced excitatory transmission and deficient inhibitory transmission would be expected to enhance vulnerability to excitotoxic cell death.
Role of astrocytic gap junctions in the propagation of neuronal death
Our finding that Cx43 protein levels in hippocampus are not detectably altered from 8 hr to 7 d after (20 min) global ischemia in mice, as assessed by Western blot analysis, is consistent with the finding that in rats Cx43 protein abundance in CA1 and CA3, assessed by immunolabeling, is not altered after ischemia but rather undergoes redistribution, forming a network of intense immunoreactive punctae in and around pyramidal neurons of the CA3 (Hossain et al., 1994). Because astrocytes survive global ischemic insults and astrocytic gap junctions remain functional in postischemic brain, they may play a role in propagation of neuronal death, inhibition of neuronal death, or both.
Recent studies support a role for interastrocytic gap junctions in the propagation and amplification of neuronal injury (Lin et al., 1998). In adult brain, astrocytes are abundantly interconnected by gap junctions; electron microscopic studies indicate that a single astrocyte can express >30,000 gap junction channels. Astrocytes communicate not only with other astrocytes but also with neighboring neurons to modulate neuronal calcium levels. Astrocytes are activated during synaptic transmission and respond by a rise in intracellular calcium concentration. In turn, astrocytic calcium signaling modulates excitatory and inhibitory transmission between neurons (Avoli et al., 1996; Newman and Zahs, 1998). Astrocytic gap junctions remain functional in postischemic brain (Lin et al., 1998), and gap junction inhibitors limit infarct size in focal ischemia (Warner et al., 1995;Rawanduzy et al., 1997; Saito et al., 1997). Astrocytic gap junctions propagate transcellular signals, which exacerbate cell injury induced by calcium overload, oxidative stress, and metabolic inhibition (Lin et al., 1998). Moreover, dying glia propagate and amplify cell death via bystander cell death in proportion to their expression of gap junctions and functional coupling. By extrapolation, these findings suggest that interastrocytic Cx43 gap junctions would propagate and amplify neuronal death after global ischemia as well.
Somewhat paradoxically, other studies indicate that interastrocytic gap junctions afford neuroprotection. Possible mechanisms underlying astrocyte-mediated neuroprotection include (1) enhanced synthesis and release of protective signaling molecules from astrocytes, (2) reduced synthesis and release of neurotoxic substances, (3) spatial buffering of K+, and (4) buffering of neuronal Ca2+. Astrocytes synthesize and release neurotrophic factors and cytokines such as nerve growth factor (Schwartz et al., 1994), tumor necrosis factor-α (Benveniste et al., 1995), and basic fibroblast growth factor (Mattson and Rychlik, 1990;Eckenstein, 1994), as well as the antioxidant enzyme catalase (Desagher et al., 1996), which act to promote neuroprotection and survival after global ischemia. Synthesis and release of neurotrophins and cytokines by astrocytes can be regulated in an activity-dependent manner (Koyama et al., 1997). In addition, astrocytes also take up neurotoxic substances such as glutamate (Anderson and Swanson, 2000). Astrocytic gap junctions appear to play a neuroprotective role in models of oxidative stress (Blanc et al., 1998). Administration of gap junction uncouplers, such as 18-α-glycyrrhetinic acid (GA), to slice cultures exacerbates oxidative stress-induced neuronal death. Together, these observations are consistent with a mechanism whereby interastrocytic gap junctions reduce neuronal vulnerability to oxidative injury by facilitating neuronal buffering of Ca2+.
In conclusion, the present study supports a role for interneuronal gap junctions in the cell-specific patterns of global ischemia-induced neuronal death. Our study shows that global ischemia induces a selective upregulation in the gap junctional proteins Cx32 and Cx36 in the vulnerable CA1 before the onset of neuronal death, as assessed by Western blot analysis. Cx32(Y/−) null mice exhibited enhanced vulnerability to brief ischemic insults, consistent with a role for neuronal or oligodendrocytic Cx32 gap junctions, or both, in neuronal survival. These findings suggest that Cx32 and Cx36 gap junctions might contribute to the survival and resistance of GABAergic interneurons, thereby defining cell-specific patterns of global ischemia-induced neuronal death.
Footnotes
This work was supported by National Institutes of Health Grants NS20752 and NS31282 (R.S.Z.) and NS07412 (to M.V.L.B.). M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience. We thank Dr. David Paul for rat Cx26, Cx32, and Cx43 cDNAs and Dr. Elliot Hertzberg for Cx26, Cx32, and Cx43 antibodies.
Correspondence should be addressed to Dr. Michael V. L. Bennett, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: mbennett@aecom.yu.edu.
