

Abstract
The neural adhesion molecule L1 mediates the axon outgrowth, adhesion, and fasciculation that are necessary for proper development of synaptic connections. L1 gene mutations are present in humans with the X-linked mental retardation syndrome CRASH (corpus callosum hypoplasia, retardation, aphasia, spastic paraplegia, hydrocephalus). Three missense mutations associated with CRASH syndrome reside in the cytoplasmic domain of L1, which contains a highly conserved binding region for the cytoskeletal protein ankyrin. In a cellular ankyrin recruitment assay that uses transfected human embryonic kidney (HEK) 293 cells, two of the pathologic mutations located within the conserved SFIGQY sequence (S1224L and Y1229H) strikingly reduced the ability of L1 to recruit 270 kDa ankyrinG protein that was tagged with green fluorescent protein (ankyrin–GFP) to the plasma membrane. In contrast, the L1 missense mutation S1194L and an L1 isoform lacking the neuron-specific sequence RSLE in the cytoplasmic domain were as effective as RSLE-containing neuronal L1 in the recruitment of ankyrin–GFP. Ankyrin binding by L1 was independent of cell–cell interactions. Receptor-mediated endocytosis of L1 regulates intracellular signal transduction, which is necessary for neurite outgrowth. In rat B35 neuroblastoma cell lines stably expressing L1 missense mutants, antibody-induced endocytosis was unaffected by S1224L or S1194L mutations but appeared to be enhanced by the Y1229H mutation. These results suggested a critical role for tyrosine residue 1229 in the regulation of L1 endocytosis. In conclusion, specific mutations within key residues of the cytoplasmic domain of L1 (Ser1224, Tyr1229) destabilize normal L1–ankyrin interactions and may influence L1 endocytosis to contribute to the mechanism of neuronal dysfunction in human X-linked mental retardation.
Keywords: neural cell adhesion molecule, L1, ankyrin, endocytosis, CRASH, mental retardation
The cell adhesion molecule L1 functions broadly in regulating the growth of axons in developing neurons and in fostering learning in the adult. L1 is localized in the plasmalemma of growth cones and processes of developing neurons, on axons of mature nonmyelinating neurons, and in glial cells (for review, see Kamiguchi et al., 1998b). L1 knock-out mice show axon guidance errors in the corticospinal tract and corpus callosum, misoriented dendrites of cortical pyramidal cells, degeneration of sensory axons, and enlarged ventricles (Cohen et al., 1997; Dahme et al., 1997;Demyanenko et al., 1999; Haney et al., 1999). Many of these features are present in humans with the X-linked mental retardation syndrome termed CRASH (corpus callosum hypoplasia, retardation, aphasia, spastic paraplegia, hydrocephalus), which results from L1 gene mutations (Fransen et al., 1997). However, the molecular basis for L1 function in normal processes of neurodevelopment and in human neurological disease is unclear.
L1 is the founding member of a subgroup of cell adhesion molecules (Holm et al., 1996). These transmembrane glycoproteins have a short cytoplasmic domain containing a conserved region capable of binding to the cytoskeletal protein ankyrin (Davis and Bennett, 1994;Brümmendorf et al., 1998). An isoform of ankyrin (440 kDa ankyrinB) colocalizes with L1 and may stabilize L1 within the axon (Scotland et al., 1998); however, the physiological role of L1–ankyrin binding is not yet defined. A correspondence in the phenotypes of ankyrinB and L1 knock-out mice in regard to dysplastic axon tracts (corpus callosum, pyramidal tracts) and ventricular dilation lends support to the significance of this interaction (Scotland et al., 1998). The cytoplasmic region of L1 also contains a neuronal-specific sequence RSLE, arising by alternative splicing, that targets L1 to axons (Kamiguchi and Lemmon, 1998). RSLE participates in receptor-mediated endocytosis of L1, which depends on clathrin and dynamin GTPase (Kamiguchi et al., 1998a). L1 endocytosis is required for the activation of a MAP kinase signaling pathway that governs neurite growth on L1 substrates (Schaefer et al., 1999; Schmid et al., 2000).
Three missense mutations associated with CRASH syndrome have been described in the cytoplasmic domain of L1 (Fig. 1). Two of these pathological missense mutations (S1224L and Y1229H) are located in the conserved SFIGQY sequence that is within a region involved in ankyrin–L1 interactions (Fransen et al., 1997; Saugier-Veber et al., 1998). The remaining missense mutation (S1194L) is in a portion of the L1 cytoplasmic region of unknown function, located midway between the RSLE sequence and the ankyrin binding domain (Fransen et al., 1994). It is not understood how these mutations perturb normal L1 properties to give rise to neurological abnormalities.
Fig. 1.

Schematic diagram of human L1 missense mutations. Human L1 is a transmembrane protein with the following domain structure: six extracellular immunoglobulin domains (Ig domains, amino acid residues 33–609) and five extracellular fibronectin type III domains (FNIII-like domains, amino acid residues 610–1113), a transmembrane domain (amino acid residues 1114–1144), and a cytoplasmic domain (amino acid residues 1145–1253). Within the cytoplasmic domain the site of the RSLEsequence (amino acid residues 1177–1180) encoded by the alternatively spliced RSLE exon is indicated. The location of each of the analyzed point mutations of L1 (R184Q, H210Q, E309K, Y784C, Y1070C, S1194L, S1224L, and Y1229H) is indicated.
We have explored the effects of the intracellular domain L1 point mutations on the interaction of L1 with ankyrin and have tested the hypothesis that ankyrin binding may stabilize L1 within the axon by decreasing the endocytosis of L1. Two of the pathological missense mutations in the L1 cytoplasmic domain (S1224L and Y1229H) reduced the ability of L1 to recruit ankyrin to the plasma membrane, whereas the remaining mutation (S1194L) had no effect on ankyrin recruitment. Interestingly, the L1 Y1229H mutant also exhibited altered regulation of L1 endocytosis. These results indicate that specific mutations of residues Ser1224 or Tyr1229 in the cytoplasmic domain disrupt L1–ankyrin binding and may influence the endocytotic machinery to regulate normal development of the nervous system.
MATERIALS AND METHODS
cDNA constructs. The cDNAs encoding human L1 with and without the RSLE exon were a gift from Dr. John Hemperly (Becton Dickinson, Research Triangle Park, NC). L1 with the RSLE exon was subcloned into the pALTER vector (Promega, Madison, WI), and point mutations in L1 corresponding to human mutations (Fig. 1) were prepared with the Altered Sites II site-directed mutagenesis kit (Promega) according to the manufacturer's specifications and were confirmed by DNA sequencing. Then the cDNAs encoding the wild-type and mutant L1 proteins were subcloned into the eukaryotic expression plasmid pcDNA3 (Invitrogen, San Diego, CA). The construct encoding a fusion protein between ankyrin and green fluorescent protein (GFP) was a gift from Dr. Vann Bennett (Duke University, NC). This construct, ankyrin–GFP, encodes the full-length rat 270 kDa ankyrinG with GFP tagged at the C terminus in the eukaryotic expression plasmid pEGFP-N1 (Clontech, Palo Alto, CA) (Zhang et al., 1998).
