

Abstract
The role of dopamine in sleep regulation and in mediating the effects of wake-promoting therapeutics is controversial. In this study, polygraphic recordings and caudate microdialysate dopamine measurements in narcoleptic dogs revealed that the wake-promoting antinarcoleptic compounds modafinil and amphetamine increase extracellular dopamine in a hypocretin receptor 2-independent manner. In mice, deletion of the dopamine transporter (DAT) gene reduced non-rapid eye movement sleep time and increased wakefulness consolidation independently from locomotor effects. DAT knock-out mice were also unresponsive to the normally robust wake-promoting action of modafinil, methamphetamine, and the selective DAT blocker GBR12909 but were hypersensitive to the wake-promoting effects of caffeine. Thus, dopamine transporters play an important role in sleep regulation and are necessary for the specific wake-promoting action of amphetamines and modafinil.
Keywords: sleep–wake cycle, modafinil, amphetamine, caffeine, hypocretin, dopamine, narcolepsy
The monoamines serotonin (5-HT) and norepinephrine (NE) (Steriade and McCarley, 1990; Jones, 2000; Siegel, 2000) and histamine (HA) (Monti, 1993; Lin et al., 1996; Steininger et al., 1999; Jones, 2000), neuropeptides including hypocretin (orexin) (Chemelli et al., 1999; Hagan et al., 1999; Lin et al., 1999), and other transmitters including acetylcholine (Steriade and McCarley, 1990; Jones, 2000; Siegel, 2000), GABA (Jones, 2000; Steriade, 2000), and adenosine (Porkka-Heiskanen et al., 1997; Portas et al., 1997) have been prominently implicated in sleep–wake regulation. In contrast, many authors have assigned only a marginal role for dopamine (DA) in sleep–wake control (Jones et al., 1973; Miller et al., 1983; Steinfels et al., 1983). Electrical activities of acetylcholine, NE, 5-HT (Steriade and McCarley, 1990), and HA (Monti, 1993; Steininger et al., 1999) neurons display robust changes across sleep–wake states that contrast with the limited alterations in firing rates of DA neurons across stages of sleep and wakefulness (Miller et al., 1983; Steinfels et al., 1983). The latter forms the basis of contemporary belief that alteration in acetylcholine, 5-HT, NE, or HA are more critically involved in regulating the cortical electroencephalogram (EEG) desynchronization characteristics of wakefulness (Steriade and McCarley, 1990; Monti, 1993; Lin et al., 1996; Steininger et al., 1999;Jones, 2000; Siegel, 2000), whereas dopaminergic activity is thought to mediate motor-related aspects of behaviors (Steinfels et al., 1983).
The lack of covariance between electrophysiological measures and sleep stages does not, however, obviate a role for dopamine in arousal state control. Indeed, the terminal release of dopamine varies in concert with arousal states (Trulson, 1985). In addition, lesions of dopaminergic cell groups in the ventral tegmentum that project to the forebrain produce marked reduction in behavioral arousal (Jones et al., 1973), and human Parkinson's disease patients, who exhibit consistent dopaminergic lesions but inconsistent alterations in other monoamines, experience severe sleep disorders (Aldrich, 2000).
Uncertainties have also persisted about the molecular bases of efficacious wake-promoting compounds, such as amphetamines and modafinil. Amphetamines block plasma membrane transporters for DA, NE, and 5-HT and inhibit the vesicular monoamine transporter (VMAT2), releasing monoamines from the synaptic vesicles into which VMAT2 pumps them (Seiden et al., 1993). Noradrenergic mechanisms have been proposed to explain the wake-promoting effects of amphetamine-like stimulants (Parkes, 1990; Fawcett and Busch, 1998). However, dopamine-specific reuptake blockers can promote wakefulness in normal and sleep-disordered narcoleptic animals better than NE transporter-selective blockers (Nishino and Mignot, 1997; Nishino et al., 1998). Furthermore, the wake-promoting effect of amphetamine is maintained after severe reduction of brain norepinephrine produced by lesions of the noradrenergic cells of the locus ceruleus in cats (Jones et al., 1977).
The mode of action of modafinil, a new wake-promoting compound used in the treatment of sleepiness associated with narcolepsy (US Modafinil in Narcolepsy Multicenter Study Group, 1998), is even more uncertain. Studies have suggested that modafinil increases wakefulness by activating α-1 noradrenergic transmission (Duteil et al., 1990;Jouvet et al., 1991) or hypothalamic cells that contain the peptide hypocretin (Chemelli et al., 1999), or that it may work by modulating GABAergic tone (Ferraro et al., 1996). To identify the molecular basis for the wake-promoting effects of amphetamines and modafinil, we studied the responses to these compounds in narcoleptic canines, a genetic model for excessive sleepiness, and in dopamine transporter (DAT) knock-out mice.
MATERIALS AND METHODS
Canine subjects. Four narcoleptic canines (mean ± SD age, 3.8 ± 2.1 years; 26.5 ± 0.6 kg) born at Stanford University were used in both polygraphic recording and microdialysis experiments. Animals were anesthetized and surgically prepared for EEG and electromyogram (EMG) recordings as described previously (Reid et al., 1996; Nishino et al., 1998). All dogs were given 3–6 weeks to recover before experiments were initiated. Dogs were kept in an 12 hr light/dark cycle (lights on 7:00 A.M.) and were fed daily at 9:00 A.M. Room temperature was maintained at 22.2 ± 1.1°C and relative humidity at 40%. All experimental procedures complied with institutional and National Institutes of Health guidelines for treatment of experimental animals and with the Policy on the Use of Animals in Neuroscience Research of the Society for Neuroscience.
Polygraphic evaluation of the wake-promoting effects of modafinil and d-amphetamine in narcoleptic canines. Intravenous injections of modafinil (5 mg/kg), d-amphetamine (0.1 mg/kg), or vehicle (DMSO, 1 ml/30 kg) were made under lights-on conditions between 10:00 A.M. and 2:00 P.M., with EEG and EMG recorded for 1 hr after injection. Recordings for active drug and vehicle baseline were performed sequentially, separated by a 48 hr washout period, and the order of injection (active drug followed by vehicle or vehicle followed by active drug) was randomized in each animal. Two recording sessions (modafinil and d-amphetamine with respective vehicle injections) were done for each animal with at least a 1 week interval between sessions. Polygraphic recordings were visually scored in 30 sec epochs as wake, drowsy, light sleep, deep sleep, rapid-eye movement (REM) sleep, or cataplexy (Nishino et al., 1998).
