

Abstract
Female rodents producing endogenous estrogens are protected from stroke damage in comparison with male counterparts. This natural protection is lost after ovariectomy or reproductive senescence. The aim of this study is to determine whether estrogen reduces early neuronal injury and cell loss after ischemia by increasing the expression of Bcl-2. Male, intact female, ovariectomized, and estrogen-repleted ovariectomized rats were subjected to middle cerebral artery occlusion, and 22 hr later the level and localization of Bcl-2 mRNA and protein were determined. The levels of post-ischemicbcl-2 mRNA and protein were increased exclusively in neurons within the peri-infarct region. Intact females and estrogen-treated castrates demonstrated increased bcl-2mRNA and protein expression compared with males and estrogen-deficient females, accompanied by a decrease in infarct size. To test the hypothesis that the neuroprotective mechanism of estrogen functions via Bcl-2, we compared ischemic outcome in male, female, and ovariectomized wild-type mice and mice overexpressing Bcl-2 exclusively in neurons. Wild-type female mice sustained smaller infarcts compared with males. Bcl-2 overexpression reduced infarct size in males, but provided no added protection in the female. Moreover, ovariectomy exacerbated infarction in wild-type females, but had no effect in Bcl-2 overexpressors. These data indicate that overexpression of Bcl-2 simulates the protection against ischemic injury conferred by endogenous female sex steroids. We concluded that estrogen rescues neurons after focal cerebral ischemia by increasing the level of Bcl-2 in peri-infarct regions and that estrogen-induced bcl-2gene expression is an important downstream component of neuronal protection in female stroke.
Keywords: estrogen, stroke, cerebral ischemia, bcl-2, neuroprotection, neuronal cell death, male, female, sex, gender differences
Women in their reproductive years are at lower risk of stroke than men. There is a large body of evidence from animal studies showing that sex steroids modulate brain damage during cerebral ischemia. Although estrogen has been widely shown to be neuro- and vasoprotective, the cellular and molecular mechanisms remain unknown (Green and Simpkins, 2000; Hurn and Macrae, 2000; Roof and Hall, 2000). The steroid has been linked to an important family of proteins, the proto-oncogene Bcl-2 family, which is known to promote cell survival in cerebral ischemia and protect against necrosis and apoptosis. Bcl-2 is expressed at low levels in adult brain (Merry and Korsmeyer, 1997), but its expression is induced in post-ischemic neurons (Honkaniemi et al., 1996; Chen et al., 1997), resulting in increased cell viability (Shimazaki et al., 1994; Chen et al., 1995). In male animals, Bcl-2 overexpression ameliorates cerebral ischemic injury (Martinou et al., 1994; Linnik et al., 1995; Kitagawa et al., 1998; Antonawich et al., 1999), whereas suppression of its expression by gene deletion (Hata et al., 1999) or antisense treatment (Chen et al., 2000) exacerbates brain damage.
In females, estrogen induces Bcl-2 and decreases Bax, resulting in suppression of cyclical cell death in normal reproductive tissue (Sabourin et al., 1994; Goodman et al., 1998). Estrogen also promotes the survival of estrogen-dependent breast cancer cells, which are characterized by high levels of Bcl-2 (Teixeira et al., 1995).
In neuronal cells, estrogen treatment increases Bcl-2 expression, which may account for steroid-induced protection from glutamate or hydrogen peroxide (Singer et al., 1998). Estrogen also increases the survival of cultured sensory neurons deprived of nerve growth factor (Patrone et al., 1999). In rat hypothalamic neurons, Bcl-2 expression varies with the estrus cycle and correlates with plasma levels of estrogen (Garcia-Segura et al., 1998). We propose that estrogen stimulates Bcl-2 expression in steroid-sensitive brain and promotes its post-ischemic expression in females, resulting in inhibition of neuronal death. We, therefore, characterized regional and cellular expression ofbcl-2 mRNA and protein after middle cerebral artery (MCA) occlusion in rats of both sexes and compared these findings with those of ovariectomized females with and without estrogen replacement. Preliminary data from these experiments have been reported previously (Alkayed et al., 1998b). To further evaluate the importance ofbcl-2 induction to neuronal salvage by estrogen, transgenic mice overexpressing bcl-2 under the control of neuron-specific enolase (NSE) promoter were also treated with MCA occlusion. We hypothesized that if estrogen acts through neuronalbcl-2 induction to inhibit neuronal death and reduce stroke damage, then genetic overexpression of this protein in neurons would protect against the deleterious effect of ovariectomy on ischemic vulnerability.
MATERIALS AND METHODS
This study was conducted in accordance with the National Institutes of Health guidelines for care and use of animals in research, and the protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University.
Experimental groups. Analysis of bcl-2 expression after stroke was conducted on four groups of sexually mature, young adult (12–14 weeks old; Harlan Sprague Dawley, Indianapolis, IN) Wistar rats: male, intact female, ovariectomized female, and estrogen-supplemented ovariectomized female rats (n = 13 rats per group). The effect of overexpression of bcl-2was examined in male and female transgenic mice overexpressingbcl-2 under the control of NSE promoter and their genetically matched C57BL/6 wild-type strain (Charles River Laboratories, Wilmington, MA). The identity of the transgenic mice was verified using dot blot with SV40 polyadenylation probe, which was part of the transgenic construct. The transgenic mice were generated by injection of the construct to C57BL/6 fertilized eggs (Farlie et al., 1995; Bernard et al., 1997). Experimental mouse groups were male, intact female, and ovariectomized female wild-type and transgenic mice (n = 8–11 per group; two groups of intact females where compared with males and ovariectomized females separately).
Ovariectomy and estrogen replacement. Ovariectomy in rats and mice was performed 1–2 weeks before the experiment, and estrogen was administered to rats via 21-d-release subcutaneous pellets containing 25 μg of 17β-estradiol (Innovative Research of America, Toledo, OH), as previously described (Rusa et al., 1999; Alkayed et al., 2000). Plasma 17β-estradiol was measured using a commercially available radioimmunoasay kit (Coat-A-Count Estradiol; Diagnostic Products, Los Angeles, CA).