Human embryonic kidney (HEK) 293 cell culture and transient transfection. Cultures of transformed HEK 293 cells were maintained in DMEM supplemented with 10% fetal calf serum (HyClone, Logan, UT), gentamycin, and kanamycin in a humidified atmosphere containing 5% CO2 at 37°C. HEK 293 cells were transfected with plasmids, using Lipofectamine (Life Technologies, Gaithersburg, MD) as described (Needham and Rozengurt, 1998). Briefly, HEK 293 cells were plated at a density of 8 × 105 cells/35 mm dish and were transfected the next day. Cultures prepared for confocal microscopy analysis of ankyrin–GFP recruitment to the plasma membrane by L1 were plated at the same density into 35 mm dishes containing a poly-d-lysine-coated glass coverslip. In the standard transfection procedure, which yielded a transfection efficiency of ≈60%, the HEK 293 cells were transfected with ankyrin–GFP (0.5 μg) and the indicated L1 plasmid DNA (0.5 μg) plus 9 μl of Lipofectamine in 1 ml of serum-free OptiMEM-1 medium (Life Technologies) per 35 mm dish for 18 hr in 5% CO2. The standard transfection procedure was optimal for cell viability after transfection. In some experiments the HEK 293 cells were plated at a density of 4 × 105 cells/35 mm dish to decrease cell density and cell–cell interaction. In additional experiments an alternative procedure was used that yielded a lower transfection efficiency (≈10%). In the alternative transfection procedure the HEK 293 cells were transfected with ankyrin–GFP DNA (2.5 μg) and L1 plasmid DNA (2.5 μg), together with 10 μl of Lipofectamine per 35 mm dish. HEK 293 cultures were analyzed for recruitment of ankyrin to the plasma membrane 18 hr after the addition of the Lipofectamine transfection medium.
B35 neuroblastoma cell culture and production of stable L1-expressing B35 cell lines. The rat B35 neuroblastoma cell line was derived from the CNS (Schubert et al., 1974) and expressed little or no detectable L1 (Schmid et al., 2000). B35 cells were maintained in DMEM supplemented with 10% fetal calf serum (HyClone), gentamycin, and kanamycin in a humidified atmosphere containing 5% CO2 at 37°C. We previously characterized B35 cell lines stably expressing exogenous human L1 (Schmid et al., 2000). For the production of cell lines stably expressing L1 point mutants, B35 cells were transfected with neomycin-selectable pcDNA3 expression plasmids encoding mutant L1, using Lipofectamine (Life Technologies). Briefly, B35 neuroblastoma cells were plated in 100 mm dishes and transfected the next day when they were ∼50% confluent. The B35 cells were transfected with 10 μg of expression plasmid DNA encoding L1 point mutants and 20 μl of Lipofectamine in 6.4 ml of serum-free OptiMEM-1 medium per 100 mm dish for 12 hr in 5% CO2. At 4 d after the addition of the Lipofectamine transfection medium, the B35 cells were passaged for clonal selection into culture medium supplemented with 500 μg/ml G418 (Mediatech, Washington, DC) and cultured for an additional 10–14 d. Then 12–24 G418-resistant clones from each transfection were screened for L1 expression by Western blot analysis. Examination of these B35 cell lines for L1 detected by indirect immunofluorescence and confocal microscopy analysis showed that one or two of the characterized clones expressed the mutant L1 at the cell surface at a level equivalent to the B35 cell line expressing wild-type L1 (Fig. 5). The stable L1-expressing B35 neuroblastoma cell lines were maintained in culture medium supplemented with 250 μg/ml G418.
Fig. 5.
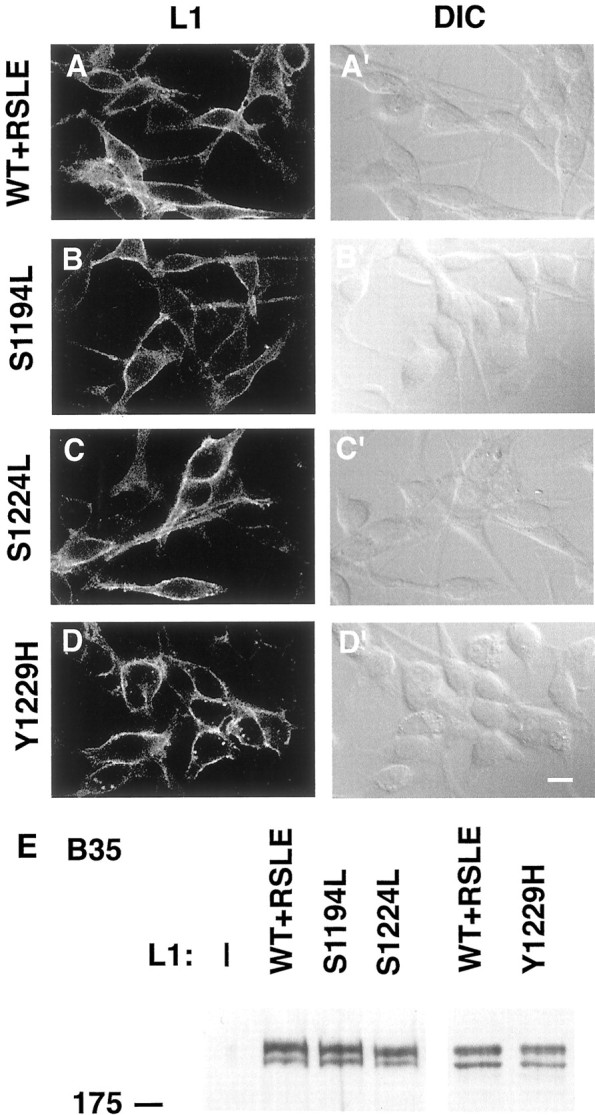
L1 wild-type and mutant constructs are expressed at equivalent levels in B35 neuroblastoma cell lines. The following stable cell lines of rat B35 neuroblastoma cells were processed for L1 indirect immunofluorescence and Western blot analysis as described in Materials and Methods: untransfected parental B35 cells (—,E); B35 cells expressing wild-type L1 (WT+RSLE, A, A′, E); or the L1 point mutants L1+S1194L (S1194L, B, B′, E), L1+S1224L (S1224L, C, C′, E), or L1+Y1229H (Y1229H, D, D′, E). A–D′, B35 cells were fixed and processed for indirect immunofluorescence staining and confocal microscopy analysis to determine relative levels of L1 expression at the cell surface. The left columnpresents confocal microscopy images of immunostained L1 (L1, A–D) and the right column presents differential interference contrast images (DIC, A′–D′). Each of thehorizontal panels (A–A′ throughD–D′) presents the same field of cells. Scale bar, 10 μm. E, B35 cells were lysed, and the aliquots were analyzed by SDS-PAGE and Western blotting with monoclonal antibodies that are specific for L1 to determine relative levels of L1 expression. The position of the 175 kDa molecular mass marker is indicated to the left of the panel.
L1 expression analysis by Western blotting. For analysis of relative levels of L1 expression the cells in 35 mm dishes were washed with buffered saline and extracted in ice-cold NP-40 lysis buffer [1% Nonidet P-40, 0.25% (w/v) sodium deoxycholate, and (in mm)50 HEPES, pH 7.4, 137 NaCl, 1 Na-EDTA, 10 NaF, 1 sodium orthovanadate, 10 p-nitrophenyl-phosphate, 1 phenylmethylsulfonyl fluoride (PMSF), and 10 β-glycero-phosphate plus 10 μg/ml leupeptin, 0.1 TIU/ml aprotinin, and 10% glycerol]. Cell lysates were clarified by centrifugation at 16,000 × g for 15 min at 4°C. The protein concentration of the clarified lysates was determined by using the bicinchoninic acid protein assay (Pierce, Rockford, IL). Clarified lysates (25 μg) in Laemmli sample buffer containing 2 mm Na-EDTA were resolved by discontinuous SDS-polyacrylamide gel electrophoresis (Laemmli, 1970), and the proteins were transferred onto nitrocellulose membranes on a semi-dry electroblot transfer. Then the membranes were blocked by incubation for 2 hr with 1% polyvinylpyrrolidone (PVP-40, Sigma), 0.05% Tween 20, and 0.02% sodium azide in TRIS-buffered saline, pH 7.4 (PVP-40 blocking buffer) (Haycock, 1993). After being blocked, the membranes were incubated with purified L1 monoclonal antibody Neuro4 at 1 μg/ml in PVP-40 blocking buffer. The monoclonal antibody Neuro4, which recognizes an extracellular domain epitope of human L1, was a generous gift from Dr. Hemperly. After being washed, the membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse IgG. After the membrane was washed, the bound immune complexes were detected by using enhanced chemiluminescence and x-ray film.
