

Abstract
The retinoblastoma tumor suppressor protein, pRb, is a key regulator of cell cycle and has been implicated in the terminal differentiation of neuronal cells. Mice nullizygous for pRb die by embryonic day 14.5 from hematopoietic and neurological defects attributed to failed differentiation (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). Previous studies by MacLeod et al. (1996)have demonstrated that the loss of p53 protects Rb-deficient CNS neurons but not peripheral nervous system (PNS) neurons from cell death. Thus, the mechanisms by which PNS neurons undergo apoptosis in response to Rb deficiency remain unknown. In view of the pivotal role of caspase 3 in the regulation of neuronal apoptosis during development, we examined its function in the execution of the wide-spread neuronal cell death induced by Rb deficiency. Our results support a number of conclusions. First, we show that caspase 3 becomes activated in all neuronal populations undergoing apoptosis. Second, caspase 3 deficiency does not extend the life span of Rb null embryos, because double null mutants exhibit high rates of liver apoptosis resulting in erythropoietic failure. Third, Rb/caspase 3 double-mutant neurons of the CNS exhibit widespread apoptosis similar to that seen in Rb mutants alone; thus caspase 3 deficiency does not protect this population from apoptosis. Finally, in contrast to the CNS, neurons of the PNS including those comprising the trigeminal ganglia and the dorsal root ganglia are protected from apoptosis in Rb/caspase 3 double-mutant embryos. Examination of the mechanistic differences between these two cell types suggest that CNS neurons may invoke other caspases to facilitate apoptosis in the absence of caspase 3. These findings suggest that PNS neurons are dependent on caspase 3 for the execution of apoptosis and that caspase 3 may serve as a key therapeutic target for neuroprotection after injury of this cell type.
Keywords: caspases, apoptosis, retinoblastoma, p53, development, peripheral nervous system
During embryogenesis, cycling neural progenitor cells in the ventricular zones commit to a neuronal fate, and as a consequence of that decision, undergo terminal mitosis and adopt a neuronal phenotype. These newly born neurons migrate through a complex environment, extend axons that find their way to targets, and ultimately recognize those targets and form synapses. A key developmental step in this process is the decision to undergo terminal mitosis. The importance of this event is underscored by the fact that failure to permanently withdraw from the cell cycle results in impaired differentiation and apoptosis (for review, see Slack and Miller, 1996). One key regulator of the cell cycle, the tumor suppressor protein, pRb, has been implicated in terminal mitosis and neuronal differentiation.
The retinoblastoma gene was the first tumor suppressor gene to be cloned and as such has been studied intensively in the field of oncogenesis (for review, see Mulligan and Jacks, 1998). Mice nullizygous for Rb die by embryonic day (E) 14.5 from hematopoietic and neurological defects attributed to failed terminal differentiation (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). By E12.5 onward, ectopic mitoses and massive cell death are observed throughout the developing nervous system, being most pronounced in sensory ganglia and the hindbrain. We have previously examined the temporal requirement for pRb during development relative to the commitment decision by introducing a “neuronal marker gene” into mice carrying a null mutation for pRb (Slack et al., 1998). This transgene consists of the neuron-specific Tα1 α-tubulin promoter (Gloster et al., 1994) driving a lacZ reporter gene (Tα1:nlacZ) that is induced as progenitor cells commit to a neuronal fate (Gloster et al., 1999). These studies demonstrated that the aberrations in the developing nervous system are widespread, and abnormal neuronal development was detected throughout the nervous system, including the olfactory epithelium, the retina, and the neocortex. On the basis of the timing of marker gene expression, it appears that pRb becomes essential immediately after commitment to a neuronal fate, and in its absence virtually all neuronal populations undergo apoptosis (Slack et al., 1998).
The mechanisms by which Rb-deficient neural precursors undergo apoptosis appear to be more complex than originally predicted. Although numerous studies have implicated p53 as the key mediator of apoptosis in the context of cell cycle deregulation (Morgenbesser et al., 1994;Ko and Prives, 1996), p53 deficiency surprisingly did not rescue all neuronal populations from cell death. Studies by MacLeod et al. (1996)demonstrated striking differences in the p53 requirement for neuronal apoptosis in Rb-deficient embryos. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining revealed that cell death in the Rb-mutant CNS but not in the PNS was suppressed in the absence of p53. Thus, the mechanisms by which cell death is evoked in the Rb-deficient peripheral nervous system remain unknown and are quite distinct from those observed in the CNS.
Previous studies have demonstrated that caspase 3 is a key determinant in naturally occurring neuronal apoptosis during embryogenesis. Embryos deficient in caspase 3 undergo embryonic or early postnatal lethality attributable to excessive cellularity and duplicated structures in the developing brain (Kuida et al., 1996). The widespread activation of caspase 3 in the developing nervous system as well as the finding that the death of sympathetic neurons deprived of NGF could be blocked by a cell-permeable pan-caspase inhibitor, bocaspartyl(OMe)-fluoromethylketone, lends further support to the importance of caspase activity in naturally occurring cell death (Deschmukh et al., 1996; Urase et al., 1998). In view of the pivotal role of caspase 3 in the regulation of neuronal apoptosis during nervous system development, we examined its function in executing the widespread neuronal cell death induced by Rb deficiency. In the present study, we show that caspase 3 is activated in all neuronal populations undergoing apoptosis, with highest levels of activity in the peripheral nervous system. The requirement for caspase 3 to execute apoptosis in the Rb-deficient nervous system is highly cell-type dependent such that caspase 3 disruption does not impair cell death in CNS neurons or cells of the developing liver. In contrast, PNS neurons exhibit a dramatic protection from programmed cell death in the absence of caspase 3. Our results indicate that caspase 3-deficient CNS neurons can induce the activation of other caspases to facilitate apoptosis. These studies suggest that caspase 3 is an essential regulator of apoptosis in PNS neurons and may serve as a key therapeutic target for maintaining the survival of injured cells in the peripheral nervous system.