Experimental stroke in rat. MCA occlusion was performed in spontaneously breathing rats under halothane anesthesia (1% in O2-enriched air via mask), as previously described (Alkayed et al., 1998a, 2000). Rats were instrumented with a femoral artery catheter for monitoring arterial blood pressure and gases. Body core and head temperatures were monitored using rectal and temporalis muscle thermoprobes. Ischemia (2 hr) was induced in the territory of the right MCA using the intraluminal filament occlusion technique. Adequacy of vascular occlusion and reperfusion was assessed by monitoring cerebral cortical perfusion over the ipsilateral parietal cortex by laser-Doppler flowmetry (Alkayed et al., 1998a, 2000). After 22 hr of recovery, the rat was deeply anesthetized (3% halothane), and a final blood sample was taken from the heart for measurement of plasma estrogen. The rat was either transcardially perfused to fix the brain or decapitated without previous fixation. Fixed brains were sectioned and used for either immunohistochemical analysis (n = 4 rats per group) of Bcl-2 expression or histological examination and infarct size measurement in adjacent sections after staining with cresyl violet (CV). Unperfused fresh brains were removed quickly and frozen and were either sectioned on a cryostat for in situ hybridization (ISH) (n= 4 rats per group) or processed as described below for simultaneous measurement of bcl-2 mRNA and protein using RNase protection assay and Western blotting (n = 5 rats per group).
Immunohistochemistry. The brains were perfusion-fixed with 4% paraformaldehyde in 0.1 m PBS, pH 7.4, post-fixed for 30 min, and transferred to 30% sucrose in phosphate buffer overnight at 4°C. The brains were cut at 40 μm on a sliding microtome. Sections were treated with 5% hydrogen peroxide, permeabilized with 0.4% Triton X-100, blocked with 10% normal swine serum and 2% bovine serum albumin, then incubated for 48 hr at 4°C with a mouse monoclonal antibody against human Bcl-2 (1:100, clone 124; Dako, Carpinteria, CA). This was followed by a 60 min incubation at room temperature with a biotinylated F(ab′)2 fragment of affinity-purified rabbit anti-mouse IgG (1:500; Dako). The bound antibody was visualized using avidin–biotin–horseradish peroxidase (Vector Elite kit; Vector Laboratories, Burlingame, CA) with diaminobenzidene. Adjacent slices were processed simultaneously as negative controls and underwent the same procedure, except for omission of the primary antibody.
Determination of stroke volume in rats. CV-stained coronal sections were photographed using a digital camera, and images were imported to a computer and analyzed using image analysis software (Inquiry; Loats Associates, Westminster, MD). Areas of pallor were traced in selected coronal slices and integrated along the rostral–caudal axis to calculate infarct volume (expressed as a percentage of ipsilateral hemisphere).
In situ hybridization. A synthetic oligonucleotide corresponding to the rat bcl-2 sequence 5′GATC CAGG TGTG CAGA TGCC GGTT CAGG TACT CAGT CATC-3 was used as a probe (Chen et al., 1997). The probe was 3′ end-labeled using terminal deoxynucleotidyl transferase and [35S]deoxyadenosine 5′-(α-thio)triphosphate to a specific activity of 5–15 × 108 cpm/μg after centrifugation through G-25 columns to remove unincorporated nucleotides. Adjacent brain slices were processed simultaneously and probed with labeled sense and unlabeled antisense probes. Coronal brain sections (20 μm) were fixed in 4% formaldehyde and treated with 0.25% acetic anhydride in 0.1m triethanolamine. Then, the slides were dehydrated and hybridized overnight at 37°C with 1–3 × 106 cpm of labeled probe in 50% formamide, 4× SSC, 1× Denhardt's solution, 10% dextran sulfate, 100 mm dithiothreitol, and 500 μg/ml salmon sperm DNA. Slides representing similar sections of control and experimental groups were serially washed (formamide, then SSC) and exposed for 21 d to Amersham β-Max film (Amersham Pharmacia Biotech, Arlington Heights, IL). Cellular localization was determined after 6 week exposure to Kodak NTB 3 liquid emulsion developed in D19. The brain slices were counter-stained with thionin.
Tissue processing for quantitative analysis of bcl-2 expression. Frozen brains were first sectioned at −20°C into seven 2 mm thick coronal slabs, which were mounted then on a cryostat for further sectioning. Representative 20 μm thin sections were stained with cresyl violet. The remaining portions (1.5 mm) of slices 2, 3, 4, and 5, which encompass the MCA territory (between +2 and −6 mm relative to bregma) were used for bcl-2 RNase protection assay and Western blotting. Slices 2 and 4 were analyzed for regional expression of bcl-2 mRNA by extracting micropunches from eight discrete regions (medial and dorsolateral somatosensory cortex and medial and lateral striatum at the level of the caudate nucleus on ipsilateral and contralateral hemispheres) using a sterile stainless-steel cannula with 1 mm internal diameter (Strauss and Jacobowitz, 1993). Punches representing the same region were pooled from five animals within each group and immediately lysed in appropriate buffer for RNase protection assay. Analysis of Bcl-2 protein expression could not be performed using the micropunch technique because preliminary experiments showed that the amount of Bcl-2 protein in micropunches was below the detection level of Western blots. Therefore, slices 3 and 5 were divided across the midline for analysis of Bcl-2 protein expression in ischemic and contralateral hemispheres.
RNase protection assay. Radio-labeled bcl-2antisense riboprobe was transcribed from a 272 bp rat bcl-2cDNA fragment (Sato et al., 1994) cloned into plasmid vector (Bluescript) under the control of T3 bacteriophage polymerase promoter to a specific activity of ∼108 cpm/μg. RNase protection was performed directly in tissue lysates (Strauss et al., 1993). Briefly, tissue punches were immediately homogenized in a commercial lysis solution (Ambion, Austin, TX) and incubated with excess bcl-2 and β-actin probes overnight. Unhybridized probes were removed by digestion with ribonuclease. Hybridized probes were visualized by autoradiography after denaturing PAGE. Images of autoradiographic bands were digitized, and optical densities of bcl-2 bands were normalized to correspondingβ-actin.