Analysis of ankyrin–GFP recruitment to the plasma membrane by wild-type and mutant L1. HEK 293 cells on glass coverslips were transfected with ankyrin–GFP and L1 expression plasmids and then were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, for 20 min; they were washed and treated with 0.1 mglycine for 5 min. After being washed, the cells were incubated with 10% normal goat serum in PBS. To label cell surface L1, we then treated the fixed cells with purified L1 monoclonal antibody Neuro4 at a concentration of 1 μg/ml. Alternatively, in some experiments an antibody directed against the C terminus of the cytoplasmic domain of L1 was used to label L1 in permeabilized transfected HEK 293 cells. In these experiments the fixed cells were permeabilized with PBS containing 0.1% Triton X-100 and 10% rabbit serum and then were treated with goat anti-L1 antibody C-20 (sc-1508, Santa Cruz Biotechnology, Santa Cruz, CA) at a concentration of 0.2 μg/ml in rabbit serum containing PBS. After incubation with the primary mouse or goat antibody against L1, the cells were washed and subjected to secondary staining with tetramethylrhodamine isothiocyanate (rhodamine)-conjugated goat anti-mouse IgG or rabbit anti-goat antibodies in PBS, respectively. The GFP/immunofluorescence experiments were recorded on a Zeiss LSM 210 laser confocal microscope at the Microscopy Service Laboratory (Dr. Robert Bagnell, Director, University of North Carolina at Chapel Hill). The Zeiss confocal microscope was equipped with a 63× Plan Apochromat oil objective, an argon laser (excitation lines 488 and 514), and differential interference contrast (DIC) optics. For each field of cells that was analyzed, DIC images were recorded, and confocal images (optical section thickness, 0.5 μm) were recorded of the rhodamine-labeled L1 by using the 514 excitation line of the laser and of the ankyrin–GFP by using the 488 excitation line of the laser. Each experiment was repeated at least three times.
Analysis of cell surface L1, antibody-induced endocytosis of L1, and localization of cytoplasmic L1 in B35 neuroblastoma cell lines.B35 neuroblastoma cell lines stably expressing wild-type L1 or L1 point mutants were plated onto poly-d-lysine-coated eight-well chamber slides (LabTek II, Nunc, Naperville, IL) at 40,000 cells per chamber 18 hr before being processed for indirect immunofluorescence. For the analysis of relative levels of cell surface L1, B35 cells were fixed and stained with the Neuro4 monoclonal antibody that recognizes an extracellular domain epitope of L1. Paraformaldehyde-fixed cells were incubated with PBS containing 10% goat serum and then were treated with purified anti-L1 Neuro4 monoclonal antibody at a concentration of 1 μg/ml. After being washed, the cells were subjected to secondary staining with fluorescein isothiocyanate (fluorescein)-conjugated goat anti-mouse IgG antibodies. As a control for the specificity of the Neuro4 mouse monoclonal IgG antibody, additional fixed cells were treated as above with the exception that purified mouse IgG was substituted for the Neuro4 monoclonal antibody.
To examine the antibody-induced endocytosis of L1 mutants in the B35 neuroblastoma cells lines, we treated living B35 cells with L1 antibody and used a procedure to label differentially the cell surface and internalized L1 antibody complexes with double-label indirect immunofluorescence and laser confocal microscopy. Purified Neuro4 monoclonal L1 antibody was added (25 μg/ml final concentration) to cultures of viable B35 cells to complex cell surface L1, and the cells were incubated in DMEM at 37° for 30 min to allow for antibody-mediated endocytosis of the cell surface L1 antibody complexes. As a control for the specificity of the Neuro4 antibody, additional live cells were treated as above with the exception that purified mouse IgG was substituted for the Neuro4 mouse monoclonal IgG antibody. To stop further endocytosis, we then cooled the cell dishes on ice. The cells were washed with DMEM at 4°C, and L1 antibody complexes remaining at the cell surface were labeled by incubation with rhodamine-conjugated goat anti-mouse IgG antibodies in DMEM for 30 min at 4°C. To block any remaining unlabeled cell surface L1 antibody complexes, we then incubated the cells with unlabeled goat anti-mouse IgG at 200 μg/ml in DMEM for 1 hr at 4°C. After an additional wash at 4°C with PBS the cells were treated with paraformaldehyde, fixing the cell surface rhodamine-labeled L1 antibody complexes, and the cells then were permeabilized with PBS containing 0.1% Triton X-100 and 10% goat serum for 1 hr. After permeabilization the internalized L1–Neuro4 antibody complexes were labeled by the addition of fluorescein isothiocyanate (fluorescein)-conjugated goat anti-mouse IgG antibodies in PBS containing 10% goat serum.
For the analysis of subcellular localization of cytoplasmic L1 in the absence of antibody-induced endocytosis, B35 cells were fixed with paraformaldehyde and permeabilized with PBS containing 0.1% Triton X-100 and 10% normal goat serum before being labeled with Neuro4 monoclonal antibody IgG antibodies. Fixed and permeabilized B35 cells were treated with purified anti-L1 Neuro4 monoclonal antibody at a concentration of 1 μg/ml and subjected to secondary staining with fluorescein-conjugated goat anti-mouse IgG antibodies. For each field of cells that was analyzed, DIC images and confocal images were recorded on the Zeiss LSM 210 laser confocal microscope. Rhodamine-labeled L1 was recorded by using the 514 excitation line, and fluorescein-labeled L1 was recorded by using the 488 excitation line of the laser. Each experiment was repeated at least three times.
RESULTS
Interaction of wild-type and mutant L1 with ankyrin
cDNAs encoding the neuronal form of L1 expressing the RSLE sequence were mutated to produce the L1 missense mutations found in patients with CRASH syndrome. These mutations included point mutations in the extracellular domain of L1 (L1+R184Q, L1+H210Q, L1+E309K, L1+Y784C, and L1+Y1070C) and point mutations in the intracellular domain of L1 (L1+S1194L, L1+S1224L, and L1+Y1229H). The alternatively spliced neuron-specific RSLE sequence (Miura et al., 1991; Takeda et al., 1996) contributes to the recognition sequence YRSL for the AP-2 adaptor protein and thus targets L1 for clathrin-mediated endocytosis (Kamiguchi et al., 1998a). cDNAs encoding wild-type L1 with and without the RSLE sequence also were used in these studies. The positions of these mutations in the human L1 transmembrane protein are shown in Figure 1 relative to the extracellular domains and the alternatively spliced RSLE sequence. Two of the human L1 cytoplasmic domain mutations, S1224L and Y1229H, are located at the N and C termini, respectively, of a highly conserved sequence, SFIGQY, which is involved in ankyrin binding (Zhang et al., 1998). The remaining pathological missense mutation (S1194L) is found in a portion of the L1 cytoplasmic region midway between the RSLE sequence and the ankyrin binding domain (Fransen et al., 1994). Although this serine is conserved in the members of the L1 subfamily of proteins (Holm et al., 1996), its possible function is not known.
To examine the interaction between wild-type and mutant L1 and ankyrin, we adopted a cellular ankyrin recruitment assay (Zhang et al., 1998), using transfected HEK 293 cells transiently expressing L1 isoforms as well as ankyrin that was tagged with GFP. No proteins cross-reactive with anti-human L1 antibodies were detected in this human cell line by Western blot (Fig. 2) nor by indirect immunofluorescence analysis (Fig.3A). Western blot analysis with the L1 monoclonal antibody Neuro4 confirmed that the exogenous wild-type L1 and L1 proteins with point mutations in the intracellular domain were expressed at equivalent levels in transfected HEK 293 cells (Fig. 2). By SDS-PAGE and Western blot analysis the wild-type and mutant L1 proteins were resolved as two bands at apparent molecular weights of ∼210 and 220 kDa. Previous reports have suggested that the higher molecular weight form of L1 is the mature glycosylated isoform predominantly expressed at the cell surface (Zisch et al., 1997;Moulding et al., 2000). In addition, indirect immunofluorescence analysis showed that the exogenous wild-type L1 and the intracellular domain mutant L1 proteins were expressed on the cell surface at approximately equivalent levels in HEK 293 cells cotransfected with a GFP-tagged 270 kDa ankyrinG protein (ankyrin–GFP) (Figs.3A–D, 4A–D).