Microdialysis measurements of the effects of modafinil andd-amphetamine on extracellular dopamine in narcoleptic canines. One to 2 months subsequent to the above described polygraphic recording sessions, microdialysis probes were implanted bilaterally into the caudate nucleus (lateral, 7.0 mm; anteroposterior, 26–28 mm; height 20 mm from stereotaxic zero) and anchored in place under isoflurane anesthesia. On each of the next 3 d, animals were connected to a dialysis inlet and perfused with artificial CSF (2.0 μl/min) for 30 min before and 60 min after intravenous injection of active drug (modafinil, 5 mg/kg; ord-amphetamine, 0.1 mg/kg) or vehicle (DMSO, 1 ml/30 kg). One injection was performed on each experimental day at 10:00 A.M., and the order of the injections was randomized in each animal. Perfusate samples were collected in 10 min bins throughout the perfusion period. DA levels in each 10 min perfusate were measured using reverse-phase HPLC (detection limit, 20 fmol) (Reid et al., 1996).
Murine subjects. Mice were bred at the National Institute on Drug Abuse, National Institutes of Health (Baltimore, MD) and shipped to Stanford University at age 3–6 months (Sora et al., 1998). DAT knock-out (−/−; n = 14; age, 240 ± 8 (±SEM) d; weight, 24 ± 1 gm), littermate heterozygous (+/−;n = 11; age, 267 ± 45 d; weight, 33 ± 1 gm), and wild-type (+/+; n = 14; age, 291 ± 25 d; weight, 32 ± 1 gm) mice were anesthetized with isoflurane (3% in medical grade oxygen) and surgically prepared for EEG and EMG recording (Van Gelder et al., 1991). After 2–3 weeks of postsurgical recovery, mice were isolated in separate compartments of a sound-attenuated stainless steel recording chamber with ad libitum food and water in an 12 hr light/dark cycle. Room temperature was maintained at 24 ± 1°C throughout experimentation. All experimental procedures complied with institutional and National Institutes of Health guidelines for treatment of experimental animals and with the Policy on the Use of Animals in Neuroscience Research of the Society for Neuroscience.
Baseline sleep study. Mice were acclimated to the recording apparatus for 3 d before the initiation of data collection. A 24 hr baseline recording of EEG, EMG, wheel-running activity, and drinking behavior was initiated 18–24 hr after a visual animal health check. The recording chamber remained sealed throughout the entire recording period.
Murine pharmacological experiments. +/+ and −/− mice were subjected to pharmacological or vehicle (0.25% methylcellulose in physiological saline) treatments beginning the day after the above described baseline recordings. Injections were delivered once per week, with each injection followed by a 6–8 d washout period, during which mice remained in the recording chamber. Each injection was preceded by a baseline 24 hr recording period and followed by a 24 hr posttreatment period, during which time recording chambers remained sealed and experimenters did not enter the room in which the chambers are located. All treatments were delivered intraperitoneally 5 hr into the daily light period (±30 min). Because the order of treatments was not consistent across animals and because some mice did not receive all treatments, sample sizes differed among treatments [sample sizes (+/+, −/−): vehicle (9, 8); methamphetamine, 2 mg/kg (14, 10); GBR12909, 20 mg/kg (14, 6); modafinil, 90 mg/kg (10, 9) and 300 mg/kg (14, 6); and caffeine, 2.5 mg/kg (10, 8), 10 mg/kg (12, 7), and 20 mg/kg (8, 0)]. Consequently, the data are treated as independent measures in statistical comparisons of treatments.
Mouse data collection and analysis. The acquisition and processing of EEG, EMG, wheel, and drink data by the SCORE algorithm are described in detail previously (Van Gelder et al., 1991). Digitized EEG (bandpass, 1–30 Hz; digitization rate, 100 Hz), integrated EMG (bandpass, 10–100 Hz), and wheel and drink signal (binary variables) were stored in 10 sec epochs. Epochs were classified as wake, REM sleep (REM), or non-REM sleep (NREM) by a pattern-matching algorithm (SCORE;Van Gelder et al., 1991). The scoring of sleep states by the SCORE algorithm was validated by visual inspection of individual epoch scores for every animal.
Long-term mouse wheel-running activity recordings. A subset of mice of each genotype (DAT +/+, n = 7; DAT +/−,n = 4; DAT −/−, n = 6) were subjected to long-term (>7 d), continuous monitoring of wheel-running activity under a 12 hr light/dark cycle. The mean daily waveform of wheel activity from each animal was calculated by averaging the number of minutes of wheel-running activity occurring within each hour of the 12 hr light/dark cycle over a 7 d period. The group mean (±SEM) of these individual values was then calculated within each genotype to determine mean group waveform.
Statistics: canine experiments. Statistical significance of the effect of modafinil and d-amphetamine on wake time in narcoleptic dogs when compared with respective vehicle sessions was determined using Student's t test. Statistical significance of the effects of modafinil and d-amphetamine on DA efflux was assessed using repeated-measures ANOVA with a grouping factor (treatment) and with Bonferroni's–Dunn's post hoccomparisons with the vehicle session.
Statistics: murine experiments. The effect of genotype on long-term wheel-running data were assessed using repeated-measures ANOVA with genotype as a factor. Effects of genotype on NREM time, REM time, wake time, mean sleep bout duration, and mean wake bout duration values from the undisturbed baseline period were assessed by one-way ANOVA with genotype as a factor. Sleep bouts are defined as episodes of sleep (NREM + REM) initiated by at least 2 consecutive 10 sec epochs of sleep and terminated by at least two 10 sec epochs of wake. Wake bouts are defined as episodes of wake initiated by at least 2 consecutive 10 sec epochs of wake and terminated by at least two 10 sec epochs of sleep.
Statistical significance of the effects of GBR12909 (20 mg/kg) and modafinil (90 mg/kg) on sleep states in mice was determined separately within each genotype. Repeated-measures ANOVA with treatment (vehicle vs drug) as a factor was performed on NREM time, REM time, and wake time during the 48 hr recording period encompassing injection. The cumulative wake-promoting effects of GBR12909 (20 mg/kg), modafinil (90 mg/kg), and vehicle were determined by subtracting the number of minutes of wake during each hour of the preinjection baseline period from the number of minutes of wake during the corresponding hour of the postinjection period on an hourly basis and cumulating these subtracted values serially over the entire 24 hr postinjection period. Repeated-measures ANOVA with treatment (vehicle vs drug) as a factor was performed on cumulated wake value separately within each genotype.