Western blotting. Fresh frozen samples were homogenized in 150 mm NaCl, 20 mm Tris, 100 mm EDTA, a mixture of protease inhibitors (2 μg/ml aprotinin, 5 μg/ml leupeptin, 2 μg/ml pepstatin with 500 μm phenylmethylsulphonylfluoride), and 1% Triton X-100. The homogenate was centrifuged at 10,000 ×g for 10 min at 4°C, and the supernatant was centrifuged further at 100,000 × g for 60 min at 4°C. The protein concentration was measured by BCA protein assay (Pierce, Rockford, IL). Then, samples of protein (200 μg) were boiled in Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min, electrophoresed on 12% SDS-polyacrylamide gels, and blotted onto polyvinylidene difluoride membrane (Bio-Rad). Blots were blocked with 5% nonfat dry milk in saline for 2 hr at room temperature and then incubated with rabbit anti-rat Bcl-2 polyclonal antibody (1:2500; overnight at 4°C; PharMingen, San Diego, CA) followed by incubation with biotinylated goat-anti-rabbit IgG and horseradish peroxidase (1:5000; 1 hr at room temperature; Kirkegaard & Perry, Gaithersburg, MD). Blots were incubated with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ) for 1 min, and signals were visualized on Hyperfilm ECL (Amersham Pharmacia Biotech) after exposure for 0.5–3.0 min. Blots and gels were stained then with Coumassie blue to estimate transfer efficiency. The amount of transferred protein was calculated for each lane (percentage of loaded amount, based on transfer ratio for each lane). To account for differences in tissue survival between experimental groups, the optical density of the Bcl-2 band was divided by the area of infarction in adjacent cresyl violet-stained sections as an estimate of the size of the penumbra. Mouse myeloblast cell lysate expressing high levels of bcl-2 (PharMingen) was included as positive control.
Experimental stroke in mice. Reversible MCA occlusion in transgenic mice and their wild-type controls was also achieved by the intraluminal filament technique (Sampei et al., 2000; M. Sawada et al., 2000). Briefly, adult (18–28 gm, 3 months of age) wild-type (C57BL/6) or transgenic mice (NSE/bcl-2 overexpressing mice) (Bernard et al., 1997) were anesthetized with halothane in O2-enriched air by face mask. Rectal and temporalis muscle temperatures were controlled at 37 ± 0.5°C throughout the experiment with heating lamps and water pads. Ischemia was induced; then the mice were awakened and evaluated for neurological deficit at 60 min. Mice with clear neurological deficits were reanesthetized for removal of the suture 90 min after onset of occlusion and were allowed to survive for 22 hr. At the end of the experiment, the mice were anesthetized and decapitated, and a final blood sample was taken from trunk blood. The brain was removed, sliced, and analyzed for infarct volume as described below. Laser-Doppler perfusion, arterial blood pressure, and blood gases were measured in separate groups of mice (Sampei et al., 2000; M. Sawada et al., 2000).
Determination of stroke volume in mice. The mouse brain was removed and sectioned into five 2-mm-thick coronal sections. Slices were incubated in 2,3,5-triphenyltetrazolium chloride (TTC) (Sampei et al., 2000; M. Sawada et al., 2000) for 10 min on each side at 37°C, fixed in 10% formalin for 24 hr, and then photographed. Images were analyzed using image analysis software (Inquiry, Loats). Infarct size was expressed as a percentage of the contralateral hemisphere after correcting for edema (M. Sawada et al., 2000).
Statistical analysis. Messenger RNA expression was estimated by RNase protection assay from the optical density (OD) ofbcl-2 mRNA band after normalization to the OD of β-actin band within the same lane. Each value represents a pooled mean of 10 micropunches extracted from the same region of five animals. Because it was not possible to detect Bcl-2 protein signal from micropunches, Western blot analysis was performed on ischemic and contralateral hemispheres of individual animals, and values are presented therefore as mean ± SEM. Differences in infarct volumes and densitometric measurements of Western blots were analyzed with one-way ANOVA and post hoc Newman–Keuls test. Laser-Doppler perfusion and physiological measurements were subjected to two-way ANOVA. The criterion for statistical significance was p < 0.05. Except for RNase protection assay, all values are reported as mean ± SEM.
RESULTS
Estrogen treatment rescues infarct size in ovariectomized females
Ischemia was induced in mice and rats as described in Materials and Methods. The physiological variables during ischemia were kept within physiological range in all treatment groups (Table1). Rectal and temporalis muscle temperatures were maintained at 37 ± 0.5°C throughout ischemia and early reperfusion. Plasma 17β-estradiol was higher in female rats (5 ± 1 pg/ml; n = 8) and ovariectomized females treated with estrogen (6 ± 3 pg/ml; n = 6), compared with males (1 ± 1 pg/ml; n = 4) and untreated ovariectomized females (0 ± 0 pg/ml; n= 6). In wild-type female mice, plasma estrogen was reduced by ovariectomy from 15 ± 1 pg/ml (n = 9) to 7 ± 2 pg/ml (n = 11) and in female transgenic mice from 11 ± 2 pg/ml (n = 21) to 4 ± 1 pg/ml (n = 10). Laser-Doppler cortical perfusion (LDP) was reduced to <45% of preocclusion baseline in rats and <20% in mice. No differences in LDP were observed between any of the rat (n = 5 per group; p > 0.05) or mouse (n = 4 per group; p > 0.05) groups.
Table 1.
Physiological variables during MCA occlusion in rats and mice
| Species | Group (n) | MAP (mmHg) | pH | PaCO2 (mmHg) | PaO2 (mmHg) | Hb (gm/dl) | Glucose (gm/dl) |
|---|---|---|---|---|---|---|---|
| Rat | M (9) | 102 ± 3 | 7.42 ± 0.01 | 43 ± 2 | 138 ± 12 | 12.5 ± 0.5 | 100 ± 9 |
| F (9) | 103 ± 4 | 7.43 ± 0.03 | 40 ± 3 | 142 ± 15 | 13.3 ± 0.7 | 87 ± 9 | |
| O (9) | 103 ± 4 | 7.42 ± 0.02 | 44 ± 4 | 161 ± 18 | 13.3 ± 0.5 | 138 ± 23 | |
| E (9) | 101 ± 3 | 7.43 ± 0.01 | 43 ± 2 | 160 ± 22 | 13.0 ± 0.3 | 99 ± 14 | |
| Mouse | F/WT (4) | 79 ± 1 | 7.34 ± 0.02 | 43 ± 3 | 140 ± 7 | ||
| O/WT (4) | 82 ± 2 | 7.35 ± 0.00 | 47 ± 1 | 139 ± 3 | |||
| F/BCL (4) | 78 ± 4 | 7.36 ± 0.02 | 40 ± 3 | 140 ± 10 | |||
| O/BCL (4) | 79 ± 4 | 7.26 ± 0.03 | 43 ± 2 | 136 ± 1 |
Values are mean ± SEM. M, Male; F, female; O, ovariectomized; E, estrogen-treated O; WT, wild-type mice; BCL, bcl-2 overexpressing mice; n, number of animals; MAP, mean arterial blood pressure; Hb, plasma hemoglobin concentration.