Fig. 2.

L1 wild-type and mutant constructs are expressed at equivalent levels in HEK 293 cells. HEK 293 cells were lysed, and aliquots were analyzed by SDS-PAGE and Western blotting with monoclonal antibodies specific for L1 to determine the relative levels of L1 expression. HEK 293 cells were transfected with the ankyrin–GFP expression plasmid and with the following human L1 expression plasmids (L1): none (—); wild-type L1 with or without the RSLE exon sequence (WT+RSLE and WT-RSLE); or the point mutants L1+S1194L (S1194L), L1+S1224L (S1224L), and L1+Y1229H (Y1229H), as indicated. The position of the 175 kDa molecular mass marker is indicated to the left of the panel.
Fig. 3.
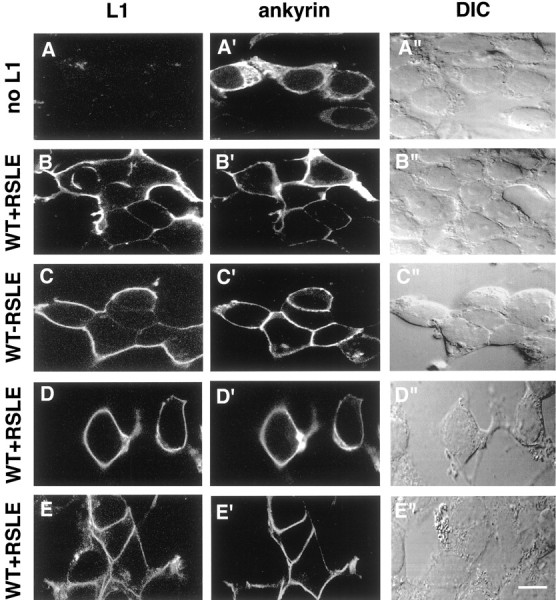
L1 recruits ankyrin to the plasma membrane independently of the L1 YRSL sequence. HEK 293 cells were cotransfected with the ankyrin–GFP expression plasmid and the following L1 expression plasmids (left labels): none (no L1, A–A"), wild-type L1 containing the RSLE exon sequence (WT+RSLE, B–B", D–D", E–E"), or wild-type L1 without the RSLE exon sequence (WT-RSLE, C–C"). At 18 hr after transfection the cells were fixed and processed for indirect immunofluorescence staining by using antibodies specific for L1 and rhodamine-labeled secondary antibodies. The cells in the horizontalpanels A–A" throughD–D" were stained with the L1 monoclonal antibody Neuro4 that recognizes an L1 extracellular domain epitope. The cells in the horizontal panel E–E" were permeabilized and stained with an L1 goat antiserum that recognizes an intracellular domain epitope of L1. The left column presents confocal microscopy images of immunostained L1 (L1, A–E), themiddle column presents confocal microscopy images of ankyrin–GFP (ankyrin, A′–E′), and the right column presents differential interference contrast images (DIC, A"–E"). Each of the horizontal panels (A–A" through E–E") presents the same field of cells. Scale bar, 10 μm.
Fig. 4.
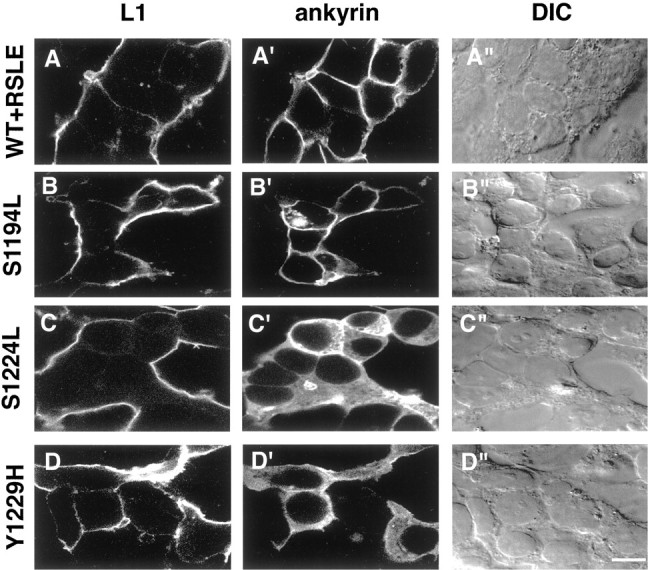
The serine residue S1224 and the tyrosine residue Y1229 in the SFIGQY sequence of L1 are crucial for the recruitment of ankyrin to the plasma membrane. HEK 293 cells were cotransfected with the ankyrin–GFP expression plasmid and the following L1 expression plasmids (left labels): wild-type L1 containing the RSLE exon sequence (WT+RSLE, A–A"), the L1 point mutants L1+S1194L (S1194L, B–B") or L1+S1224L (S1224L, C–C"), or L1+Y1229H (Y1229H, D–D"). At 18 hr after transfection the cells were fixed and processed for indirect immunofluorescence staining by using antibodies specific for L1 and rhodamine-labeled secondary antibodies. The left columnpresents confocal microscopy images of immunostained L1 (L1, A–D), the middle column presents confocal microscopy images of ankyrin–GFP (ankyrin, A′–D′), and the right column presents differential interference contrast images (DIC, A"–D"). Each of thehorizontal panels (A–A" throughD–D") presents the same field of cells. Scale bar, 10 μm.
In the cellular ankyrin recruitment assay the interaction of ankyrin–GFP with L1 was detected indirectly in HEK 293 cells by the recruitment of ankyrin–GFP to the cytoplasmic domain of L1 in the plasma membrane. When HEK 293 cells were transfected with an ankyrin–GFP expression plasmid in the absence of L1, no L1 was detected in HEK 293 cells by indirect immunofluorescence with the L1 monoclonal antibody Neuro4 (Fig. 3A), and the ankyrin–GFP remained cytoplasmic, as indicated by the diffuse intracellular distribution and nuclear exclusion of the GFP fluorescence (Fig.3A′). Previous studies had demonstrated that HEK 293 cells express endogenous spectrin and endogenous ankyrin, which both are localized predominantly at the plasma membrane (Zhang et al., 1998). In agreement with the previously reported results, endogenous spectrin is insufficient in amount to recruit exogenous ankyrin–GFP to the plasma membrane (Fig. 3A′). In contrast, when the HEK 293 cells were cotransfected with ankyrin–GFP and wild-type L1+RSLE, the ankyrin–GFP became localized to the plasma membrane (Fig.3B′). As shown in Figure 3B–D, sites on L1-expressing cells that were not in contact with other cells or that were in contact with cells not expressing L1 were labeled readily with the monoclonal antibody Neuro4, which recognizes an extracellular domain epitope of L1. However, sites of cell–cell adhesion between transfected HEK 293 cells expressing L1 were, in general, poorly labeled with this antibody. We obtained identical results by using a polyclonal rabbit antibody that was directed against the extracellular domain of L1 (data not shown). When HEK 293 cells expressing wild-type L1 were permeabilized and stained with a goat antiserum recognizing an intracellular domain epitope of L1, plasma membrane sites of apposition between L1-expressing cells were labeled as readily as plasma membrane sites not in apposition to L1-expressing cells (Fig.3E). Therefore, these results suggested that the apparent lack of localization of L1 at sites of cell–cell adhesion between L1-expressing HEK 293 cells stained with the Neuro4 antibody was an artifact caused by antibody inaccessibility to sites of L1–L1trans-interaction. When HEK 293 cells expressing wild-type L1 were stained with this intracellular domain epitope antibody, there was a high degree of colocalization of plasma membrane L1 and ankyrin–GFP, as shown in Figure 3E,E′. In conjunction, these results suggested that the cytoplasmic domain of wild-type L1 provided an effective membrane-binding site for recruitment of 270 kDa ankyrinG to the plasma membrane. Because the intracellular domain epitope L1 antibody also stained cytoplasmic L1 in permeabilized cells, the plasma membrane-localized L1 often was obscured in HEK 293 cells, which generally expressed high levels of L1 throughout the cell. Therefore, the ankyrin recruitment assay data that used the monoclonal Neuro4 L1 antibody against an extracellular domain epitope to localize cell surface L1 in L1-expressing cells is presented.