MATERIALS AND METHODS
Transgenic mice
The Rb-deficient transgenic mice originally generated by Jacks et al. (1992) were purchased from The Jackson Laboratories (Bar Harbor, ME) and maintained on a C57BL6 genetic background. Mice were genotyped by PCR, as described previously (Jacks et al., 1992). Rb mice were interbred with transgenic mice carrying a null mutation for caspase 3 (Cregan et al., 1999; Keramaris et al., 2000) that were obtained from Dr. D. W. Nicholson (Merck Frosst Canada). The phenotype of the caspase 3-deficient mice used in this study was similar to those described previously (Kuida et al., 1996; Woo et al., 1999). All caspase 3-deficient mice were maintained on C57BL6 background to maintain genetic uniformity. Genotyping was performed by PCR in the usual PCR reaction buffer containing 0.25 mmMgCl2 and 5% DMSO (Cregan et al., 1999). The primers for the wild-type caspase 3 alleles were 5′-CTAAGTT- AACCAAACTGAGCACCGA-3′ (sense) and 5′-ATGAATGAAGG- CAGCATAGTACTCC-3′ (antisense). For the detection of the targeted allele, the same sense and the following antisense primer was used: 5′-GTCGATCCACTAGTTCTAGAGCGGC-3′. Conditions were set as follows: 94°C for 2 min (1 cycle); 94°C for 30 sec; 60°C for 1 min, 72°C for 1 min, (30 cycles); 72°C for 5 min (1 cycle). Mice deficient for both Rb and caspase 3 were generated by mating mice heterozygous for Rb and caspase 3, from which [Rb+/−:caspase 3+/−] progeny were mated to produce [Rb−/−:caspase 3−/−] double nullizygous mice. Embryos were removed at E13.5 d of gestation, and tissue samples of individual embryos were taken for genotyping before fixation in 4% paraformaldehyde for 4 hr. After washing in PBS, embryos were cryoprotected in 10% sucrose followed by fast freezing. The tissue was sectioned at 14 μm followed by immunostaining or TUNEL assay. Embryos were not used at later time points because of the high level of lethality found at E14.5 onward.
Determination of cell death
TUNEL staining. For TUNEL staining, frozen sections were treated with acetone/methanol (1:1) for 1 min followed by three washes with PBS. Sections were then incubated for 1 hr at 37°C with 75 μl of a mixture (Roche Diagnostics, Mississauga, ON) consisting of 0.5 μl terminal deoxynucleotide transferase (TdT), 0.95 μl biotin-16-dUTP, 6.0 μl CoCl2, 15.0 μl 5× TdT buffer, and 52.55 μl distilled water. After three washes in PBS, sections were incubated with a streptavidin CY2 (Jackson Immunoresearch Laboratories, West Grove, PA). After three washes, sections were counterstained with Hoechst and examined with a Zeiss Axioskop fluorescent microscope. For cell counting the following regions in the nervous system of [wild type], [Rb−/−], [caspase 3−/−], [Rb−/−:caspase 3 −/−] littermates were selected: forebrain at the medial aspect of the ganglionic eminence; hindbrain at the caudal pons adjacent to the fourth ventricle, trigeminal ganglion (TG), caudal, and rostral dorsal root ganglion (DRG). At 40× magnification, images were captured using Northern Eclipse software, and a defined 100 μm2area at the center of the ganglia was counted for each specimen. The data were expressed as the number of TUNEL-positive cells as a percentage of the total cell count as determined by Hoechst.
Fluoro-jade staining. To evaluate neuronal degeneration including both apoptotic and necrotic modes of cell death, Fluoro-jade labeling was used (Schmued et al., 1997; Noraberg et al., 1999). Fluoro-jade staining was performed on sections immunolabeled for ΔC-amyloid-β-precursor protein (ΔC-APP) (see Immunostaining for details). Briefly, sections were washed in three changes of PBS (10 min each) after immunolabeling for ΔC-APP, followed by a brief 1 min rinse in distilled water. The sections were then stained with 0.00001% Fluoro-jade in 0.1% acetic acid for 20 min at room temperature. After washing in three changes of distilled water (1 min each), the slides were dried on a slide warmer, and coverslips were mounted with D.P.X. neutral mounting media (Sigma-Aldrich Canada, Oakville, ON).
Immunostaining
Caspase activation. Active caspase 3 was detected immunohistochemically by using three different antibodies. These antibodies were anti-caspase 3 from PharMingen (San Diego, CA) and anti-neoepitope and anti-active caspase 3 antibody from Merck Frosst. Among the three antibodies, the former two selectively recognize the p17 fragment of caspase 3, the larger subunit of the active enzyme, whereas the third was directed against the catalytically active (p17/p12) conformer. Fresh-frozen sections (14 μm) were incubated with the primary antibodies [anti-caspase 3 (PharMingen), 1:1000; anti-neoepitope (Merck Frosst), 1:1000; and anti-active caspase-3 (Merck Frosst), 1:2000] at 4°C for 48 hr. After three washes with PBS (10 min each), the sections were incubated with CY3-labeled donkey anti-rabbit IgG (1:800; Amersham, Buckinghamshire, UK) for 2 hr at room temperature.
Detection of the caspase-cleaved fragment of APP in apoptotic neurons. A polyclonal antibody was used for the detection of the neoepitope, designated as ΔC-APP, which is generated by caspase-mediated cleavage of APP and has been detected in neurons undergoing apoptosis by multiple death stimuli (Gervais et al., 1999). The αΔCCsp-APP antibody was confirmed to be highly specific for the neo epitope generated by caspase cleavage of APP on the basis of the following experiments. (1) ELISA titer was >2000-fold selective for the neo epitope (ΔC-APP) versus the peptide containing the sequence corresponding to intact APP; (2) biosynthetic APP that was truncated at the D720 caspase site but not the intact APP could be immunoprecipitated by the antibody; and (3) the antibody was unable to immunoprecipitate intact APP from nonapoptotic NT2 cells, but after induction of apoptosis the ΔC-APP caspase cleavage product was efficiently immunoprecipitated from these cells (Gervais et al., 1999). Fresh-frozen sections were incubated with the α-ΔCcsp-APP antibody at a dilution of 1:500 overnight at 4°C followed by a CY3 donkey anti-rabbit IgG (as above).
Protein gene product 9.5. A monoclonal antibody directed against protein gene product 9.5 (PGP 9.5) was used as a neuronal marker, staining neuronal cell bodies and axons of neurons in the CNS, periphery, small nerve fibers in the peripheral tissues, neuroendocrine cells in the pituitary, thyroid, and pancreas (Wilson et al., 1988). The polyclonal antibody to PGP 9.5 (Cedarlane Laboratories, Hornby, ON) was diluted at 1:500 followed by a CY3 anti-rabbit secondary.
B-III tubulin. Class III β-tubulin was used as an early neuronal marker that is normally induced at the time of neuronal commitment (Gloster et al., 1994). A monoclonal antibody directed against this protein described previously (Caccamo et al., 1989) was a gift from Dr. David Brown (University of Ottawa). The hybridoma supernatant was diluted at 1:10 in 5% goat serum and incubated at 4°C for 24 hr. After three washes in PBS, a secondary antibody, Alexa fluor 488 anti-mouse (1:2000; Molecular Probes, Eugene, OR) was applied for 1 hr at room temperature.
Trk A. A polyclonal antibody directed against Trk A [kindly supplied by Dr. Louis Riechardt (Clary et al., 1994)] was used as a differentiation marker for peripheral neurons at 1:200 followed by a CY2 goat anti-rabbit secondary (1:500; Jackson ImmunoResearch Laboratories).