Estrogen replacement in ovariectomized rats resulted in the reduction in infarct size after MCA occlusion (Fig.1). As previously reported in this model (Rusa et al., 1999), estrogen replacement reduced infarct from 61 ± 6% in ovariectomized females (n = 4) to 37 ± 16% (n = 4) in estrogen-supplemented ovariectomized females. Figure 1 also demonstrates the location of brain areas sampled for regional analysis of bcl-2 mRNA expression using RNase protection assay.
Fig. 1.
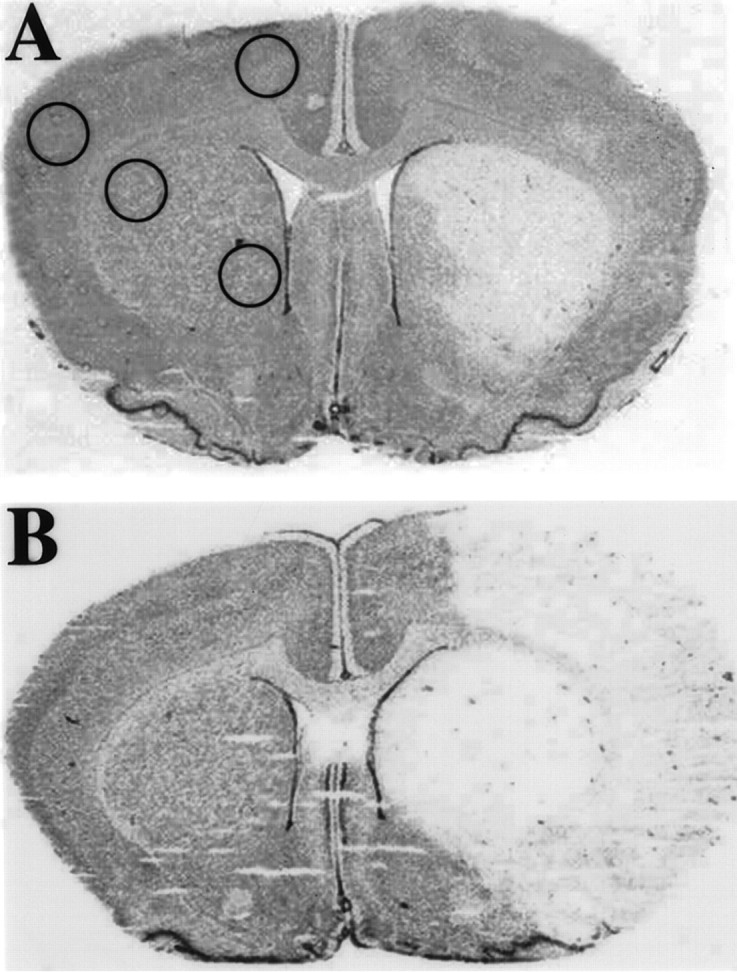
Cresyl violet-stained coronal brain sections from estrogen-treated (A) and untreated (B) ovariectomized female rats after stroke. The area of pallor delineates the “core” of cerebral infarction, and the area immediately surrounding that core represents the “penumbra” of ischemic injury. Estrogen treatment resulted in 40% reduction in cerebral infarct (n = 4 per group).Circles delineate areas from which tissue micropunches were extracted for bcl-2 mRNA quantification using RNase protection assay.
Bcl-2 expression after stroke in rats
We used in situ hybridization to study bcl-2mRNA expression in brain slices 22 hr after MCA occlusion (Fig.2). Bcl-2 mRNA expression was undetectable in the uninjured hemisphere and in the core of ischemic injury. In contrast, bcl-2 mRNA expression was highly induced in cells within the ischemic penumbra. A similar pattern ofbcl-2 mRNA expression was evident in all experimental groups (n = 4 per group). Bcl-2 mRNA levels were then quantified in selected areas (as shown in Fig. 1) within the peri-infarct region using RNase protection assay. Induction ofbcl-2 expression in females and estrogen-treated ovariectomized females was apparent in dorsolateral cerebral cortex and lateral striatum of the ischemic hemisphere (Fig.3). The level of bcl-2transcripts in post-ischemic lateral striatum was six times higher in females compared with males (Fig. 3A; n = 5 pooled animals). However, the amount of bcl-2 mRNA in ovariectomized females was reduced to a level similar to that of males. Estrogen replacement resulted in a twofold increase in bcl-2mRNA expression after stroke. Similar to the expression pattern observed in lateral striatum, bcl-2 expression was higher in the cortex of females than that in males (50%; n = 5 pooled animals) (Fig. 3B). The difference in expression disappeared after ovariectomy (n = 5 pooled animals) and was restored by estrogen replacement (n = 5 pooled animals).
Fig. 2.
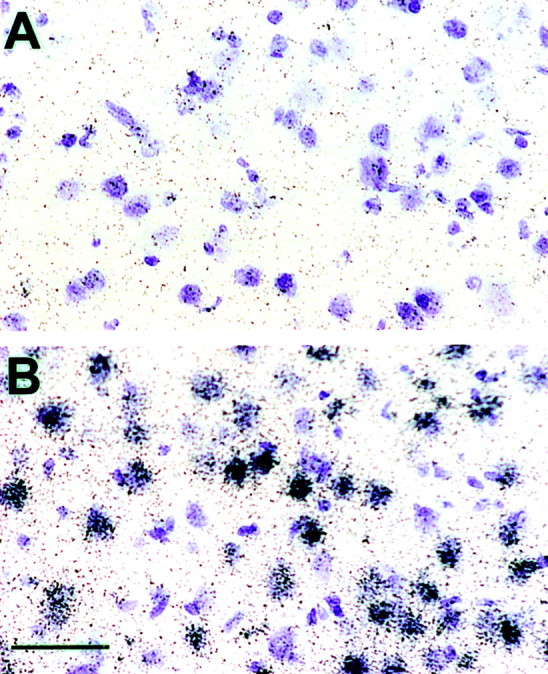
Bcl-2 in situ hybridization of female rat brain after stroke. Rat brains (n = 4 in each of 4 groups) were frozen 22 hr after 2 hr MCA.Bcl-2 mRNA is visualized as dark grainsover thionin-stained cells (purple). Labeled cells are abundant in the peri-infarct region (B), but not in uninjured (contralateral) hemisphere (A). Control sections hybridized with radiolabeled sense and with excess unlabeled antisense probes were negative for bcl-2 mRNA signal. Photomicrographs were taken at 100× magnification. Scale bar, 50 μm.