Cotransfection of wild-type L1 without the RSLE sequence, together with ankyrin–GFP, resulted in the recruitment of ankyrin–GFP to the plasma membrane in a manner that was indistinguishable from that seen in HEK 293 cells cotransfected with wild-type L1 with the RSLE sequence (Fig.3C′). This result suggested that the neuronal-specific RSLE sequence was not required for the interaction of L1 and ankyrin.
Cotransfection of L1 and ankyrin–GFP into HEK 293 cells by the standard transfection procedure resulted in a high percentage of transfected cells (≈60%), such that HEK 293 cells expressing L1 were most often in contact with other L1-expressing cells. This made it difficult to determine whether L1–L1 trans-interaction was required for effective L1-mediated recruitment of ankyrin to the plasma membrane. However, when an alternate transfection procedure with a lower transfection efficiency (≈10%) was used, most HEK 293 cells expressing L1 were isolated from other L1-expressing cells. Wild-type L1 effectively recruited ankyrin–GFP to the plasma membrane in these HEK 293 cells that were isolated from other L1-expressing cells (data not shown). These results suggested that homophilic L1trans-interaction was not required for the recruitment of ankyrin to the plasma membrane by L1 in this assay. Furthermore, when HEK 293 cells were plated at a lower cell density before transfection by the standard procedure, many of the L1-expressing cells did not contact any other cells. As shown in Figure 3D′, wild-type L1 effectively recruited ankyrin–GFP to the plasma membrane in these HEK 293 cells in the absence of interaction with other cells. These results suggested that neither heterophilic L1trans-interactions nor cell–cell adhesion was required for the recruitment of ankyrin to the plasma membrane by L1 in this assay.
When HEK 293 cells were cotransfected with ankyrin–GFP and L1 expressing point mutations in the extracellular domain (L1+R184Q, L1+H210Q, L1+E309K, L1+Y784C, and L1+Y1070C), Western blot and indirect immunofluorescence analysis suggested that approximately equivalent levels of the mutant L1 and wild-type L1 proteins were expressed in total and at the cell surface (data not shown). HEK 293 cells expressing these extracellular domain L1 point mutants efficiently recruited ankyrin–GFP to the plasma membrane whether or not they were in contact with other L1-expressing cells (data not shown). These results suggested that the recruitment of ankyrin to the plasma membrane by L1 in this assay was independent of the L1 extracellular domain interactions affected by these extracellular domain mutations associated with CRASH syndrome. Furthermore, because the mutations R184Q and H210Q previously had been shown to reduce L1–L1 trans-interactions substantially (Zhao and Siu, 1996; De Angelis et al., 1999), these results confirm the independence of L1–L1 trans-interaction and L1–ankyrin interaction in this assay.
In striking contrast to the extracellular domain mutants, the L1 cytoplasmic domain mutant S1224L failed to recruit ankyrin to the plasma membrane. When HEK 293 cells were cotransfected with ankyrin–GFP and the L1 point mutant L1+S1224L, the ankyrin–GFP remained diffusely distributed throughout the cytoplasm (Fig.4C′). Another L1 cytoplasmic domain mutant, L1+Y1229H, also failed to recruit ankyrin to the plasma membrane (Fig. 4D′). On the other hand, a third mutation of L1 in the cytoplasmic domain, L1+S1194L, effectively recruited ankyrin–GFP to the plasma membrane (Fig.4B′). These results suggest that, for optimal interaction between L1 and ankyrin, the amino acid residues Ser1224 and Tyr1229 of L1 are crucial. Thus, the L1 S1224L and Y1229H point mutations associated with the human CRASH syndrome appeared intrinsically defective in a cellular function involving ankyrin binding to the L1 cytoplasmic domain.
Endocytosis of wild-type L1 and the mutants L1+S1194L, L1+S1224L, and L1+Y1229H in B35 neuroblastoma cell lines
Rat B35 neuroblastoma is a CNS-derived cell line, which exhibits neuronal properties including membrane excitability and expression of enzymes involved in neurotransmitter metabolism (Schubert et al., 1974). B35 neuroblastoma cells were used for the analysis of L1 mutations because they do not express detectable levels of endogenous L1 and are transfected more easily than primary neurons. Furthermore, L1 was shown to undergo antibody-mediated endocytosis in B35 cells that stably express exogenous L1 encoding the RSLE exon (B35-L1+RSLE) (Schmid et al., 2000). To compare the ability of L1 mutants to undergo antibody-mediated endocytosis, we established B35 cell lines stably expressing L1 cytoplasmic domain mutations: S1194L (B35-L1+S1194L), S1224L (B35-L1+S1224L), or Y1229H (B35-L1+Y1229H). Analysis of these B35 cell lines via indirect immunofluorescence with the monoclonal antibody Neuro4 and laser confocal microscopy confirmed that the exogenous wild-type L1+RSLE and mutant L1 proteins were expressed at approximately equivalent levels at the cell surface (Fig.5A–D). Western blot analysis with the L1 monoclonal antibody Neuro4 confirmed that the exogenous wild-type L1 and L1 proteins with point mutations in the intracellular domain were expressed at equivalent levels in the stably transfected B35 cells (Fig. 5E). By SDS-PAGE and Western blot analysis the wild-type and mutant L1 proteins expressed in the B35 cells were resolved as two bands at apparent molecular weights of ∼210 and 220 kDa. No endogenous protein cross-reactive with the anti-human L1 monoclonal antibody was detected in the parental B35 cell line by indirect immunofluorescence (data not shown) nor by Western blotting (Fig. 5E).
To examine the antibody-induced endocytosis of L1 mutants in the B35 neuroblastoma cell lines, we treated live B35 cells with L1 antibody and used a procedure to label differentially the cell surface and internalized L1 antibody complexes with double-label indirect immunofluorescence and laser confocal microscopy. On treatment of living B35 cells stably expressing wild-type L1 (B35-L1+RSLE) with the L1 monoclonal antibody Neuro4, L1 antibody complexes became internalized into vesicles within the cytoplasm of the B35 cells (Fig.6A′). Additional L1 antibody complexes remained localized at the cell surface (Fig.6A). When B35-L1+S1194L, B35-L1+S1224L, or B35-L1+Y1229H cells were treated with Neuro4, L1 antibody complexes also were internalized effectively into vesicles within the cytoplasm of the B35 cells (Fig. 6B′,C′,D′, respectively), with additional L1 antibody complexes remaining at the cell surface (Fig.6B–D). When B35-L1+S1194L cells were treated with Neuro4, L1 antibody complexes were internalized in a manner that was indistinguishable from the internalization of wild-type L1 in B35-L1+RSLE cells (Fig. 6B′), suggesting that Ser1194 was not required for the endocytosis of L1. Because L1+S1224L and L1+Y1229H did not recruit ankyrin–GFP to the plasma membrane, these results suggested that L1–ankyrin interaction was not required for endocytosis. Interestingly, when B35 cells stably expressing L1+Y1229H derived from two independently isolated clones were treated to induce the antibody-mediated endocytosis of L1, the vesicles containing endocytosed L1 antibody complexes were consistently larger and brighter than those found in the B35 cell line stably expressing wild-type L1+RSLE. Because the exogenous wild-type L1+RSLE and mutant L1 proteins were expressed at approximately equivalent levels at the cell surface of the B35 cells, these results suggested that antibody-induced endocytosis was enhanced by the Y1229H mutation.