Western blot analysis
Tissue was extracted in lysis buffer (50 mm HEPES, pH 7.8, 250 mm KCl, 0.1 m EDTA, 0.1m EGTA, 10% glycerol, 0.1% NP-40, 1.0 mm DTT, 0.5 mm PMSF, 5 μg/ml aprotinin, 2 μg/ml leupeptin, 0.4 mm sodium vanadate), and aliquots containing 30 μg of protein were separated on a 10% acrylamide gel and transferred to a nitrocellulose membrane. After blocking for 2 hr with 5% skim milk, membranes were incubated for 1 hr with a goat polyclonal antibody directed against caspase 2, actin (Santa Cruz Biotechnologies, Santa Cruz, CA), or rabbit polyclonal antibodies against caspases 6 and 7 (StressGen Biotechnologies, Victoria, British Columbia). After three washes with TPBS (25 mmNa2HPO4, 5 mmNaH2PO4, 0.9% NaCl, 0.1% Tween 20), membranes were incubated for 1 hr at 25°C with the appropriate secondary antibody, washed five times for 5 min each in TPBS, and then developed by an enhanced chemoluminescence system according to the manufacturers instructions (Perkin-Elmer, Boston, MA). To verify the identify of caspase 2, a competitor peptide (Santa Cruz) was used according to the manufacturer's instructions.
RESULTS
Caspase 3 is induced in the Rb-deficient nervous system
To determine whether caspase 3 plays any role in the widespread neuronal apoptosis in the Rb-deficient nervous system, we first asked whether caspase 3 is activated in neuronal populations undergoing cell death. Caspase 3 activation was examined using three different antibodies that selectively recognize the products of caspase 3 activation: p17 fragment or the active (p17/p12) tetramer (Nicholson et al., 1995; Cohen, 1997; Rasper et al., 1998). Rb-deficient embryos exhibited a striking increase in the activated species of caspase 3 that appeared throughout the developing nervous system in regions where there were high rates of neuronal apoptosis (Fig.1). PNS neurons lacking Rb exhibited a high proportion of active caspase 3-positive cells that was particularly striking in the DRGs (Fig. 1C). Similarly, the CNS also contained many positive cells for active caspase 3 in regions undergoing high levels of apoptosis (Fig.1G). Immunoreactivity for active caspase 3 was also observed in a small number of CNS and PNS neurons in the healthy wild-type littermates demonstrating caspase 3 involvement in developmental apoptosis (Fig. 1A,E). Caspase 3 activation, however, was not detectable in tissue derived from caspase 3 null and Rb/caspase 3 double knock-out littermates (Fig.1B,D,F,H). The results of these studies suggest that caspase 3 might play an important role in regulating the widespread apoptosis in the nervous system of Rb-deficient mice. We therefore asked whether caspase 3 is essential for the execution of apoptosis in Rb-deficient neurons.
Fig. 1.
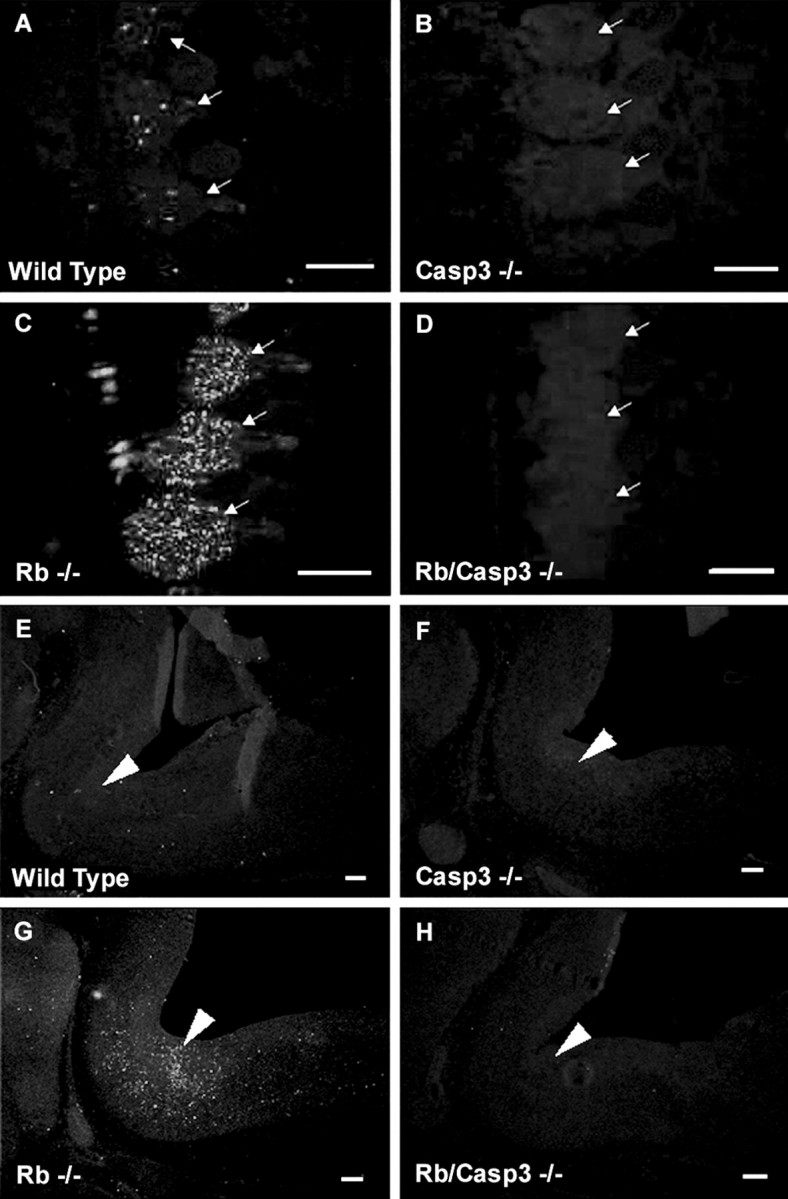
Caspase 3 is activated in the Rb-deficient mouse nervous system. Active caspase 3 immunostaining of DRG (A–D) and hindbrain (E–H) from E13.5 embryos is shown. Extensive caspase 3 activation is seen throughout the developing nervous system of the Rb−/− embryo (C,G) and is present at low levels in wild-type tissue (A, E) and undetectable in caspase 3−/− (B, F) or Rb−/−/caspase 3−/− (D, H) samples. Arrows indicate DRGs; arrowheadspoint to the pons. Scale bars, 300 μm.
Caspase 3 deficiency rescues the peripheral nervous system defect in Rb-deficient mice
Mice carrying a null mutation for pRb were interbred with mice heterozygous for a targeted mutation for caspase 3. The progeny of these mice (Rb+/−:caspase 3+/−) were viable and fertile and were then interbred to generate double null embryos (Rb−/−:caspase 3−/−). The frequencies of the genotypes obtained from these crosses are shown in Table 1. Embryos were routinely examined at E13.5, which represents the time at which apoptosis was detectable in virtually all neuronal populations. At time points later than E14, a large proportion of Rb or Rb/caspase 3-deficient embryos were already dead. Gross morphological comparison of Rb-deficient and Rb/caspase 3-deficient embryos did not reveal any striking morphological differences other than those previously described for Rb or caspase 3 null embryos alone (data not shown). Furthermore, caspase 3 deficiency did not rescue the hematopoietic defect manifest in Rb deficiency, as evident by the pale color caused by massive apoptosis in the developing liver (see below). Rb/caspase 3-deficient embryos also exhibited the typical hunchback appearance caused by swelling in the region of the fourth ventricle, as described previously for the Rb knockout (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). Thus, gross examination of Rb/caspase 3-deficient embryos revealed embryonic defects typically seen by E13.5 in Rb deficiency alone, and survival was not extended because of the hematopoietic failure in these embryos.