The net wake-promoting effects of all pharmacological treatments and of vehicle injection over the 5 hr immediately after injection were quantified in the form of cumulated wake: the difference in minutes of wake between the 5 hr immediately after injection and the corresponding 5 hr of the baseline day. Two-way ANOVA, with genotype as one factor and treatment (pharmacological compound vs vehicle) as the other, was performed separately for each compound to determine whether there was a significant effect of genotype or of treatment on cumulated wake.
RESULTS
Modafinil increases cerebral dopamine release and wakefulness in a hypocretin-independent manner
To address the possibility that dopamine mediates the wake-promoting effect of modafinil and amphetamine, in vivodialysis was performed in freely moving narcoleptic canines. Systemic administration of modafinil (5 mg/kg, i.v.) andd-amphetamine (0.1 mg/kg, i.v.) significantly increased time spent awake. The most pronounced effect on wakefulness was observed during the first hour after injection (p < 0.01 for both treatments, compared with the respective vehicle treatment), and the wake-promoting effects of modafinil and amphetamine were equipotent. Thus, respective doses of modafinil (5 mg/kg) and amphetamine (0.1 mg/kg) were administered intravenously to narcoleptic animals while DA release was continuously monitored in striatal extracellular fluid (Fig. 1). A quantitatively similar significant increase in extracellular DA concentration was observed with equipotent doses of either amphetamine or modafinil (p < 0.01; repeated-measures ANOVA). These observations indicate that hypocretin receptor 2 is not required for the wake-promoting effects of modafinil and amphetamine or for their effects on dopaminergic transmission.
Fig. 1.

Effect of modafinil andd-amphetamine on wakefulness and caudate DA efflux in narcoleptic hypocretin receptor 2 mutant dogs. Systemic administration of modafinil (5 mg/kg, i.v.) and d-amphetamine (0.1 mg/kg, i.v.) equipotently increased time spent awake. The most pronounced effect on wakefulness was observed during the first hour after injection [inset; *p < 0.01 for both treatments compared with the respective vehicle treatment (white bars) by Student's t test;n = 4 per group; mean ± SEM]. Microdialysis experiments demonstrated that administration of amphetamine and modafinil significantly increased extracellular DA levels (p < 0.05;d-amphetamine or modafinil relative to vehicle treatment; repeated-measures ANOVA with Bonferroni's–Dunn'spost hoc comparisons; n = 4 per group; mean ± SEM). The baseline DA concentrations (at time 0) for modafinil and d-amphetamine sessions were 17.2 ± 3.1 and 17.6 ± 3.5 nm (mean ± SEM), respectively, and did not differ statistically among treatments.VEH, 1 ml of 100% DMSO; d-AMP,d-amphetamine.
DAT knock-out mice exhibit normal circadian patterns of inactivity and activity
DAT knock-out homozygote (−/−), heterozygote (+/−), and wild-type littermates (+/+) displayed robust rhythms of activity that were entrained to the 12 hr light/dark cycle, with quiescence during the light phase and wheel running during the dark phase (Fig.2). A moderate increase in wheel-running activity was observed in DAT knock-out mice only during the latter portion of the dark period (Fig. 2).
Fig. 2.
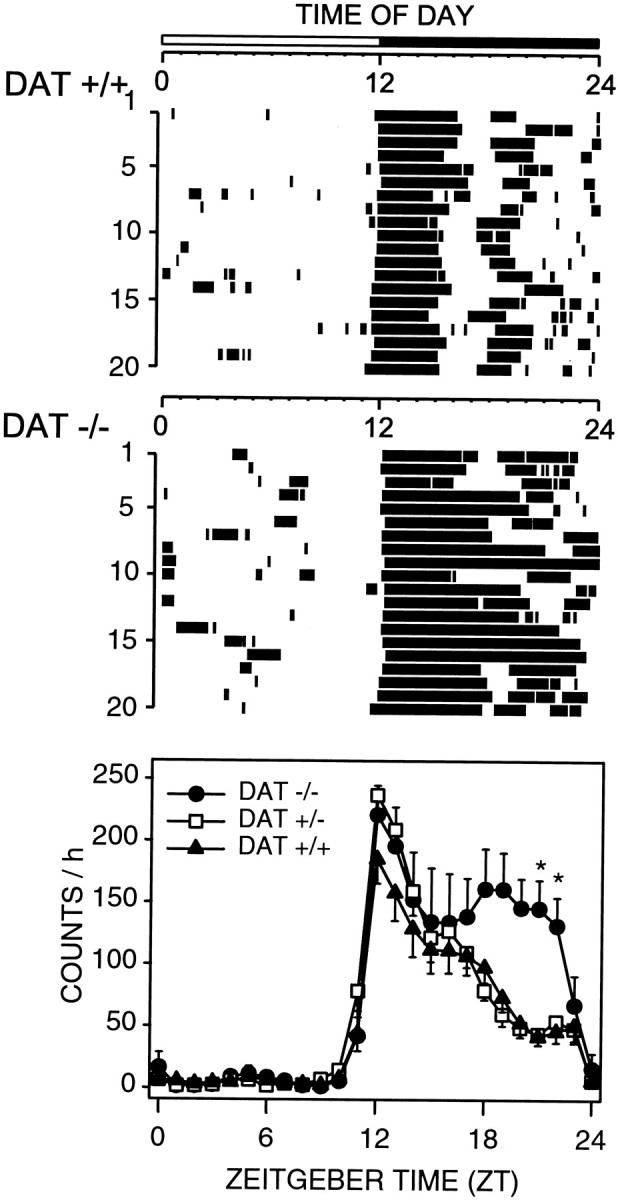
Entrained circadian rhythms of wheel-running activity in wild-type (DAT +/+) and homozygous DAT knock-out (DAT −/−) mice. Representative raster plots (top 2 panels) show nocturnal consolidation of wheel-running and a lack of wheel-running activity during the light phase in both DAT +/+ and DAT −/− mice. Group mean ± SEM waveforms are shown for DAT +/+ (filled triangles; n = 7), DAT −/− (filled circles; n= 6), and heterozygous DAT +/− (open squares;n = 4) mice in the bottom panel.Tick marks in the raster plots denote when wheel-running activity was present in 5 min bins. Light–dark bar at the top denotes lighting schedule. Twenty-four hour activity patterns of DAT −/− mice differ significantly from those of DAT +/+ mice (p < 0.02; two-way repeated-measures ANOVA), *p < 0.05; DAT−/− versus DAT +/+; Bonferroni's–Dunn's post hoccomparisons.