REFERENCES
- 1.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 2.Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avoli M, Kohling R, Barbarosie M. Anoxia blocks the presynaptic control of GABA release at inhibitory terminals in the rat hippocampus. Neuroscience. 1996;75:999–1002. doi: 10.1016/0306-4522(96)00429-0. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MV, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: new tools, new answers, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- 5.Benveniste EN, Tang LP, Law RM. Differential regulation of astrocyte TNF-α expression by the cytokines TGF-β, IL-6 and IL-10. Int J Dev Neurosci. 1995;13:341–349. doi: 10.1016/0736-5748(94)00061-7. [DOI] [PubMed] [Google Scholar]
- 6.Beyer EC, Paul DL, Goodenough DA. Connexin 43: a protein from rat heart homologous to a gap junction protein from liver. J Cell Biol. 1987;105:2621–2629. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanc EM, Bruce-Keller AJ, Mattson MP. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J Neurochem. 1998;70:958–970. doi: 10.1046/j.1471-4159.1998.70030958.x. [DOI] [PubMed] [Google Scholar]
- 8.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 9.Bukauskas FF, Jordan K, Bukauskiene A, Bennett MV, Lampe PD, Laird DW, Verselis VK. Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc Natl Acad Sci USA. 2000;97:2556–2561. doi: 10.1073/pnas.050588497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Condorelli DF, Parenti R, Spinella F, Trovato SA, Belluardo N, Cardile V, Cicirata F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci. 1998;10:1202–1208. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- 12.Condorelli DF, Belluardo N, Trovato-Salinaro A, Mudo G. Expression of Cx36 in mammalian neurons. Brain Res Rev. 2000;32:72–85. doi: 10.1016/s0165-0173(99)00068-5. [DOI] [PubMed] [Google Scholar]
- 13.Dermietzel R, Spray DC. From neuro-glue (“Nervenkitt”) to glia: a prologue. Glia. 1998;24:1–7. doi: 10.1002/(sici)1098-1136(199809)24:1<1::aid-glia1>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 14.Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC, Willecke K. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci USA. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci. 1996;16:2553–2562. doi: 10.1523/JNEUROSCI.16-08-02553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckenstein FP. Fibroblast growth factors in the nervous system. J Neurobiol. 1994;25:1467–1480. doi: 10.1002/neu.480251112. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda T, Kosaka T. Gap junctions linking the dendritic network of GABAergic interneurons in the hippocampus. J Neurosci. 2000;20:1519–1528. doi: 10.1523/JNEUROSCI.20-04-01519.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature. 1999;402:72–75. doi: 10.1038/47029. [DOI] [PubMed] [Google Scholar]
- 19.Giaume C, Fromaget C, el Aoumari A, Cordier J, Glowinski J, Gros D. Gap junctions in cultured astrocytes: single-channel currents and characterization of channel-forming protein. Neuron. 1991;6:133–143. doi: 10.1016/0896-6273(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 20.Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- 21.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 22.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hossain MZ, Peeling J, Sutherland GR, Hertzberg EL, Nagy JI. Ischemia-induced cellular redistribution of the astrocytic gap junctional protein connexin43 in rat brain. Brain Res. 1994;652:311–322. doi: 10.1016/0006-8993(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 24.Hsu M, Buzsaki G. Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci. 1993;13:3964–3979. doi: 10.1523/JNEUROSCI.13-09-03964.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 26.Kirino T, Robinson HP, Miwa A, Tamura A, Kawai N. Disturbance of membrane function preceding ischemic delayed neuronal death in the gerbil hippocampus. J Cereb Blood Flow Metab. 1992;12:408–417. doi: 10.1038/jcbfm.1992.58. [DOI] [PubMed] [Google Scholar]
- 27.Koyama S, Haruyama T, Kobatake E, Aizawa M. Electrically induced NGF production by astroglial cells. Nat Biotechnol. 1997;15:164–166. doi: 10.1038/nbt0297-164. [DOI] [PubMed] [Google Scholar]
- 28.Kunzelmann P, Blumcke I, Traub O, Dermietzel R, Willecke K. Coexpression of connexin-45 and -32 in oligodendrocytes of rat brain. J Neurocytol. 1997;26:17–22. doi: 10.1023/a:1018555207379. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Hertzberg EL, Nagy JI. Connexin32 in oligodendrocytes and association with myelinated fibers in mouse and rat brain. J Comp Neurol. 1997;379:571–591. doi: 10.1002/(sici)1096-9861(19970324)379:4<571::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 30.Li WE, Ochalski PA, Hertzberg EL, Nagy JI. Immunorecognition, ultrastructure and phosphorylation status of astrocytic gap junctions and connexin43 in rat brain after cerebral focal ischaemia. Eur J Neurosci. 1998;10:2444–2463. doi: 10.1046/j.1460-9568.1998.00253.x. [DOI] [PubMed] [Google Scholar]