Fig. 6.
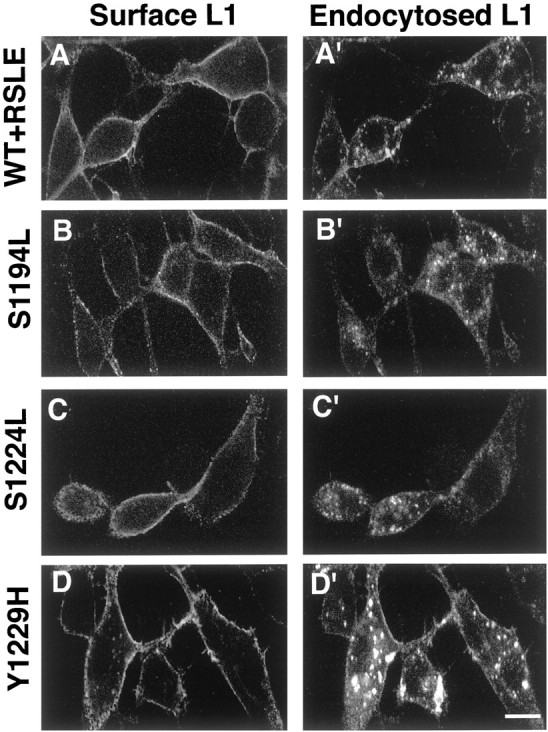
Antibody-induced endocytosis of wild-type and mutant L1 in B35 neuroblastoma cell lines. B35 cell lines stably expressing wild-type L1 (WT+RSLE, A, A′) or the L1 point mutants S1194L (B, B′),S1224L (C, C′), or Y1229H(D, D′) were treated with Neuro4 monoclonal antibodies against an extracellular epitope of L1 to induce the endocytosis of L1. The cells were processed to localize differentially the cell surface and endocytosed L1 by double immunofluorescence staining and confocal microscopy as described in Materials and Methods. Left column (Surface L1, A–D), Stable L1-transfected B35 cell lines stained for cell surface L1 with rhodamine-labeled secondary antibodies. Right column (Endocytosed L1, A′–D′), Stable L1-transfected B35 cell lines stained for internalized L1 with fluorescein-labeled secondary antibodies. Each of the horizontal panels (A, A′ throughD, D′) presents the same field of cells. Scale bar, 10 μm.
To investigate the possibility that the L1 mutant Y1229H might exhibit increased basal endocytosis in the absence of L1 antibody treatment, we fixed and permeabilized the L1-expressing B35 cell lines before treatment with the Neuro4 L1 antibody to analyze the localization of intracellular L1. Strikingly, in permeabilized B35 cells stably expressing L1+Y1229H derived from two independently isolated clones, large L1-containing vesicles were evident within the cytoplasm (Fig.7D). These vesicles appeared similar to the L1-containing vesicles internalized after the treatment of living B35 cells stably expressing L1+Y1229H with the L1 monoclonal antibody Neuro4 (Fig. 6D′). However, they were generally not as large nor as numerous as those L1-containing vesicles internalized after L1 antibody treatment of B35 cells expressing L1+Y1229H. In contrast, when permeabilized B35-L1+RSLE cells, B35-L1+S1194L cells, or B35-L1+S1224L cells were stained with Neuro4, L1 was localized diffusely within the cytoplasm of the B35 cells and was not apparent within cytoplasmic vesicles (Fig. 7A–C). Interestingly, as shown in the DIC images, the B35-L1+Y1229H cells exhibited more vesicles than the other B35-L1-expressing cell lines (Fig. 7A′–D′).
Fig. 7.
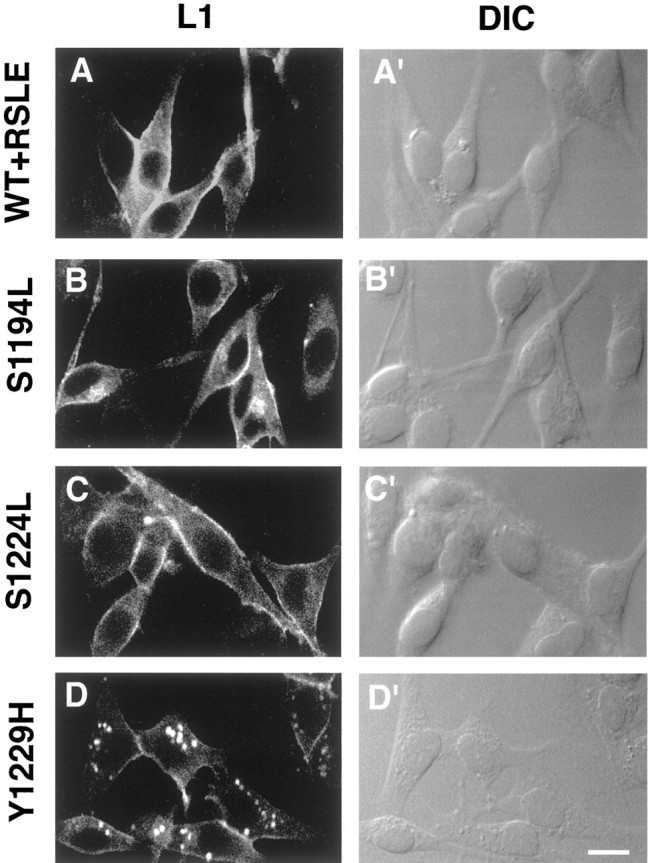
Localization of cytoplasmic wild-type and mutant L1 in B35 neuroblastoma cell lines. B35 cell lines stably expressing wild-type L1 (WT+RSLE, A, A′) or the L1 point mutantsS1194L (B, B′), S1224L(C, C′), or Y1229H (D, D′) were fixed and permeabilized before treatment with Neuro4 monoclonal antibodies and were processed for immunofluorescence staining and confocal microscopy as described in Materials and Methods. Theleft column presents confocal microscopy images of immunostained L1 (L1, A–D), and the right column presents differential interference contrast images (DIC, A′–D′). Each of the horizontal panels (A, A′ through D, D′) presents the same field of cells. Scale bar, 10 μm.
DISCUSSION
We have explored the effects of naturally occurring point mutations in the cytoplasmic domain of L1 on its ability to recruit ankyrin and undergo endocytosis. Two missense mutations located within the cytoplasmic domain of L1 (S1224L and Y1229H) strikingly reduced the ability of L1 to recruit ankyrin to the plasma membrane, whereas the remaining S1194L mutation had no effect. These results demonstrate that the L1 S1224L and Y1229H point mutations associated with the human CRASH syndrome are defective in cellular functions involving ankyrin binding to the L1 cytoplasmic domain.
L1, a member of the immunoglobulin superfamily, is the founding member of a subgroup of cell adhesion molecules including L1, CHL1, neurofascin, Nr-CAM, Ng-CAM, and Drosophila neuroglian (Holm et al., 1996). Each of these members of the L1 subfamily contains a short, highly conserved cytoplasmic region within which is an ankyrin binding site. Two of the human L1 cytoplasmic domain mutations (S1224L and Y1229H) are located at the N and C termini, respectively, of a highly conserved sequence in the cytoplasmic region, SFIGQY, which is involved in ankyrin binding (for review, see Zhang et al., 1998). Our study demonstrates for the first time that the Ser1224 residue in the SFIGQY sequence is crucial for ankyrin binding and that a serine-to-leucine change disrupts ankyrin–L1 interactions. Our study also revealed that the L1 Y1229H mutation resulted not only in the loss of ankyrin binding but also in the increase of L1 endocytosis. Loss of ankyrin binding to the L1 Y1229H mutant is in accord with studies of neurofascin (Zhang et al., 1998), which showed that an equivalent tyrosine-to-histidine mutation within the SFIGQY sequence results in loss of ankyrin binding. Because phosphorylation of the tyrosine residue of SFIGQY by an unidentified tyrosine kinase has been shown to regulate the interaction between L1 family members and ankyrin negatively (Garver et al., 1997;Tuvia et al., 1997), our result demonstrating that the L1 Ser1224 residue is crucial for ankyrin interaction raises the possibility that this serine also may be phosphorylated to regulate L1 interaction with ankyrin negatively.