Sleep architecture is altered in DAT knock-out mice in undisturbed baseline conditions
When studied in undisturbed baseline conditions, mice of each genotype displayed sleep and wakefulness that were normal by several electrophysiological criteria, including EEG spectral analyses. However, sleep architecture differed considerably among mice of the three genotypes (Table1). Mice lacking both copies of the DAT gene displayed a threefold increase in wake bout duration compared with wild-type littermates (p < 0.017; post hoc comparisons). Heterozygous (+/−) mice exhibited wake bout durations that were intermediate of DAT knock-out and wild-type (Fig.3). NREM sleep as a percentage of recording time and mean sleep bout duration were reduced in DAT knock-out mice relative to wild-type mice (Table 1).
Table 1.
Sleep in wild-type, DAT +/−, and DAT −/− mice
| DAT genotype | NREM time (%) | REM time (%) | Wake time (%) | Mean sleep bout (min) | ||||
|---|---|---|---|---|---|---|---|---|
| Light | Dark | Light | Dark | Light | Dark | Light | Dark | |
| +/+ (11, 3) | 50.3 ± 1.9 | 34.7 ± 1.9 | 8.1 ± 0.7 | 4.5 ± 0.7 | 41.6 ± 2.3 | 60.7 ± 2.0 | 4.8 ± 0.6 | 2.9 ± 0.3 |
| +/− (9, 2) | 49.7 ± 2.3 | 35.2 ± 2.9 | 8.1 ± 0.5 | 3.8 ± 0.8 | 42.2 ± 2.4 | 61.1 ± 3.2 | 6.2 ± 1.1 | 4.0 ± 0.5 |
| −/− (11, 3) | 41.3 ± 2.6* | 29.0 ± 2.6 | 6.4 ± 0.7 | 3.9 ± 0.7 | 52.3 ± 2.7* | 67.1 ± 2.9 | 2.9 ± 0.3 | 2.3 ± 0.3 |
Data (mean ± SEM) were obtained over a 24 hr baseline period in which the animals were undisturbed. Numbers in parentheses indicate number of male and female mice, respectively. Sex differences were not significant (data not shown). NREM time, REM time, and wake time are reported as percentage of recording time. Sleep bout duration is in minutes. One-way ANOVA indicated significant genotype effects [p < 0.014, df = (2,36)] on NREM time, wake time, and mean sleep bout during the light portion of the cycle.
p < 0.05 versus wild type; Bonferroni's–Dunns post hoc comparisons.
Fig. 3.

Mean ± SEM wake bout duration during 24 hr baseline recordings in wild-type (+/+; n = 14), heterozygous (+/−; n = 11), and homozygous DAT knock-out (n = 14) mice. One-way ANOVA indicated significant genotype effect [p < 0.014; df = (2,36)]. *p < 0.05 versus DAT +/+; Student's t test.
The wake-promoting effects of classical stimulants and the previously undefined stimulant modafinil are abolished in DAT knock-out mice
To verify that inhibition of DAT function is capable of producing increased wakefulness in mice, we examined the effect of GBR12909, a highly specific DAT inhibitor, during the fifth hour of the daily light period, when the majority of time is spent sleeping. A dose of GBR12909 that resulted in a 67.77 ± 12 (±SEM) min increase in wake time in wild-type mice over a 5 hr period (p < 0.001; treatment × time interaction) (Fig.4; see Fig. 6) failed to increase wake in the DAT knock-out mouse (p > 0.1) (Fig. 4; see Fig. 6). This result confirms that the wake-promoting effect of GBR12909 requires functioning dopamine transporters.
Fig. 4.

Effect of GBR12909 (20 mg/kg) on sleep states in wild-type (+/+) and DAT knock-out (−/−) mice. Data are the percentages of time (mean ± SEM in 2 hr bins) in NREM, REM, and wake in the 24 hr pretreatment and 24 hr posttreatment periods. Open circles, Vehicle (0.25% methylcellulose vehicle); filled circles, GBR12909 (20 mg/kg).Arrows indicate injection. Light–dark bars at the bottom of thepanels indicate lighting schedule. Two-way repeated-measures ANOVA indicated postinjection treatment × time interactions in DAT +/+ (p < 0.001 for wake; p < 0.003 for NREM; p < 0.011 for REM) but not DAT −/− (p > 0.1; all states) mice. Sample sizes (+/+, −/−): vehicle (9, 8); GBR12909 (14, 6).
Fig. 6.

Cumulative effects of GBR12909 (20 mg/kg) and of modafinil (90 mg/kg) on wake in wild-type (+/+) and DAT knock-out (−/−) mice. The cumulative change in wake time (mean ± SEM) in the posttreatment period relative to the corresponding period of the baseline 24 hr pretreatment period is shown for vehicle (open circles) and drug treatment (filled circles). Note large increase in wakefulness in DAT +/+ but not DAT −/− animals. Two-way repeated-measures ANOVA within each genotype indicated significant treatment effect and treatment × time interaction in DAT +/+ but not in DAT −/−. *p < 0.05 drug versus vehicle; Bonferroni's–Dunn's post hoc comparisons. Sample sizes (+/+, −/−): vehicle (9, 8); GBR12909, (14, 6); modafinil (10, 9).
The effect of modafinil on sleep architecture in wild-type and DAT knock-out animals is shown in Figure 5, and its wake-promoting effect is quantified in Figure6. These data clearly demonstrate a striking wake-promoting effect of modafinil in wild-type mice that is abolished in DAT knock-out mice. Both modafinil and GBR12909 significantly increase wake time by at least 60 min over a 5 hr period in wild-type mice (p < 0.001; repeated-measures ANOVA) (Fig. 6). A significant but minor (<10 min) decrease in REM sleep was seen in the DAT knock-out after modafinil treatment (Fig. 5). However, the absence of a significant change in wake time in DAT knock-out mice over the 24 hr posttreatment period (p > 0.1; repeated-measures ANOVA) (Fig. 6) confirms that DAT is required for the wake-promoting effects of modafinil and GBR12909.