Table 1.
Genotype frequency of offspring from interbreeding Rb+/−/CASP3+/− mice
| Genotype of offspring (expected frequency, %) | Number of E13.5 embryos | Frequency (%) | |
|---|---|---|---|
| RB+/+/CASP3+/+ | (6.25) | 11 | 6.9 |
| RB+/+/CASP3+/− | (12.5) | 20 | 12.5 |
| RB+/+/CASP3−/− | (6.25) | 10 | 6.3 |
| RB+/−/CASP3+/+ | (12.5) | 21 | 13.1 |
| RB+/−/CASP3+/− | (25) | 46 | 28.7 |
| RB+/−/CASP3−/− | (12.5) | 18 | 11.2 |
| RB−/−/CASP3+/+ | (6.25) | 5 | 3.1 |
| RB−/−/CASP3+/− | (12.5) | 22 | 13.7 |
| RB−/−/CASP3−/− | (6.25) | 7 | 4.4 |
E13.5 embryos (n = 160) were examined.
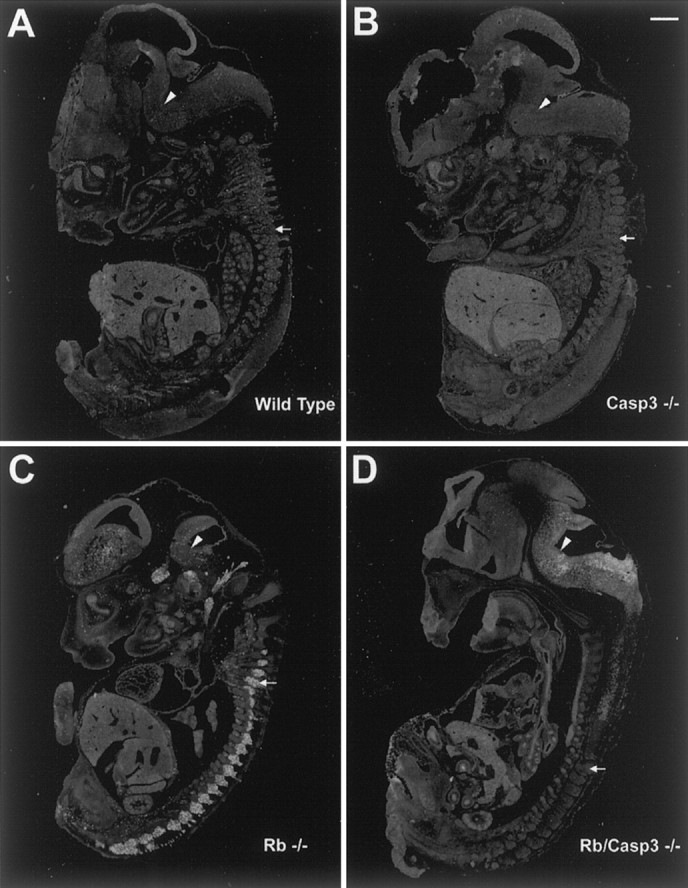
To determine whether any of the Rb-deficient cell populations were protected from apoptosis in the absence of caspase 3, littermates of the following genotypes were examined: [wild type]; [caspase 3−/−]; [ Rb−/−]; and [Rb−/−:caspase 3−/−]. Embryos were removed at E13.5, and tissue was sectioned and stained with the TUNEL reagent for the detection of apoptotic cells (Fig.2). Wild-type and caspase 3-deficient embryos exhibited very low levels of TUNEL-positive cells (Fig.2A,B), whereas Rb-deficient embryos exhibited intense TUNEL staining in the liver, CNS (including the forebrain, hindbrain, and spinal cord), and the peripheral nervous system (Fig. 2C). This is the typical phenotype of Rb-deficient embryos (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). In contrast, examination of the Rb null phenotype on a caspase 3-deficient background revealed a striking protection of a very specific cell population (Fig. 2D). Consistent with the pale color of Rb/caspase 3 null embryos, there was intense apoptosis in the liver, indicating that the hematopoeitic defect that causes the early death of the embryo was not rescued by the absence of caspase 3. Similarly, the CNS including the hindbrain and forebrain structures exhibited massive apoptosis at levels similar to Rb-deficient embryos. Thus, apoptosis in structures including the liver and the CNS could not be rescued by the absence of caspase 3. In contrast, neurons of the peripheral nervous system were significantly protected from cell death as evident by the absence of TUNEL-positive cells in the DRGs despite the advanced phenotype of this particular E13.5 Rb/caspase 3 double null embryo. Note that there are very few apoptotic cells in the DRGs, yet the levels of apoptosis in the liver and hindbrain are striking in this particular embryo. Although these experiments suggest that the absence of caspase 3 protects neurons of the peripheral nervous system from apoptosis, a closer examination of the structures was performed.
Fig. 2.

Caspase 3 deficiency protects neurons of the PNS but not the CNS or liver from apoptosis induced by Rb deficiency. Sections from wild type (A), caspase 3−/− (B), Rb−/− (C), and Rb−/−/caspase 3−/− (D) were stained for TUNEL, and whole embryo images were captured. Arrowsindicate rostral and caudal DRGs. Arrowheads indicate hindbrain. Scale bar, 1.5 mm.
To assess the progression of neuronal apoptosis in the absence of caspase 3, specific structures were examined in detail including (1) the forebrain at the medial aspect of the ganglionic eminence, (2) the hindbrain at the caudal pons adjacent to the fourth ventricle, (3) the TGs, and (4) the rostral and caudal DRGs. Sections were stained with a neuronal marker PGP 9.5 (Wilson et al., 1988) to identify the ganglia in the peripheral nervous system and then labeled with two markers for cell death, including the TUNEL reagent for detection of apoptotic cells and Fluoro-jade (Schmued et al., 1997; Noraberg et al., 1999), for the detection of both apoptotic and necrotic cells. The number of TUNEL-positive cells was counted in these specific regions and expressed as a percentage of total cell count in the field. The cell counts for each region are summarized in Figure3.
Fig. 3.

Caspase 3 deficiency results in decreased TUNEL labeling in the PNS but not the CNS of the Rb−/− mouse embryo. TUNEL-positive cells in the hindbrain, trigeminal ganglion, and caudal and rostral DRGs were quantified by examination at 40× magnification. The mean is expressed as a percentage of TUNEL-positive cells/total cell number as determined by Hoechst staining within a 100 μm2 area for each group. Counts were obtained from three independent embryos (n = 3), and error bars indicate SE. *p < 0.001.