- 31. Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci 1 1998. 494 500 [Erratum (1998) 1:743] [DOI] [PubMed] [Google Scholar]
- 32.Mattson MP, Rychlik B. Glia protect hippocampal neurons against excitatory amino acid-induced degeneration: involvement of fibroblast growth factor. Int J Dev Neurosci. 1990;8:399–415. doi: 10.1016/0736-5748(90)90073-b. [DOI] [PubMed] [Google Scholar]
- 33.Nelles E, Butzler C, Jung D, Temme A, Gabriel HD, Dahl U, Traub O, Stumpel F, Jungermann K, Zielasek J, Toyka KV, Dermietzel R, Willecke K. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc Natl Acad Sci USA. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Brien J, al-Ubaidi MR, Ripps H. Connexin 35: a gap-junctional protein expressed preferentially in the skate retina. Mol Biol Cell. 1996;7:233–243. doi: 10.1091/mbc.7.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Brien J, Bruzzone R, White TW, al-Ubaidi MR, Ripps H. Cloning and expression of two related connexins from the perch retina define a distinct subgroup of the connexin family. J Neurosci. 1998;18:7625–7637. doi: 10.1523/JNEUROSCI.18-19-07625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opitz T, Grooms SY, Bennett MV, Zukin RS. Remodeling of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit composition in hippocampal neurons after global ischemia. Proc Natl Acad Sci USA. 2000;97:13360–13365. doi: 10.1073/pnas.97.24.13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paul DL. Molecular cloning of cDNA for rat liver gap junction protein. J Cell Biol. 1986;103:123–134. doi: 10.1083/jcb.103.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA 89 1992. 10499 10503 [Erratum (1993) 90:780] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 42.Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- 43.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 44.Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci USA. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rawanduzy A, Hansen A, Hansen TW, Nedergaard M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg. 1997;87:916–920. doi: 10.3171/jns.1997.87.6.0916. [DOI] [PubMed] [Google Scholar]
- 46.Saito R, Graf R, Hubel K, Fujita T, Rosner G, Heiss WD. Reduction of infarct volume by halothane: effect on cerebral blood flow or perifocal spreading depression-like depolarizations. J Cereb Blood Flow Metab. 1997;17:857–864. doi: 10.1097/00004647-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz JP, Nishiyama N, Wilson D, Taniwaki T. Receptor-mediated regulation of neuropeptide gene expression in astrocytes. Glia. 1994;11:185–190. doi: 10.1002/glia.440110212. [DOI] [PubMed] [Google Scholar]
- 50.Sohl G, Degen J, Teubner B, Willecke K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998;428:27–31. doi: 10.1016/s0014-5793(98)00479-7. [DOI] [PubMed] [Google Scholar]
- 51.Srinivas M, Rozental R, Kojima T, Dermietzel R, Mehler M, Condorelli DF, Kessler JA, Spray DC. Functional properties of channels formed by the neuronal gap junction protein connexin 36. J Neurosci. 1999;19:9848–9855. doi: 10.1523/JNEUROSCI.19-22-09848.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka H, Grooms SY, Bennett MVL, Zukin RS. The AMPAR subunit GluR2: still front and center-stage. Brain Res. 2000;886:190–207. doi: 10.1016/s0006-8993(00)02951-6. [DOI] [PubMed] [Google Scholar]
- 54.Venance L, Rozov A, Blatow M, Burnashev N, Feldmeyer D, Monyer H. Connexin expression in electrically coupled postnatal rat brain neurons. Proc Natl Acad Sci USA. 2000;97:10260–10265. doi: 10.1073/pnas.160037097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vissavajjhala P, Janssen WG, Hu Y, Gazzaley AH, Moran T, Hof PR, Morrison JH. Synaptic distribution of the AMPA-GluR2 subunit and its colocalization with calcium-binding proteins in rat cerebral cortex: an immunohistochemical study using a GluR2-specific monoclonal antibody. Exp Neurol. 1996;142:296–312. doi: 10.1006/exnr.1996.0199. [DOI] [PubMed] [Google Scholar]
- 56.Warner DS, Ludwig PS, Pearlstein R, Brinkhous AD. Halothane reduces focal ischemic injury in the rat when brain temperature is controlled. Anesthesiology. 1995;82:1237–1245. doi: 10.1097/00000542-199505000-00019. [DOI] [PubMed] [Google Scholar]
- 57.Xie WQ, Rothblum LI. Rapid, small-scale RNA isolation from tissue culture cells. Biotechniques. 1991;11:324–327. [PubMed] [Google Scholar]