Fig. 5.

Effect of modafinil (90 mg/kg) on sleep states in wild-type (+/+) and DAT knock-out (−/−) mice. Data are the percentages of time (mean ± SEM in 2 hr bins) in NREM, REM, and wake in the 24 hr pretreatment and 24 hr posttreatment periods. Open circles, Vehicle (0.25% methylcellulose vehicle); filled circles, modafinil (90 mg/kg).Arrows indicate injection. Light–darkbars at the bottom of the panels indicate lighting schedule. Two-way repeated-measures ANOVA indicated postinjection treatment × time interactions for wake, NREM, and REM sleep in both DAT +/+ (p < 0.001 for wake, NREM, and REM) and DAT −/− (p = 0.007, 0.004, and 0.007, respectively) mice, but increased wake after modafinil administration was only observed in DAT +/+ mice. Sample sizes (+/+, −/−): vehicle (9, 8); modafinil (10, 9).
To further characterize the role of dopaminergic components in the effects of stimulants, the wake-promoting effects of methamphetamine were also examined. When wild-type mice were treated with methamphetamine at the time of their maximal sleep tendency, 5 hr after lights on, wakefulness was enhanced. Thus, modest doses of methamphetamine increase wake time by >60 min over a 5 hr period (Fig.7). Increased wake was associated with reductions in both NREM and REM sleep times in wild-type animals (data not shown). In sharp contrast, methamphetamine did not increase wakefulness in DAT knock-out mice (Fig. 7). The absence of effect could not be explained by the overall sleep reductions noted in DAT knock-out animals because the baseline wake time (∼40–50%/hr) did not differ significantly between genotypes in the latter portion of the day (see baseline data in Fig. 4 for circadian waveform).
Fig. 7.

Response to wake-promoting therapeutics in DAT knock-out (−/−; open bars) and wild-type (filled bars) mice. Data are reported as the cumulative change in time awake (mean ± SEM) 5 hr after treatment relative to corresponding baseline 24 hr earlier. Two-way ANOVA indicated significant genotype × treatment interaction in comparisons of individual pharmacological treatments versus vehicle (p < 0.05, all treatments).VEH, 0.25% methylcellulose vehicle;METH, methamphetamine; GBR, GBR12909.1p < 0.001;2p < 0.025 between groups;3p < 0.002 relative to vehicle; Student's t test. Sample sizes (+/+, −/−): vehicle (9, 8); methamphetamine, 2 mg/kg (14, 10); GBR12909, 20 mg/kg (14, 6); modafinil, 90 mg/kg (10, 9) and 300 mg/kg (14, 6); caffeine, 2.5 mg/kg (10, 8), 10 mg/kg (12, 7), and 20 mg/kg (8, 0).
DAT knock-out mice are hypersensitive to the wake-promoting effect of caffeine
Caffeine, a widely used wakefulness-enhancing agent, retained full ability to enhance wakefulness in the DAT knock-out. In fact, these mice were hypersensitive to the wake-promoting effect of caffeine, exhibiting a threefold to fivefold greater increase in wake time relative to wild-type mice in response to 2.5 or 10 mg/kg doses (Fig.7).
DISCUSSION
Dopamine has been thought to play a minor role in sleep–wake regulation, yet compounds that block DAT or enhance dopamine release potently promote wakefulness. Such compounds are commonly used to treat the sleep disorder narcolepsy. Using a multidisciplinary approach, we now show that amphetamine-like compounds and modafinil require DAT for their wake-promoting effects.
Recent experiments have shown that hypocretin-containing neurons express Fos-immunoreactivity after modafinil treatment (Chemelli et al., 1999), leading the authors to speculate that modafinil may increase wakefulness by activating hypocretin-containing cells. Our results now indicate that intact hypocretin transmission is not required for the wake-promoting effect of modafinil. Indeed, this compound increases wakefulness in both hypocretin receptor 2-deficient canines (Fig. 1) and in hypocretin-deficient human patients (Peyron et al., 2000). The finding that modafinil and amphetamine induce similar increases in dopamine release at equipotent wake-promoting doses rather suggests that increased dopaminergic transmission mediates the effects of both compounds. Together with the previously observed binding of amphetamine-like compounds (Nishino et al., 1998) and modafinil (Mignot et al., 1994) to DAT, these observations in the narcoleptic dog support the hypothesis that both modafinil and amphetamine promote wakefulness primarily by increasing dopaminergic tone and not by stimulating hypocretin transmission.
DAT knock-out mice have proven to be a powerful tool to help dissect the molecular mechanisms for the effects of nonselective compounds that interact with multiple monoaminergic transmitter systems. For example, studies of DAT knock-out mice have implicated cocaine and amphetamine interactions with DAT in cocaine- and amphetamine-induced locomotor stimulation (Giros et al., 1996; Gainetdinov et al., 1999) but not in cocaine reward (Sora et al., 1998). In the current study, this approach was used to demonstrate dramatic baseline sleep–wake abnormalities and a total absence of wake promotion after treatment with GBR12909, methamphetamine, and modafinil. These results are consistent with dopamine mediation of wake-promoting therapeutics (with the notable exception of caffeine).