In addition to the ankyrin binding domain, the cytoplasmic region of L1 contains a neuronal-specific alternatively spliced sequence (Y)-RSLE, which is homologous to the YRRL sequence that is a favored motif for recognition by the clathrin adaptor AP-2 (Boll et al., 1996). Our study demonstrates that L1 recruits ankyrin to the plasma membrane independently of the RSLE sequence. Because the deletion of the RSLE sequence previously was shown to block the clathrin-mediated endocytosis of L1 (Kamiguchi et al., 1998a), the binding of ankyrin to L1 did not appear to be dependent on L1 interaction with the AP-2 adaptor protein nor on clathrin-mediated endocytosis. Other studies have demonstrated that the activation of a MAP kinase signaling pathway mediates neurite outgrowth on L1 (Schmid et al., 2000) and that clathrin-mediated endocytosis is required for L1-mediated MAP kinase activation (Schaefer et al., 1999; Schmid et al., 2000). Because L1 and the 440 kDa isoform of ankyrinB are colocalized in unmyelinated axons (Chan et al., 1993) and L1 expression is greatly reduced in premyelinated axon tracts of ankyrinB (−/−) mice, the 440 kDa isoform of ankyrinB has been suggested to collaborate in L1 function by stabilizing L1 within the axon (Scotland et al., 1998). Taking into account these findings, we suggest that ankyrin-mediated tethering of L1 to the spectrin cytoskeleton could stabilize L1 within the axon and negatively regulate L1-mediated signal transduction by decreasing the endocytosis of L1.
To test the hypothesis that the interaction of L1 and ankyrin may stabilize L1 within the axon by decreasing the endocytosis of L1, we have investigated the endocytosis of the cytoplasmic L1 missense mutants that are associated with CRASH syndrome. Treatment of living cells stably expressing wild-type L1 or L1 cytoplasmic domain mutants with L1 antibody resulted in the internalization of L1 antibody complexes into vesicles. In B35-L1+S1194L cells, L1 antibody complexes were internalized in a manner that was indistinguishable from the internalization of wild-type L1 in B35-L1+RSLE cells, which suggested that Ser1194 was not required for endocytosis of L1. Because the S1224L and Y1229H L1 mutants did not interact effectively with ankyrin, the demonstration that L1+S1224L and L1+Y1229H antibody complexes were internalized effectively suggested that ankyrin–L1 binding was not necessary for endocytosis. However, antibody-induced endocytosis of the L1 Y1229H mutant resulted in internalized L1-containing vesicles that were larger and brighter than those found in cells expressing wild-type L1 or the L1 mutants S1194L or S1224L. Strikingly, the L1 Y1229H mutant was localized in large cytoplasmic vesicles in cells stably expressing this mutant even in the absence of L1 antibody-induced endocytosis, whereas L1 was localized diffusely throughout the cytoplasm in cells stably expressing wild-type L1 or the S1194L and S1224L L1 mutants. Because the exogenous wild-type L1+RSLE and mutant L1 proteins were expressed at equivalent total levels in B35 cells, the large cytoplasmic L1-containing vesicles found only in the B35-L1+Y1229H cells were unlikely to be attributable to overexpression of L1+Y1229H. In addition, because the exogenous wild-type L1+RSLE and mutant L1 proteins were expressed at approximately equivalent amounts at the cell surface in B35 cells, the large cytoplasmic L1-containing vesicles found only in the B35-L1+Y1229H cells were unlikely to be attributable to a decrease in trafficking of L1+Y1229H to the cell surface. Therefore, the increase in vesicular L1+Y1229H in the absence of antibody-induced endocytosis was consistent with an interpretation that the Y1229H mutant L1 undergoes some degree of increased constitutive endocytosis. These results suggested that the Y1229H mutation may decrease the regulatory constraints on endocytosis of L1 and thereby increase L1 endocytosis. In view of the observations that the L1 S1224L mutant also failed to interact with ankyrin but did not display alterations in endocytosis, the possible modulatory effect of the Y1229H mutation on L1 endocytosis cannot be attributable to only a failure of L1 to interact with ankyrin. For example, phosphorylation of tyrosine 1229 within the SFIGQY motif also may be involved in the endocytotic cycling of L1. An alternative explanation is that the L1+Y1229H mutation may not affect endocytosis but may shunt newly synthesized mutant L1 directly into a large vesicular endocytic compartment (Fig. 7) that also is reached by L1+Y1229H from the cell surface after antibody-mediated endocytosis (Fig. 6).
Our investigation showing differences in cellular functioning among the three known intracellular domain point mutations of L1 associated with CRASH syndrome (S1194L, S1224L, and Y1229H) suggests that cytoplasmic domain mutations may cause pathological defects by different cellular mechanisms. The L1 Y1229H mutation results in alterations in ankyrin binding and in endocytosis, the L1 S1224L mutation results only in alterations in ankyrin binding, and the L1 S1194L mutation affects neither ankyrin binding nor endocytosis. Abnormalities in nervous system development associated with the L1 mutations S1224L and Y1229H may result from reduced ankyrin binding with or without endocytic defects, whereas that of the S1194L mutation is most likely independent of these cellular functions. The widespread dysgenesis of axonal tracts in ankyrinB (Scotland et al., 1998) and L1 knock-out mice (Cohen et al., 1997; Dahme et al., 1997; Demyanenko et al., 1999; Haney et al., 1999) underscores the importance of L1–ankyrin interactions for normal development.
Although some regions of cell–cell adhesion between L1-expressing cells appeared to accumulate both L1 and ankyrin–GFP, our results showed that L1 was effective at recruiting ankyrin to the plasma membrane in cells that were not in contact with other cells. In addition, L1 mutations in the extracellular domain (R184Q, H210Q, E309K, Y784C, and Y1070C) effectively recruited ankyrin to the plasma membrane. The L1 R184Q and H210Q mutations previously have been shown to disrupt L1–L1 trans-interactions substantially (Zhao and Siu, 1996; De Angelis et al., 1999). Furthermore, these two mutations as well as the mutations E309K and Y1070C previously have been shown to alter L1–heterophilic interactions with the L1-binding partners F11/F3/contactin and axonin I/TAG1 (De Angelis et al., 1999). Thus the recruitment of ankyrin to the plasma membrane by L1 appeared to be independent of homophilic and heterophilic L1trans-interactions and cell–cell adhesion. This result was in agreement with previous studies with ankyrin–GFP that showed neurofascin recruitment of ankyrin–GFP to the plasma membrane independently of extracellular interactions (Zhang et al., 1998). On the contrary, studies using Drosophila S2 cell lines stably expressing human L1 have indicated that Drosophila ankyrin selectively associates with the plasma membrane at sites of cell–cell contact (Hortsch et al., 1998). Relative levels of expression of L1 and ankyrin in the transfected HEK 293 cells and in the stable S2 cell lines might not be equivalent and could contribute to the observed differences. It is conceivable that the high levels of expression of L1 at the plasma membrane reached after the transient transfection of HEK 293 cells might mimic the clustering of L1 found at sites of cell–cell contact in the stable S2 cell lines. Alternatively, intrinsic differences between HEK 293 cells and Drosophila S2 cells or mammalian ankyrinG and Drosophila ankyrin might contribute.
Our observations that the L1+R184Q mutation is expressed at normal levels at the cell surface in transiently transfected HEK 293 cells agree with the observation reported by De Angelis et al. (1999) that L1+R184Q is expressed at normal levels at the cell surface in transiently transfected COS cells. In contrast, Moulding et al. (2000)have reported that the R184Q mutation substantially reduced the cell surface expression of L1. The discrepancy between these observations may be attributable to experimental differences. We examined the cell surface expression of L1 in HEK 293 cells that had been transiently transfected with plasmid DNA expression vectors, whereas Moulding et al. (2000) examined the cell surface expression of L1 in astrocytes, neurons, and COS cells that were infected with herpes simplex virus vectors. It is possible that different levels of L1 expression in transiently transfected HEK 293 cells relative to virally infected astrocytes mask a defect in protein processing and trafficking that is seen after expression of virally derived L1 gene products in astrocytes.