Fig. 3.

Analysis of bcl-2 mRNA expression in male (M), intact female (F), ovariectomized female (O), and estrogen-treated O (E) rats at 22 hr after 2 hr MCA occlusion in rat. RNase protection assay was performed directly on micropunches that were extracted from selected areas within the striatum (A) and the cerebral cortex (B) as defined in Figure 1. Each value represents the mean of 10 pooled micropunches (∼25–30 mg) that were extracted from the same region of five animals per group. Bcl-2mRNA expression was higher in F and Ethan in M and O in the cerebral cortex and striatum.
The distribution of Bcl-2 protein expression mirrored the distribution of bcl-2 mRNA (Fig. 4). No expression of Bcl-2 protein could be detected in the uninjured contralateral hemisphere (Fig. 4C). In agreement with the ISH results, Bcl-2 expression was highly induced in the peri-infarct, or penumbral, region (Fig. 4A,B). Microscopic examination revealed that Bcl-2 was found almost exclusively in neurons, restricted to the peri-nuclear cytoplasm and neuronal processes and absent in white matter (Fig.4B). Bcl-2 protein expression was increased in the ischemic hemisphere in all experimental groups (Fig.5A). In the contralateral hemisphere, Bcl-2 expression was undetectable in males (M) and ovariectomized females (O), but was upregulated in intact females (F) and estrogen-treated castrates (E). In agreement with the RNase protection assay results, post-ischemic expression of Bcl-2 protein in the ischemic hemisphere was >60% higher in females than males (Fig.5B; n = 5 each). Ovariectomy reduced Bcl-2 expression in females to the level seen in males, and estrogen treatment increased Bcl-2 protein expression in ovariectomized animals by almost three-fold (n = 5 each).
Fig. 4.

Immunohistochemical analysis of Bcl-2 expression at 22 hr after 2 hr MCA occlusion in the rat. A is a 10× magnification of a segment of a cresyl violet-stained coronal brain section at the level of the dorsal hippocampus. The area within the cerebral cortex in A with staining hypointensity, or “pallor,” represents tissue infarction, and thesquare at the edge of the infarct depicts the location from which the photomicrographs in B andD were taken. B and Drepresent the peri-infarct region within the cerebral cortex in the presence (B) and absence (D) of the primary antibody. Crepresents the corresponding area in the contralateral, uninjured hemisphere in the presence of the primary antibody. Bcl-2 immunoreactivity was detected in cell bodies and processes of large pyramidal neurons within the peri-infarct region (B). No immunoreactivity to Bcl-2 was apparent in the uninjured hemisphere (C). Background staining in D is caused by endogenous peroxidase activity in ischemic tissue. Scale bar, 50 μm.
Fig. 5.

Expression of Bcl-2 protein at 22 hr after 2 hr MCA occlusion in rat. A, Western blot analysis of Bcl-2 expression in contralateral (C) and ischemic (I) hemispheres in male (lanes 2C and 2I, respectively), intact female (3C, 3I), ovariectomized female (4C, 4I), and estrogen-supplemented ovariectomized female rats (5C,5I). Bcl-2 protein was identified as a 26 kDa protein in the presence of mouse myeloblast cell lysate expressing a high level of Bcl-2 as positive control (lane 1). Bcl-2 protein was induced by ischemia in all groups. Estrogen treatment and female sex were associated with increased Bcl-2 protein expression in ischemic and contralateral hemispheres. B, Western blot analysis of Bcl-2 expression within the ischemic hemisphere after stroke in male (M; n = 5), intact female (F; n = 5), ovariectomized female (O; n = 5), and estrogen-supplemented O (E;n = 5) rats. Bcl-2 expression was estimated from the optical density (OD) of bcl-2 band after normalization to the amount of transferred protein (micrograms) and the area of pallor (square millimeters) in adjacent sections after staining with cresyl violet. * denotes a statistically significant difference from both M and O(p < 0.05, ANOVA).
Effect of bcl-2 transgene in mice
The effect of bcl-2 overexpression on infarct size was studied in transgenic mice overexpressing bcl-2 in their neurons. Male mice overexpressing bcl-2 were markedly protected from ischemic injury compared with wild-type males (>50% reduction in infarct size from 46 ± 6% to 23 ± 5% of ipsilateral hemisphere; p < 0.05; n = 8 per group). In contrast to males, female transgenic mice were not protected against ischemic brain injury, compared with wild-type females (infarct size of 23 ± 5% vs 28 ± 6%, respectively; p > 0.05; n = 8 per group). In agreement with our previous finding in the rat, infarct size was larger in wild-type males versus females (46 ± 6% vs 28 ± 6%; p < 0.05; n = 8 per group). No gender difference, however, was observed in bcl-2 transgenic mice (23 ± 5% in males vs 23 ± 5% in females;p > 0.05; n = 8 per group) (Fig.6). Ovariectomy increased infarct size after focal cerebral ischemia in wild-type females by >35% (n = 10 per group; p < 0.05). However, female mice overexpressing Bcl-2 were resistant to the deleterious effect of ovariectomy, because infarct size was not different between intact and ovariectomized female transgenic mice (n = 10–11 per group; p = 0.5) (Fig.7).
Fig. 6.

Comparison between the infarct size of male and female wild-type (WT) mice and transgenic mice overexpressing Bcl-2 in neurons (BCL-2) 22 hr after 90 min MCA occlusion. Cerebral infarct, identified in coronal brain sections by TTC staining, was smaller in male but not female transgenic mice compared with WT mice. Values are mean ± SEM of eight animals. * denotes statistically significant difference from WT males.
Fig. 7.

Comparison between the infarct size of intact (F) and ovariectomized (O) wild-type mice (WT) and mice overexpressing Bcl-2 (BCL2) 22 hr after 90 min MCA occlusion. Infarct size was 35% larger in ovariectomized WT female mice compared with intact females but was not different between ovariectomized and intact BCL2 females. Values are mean ± SEM of 10–11 animals. * denotes statistically significant difference from other groups (p < 0.05; ANOVA).