Although there were few TUNEL-positive cells in wild-type and caspase 3-deficient hindbrains (Fig.4A–D), Rb-deficient brains exhibited many apoptotic cells, and this enhanced rate of apoptosis was not affected by the absence of caspase 3 (Fig.4E–H). Examination of forebrains in wild-type, Rb-deficient, caspase 3-deficient, and the double null littermates produced similar results (data not shown). These results demonstrate that Rb-deficient CNS neurons lacking caspase 3 are not protected from programmed cell death and that these cells exhibit DNA fragmentation as detected by TUNEL labeling that is typical of apoptosis.
Fig. 4.
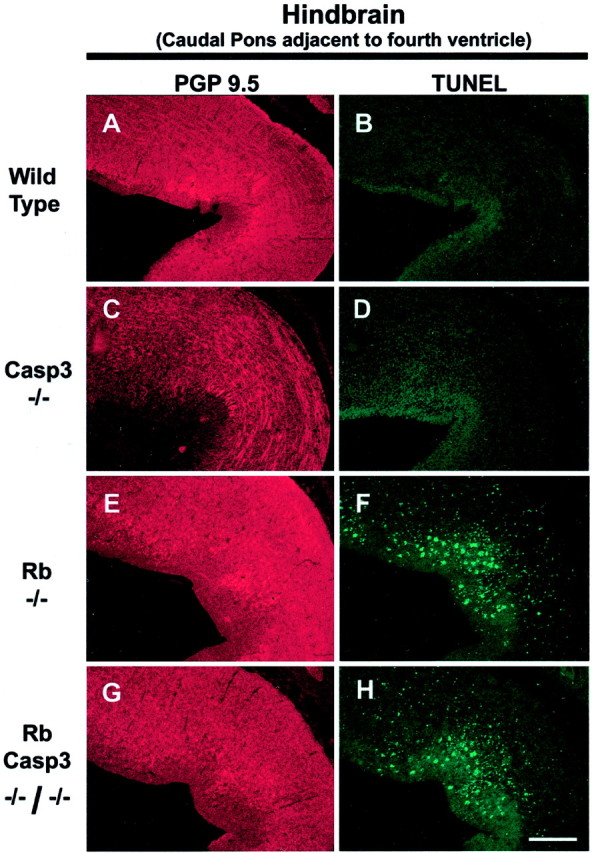
Caspase 3 deficiency does not rescue CNS neurons of Rb-deficient embryos from apoptosis. Frozen sections of wild-type (A, B), caspase 3−/− (C, D), Rb−/− (E, F), and Rb−/−/caspase 3−/− (G, H) E13.5 mouse brain were stained for TUNEL. Scale bar, 150 μm.
Although cells of the CNS exhibited widespread cell death despite the absence of caspase 3, neurons of the peripheral nervous system appeared to be protected from apoptosis. Massive cell death in the developing DRGs and TGs is one of the hallmarks of the Rb phenotype (Lee et al., 1992, 1994). In the absence of both Rb and caspase 3, a dramatic protection from apoptosis was found (Fig.5M–P), such that these ganglia remained intact with very few apoptotic cells. Specifically, in the absence of Rb alone, 55 ± 8% of the cells in the TGs are TUNEL positive, whereas DRGs were found to be 53 ± 9% TUNEL positive (Fig. 5I–L). When both Rb and caspase 3 are absent, the rate of neuronal cell death is dramatically reduced to 2.5 ± 0.3% in TGs and 2.5 ± 0.6% in DRGs (Fig.5M–P). These findings suggest that caspase 3-deficient DRGs and TGs are protected from neuronal cell death induced by the loss of Rb. Thus, histological examination and cell counting within specific regions of the Rb-deficient embryo reveal high rates of cell death throughout the entire nervous system. In contrast, mice deficient for both caspase 3 and Rb exhibit massive cell death in the liver and the CNS; however, apoptosis in the peripheral nervous system appears to be abated, exhibiting rates similar to those observed in wild-type littermates.
Fig. 5.
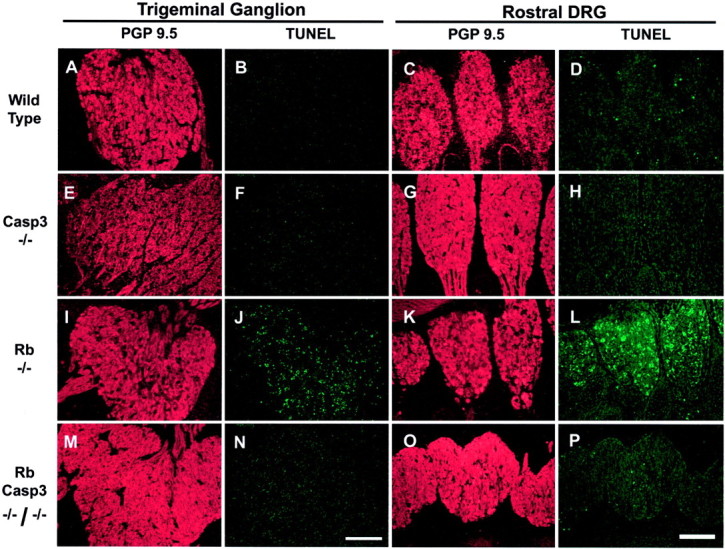
Caspase 3 is required for apoptosis of the Rb-deficient trigeminal and dorsal root ganglia neurons. Frozen sections from wild type (A–D), caspase 3−/− (E–H), Rb−/− (I–L), and Rb−/−/caspase 3−/− (M–P) were stained for TUNEL and counterstained with PGP 9.5 to view neuronal cell bodies and axons.Panels on left show Trigeminal Ganglion, and panels onright show Rostral DRG. Scale bars, 160 μm.
Although the peripheral nervous system, including the DRGs and the trigeminal ganglia, exhibits TUNEL staining at levels similar to wild type, one possibility is that these cells may nevertheless die by a more necrotic mechanism because of the absence of caspase 3, and this caspase-independent mechanism may not be readily detectable by TUNEL staining. To rule out this possibility, embryos were stained with Fluoro-jade for the detection of all dying cells, including necrotic as well as apoptotic death pathways (Schmued et al., 1997; Noraberg et al., 1999). To confirm protection from cell death in the peripheral nervous system, sections from wild-type, Rb−/−, caspase 3−/−, and Rb/caspase 3 double null were double stained for caspase activation and Fluoro-jade to identify dying cells. Caspase activity was detected using a polyclonal antibody directed against the neoepitope, designated ΔC-APP. Sections were then double labeled with Fluoro-jade to identify dying cells. The results in Figure6 demonstrate that Rb-deficient DRGs exhibit abundant caspase activity coincident with striking positive staining for Fluoro-jade suggesting that most of the neurons of Rb-deficient DRGs are undergoing cell death (Fig.6A,B). In contrast, DRGs from Rb/Caspase 3 double null embryos exhibit no detectable levels of ΔC-APP and little positive staining for Fluoro-jade (Fig.6C,D). These staining levels are equivalent to that detected in healthy wild-type or caspase 3-deficient littermates (data not shown). Thus, consistent with the results from TUNEL staining, Rb-deficient DRGs appear to be protected from apoptosis when caspase 3 is absent.