In conclusion, the results suggest that the L1 cytoplasmic domain participates in multiple cellular functions that are necessary for normal development of the nervous system and that these functions may be affected differentially by separate mutations occurring in the X-linked mental retardation syndrome, CRASH.
Footnotes
This research was supported by National Institutes of Health Grants HD 35170, NS 26620, and AA 11605 to P.F.M. and by a fellowship from the Imperial Cancer Research Fund and a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression to L.K.N. We are grateful to Steven Baragona for assistance in producing the L1 mutants and to Bentley Midkiff for producing the B35–L1 mutant cell lines. We thank Dr. John Hemperly for providing the Neuro4 antibody and L1 plasmids as well as Dr. Vann Bennett for providing the ankyrin–green fluorescent protein plasmid and for helpful discussions.
Correspondence should be addressed to Dr. Patricia F. Maness, Department of Biochemistry CB#7260, University of North Carolina, Chapel Hill, NC 27599-7260. E-mail: srclab@med.unc.edu.
REFERENCES
- 1.Boll W, Ohno H, Songyang Z, Rapoport I, Cantley LC, Bonifacino JS, Kirchhausen T. Sequence requirements for the recognition of tyrosine-based endocytic signals by clathrin AP-2 complexes. EMBO J. 1996;15:5789–5795. [PMC free article] [PubMed] [Google Scholar]
- 2.Brümmendorf T, Kenwrick S, Rathjen FG. Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol. 1998;8:87–97. doi: 10.1016/s0959-4388(98)80012-3. [DOI] [PubMed] [Google Scholar]
- 3.Chan W, Kordeli E, Bennett V. 440 kDa ankyrinB: structure of the major developmentally regulated domain and selective localization in unmyelinated axons. J Cell Biol. 1993;123:1463–1473. doi: 10.1083/jcb.123.6.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1997;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 5.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 6.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- 7.De Angelis E, MacFarlane J, Du JS, Yeo G, Hicks R, Rathjen FG, Kenwrick S, Brümmendorf T. Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 1999;18:4744–4753. doi: 10.1093/emboj/18.17.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knock-out mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fransen E, Schrander-Stumpel C, Vits L, Coucke P, Van Camp G, Willems PJ. X-linked hydrocephalus and MASA syndrome present in one family are due to a single missense mutation in exon 28 of the L1CAM gene. Hum Mol Genet. 1994;3:2255–2256. doi: 10.1093/hmg/3.12.2255. [DOI] [PubMed] [Google Scholar]
- 10.Fransen E, Van Camp G, Vits L, Willems PJ. L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum Mol Genet. 1997;6:1625–1632. doi: 10.1093/hmg/6.10.1625. [DOI] [PubMed] [Google Scholar]
- 11.Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haney CA, Sahenk Z, Li C, Lemmon VP, Roder J, Trapp BD. Heterophilic binding of L1 on unmyelinated sensory axons mediates Schwann cell adhesion and is required for axonal survival. J Cell Biol. 1999;146:1173–1184. doi: 10.1083/jcb.146.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haycock JW. Polyvinylpyrrolidone as a blocking agent in immunochemical studies. Anal Biochem. 1993;208:397–399. doi: 10.1006/abio.1993.1068. [DOI] [PubMed] [Google Scholar]
- 14.Holm J, Hillenbrand R, Steuber V, Bartsch U, Moos M, Lubbert H, Montag D, Schachner M. Structural features of a close homologue of L1 (CHL1) in the mouse: a new member of the L1 family of neural recognition molecules. Eur J Neurosci. 1996;8:1613–1629. doi: 10.1111/j.1460-9568.1996.tb01306.x. [DOI] [PubMed] [Google Scholar]
- 15.Hortsch M, O'Shea KS, Zhao G, Kim F, Vallejo Y, Dubreuil RR. A conserved role for L1 as a transmembrane link between neuronal adhesion and membrane cytoskeleton assembly. Cell Adhes Commun. 1998;5:61–73. doi: 10.3109/15419069809005599. [DOI] [PubMed] [Google Scholar]
- 16.Kamiguchi H, Lemmon V. A neuronal form of the cell adhesion molecule L1 contains a tyrosine-based signal required for sorting to the axonal growth cone. J Neurosci. 1998;18:3749–3756. doi: 10.1523/JNEUROSCI.18-10-03749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998a;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V. Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci. 1998b;21:97–125. doi: 10.1146/annurev.neuro.21.1.97. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Miura M, Kobayashi M, Asou H, Uyemura K. Molecular cloning of cDNA encoding the rat neural cell adhesion molecule L1. Two L1 isoforms in the cytoplasmic region are produced by differential splicing. FEBS Lett. 1991;289:91–95. doi: 10.1016/0014-5793(91)80915-p. [DOI] [PubMed] [Google Scholar]
- 21.Moulding HD, Martuza RL, Rabkin SD. Clinical mutations in the L1 neural cell adhesion molecule affect cell surface expression. J Neurosci. 2000;20:5696–5702. doi: 10.1523/JNEUROSCI.20-15-05696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Needham LK, Rozengurt E. G-α12 and G-α13 stimulate Rho-dependent tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130 Crk-associated substrate. J Biol Chem. 1998;273:14626–14632. doi: 10.1074/jbc.273.23.14626. [DOI] [PubMed] [Google Scholar]
- 23.Saugier-Veber P, Martin C, Le Meur N, Lyonnet S, Munnich A, David A, Henocq A, Heron D, Jonveaux P, Odent S, Manouvrier S, Moncla A, Morichon N, Philip N, Satge D, Tosi M, Frebourg T. Identification of novel L1CAM mutations using fluorescence-assisted mismatch analysis. Hum Mutat. 1998;12:259–266. doi: 10.1002/(SICI)1098-1004(1998)12:4<259::AID-HUMU7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 25.Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- 27.Scotland P, Zhou D, Benveniste H, Bennett V. Nervous system defects of ankyrinB (−/−) mice suggest functional overlap between the cell adhesion molecule L1 and 440 kDa ankyrinB in premyelinated axons. J Cell Biol. 1998;143:1305–1315. doi: 10.1083/jcb.143.5.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda Y, Asou H, Murakami Y, Miura M, Kobayashi M, Uyemura K. A non-neuronal isoform of cell adhesion molecule L1: tissue-specific expression and functional analysis. J Neurochem. 1996;66:2338–2349. doi: 10.1046/j.1471-4159.1996.66062338.x. [DOI] [PubMed] [Google Scholar]
- 29.Tuvia S, Garver TD, Bennett V. The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin binding activity and patterns of cell segregation. Proc Natl Acad Sci USA. 1997;94:12957–12962. doi: 10.1073/pnas.94.24.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Davis JQ, Carpenter S, Bennett V. Structural requirements for association of neurofascin with ankyrin. J Biol Chem. 1998;273:30785–30794. doi: 10.1074/jbc.273.46.30785. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Siu CH. Differential effects of two hydrocephalus/MASA syndrome-related mutations on the homophilic binding and neuritogenic activities of the cell adhesion molecule L1. J Biol Chem. 1996;271:6563–6566. doi: 10.1074/jbc.271.12.6563. [DOI] [PubMed] [Google Scholar]
- 32.Zisch AH, Stallcup WB, Chong LD, Dahlin-Huppe K, Voshol J, Schachner M, Pasquale EB. Tyrosine phosphorylation of L1 family adhesion molecules: implication of the Eph kinase Cek5. J Neurosci Res. 1997;47:655–665. doi: 10.1002/(sici)1097-4547(19970315)47:6<655::aid-jnr12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]