The present results indicate that, in the lifelong absence of DAT, methamphetamine actions at other sites are not sufficient to promote wakefulness. These results must be interpreted in light of the primary effect of DAT knock-out but also in light of the possibility that adaptations in the knock-out could disable normal wake-promoting actions of amphetamine-like compounds at other monoaminergic sites. In fact, the adaptations identified in the DAT knock-out mouse appear to leave serotonin and norepinephrine systems either intact or supersensitive to blockade. Gainetdinov et al. (1999) have interpreted locomotor responses in the DAT knock-out to indicate that serotonergic transmission is intact in the DAT knock-out, so that amphetamine has a serotonin-mediated calming effect in these mice. Recently, we have identified enhanced reward responses to selective norepinephrine and serotonin blockers in DAT knock-outs (F. S. Hall, I. Sora, X. F. Li, and G. R. Uhl, unpublished results). No neurochemical assessment or change in gene expression identified to date suggests that serotonin or norepinephrine transmission is disabled in these mice (Fumagalli et al., 1998; Q. Liu, F. S. Hall, H. P. Lesch, D. Murphy, I. Sora, and G. R. Uhl, unpublished results). Furthermore, even if some occult compromise in noradrenergic transmission were present in DAT knock-outs, it would be unlikely to be the primary cause for the effects observed here because the wake-promoting effect of amphetamine is maintained after even severe reductions in brain norepinephrine produced by large noradrenergic lesions (Jones et al., 1977). Heightened responsiveness to amphetamine occurs in norepinephrine transporter knock-out mice (Xu et al., 2000). Other evidence supports a dopaminergic link to wake-promoting effects of psychostimulants. The wake-promoting effects of amphetamine and its derivatives correlate with the magnitude of increase in extracellular dopamine produced by these compounds and not with increased extracellular norepinephrine (Kanbayashi et al., 2000). The highly specific DAT inhibitor GBR12909 (Heikkila and Manzino, 1984) promotes wake in wild-type mice (Fig. 4). Together, these observations suggest that blockade of DAT, independent from that of norepinephrine, provides the basis for the wake-promoting effect of amphetamine and its derivatives.
Increased consolidation of wakefulness and reduced NREM sleep time in DAT knock-out mice relative to wild-type littermates parallel those reported previously for locomotion in DAT knock-outs (Giros et al., 1996; Gainetdinov et al., 1999), tempting one to speculate that the observed sleep–wake effects are secondary to increased locomotor activity. However, the sleep abnormalities identified in the DAT knock-out mice were observed under undisturbed baseline conditions, regardless of whether they exhibited locomotor wheel-running activity or not. In addition, hyperactivity has been reported in the DAT knock-out only in novel environments and not in home cage environments (Giros et al., 1996; Gainetdinov et al., 1999). Indeed, wheel-running data from the DAT knock-out (Fig. 2) suggest that these mice are hyperactive only in novel environments, in contrast to home cage conditions.
Similarly, there are reasons to believe that the wake-promoting effects of dopaminergic stimulants are independent from their locomotor effects. Modafinil and low-dose GBR12909 promote wakefulness without disproportionately increasing locomotor activity in normal dogs (Shelton et al., 1995) and rats (Edgar and Seidel, 1997). The wake-promoting doses of amphetamine used in this study are one-fifth of those typically used to elicit hyperactivity (Giros et al., 1996;Gainetdinov et al., 1999) and stereotypies (Grisel et al., 1997). This differential potency is consistent with clinical experience suggesting increased wakefulness at doses lower than those that produce locomotor activation and inconsistent with the notion that locomotor effects are the primary reason for amphetamine-induced wakefulness. Because locomotor activation and stereotypies are mediated primarily by dopaminergic projections to the nucleus accumbens and dorsal striatum, respectively (Creese, 1983; Koob, 2000), the present model suggests that other projections more sensitive to the effects of DAT inhibition [e.g., cortical and/or basal forebrain projections (Koob, 2000)] may be involved in the promotion of wakefulness. Dopamine turnover is much higher in the prefrontal cortex than in the striatum (Bannon et al., 1981), indicating a greater regulatory role for DAT, and therefore the potential for greater sensitivity to DAT blockers, in the cortex. The fact that activation of the ventral tegmental dopaminergic system has been shown to increase EEG power in the high-frequency range (Montaron et al., 1982), a frequency reflecting cortical activation independently of locomotor activity (Maloney et al., 1997), may suggest the involvement of direct dopaminergic projections to the cortex. Nonetheless, we cannot exclude the possibility of some overlap in the circuitry that regulates higher-order motor control and that which modulates sleep.
Another mechanism that may contribute to increased wake in the DAT knock-out is the regulation of growth hormone-releasing hormone (GHRH). Dopamine receptors in the hypothalamus inhibit hypothalamic GHRH release (West et al., 1997). Because GHRH promotes sleep (Ehlers et al., 1986), an inverse relationship would be expected between dopaminergic tone and sleep time. In DAT knock-out mice, GHRH is chronically low, with concomitant small body size (Bosse et al., 1997). Impairment of the somatotropic axis could thus reduce sleep in DAT knock-out mice. This hypothesis is supported by the fact that mice with a genetically engineered deficiency in GHRH, resulting in dwarfism, exhibit significantly less NREM sleep than wild-type control mice (Zhang et al., 1996).
The mechanism by which caffeine induces wake is not certain, but no direct action on DAT has been reported for this compound. Thus, its wake-promoting effect in the DAT knock-out serves as a positive control, indicating that the response to a wake-promoting compound that acts at a site other than DAT is intact in the DAT knock-out. Caffeine has a number of pharmacological effects, yet antagonism of adenosine receptors by caffeine is the only known central pharmacological effect that occurs in the range of doses produced by voluntary caffeine intake (for review, see Fredholm et al., 1999). Still, actions at other sites cannot be ruled out. Hypersensitivity to caffeine in DAT knock-out mice may reflect adenosine–dopamine interactions in sleep regulation, similar to those implicated in brain locomotor control systems (Garrett and Griffiths, 1997). Reduced dopaminergic signaling produced by dopamine D2 receptor knock-out is associated with a reduced locomotor response to caffeine and with reduced adenosine A2A receptor-dependent cAMP signaling (Zahniser et al., 2000). This observation suggests that adenosine–dopamine interactions are important in determining the stimulant effect of caffeine.
Our finding that a DAT gene deletion alters baseline sleep and responsiveness to the major therapeutic wake-promoting agents has important clinical applications. Severe and often untreated sleep disorders are common in patients with dopaminergic dysfunction caused by Parkinson's disease and Huntington's chorea (Wiegand et al., 1991;Aldrich, 2000). Dopamine metabolism and receptor abnormalities also occur in disorders of excessive daytime sleepiness, such as narcolepsy (Nishino and Mignot, 1997), and in normal aging. The current data, combined with observations that specific DAT gene polymorphic markers (Gill et al., 1997) and sleep disorders (Corkum et al., 1999) are associated with attention deficit hyperactivity disorder, suggest that human variants at the DAT gene locus could predispose to vulnerability to sleep–wake disorders. Finally, our observations with DAT knock-outs provide a new major target for development of more efficacious wake-promoting drugs. The clinical safety profile, low abuse potential, and clinical success of modafinil suggest that selective DAT inhibitors can have useful clinical applications and low side effect profiles when compared with classical amphetamine-like stimulants. Because amphetamine-like compounds are now prescribed to millions of patients with a wide variety of sleep and psychiatric disorders, the utility of highly selective DAT inhibitors may deserve reconsideration.