To determine whether the improvement in stroke damage observed in bcl-2 male mice could be explained by differences in intra-ischemic perfusion, we measured regional cerebral blood flow using quantitative [14C]iodoantipyrine autoradiography, as previously described (Sampei et al., 2000; M.Sawada et al. 2000). Intraocclusion cortical or striatal blood flow rates were not higher in bcl-2 versus WT mice (data not shown). Therefore, it is unlikely that differences in vascular anatomy or enhanced intra-ischemic vasodilation could account for the protection observed in male bcl-2 overexpressing mice.
DISCUSSION
We have demonstrated the following in this study: (1) Bcl-2 expression is selectively induced in neurons after stroke within peri-infarct regions; (2) females and estrogen-treated castrates exhibit increased levels of bcl-2 mRNA and protein compared with estrogen-deficient animals, which is accompanied by smaller infarcts; (3) Bcl-2 overexpression reduces infarct size in male animals, but provides no additional protection in females; and (4) neuronal overexpression of Bcl-2 prevents the increase in vulnerability to cerebral ischemia associated with ovariectomy. These findings suggest that bcl-2 functions downstream of estrogen in protecting neurons from ischemic insult. We concluded that estrogen rescues neurons after focal cerebral ischemia by increasing the level of Bcl-2 that ordinarily occurs in potentially salvageable, penumbral tissue.
The protective effect of estrogen in ischemic stroke has received much attention recently (Green and Simpkins, 2000; Hurn and Macrae, 2000;Roof and Hall, 2000), but the mechanism by which it protects from cell death has remained elusive. Although estrogen is vasoactive in the cerebral circulation, vascular mechanisms cannot fully account for the neuroprotective properties of the hormone. Quantification of regional cerebral blood flow during ischemia revealed no difference in cerebral perfusion between estrogen-replaced and estrogen-deficient animals, despite steroid-induced reduction of ischemic damage (Rusa et al., 1999; Alkayed et al., 2000). Estrogen possesses multiple cytoprotective properties that could preserve neurons undergoing ischemic stress. Based on published work of others that linked estrogen to Bcl-2 in non-neural tissue, we tested the hypothesis that one important mechanism for the neuroprotection of estrogen in cerebral ischemia is the induction of post-ischemic Bcl-2 expression. Our finding that bcl-2 mRNA and Bcl-2 protein in uninjured, contralateral hemisphere were almost undetectable but were highly increased in peri-infarct neurons, is consistent with numerous previous reports (Castren et al., 1994; Chen et al., 1995; Pezzella and Gatter, 1995; Honkaniemi et al., 1996; Merry and Korsmeyer, 1997). Although not the focus of this study, basal levels of Bcl-2 expression appeared to be gender- and estrogen-dependent. In males and ovariectomized females, basal levels of Bcl-2 were quite low as compared with that of intact females and hormone-replaced ovariectomized females. This observation is consistent with a previous report of estrogen-dependent Bcl-2 expression in normal brain (Garcia-Segura et al., 1998).
We have observed that Bcl-2 was expressed in the cytoplasm surrounding the nuclei and in neuronal processes. Bcl-2 expression in the peri-infarct region may represent an active survival mechanism for neuron (Isenmann et al., 1998). Bcl-2 mRNA was quantified by RNase protection assay in ischemic cortex and striatum, and these findings were confirmed at the protein level by Western blot analysis. Our observation that estrogen increases bcl-2 mRNA levels after ischemia is in contrast to a single report indicating thatbcl-2 mRNA level did not increase after stroke or with estrogen treatment (Dubal et al., 1999). Instead, these authors found that ovariectomy was associated with post-ischemic reduction inbcl-2 mRNA, which could be prevented by estrogen. The discrepancy between the two studies may be related to differences in regional sampling (dorsolateral vs dorsomedial cerebral cortex), ischemic model (permanent vs transient ischemia), or analytical technique (quantitative RNase protection assay vs semi-quantitative reverse transcription-PCR). Furthermore, we have shown that the levels of both bcl-2 mRNA and Bcl-2 protein were increased after ischemia in intact females as well as estrogen-treated female castrates.
To further explore the relationship between estrogen-induced Bcl-2 expression and its associated reduction in early neuronal loss and infarct, we compared ischemic outcome in wild-type mice and transgenic mice overexpressing neuronal Bcl-2. Neuronal populations in these mice are resistant to developmental cell death (Bernard et al., 1997) and to cell death induced by axotomy, neurotrophic factor deprivation (Farlie et al., 1995), and a variety of cytotoxic agents (Offen et al., 1998;Heaton et al., 1999). We tested the hypothesis that, in contrast to the wild type, female Bcl-2 overexpressors are unaffected by loss of estrogen after ovariectomy. Specifically, we tested the hypothesis that ovariectomy exacerbates ischemic injury in wild-type but not in Bcl-2 transgenic mice. Ovariectomized wild-type mice sustained larger infarcts compared with intact females, which is in agreement with our earlier reports of exacerbated ischemic injury in female rats after ovariectomy or after loss of natural estrogens during reproductive senescence (Alkayed et al., 1998, 2000). In contrast, ovariectomized and intact female Bcl-2 transgenic mice sustained infarcts of similar size, suggesting that overexpression of Bcl-2 prevents the increase in ischemic vulnerability that we observed with ovariectomy in the wild-type females. Moreover, male mice were markedly protected by Bcl-2 overexpression, which is in agreement with some reports (Martinou et al., 1994), but in contrast to others (Wiessner et al., 1999; De Bilbao et al., 2000) using male transgenic mice. The discrepancies could be attributable to differences in animal models (transient versus permanent ischemia), the genetic background and/or differences in the levels, distribution (regional and cellular), and timing (prenatal vs postnatal) of bcl-2 expression in different transgenic lines. In contrast to males, there was no difference in our study between wild-type and transgenic female mice in infarct size after stroke, suggesting that endogenous estrogens and Bcl-2 act in series. This is a unique observation in itself because genetic manipulation of anti-ischemic molecules is usually assumed to produce the same result regardless of sex. The observation could mean that Bcl-2-mediated mechanisms of neuroprotection selectively benefit male brain, which seems unlikely. Alternatively, the effects of Bcl-2 overexpression may not be obvious in female brain because estrogen has already increased bcl-2, and so no further benefit can be appreciated by genetic overexpression of Bcl-2. This would be consistent with our observation that ovariectomized females do benefit from Bcl-2 in a manner similar to males. Accordingly, we interpret the data to mean that estrogen subverts the benefit that can otherwise be obtained by bcl-2 overexpression, and one major mechanism by which estrogen reduces brain injury is attributable to its ability to induce bcl-2.