Fig. 6.

Fluoro-jade labeling demonstrates caspase 3 requirement for neuronal cell death in the Rb-deficient peripheral nervous system. Shown are ΔC-APP (A, C) and Fluoro-jade (B, D) double labeling of degenerating DRG neurons in Rb−/− (A, B) and Rb/caspase 3 double knock-out embryos (C, D). Arrows indicate DRGs. Scale bar, 260 μm.
Caspase 3 deficiency restores pan-neuronal gene expression in DRGs of Rb null embryos
Although our data so far suggest that neurons of the peripheral nervous system are protected from apoptosis, we questioned whether the surviving cells expressed neuronal differentiation markers. Previous studies indicated that Rb-deficient DRGs exhibit reduced expression of neuronal genes such as β-II tubulin and Trk A (Lee et al., 1994). We therefore examined the expression of these markers to determine whether the absence of caspase 3 could restore pan-neuronal gene expression in Rb-deficient peripheral neurons. The antibody directed against the nerve growth factor receptor, Trk A, stained peripheral neurons in wild-type and caspase 3-deficient embryos (Fig.7A,B). As described previously, Rb-deficient DRGs exhibit very low levels of Trk A immunostaining (Fig. 7C) relative to wild-type littermates (Fig. 7A). In contrast, Trk A immunoreactivity was restored to wild-type levels in Rb/Caspase 3 double null embryos (Fig. 7D). Similar results were obtained for β-III tubulin, whereby Rb-deficient embryos exhibited very little β-III tubulin immunoreactivity in the DRGs relative to wild-type littermates. Consistent with results obtained with Trk A, β-III tubulin immunoreactivity was restored to wild-type levels when both Rb and caspase 3 were absent (data not shown). Thus, examination of pan-neuronal gene expression indicates that the absence of caspase 3 in the Rb-deficient peripheral nervous system not only reduces apoptosis but also allows cells to continue to differentiate, consistent with the interpretation that caspase 3 deficiency rescues the apoptotic phenotype in the Rb-deficient peripheral nervous system.
Fig. 7.
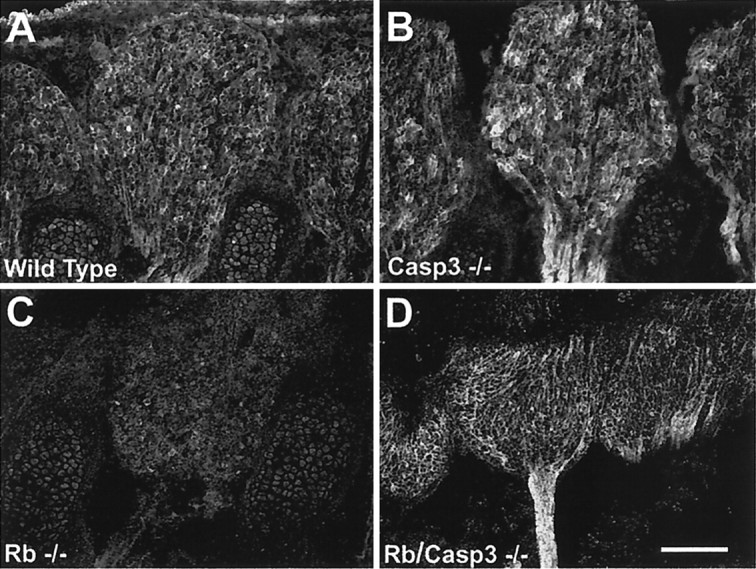
Rb−/−:caspase 3−/− double mutants exhibit normal levels of Trk A expression. Trk A is highly expressed in wild-type (A) and caspase 3−/− (B) DRGs by E13.5. This staining is dramatically reduced in Rb-deficient DRG neurons (C); however, Trk A staining is restored to wild-type levels when caspase 3 is also absent (Rb/Casp3−/−) (D). Scale bar, 150 μm.
Rb/caspase 3-deficient CNS neurons exhibit caspase activity
Because our data demonstrate that the mechanisms regulating neuronal cell death in the CNS are significantly different from those of the PNS, we sought to determine the molecular mechanisms involved. We questioned whether there may be any difference in compensatory caspase activation between the different regions of the developing nervous system by examining the appearance of a caspase cleavage product, ΔAPP. Because of the very limiting amounts of CNS and PNS tissue available from Rb/caspase 3 double mutants, APP cleavage was monitored immunohistochemically (Figs. 8, 9). Wild-type embryos exhibited low levels of the product ΔC-APP in the developing nervous system consistent with the ongoing naturally occurring cell death that occurs at this time (Figs.8A, 9A,E). Slightly less cleavage of ΔC-APP was detected in the caspase 3 null embryos consistent with the impairment of developmental cell death reported previously (Kuida et al., 1996) (Figs. 8B,9B,F); however, it should be noted that the cleavage product of APP was also detected when caspase 3 was absent. This indicates that APP can be cleaved by other caspases in the absence of caspase 3. Rb-deficient embryos showed extensive cleavage of APP throughout the developing nervous system (Figs.8C, 9C,G). In the Rb/caspase 3 double null embryos, very little cleavage product was detected in the PNS, suggesting that there was no detectable caspase cleavage of APP (Figs.8D, 9H). In contrast, neurons of the CNS exhibited intense positive staining for ΔC-APP (Figs.8D, 9D), consistent with the interpretation that CNS neurons may contain other caspase activity that is sufficient to execute cell death. To determine whether CNS cells do indeed contain additional caspases that may become activated in the absence of caspase 3, Western analysis was performed using CNS tissue derived from littermates of all four genotypes (wild type, Rb−/−, caspase 3−/−, and Rb/caspase 3 double null). Using antibodies directed against caspases 2, 6, and 7, the proteins bands corresponding to the proenzymes were readily detectable in tissue derived from all genotypes (Fig. 10). Although there was no compensatory upregulation of any of these caspases, caspase 2 did exhibit bands corresponding to the intermediate N-terminal cleavage product of ∼35–38 kDa specifically in the Rb/caspase 3-deficient extracts. The specificity of these bands was confirmed by competition with the N-terminal peptide. These results indicate that the caspase 2 proenzyme was being cleaved and activated in CNS tissue lacking caspase 3. In contrast to CNS tissue, no immunostaining for caspase cleavage product ΔC-APP could be detected in either TGs or DRGs (Figs. 8, 9). These results suggest that peripheral neurons such as those of the DRG and TG are dependent on caspase 3 to execute apoptosis.