Footnotes
This research was supported in part by National Institutes of Health Grants HL61594 and HL64243 (D.M.E.), MH12244 (J.P.W.), MH01600 (S.N.), and NS-23724 (E.M.), and Air Force Office of Scientific Research Grant F49620-95-1-0388 (D.M.E.). We are grateful to Wesley Seidel for advice on data analysis and to Laura Alexandre, Ronny Tjon, Humberto Garcia, and Dr. Takashi Kanbayashi for technical assistance. We are especially grateful to Dr. William C. Dement for inspiration and continued support of the basic sleep research program at Stanford University.
Correspondence should be addressed to Dr. Jonathan P. Wisor, Sleep Disorders Research Center, Stanford University School of Medicine, 701 Welch Road #2226, Palo Alto, CA 94304. E-mail: jwisor@stanford.edu.
REFERENCES
- 1.Aldrich M. Parkinsonism. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Saunders; Philadelphia: 2000. pp. 1051–1057. [Google Scholar]
- 2.Bannon MJ, Bunney EB, Roth RH. Mesocortical dopamine neurons: rapid transmitter turnover compared to other brain catecholamine systems. Brain Res. 1981;218:376–382. [PubMed] [Google Scholar]
- 3.Bosse R, Fumagalli F, Jaber M, Giros B, Gainetdinov RR, Wetsel WC, Missale C, Caron MG. Anterior pituitary hypoplasia and dwarfism in mice lacking the dopamine transporter. Neuron. 1997;19:127–138. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- 4.Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammel T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 5.Corkum P, Moldofsky H, Hogg-Johnson S, Humphries T, Tannock R. Sleep problems in children with attention-deficit/hyperactivity disorder: impact of subtype, comorbidity, and stimulant medication. J Am Acad Child Adolesc Psychiatry. 1999;38:1285–1293. doi: 10.1097/00004583-199910000-00018. [DOI] [PubMed] [Google Scholar]
- 6.Creese I. Stimulants, neurochemical, behavioral, and clinical perspectives. Raven; New York: 1983. [Google Scholar]
- 7.Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E. Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol. 1990;180:49–58. doi: 10.1016/0014-2999(90)90591-s. [DOI] [PubMed] [Google Scholar]
- 8.Edgar DM, Seidel WF. Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J Pharmacol Exp Ther. 1997;283:757–769. [PubMed] [Google Scholar]
- 9.Ehlers CL, Reed TK, Henriksen SJ. Effects of corticotropin-releasing factor and growth hormone-releasing factor on sleep and activity in rats. Neuroendocrinology. 1986;42:467–474. doi: 10.1159/000124489. [DOI] [PubMed] [Google Scholar]
- 10.Fawcett JF, Busch KA. Stimulants in psychiatry. In: Schatzberg AF, Nemeroff CB, editors. The American Psychiatric Press textbook in psychopharmacology, Ed 4. American Psychiatric; Washington, DC: 1998. pp. 503–522. [Google Scholar]
- 11.Ferraro L, Tanganelli S, O'Connor WT, Antonelli T, Rambert F, Fuxe K. The vigilance promoting drug modafinil increases dopamine release in the rat nucleus accumbens via the involvement of a local GABAergic mechanism. Eur J Pharmacol. 1996;306:33–39. doi: 10.1016/0014-2999(96)00182-3. [DOI] [PubMed] [Google Scholar]
- 12.Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- 13.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- 15.Garrett BE, Griffiths RR. The role of dopamine in the behavioral effects of caffeine in animals and humans. Pharmacol Biochem Behav. 1997;57:533–541. doi: 10.1016/s0091-3057(96)00435-2. [DOI] [PubMed] [Google Scholar]
- 16.Gill M, Daly G, Heron S, Hawi Z, Fitzgerald M. Confirmation of association between attention deficit hyperactivity disorder and a dopamine transporter polymorphism. Mol Psychiatry. 1997;2:311–313. doi: 10.1038/sj.mp.4000290. [DOI] [PubMed] [Google Scholar]
- 17.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 18.Grisel JE, Belknap JK, O'Toole LA, Helms ML, Wenger CD, Crabbe JC. Quantitative trait loci affecting methamphetamine responses in BXD recombinant inbred mouse strains. J Neurosci. 1997;17:745–754. doi: 10.1523/JNEUROSCI.17-02-00745.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Shah AS, Hatcher JP, Hatcher PD, Jones DN, Smith MI, Piper DC, Hunter AJ, Porter RA, Upton N. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA. 1999;96:10911–10916. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heikkila RE, Manzino L. Behavioral properties of GBR 12909, GBR 13069 and GBR 13098: specific inhibitors of dopamine uptake. Eur J Pharmacol. 1984;103:241–248. doi: 10.1016/0014-2999(84)90483-7. [DOI] [PubMed] [Google Scholar]
- 21.Jones BE. Basic mechanisms of sleep–wake states. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Saunders; Philadelphia: 2000. pp. 134–154. [Google Scholar]
- 22.Jones BE, Bobillier P, Pin C, Jouvet M. The effects of lesions of catecholamine-containing neurons upon monoamine content of the brain and EEG and behavioral waking in the cat. Brain Res. 1973;58:157–177. doi: 10.1016/0006-8993(73)90830-5. [DOI] [PubMed] [Google Scholar]
- 23.Jones BE, Harper ST, Halaris AE. Effects of locus coeruleus lesions upon cerebral monoamine content, sleep–wakefulness states and the response to amphetamine in the cat. Brain Res. 1977;124:473–496. doi: 10.1016/0006-8993(77)90948-9. [DOI] [PubMed] [Google Scholar]
- 24.Jouvet M, Albarede JL, Lubin S, Meyrignac C. Noradrenaline and cerebral aging. Encephale. 1991;17:187–195. [PubMed] [Google Scholar]
- 25.Kanbayashi T, Honda K, Kodama T, Mignot E, Nishino S. Implications of dopaminergic mechanisms in the wake-promoting effects of amphetamine: a study of d- and l-derivatives in canine narcolepsy. Neuroscience. 2000;99:651–659. doi: 10.1016/s0306-4522(00)00239-6. [DOI] [PubMed] [Google Scholar]