Our data also indicate that the protection achieved by upregulating endogenous Bcl-2 with estrogen is limited by the location of neurons expressing Bcl-2 and the level of expression of Bcl-2 in these neurons. Bcl-2 expression in neurons outside the penumbra has no effect on infarct size because of the fact that, from the perspective of stroke, Bcl-2 is important only in injured and potentially salvageable neurons such as those within the peri-infarct region. Furthermore, increasing the level of expression within penumbral neurons beyond the level achieved by estrogen provides no further protection. This is in agreement with our previous observation that the increase in number of neurons between wild-type and NSE-bcl-2 transgenic mice is similar in the homozygous and heterozygous (Heaton et al., 1999).
The present data do not determine the molecular mechanism by which estrogen induces Bcl-2 expression, particularly if the induction of Bcl-2 is mediated via the estrogen receptors (ER). Pharmacological blockade of ER with ICI182,780 exacerbates injury after stroke in female mice (M. Sawada et al., 2000). The effect, however, does not appear to be mediated via ERα, because deletion of the gene for this receptor subtype does not exacerbate ischemic brain injury (Sampei et al., 2000). In contrast, ERβ may be important in estrogen-mediated upregulation of Bcl-2 because ERβ is present in cortical regions that bind estrogen and are protected from ischemia by estrogen (Dubal et al., 1999).
Regulation of Bcl-2 by estrogen is at least in part at the level of the transcription because both bcl-2 mRNA and protein were increased. Estrogen regulates gene transcription via binding to the estrogen response element (ERE) or the activator protein-1 (AP-1)-binding DNA motif. When activated by 17β-estradiol, ERβ can either enhance gene transcription via binding to ERE or suppress transcription by inhibiting binding of AP-1 proteins, such as c-Fos and c-Jun, to AP-1 site (H. Sawada et al., 2000). Analysis ofbcl-2 gene promoter revealed no perfect ERE consensus sequences. However, the bcl-2 gene promoter contains multiple GC-rich elements that constitute binding sites for the transcriptional factor Sp-1. Expression of bcl-2 is induced by estrogen via ER-Sp-1 protein interactions in which estrogen promotes ER/Sp-1 complex formation followed by Sp-1 binding and activation of DNA transcription (Dong et al., 1999). Recently, two consensus ERE sequences have been identified within the coding sequences of the bcl-2 gene that induce bcl-2mRNA expression in the absence of its own putative promoter (Perillo et al. 2000). Bcl-2 transcription could also be upregulated indirectly; for example, bcl-2 transcription in neuronal cells is activated by the neuroprotective Brn-3a transcription factor (Smith et al., 1998), which also interacts with estrogen receptors to regulate transcriptional activity via an ERE (Budhram-Mahadeo et al., 1998). Similarly, bcl-2 expression is induced by phosphorylated cAMP-responsive element binding protein (CREB), which is also activated in neurons by estrogen (Panickar et al., 1997;Segal and Murphy, 1998). Nonconsensus estrogen-responsive motifs within the bcl-2 promoter sequence have also been identified and shown to bind ATF-1 and CREB-1 transcription factors (Dong et al., 1999).
In conclusion, estrogen increases the level of Bcl-2 after an ischemic insult, thus using this mechanism to reduce early neuronal injury and cell loss.
Footnotes
This work was supported by National Institutes of Health Grants NS20020 and NS33668. We thank Dr. Susan K. McCune, Dr. Marie J. Hardwick, and Dr. Robert Clark for valuable help and advice.
Correspondence should be addressed to Dr. Nabil J. Alkayed, Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine, 600 N. Wolfe Street, Blalock 1404-B, Baltimore, MD 21287. E-mail: nalkayed@jhmi.edu.
REFERENCES
- 1.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998a;29:159–165. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- 2.Alkayed NJ, Sharpe LG, McCune SK, Crain BJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection is associated with higher postischemic expression of bcl-2 mRNA in rat brain. Soc Neurosci Abstr. 1998b;24:517. [Google Scholar]
- 3.Alkayed NJ, Murphy JM, Traystman RJ, Hurn PD. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31:161–168. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- 4.Antonawich FJ, Federoff HJ, Davis JN. Bcl-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp Neurol. 1999;156:130–137. doi: 10.1006/exnr.1998.7004. [DOI] [PubMed] [Google Scholar]
- 5.Bernard R, Farlie P, Bernard O. NSE-bcl-2 transgenic mice, a model system for studying neuronal death and survival. Dev Neurosci. 1997;19:79–85. doi: 10.1159/000111188. [DOI] [PubMed] [Google Scholar]
- 6.Budhram-Mahadeo V, Parker M, Latchman DS. POU transcriptional factors Brn-3a and Brn-3b interact with the estrogen receptor and differentially regulate transcriptional activity via an estrogen response element. Mol Cell Biol. 1998;18:1029–1041. doi: 10.1128/mcb.18.2.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castren E, Ohga Y, Berzaghi MP, Tzimagiorgis G, Thoenen H, Lindholm D. Bcl-2 messenger RNA is localized in neurons of the developing and adult rat brain. Neuroscience. 1994;61:165–177. doi: 10.1016/0306-4522(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Graham SH, Chan PH, Lan J, Zhou RL, Simon RP. Bcl-2 is expressed in neurons that survive focal ischemia in the rat. NeuroReport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- 9. Chen J, Graham SH, Nakayama M, Zhu RL, Jin K, Stelter RA, Simon RP. Apoptosis repressor genes Bcl-2 and Bcl-x-long are expressed in the rat brain following brain global ischemia. J Cereb Blood Flow Metab 17 1997. 2 10 [Erratum (1998) 18:931] [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Simon RP, Nagayama T, Zhu R, Loeffert JE, Watkins SC, Graham SH. Suppression of endogenous bcl-2 expression by antisense treatment exacerbates ischemic neuronal death. J Cereb Blood Flow Metab. 2000;20:1033–1039. doi: 10.1097/00004647-200007000-00002. [DOI] [PubMed] [Google Scholar]
- 11.De Bilbao F, Guarin E, Nef P, Vallet P, Giannakopoulos P, Dubois-Dauphin M. Cell death is prevented in thalamic fields but not in injured neocortical areas after permanent focal ischaemia in mice overexpressing the anti-apoptotic protein Bcl-2. Eur J Neurosci. 2000;12:921–934. doi: 10.1046/j.1460-9568.2000.00984.x. [DOI] [PubMed] [Google Scholar]