Fig. 8.
Cleavage of the caspase substrate, APP, in E13.5 mouse embryos. Shown is immunodetection of ΔC-APP in wild-type (A), caspase 3−/− (B), Rb−/− (C), and Rb−/−/caspase 3−/− (D) E13.5 mouse embryos. The caspase 3 cleavage product, ΔC-APP, is detected in all neuronal populations undergoing apoptosis in the Rb null mouse (C). ΔC-APP is detected in the CNS but is significantly reduced in the PNS of Rb−/−/caspase 3−/− compound mutant embryos. Arrowsindicate DRGs. Arrowheads indicate hindbrain. Scale bar, 1.5 mm.
Fig. 9.

APP is cleaved in the CNS but not PNS neurons of Rb/caspase 3 double null mutants. Shown is immunodetection of cleaved caspase substrate, ΔAPP, in hindbrain (A–D) and DRGs (E–H) from E13.5 embryos.A, E, Wild type; B, F, caspase 3−/−; C, G, Rb−/−;D, H, Rb−/−/caspase 3−/−. Extensive ΔAPP is seen throughout the developing nervous system of the Rb−/− embryo, including hindbrain (C) and DRGs (G), and is present at low levels in corresponding wild-type tissue (A, E). ΔAPP is abundant in Rb−/−/caspase 3−/− hindbrain (D), showing compensatory caspase activation, but is undetectable above control levels in Rb−/−/caspase 3−/− DRGs (H), showing the lack of compensatory caspase-mediated ΔAPP cleavage. Scale bars, 300 μm.
Fig. 10.

Expression of other caspases in CNS tissue. CNS tissue obtained from embryos of the following genotypes, wild type, caspase 3−/−, Rb−/−, and Rb/caspase 3 double null, was extracted from forebrain (For.) and hindbrain (Hind.) and separated by SDS-PAGE. Protein was transferred to nitrocellulose and probed with antibodies directed against caspase 2 (Pro-Casp2), 6 (Pro-Casp6), and 7 (Pro-Casp7) as well as actin. A competing peptide was used to confirm specificity of caspase 2 bands and its cleavage product.
DISCUSSION
The results of these studies support a number of conclusions. First, caspase 3 becomes activated in neuronal populations undergoing apoptosis in Rb-deficient embryos. Second, caspase 3 deficiency does not extend the life span of the Rb null embryo, because double null mutants exhibit high rates of apoptosis in the developing liver and the CNS. Third, loss of caspase 3 results in the protection of a very specific neuronal population, in particular neurons of the trigeminal ganglia and the dorsal root ganglia that were protected from apoptosis induced by Rb disruption. Finally, our results using an antibody directed against the cleaved caspase substrate ΔAPP suggest that neurons of the CNS are capable of activating other caspases when caspase 3 is absent, whereas no such activity was found in the PNS. Thus, neurons of the peripheral nervous system may be dependent on caspase 3 for the execution of apoptosis.
The Rb-deficient embryo dies between E13.5 and E14.5, exhibiting neurological as well as hematopoietic defects (Clarke et al., 1992;Jacks et al., 1992; Lee et al., 1992). The widespread apoptosis in virtually all neuronal populations examined provides an ideal model for the study of apoptosis in the developing nervous system. Presently, the signal triggering apoptosis of neurons in Rb null embryos remains poorly understood. Although cell cycle deregulation can cause apoptosisin vivo, presumably as a result of enhanced proliferation, such a defect does not cause neuronal cell death in vitrowhere growth factors are present in unlimited supply (Slack et al., 1998; Callaghan et al., 1999); thus the precise trigger evoking neuronal cell death in vivo remains unclear. Enhanced proliferation of neural progenitors causing an intrinsic delay in differentiation may lead to cell death in vivo, where differentiation cues are precisely timed and growth factors are limiting. The limitation of trophic support in the Rb-deficient nervous system has been suggested previously (Lee et al., 1994; Maandag et al., 1994; Williams et al., 1994); thus apoptosis in Rb null mice may represent a model for naturally occurring cell death during development.
The molecular mechanism evoking cell death in the Rb-deficient nervous system appears to be quite complex and depends on the neuronal cell type being examined. P53 has been implicated as the molecular switch triggering the demise of Rb-deficient cells in the developing lens (Morgenbesser et al., 1994); however, the mechanisms regulating neuronal apoptosis appear to be less clear. The generation of p53/Rb double null embryos did not result in global neuroprotection nor was the life span of the embryos increased. Although there was significant protection in the CNS, no such protection was observed in the peripheral nervous system (MacLeod et al., 1998). These studies suggest that the molecular mechanisms regulating neuronal apoptosis in the peripheral nervous system are clearly distinct from those of the CNS and are presently unknown.
Interestingly, a recent study examining the role of caspase 3 in the demise of Bcl-XL-deficient neurons has revealed significant protection in all neuronal populations, including the CNS (Roth et al., 2000). The Bcl-XL null phenotype is strikingly similar to that of the Rb knock-out, exhibiting widespread neuronal apoptosis and early embryonic lethality caused by failed liver hematopoiesis. At E12.5, however, caspase 3 deficiency appears to protect all neuronal populations; even the large apoptotic clusters appearing in the Bcl-XL deficient CNS were absent when caspase 3 was also disrupted. In contrast, in the present studies, the absence of caspase 3 resulted in the protection of only PNS neurons but not those of the CNS. This suggests that although the Bcl-XL and Rb null mice exhibit similar phenotypes, the molecular pathways induced by Rb deficiency in the CNS may differ significantly from those triggered by the absence of Bcl-XL.
Because of the importance of caspase 3 in regulating neuronal cell death in development, we asked whether it plays a pivotal role in neuronal apoptosis in response to Rb deficiency. Our results demonstrate that most of the cell populations, in particular, the liver and neurons of the CNS, do not require caspase 3 to execute apoptosis or even DNA fragmentation (Fig. 3). Previous studies have indicated that p53 is a key determinant in neuronal apoptosis occurring in the CNS (MacLeod et al., 1996). We have shown previously that cells induced to die by a p53-dependent mechanism or by direct upregulation of p53 exhibit high levels of caspase 3 activation. Although caspase 3 appears to be a key component of the p53-mediated cell death pathway, its absence delays but does not protect such neurons from apoptosis (Cregan et al., 1999; Keramaris et al., 2000). Similar results from other groups have demonstrated that despite the involvement of caspases in neuronal cell death, pharmacological blockers of caspase activity could not protect CNS neurons from apoptosis (Miller et al., 1997; Johnson et al., 1998). One possibility is that CNS neurons may be capable of activating compensatory caspases. Recent studies examining Fas-mediated apoptosis have demonstrated alternative caspase activation in caspase 3 null hepatocytes that is not normally found in wild-type cells (Zheng et al., 2000). Although there was no difference in levels of caspase transcripts, biochemical analysis revealed that caspase 3 null hepatocytes activate caspase 6 and 7 preceding cell death. Thus it is likely that the lack of protection by caspase 3 disruption in hepatocytes may be accounted for by this compensatory caspase activation.