- 26.Koob GF. Stimulants: basic mechanisms and pharmacology. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Saunders; Philadelphia: 2000. pp. 419–428. [Google Scholar]
- 27.Lin JS, Hou Y, Sakai K, Jouvet M. Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci. 1996;16:1523–1537. doi: 10.1523/JNEUROSCI.16-04-01523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 29.Maloney KJ, Cape EG, Gotman J, Jones BE. High-frequency gamma electroencephalogram activity in association with sleep–wake states and spontaneous behaviors in the rat. Neuroscience. 1997;76:541–555. doi: 10.1016/s0306-4522(96)00298-9. [DOI] [PubMed] [Google Scholar]
- 30.Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep. 1994;17:436–437. doi: 10.1093/sleep/17.5.436. [DOI] [PubMed] [Google Scholar]
- 31.Miller JD, Farber J, Gatz P, Roffwarg H, German DC. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat. Brain Res. 1983;273:133–141. doi: 10.1016/0006-8993(83)91101-0. [DOI] [PubMed] [Google Scholar]
- 32.Montaron MF, Bouyer JJ, Rougeul A, Buser P. Ventral mesencephalic tegmentum (VMT) controls electrocortical beta rhythms and associated attentive behaviour in the cat. Behav Brain Res. 1982;6:129–145. doi: 10.1016/0166-4328(82)90010-9. [DOI] [PubMed] [Google Scholar]
- 33.Monti JM. Involvement of histamine in the control of the waking state. Life Sci. 1993;53:1331–1338. doi: 10.1016/0024-3205(93)90592-q. [DOI] [PubMed] [Google Scholar]
- 34.Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol. 1997;52:27–78. doi: 10.1016/s0301-0082(96)00070-6. [DOI] [PubMed] [Google Scholar]
- 35.Nishino S, Mao J, Sampathkumaran R, Shelton J, Mignot E. Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Res Online. 1998;1:49–61. [PubMed] [Google Scholar]
- 36.Parkes JD. Central nervous system stimulant drugs. In: Thorpy MJ, editor. Handbook of sleep disorders. Dekker; New York: 1990. pp. 755–778. [Google Scholar]
- 37.Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 38.Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW. Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience. 1997;79:225–235. doi: 10.1016/s0306-4522(96)00640-9. [DOI] [PubMed] [Google Scholar]
- 40.Reid MS, Tafti M, Nishino S, Sampathkumaran R, Siegel JM, Mignot E. Local administration of dopaminergic drugs into the ventral tegmental area modulates cataplexy in the narcoleptic canine. Brain Res. 1996;733:83–100. doi: 10.1016/0006-8993(96)00541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 42.Shelton J, Nishino S, Vaught J, Dement WC, Mignot E. Comparative effects of modafinil and amphetamine on daytime sleepiness and cataplexy of narcoleptic dogs. Sleep. 1995;18:817–826. [PubMed] [Google Scholar]
- 43.Siegel JM. Brainstem mechanisms generating REM sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice in sleep medicine. Saunders; Philadelphia: 2000. pp. 112–133. [Google Scholar]
- 44.Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, Lesch KP, Murphy DL, Uhl GR. Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci USA. 1998;95:7699–7704. doi: 10.1073/pnas.95.13.7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinfels GF, Heym J, Strecker RE, Jacobs BL. Behavioral correlates of dopaminergic unit activity in freely moving cats. Brain Res. 1983;258:217–228. doi: 10.1016/0006-8993(83)91145-9. [DOI] [PubMed] [Google Scholar]
- 46.Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep–waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 1999;840:138–147. doi: 10.1016/s0006-8993(99)01648-0. [DOI] [PubMed] [Google Scholar]
- 47.Steriade M. Brain electrical activity and sensory processing during waking and sleep states. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Saunders; Philadelphia: 2000. pp. 93–111. [Google Scholar]
- 48.Steriade M, McCarley RW. Brainstem control of wakefulness and sleep. Plenum; New York: 1990. [Google Scholar]
- 49.Trulson M. Simlutaneous recording of substantia nigra neurons and voltammetric release of dopamine in the caudate of behaving cats. Brain Res Bull. 1985;15:221–223. doi: 10.1016/0361-9230(85)90140-6. [DOI] [PubMed] [Google Scholar]
- 50.US Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol. 1998;43:88–97. doi: 10.1002/ana.410430115. [DOI] [PubMed] [Google Scholar]
- 51.Van Gelder RN, Edgar DM, Dement WC. Real-time automated sleep scoring: validation of a microcomputer-based system for mice. Sleep. 1991;14:48–55. doi: 10.1093/sleep/14.1.48. [DOI] [PubMed] [Google Scholar]
- 52.West CR, Lookingland KJ, Tucker HA. Regulation of growth hormone-releasing hormone and somatostatin from perifused, bovine hypothalamic slices. II. Dopamine receptor regulation. Domest Anim Endocrinol. 1997;14:349–357. doi: 10.1016/s0739-7240(97)00031-3. [DOI] [PubMed] [Google Scholar]
- 53.Wiegand M, Moller AA, Lauer CJ, Stolz S, Schreiber W, Dose M, Krieg JC. Nocturnal sleep in Huntington's disease. J Neurol. 1991;238:203–208. doi: 10.1007/BF00314781. [DOI] [PubMed] [Google Scholar]
- 54.Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang YM, Caron MG. Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci. 2000;3:465–471. doi: 10.1038/74839. [DOI] [PubMed] [Google Scholar]
- 55.Zahniser NR, Simosky JK, Mayfield RD, Negri CA, Hanania T, Larson GA, Kelly MA, Grandy DK, Rubinstein M, Low MJ, Fredholm BB. Functional uncoupling of adenosine A(2A) receptors and reduced response to caffeine in mice lacking dopamine D2 receptors. J Neurosci. 2000;20:5949–5957. doi: 10.1523/JNEUROSCI.20-16-05949.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Obal F, Fang J, Collins BJ, Krueger JM. Non-rapid eye movement sleep is suppressed in transgenic mice with a deficiency in the somatotropic system. Neurosci Lett. 1996;220:97–100. doi: 10.1016/s0304-3940(96)13232-8. [DOI] [PubMed] [Google Scholar]