- 12.Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem. 1999;274:32099–32107. doi: 10.1074/jbc.274.45.32099. [DOI] [PubMed] [Google Scholar]
- 13.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard O. Bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA. 1995;92:4397–4401. doi: 10.1073/pnas.92.10.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates bcl-2 expression in adult brain neurons. NeuroReport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- 16.Goodman SB, Kugu K, Chen SH, Preutthipan S, Tilly KI, Tilly JL, Dharmarajan AM. Estradiol-mediated suppression of apoptosis in the rabbit corpus luteum is associated with a shift in expression of bcl-2 family members favoring cellular survival. Biol Reprod. 1998;59:820–827. doi: 10.1095/biolreprod59.4.820. [DOI] [PubMed] [Google Scholar]
- 17.Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 18.Hata R, Gillardon F, Michaelidis TM, Hossmann KA. Targeted disruption of the bcl-2 gene in mice exacerbates focal ischemic brain injury. Metab Brain Dis. 1999;14:117–124. doi: 10.1023/a:1020709814456. [DOI] [PubMed] [Google Scholar]
- 19.Heaton MB, Moore DB, Paiva M, Gibbs T, Bernard O. Bcl-2 overexpression protects the neonatal cerebellum from ethanol neurotoxicity. Brain Res. 1999;817:13–18. doi: 10.1016/s0006-8993(98)01173-1. [DOI] [PubMed] [Google Scholar]
- 20.Honkaniemi J, Massa SM, Breckinridge M, Sharp FR. Global ischemia induces apoptosis-associated genes in hippocampus. Brain Res Mol Brain Res. 1996;42:79–88. doi: 10.1016/s0169-328x(96)00121-0. [DOI] [PubMed] [Google Scholar]
- 21.Hurn PD, Macrae M. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Isenmann S, Stoll G, Schroeter M, Krajewski S, Reed JC, Bahr M. Differential regulation of Bax, Bcl-2, and Bcl-X proteins in focal cortical ischemia in the rat. Brain Pathol. 1998;8:49–62. doi: 10.1111/j.1750-3639.1998.tb00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou JC, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- 24.Linnik MD, Zahos P, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke. 1995;26:1670–1674. doi: 10.1161/01.str.26.9.1670. [DOI] [PubMed] [Google Scholar]
- 25.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 26.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 27.Offen D, Beart PM, Cheung NS, Pascoe CJ, Hochman A, Gorodin S, Melamed E, Bernard R, Bernard O. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Proc Natl Acad Sci USA. 1998;95:5789–5794. doi: 10.1073/pnas.95.10.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panickar KS, Guan G, King MA, Rajakumar G, Simpkins JW. 17beta-estradiol attenuates CREB decline in the rat hippocampus following seizure. J Neurobiol. 1997;33:961–967. [PubMed] [Google Scholar]
- 29.Patrone C, Andersson S, Korhonen L, Lindholm D. Estrogen receptor-dependent regulation of sensory neuron survival in developing dorsal root ganglion. Proc Natl Acad Sci USA. 1999;96:10905–10910. doi: 10.1073/pnas.96.19.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20:2890–2901. doi: 10.1128/mcb.20.8.2890-2901.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pezzella F, Gatter K. What is the value of bcl-2 protein detection for histopathologists? Histopathology. 1995;26:89–93. doi: 10.1111/j.1365-2559.1995.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 32.Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- 33.Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. Estrogen replacement therapy is neuroprotective in experimental stroke. Stroke. 1999;30:1665–1670. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- 34.Sabourin JC, Martin A, Baruch J, Truc JB, Gompel A, Poitout P. bcl-2 expression in normal breast tissue during the menstrual cycle. Int J Cancer. 1994;59:1–6. doi: 10.1002/ijc.2910590102. [DOI] [PubMed] [Google Scholar]
- 35.Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-α-deficient mice. Stroke. 2000;31:738–744. doi: 10.1161/01.str.31.3.738. [DOI] [PubMed] [Google Scholar]
- 36.Sato T, Irie S, Krajewski S, Reed JC. Cloning and sequencing of a cDNA encoding the rat bcl-2 protein. Gene. 1994;140:291–292. doi: 10.1016/0378-1119(94)90561-4. [DOI] [PubMed] [Google Scholar]
- 37.Sawada H, Ibi M, Kihara T, Urushitani M, Honda K, Nakanishi M, Akaike A, Shimohama S. Mechanisms of antiapoptotic effects of estrogens in nigral dopaminergic neurons. FASEB J. 2000;14:1202–1214. doi: 10.1096/fasebj.14.9.1202. [DOI] [PubMed] [Google Scholar]
- 38.Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mice. J Cereb Blood Flow Metab. 2000;20:112–118. doi: 10.1097/00004647-200001000-00015. [DOI] [PubMed] [Google Scholar]
- 39.Segal M, Murphy DD. CREB activation mediates plasticity in cultured hippocampal neurons. Neural Plast. 1998;6:1–7. doi: 10.1155/NP.1998.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimazaki K, Ishida A, Kawai N. Increase in bcl-2 oncoprotein and the tolerance to ischemia-induced neuronal death in the gerbil hippocampus. Neurosci Res. 1994;20:95–99. doi: 10.1016/0168-0102(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 41.Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen protection in NT2 neurons. NeuroReport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- 42.Smith MD, Ensor EA, Coffin RS, Boxer LM, Latchman DS. Bcl-2 transcription from the proximal P2 promoter is activated in neuronal cells by the Brn-3a POU family transcription factor. J Biol Chem. 1998;273:16715–16722. doi: 10.1074/jbc.273.27.16715. [DOI] [PubMed] [Google Scholar]
- 43.Strauss KI, Jacobowitz DM. Quantitative measurement of calretinin and β-actin mRNA in rat brain micropunches without prior isolation of RNA. Brain Res Mol Brain Res. 1993;20:229–239. doi: 10.1016/0169-328x(93)90045-q. [DOI] [PubMed] [Google Scholar]
- 44.Teixeira C, Reed JC, Pratt MAC. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 45.Wiessner C, Allegrini PR, Rupalla K, Sauer D, Oltersdorf T, McGregor AL, Bischoff S, Bottiger BW, van der Putten H. Neuron-specific transgene expression of Bcl-XL but not Bcl-2 genes reduced lesion size after permanent middle cerebral artery occlusion in mice. Neurosci Lett. 1999;268:119–122. doi: 10.1016/s0304-3940(99)00392-4. [DOI] [PubMed] [Google Scholar]