Our results indicate that the molecular determinants that regulate apoptosis in the peripheral nervous system are distinct from those of the CNS. Unlike CNS neurons, our results show that neurons of the DRG and TG are protected from apoptosis when caspase 3 is inactivated by homologous recombination. Although CNS neurons continue to undergo apoptosis, neurons of the PNS continue their development, as evident by the continued expression of TrkA and βIII-tubulin, until the death of the embryos resulting from hematopoietic failure. These data are consistent with the interpretation that PNS neurons are protected from apoptosis in the absence of caspase 3. Because compensatory caspase activation has been demonstrated in hepatocytes (Zheng et al., 2000), we asked whether this might account for the differences in neuroprotection observed in the Rb/caspase 3-deficient nervous system. Tissue from Rb/caspase 3 double null nervous system and corresponding littermates (wild type, Rb null, and caspase 3 null) was examined for caspase activation immunohistochemically using an antibody directed against the caspase cleaved substrate ΔC-APP, as described previously. In the absence of caspase 3, Rb-deficient neurons of the CNS exhibited intense staining for the caspase cleavage product ΔC-APP, whereas no such cleavage product was found in peripheral neurons. This suggests that CNS neurons may activate other caspases to execute apoptosis, although neurons of the periphery lack this activity. Indeed, Western analysis revealed the presence of additional caspases in CNS tissue, including caspases 2, 6, and 7. Caspase 2 exhibited the typical intermediate N-terminal cleavage product of ∼35–38 kDa seen after activation specifically in the Rb/caspase 3 double knock-out. This suggests that caspase 2 may become activated and cleave substrates common to caspase 3 to execute apoptosis in CNS neurons. Indeed previous studies have shown that (1) the caspases 2, 3, and 7 have similar activities and are capable of cleaving common substrates such as DEVD (Thornberry et al., 1997), and (2) caspase 2 and 3 activation was found in cortical neurons induced to die by camptothecin, a p53-dependent process (Stefanis et al., 1999;Keramaris et al., 2000).
In summary, our results clearly demonstrate that different neuronal subpopulations evoke distinct death mechanisms despite a similar death stimulus. The ability of CNS neurons to induce caspase activation as evident by ΔC-APP cleavage may account for the lack of protection in the absence of caspase 3. Taken together, our results indicate that PNS neurons, in particular those comprising the DRGs and TGs, may be dependent on caspase 3 activity to execute neuronal cell death, implicating caspase 3 as a key target for therapeutic intervention in the treatment of injured peripheral neurons.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to R.S.S. R.S.S is a CIHR Scholar, and D.S.P. is a Glaxo Wellcome Professor. K.L.F. is supported by a studentship from CIHR, and J.L.V. is supported by a fellowship from the Canadian Stroke Network (CSN). We are indebted to Drs. Freda Miller, Sean Cregan, and William Staines for critical review of this manuscript.
M.T.W.S. and J.G.M. contributed equally to this work.
Correspondence should be addressed to R. S. Slack, Neuroscience Research Institute, University of Ottawa, 451 Smyth Road, Ottawa, Ontario, K1H-8M5, Canada. E-mail: rslack@uottawa.ca.
REFERENCES
- 1.Caccamo D, Katsetos CD, Herman MM, Frankfurter A, Collins VP, Rubinstein LJ. Immunohistochemistry of a spontaneous murine ovarian teratoma with neuroepithelial differentiation. Lab Invest. 1989;60:390–398. [PubMed] [Google Scholar]
- 2.Callaghan DA, Dong L, Callaghan SM, Hou YX, Dagnino L, Slack RS. Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F1 and 3 activity. Dev Biol. 1999;207:257–270. doi: 10.1006/dbio.1998.9162. [DOI] [PubMed] [Google Scholar]
- 3.Clarke AR, Maandag ER, Vam Roon M, Van der Lugt NMT, Van der Valk M, Hooper MI, Berns A, Te Reile H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 4.Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;18:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deschmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 9.Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The T alpha 1 alpha-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gloster A, El-Bizri H, Rogers D, Miller FD. Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(sici)1096-9861(19990301)405:1<45::aid-cne4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 11.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 12.Johnson MD, Xiang H, London S, Kinoshita Y, Knudson M, Mayber M, Korsmeyer SJ, Morrison RS. Evidence for involvement of bax and p53, but not caspases, in radiation-induced cell death of cultures postnatal hippocampal neurons. J Neurosci Res. 1998;54:721–733. doi: 10.1002/(SICI)1097-4547(19981215)54:6<721::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.Keramaris E, Stafanis L, MacLaurin JG, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Involvement of caspase 3 in the death of cortical neurons evoked by DNA damage. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 14.Ko JL, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 15.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 16.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 17.Lee EY, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- 18.Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O'Brien J, van Roon M, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacLeod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 20.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Krosmeyer SJ, Johnson EM. Bax deletion further orders the cell death pathway in cellebar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- 22.Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 1998;14:223–229. doi: 10.1016/s0168-9525(98)01470-x. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson DW, AA, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 24.Noraberg J, Kristensen BW, Zimmer J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc. 1999;3:278–290. doi: 10.1016/s1385-299x(98)00050-6. [DOI] [PubMed] [Google Scholar]
- 25.Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW. Cell death attenuation by “Usurpin,” a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–288. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 26.Roth KA, Kuan C, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase 3 and Bcl-XL in developmental programmed cell death. Proc Natl Acad Sci USA. 2000;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmued LC, Albertson C, Slikker W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 28.Slack RS, Miller FD. The role of the retinoblastoma gene in mouse neural development. Dev Genet. 1996;18:81–91. doi: 10.1002/(SICI)1520-6408(1996)18:1<81::AID-DVG9>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 29.Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD. A critical temporal requirement for the pRb family during neuronal determination. J Cell Biol. 1998;140:1497–1509. doi: 10.1083/jcb.140.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase: dependent and independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 32.Urase K, Fujita E, Miho Y, Kouroku Y, Mukasa T, Yagi Y, Momoi MY, Momoi T. Detection of activated caspase-3 (CPP32) in the vertebrate nervous system during development by a cleavage site-directed antiserum. Brain Res Dev Brain Res. 1998;111:77–87. doi: 10.1016/s0165-3806(98)00124-2. [DOI] [PubMed] [Google Scholar]
- 33.Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson PO, Barber PC, Hamid QA, Power BF, Dhillon AP, Rode J, Day IN, Thompson RJ, Polak JM. The immunolocalization of protein gene product 9.5 using rabbit polyclonal and mouse monoclonal antibodies. Br J Exp Pathol. 1988;69:91–104. [PMC free article] [PubMed] [Google Scholar]
- 35.Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, Senaldi G, Howard T, Lowe SW, Mak TW. Essential contribution of caspases 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1999;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase 9 or caspase 3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]