

Abstract
Interactions between amyloid β-protein (Aβ) and lipids have been suggested to play important roles in the pathogenesis of Alzheimer's disease. However, the molecular mechanism underlying these interactions has not been fully understood. We examined the effect of Aβ on lipid metabolism in cultured neurons and astrocytes and found that oligomeric Aβ, but not monomeric or fibrillar Aβ, promoted lipid release from both types of cells in a dose- and time-dependent manner. The main components of lipids released after the addition of Aβ were cholesterol, phospholipids, and monosialoganglioside (GM1). Density-gradient and electron microscopic analyses of the conditioned media demonstrated that these Aβ and lipids formed particles and were recovered from the fractions at densities of ∼1.08–1.18 g/ml, which were similar to those of high-density lipoprotein (HDL) generated by apolipoproteins. The lipid release mediated by Aβ was abolished by concomitant treatment with Congo red and the PKC inhibitor, H7, whereas it was not inhibited withN-acetyl-l-cysteine. These Aβ-lipid particles were not internalized into neurons, whereas HDL-like particles produced by apolipoprotein E were internalized. Our findings indicate that oligomeric Aβ promotes lipid release from neuronal membrane, which may lead to the disruption of neuronal lipid homeostasis and the loss of neuronal function.
Keywords: amyloid β-protein, cholesterol release, phospholipid, high-density lipoprotein, cultured neurons, Alzheimer's disease
The mechanism underlying the initiation of the clinicopathological process in Alzheimer's disease (AD) is assumed to be the age-related aggregation of amyloid β-protein (Aβ) (Selkoe, 1994; Esiri et al., 1997). This assumption has been supported in part by the findings that highly aggregated Aβ fibrils, but not Aβ monomers, induce neurodegeneration (Mattson et al., 1993; Pike et al., 1993; Lorenzo and Yankner, 1994). This assumption has also been challenged by recent evidence indicating that Aβ oligomers also play an important role in AD pathogenesis (Walsh et al., 1997; Hartley et al., 1999) and that neurodegeneration is induced in mouse brain without amyloid plaque formation (Chui et al., 1999;Hsia et al., 1999).
The role of lipid metabolism in the pathogenesis of AD has been highlighted by the finding that apolipoprotein E (apoE) epsilon 4 is a strong risk factor for the development of AD (Corder et al., 1993;Saunders et al., 1993; Strittmatter et al., 1993). The findings that monosialoganglioside (GM1)-bound Aβ is the initially deposited Aβ species in AD brain (Yanagisawa et al., 1995), and that amyloid fibril formation is induced by Aβ binding to membrane vesicles containing ganglioside (Choo-Smith et al., 1997) and phospholipids (Terzi et al., 1995), suggest that interactions of Aβ with lipids play a crucial role in the pathogenesis of AD. In addition, it has been reported that Aβ modulates cholesterol metabolism in the plasma membrane (Liu et al., 1998) and membrane functions by altering the physicochemical properties of membrane constituents including lipids (Muller et al., 1995; Mason et al., 1996; McLaurin and Chakrabartty, 1996; Avdulov et al., 1997). Previous studies have shown that the properties of this interaction of Aβ with lipids are dependent on the aggregation state of Aβ (Avdulov et al., 1997; Mason et al., 1999). These lines of evidence indicate that Aβ interacts with neuronal membranes, disrupting its lipid environment. The interactions of Aβ with lipids have also been demonstrated in physiological conditions; Aβ has been shown to be associated with high-density lipoproteins (HDLs) in the cerebrospinal fluid (Koudinov et al., 1996; Fagan et al., 2000) and human plasma (Koudinov et al., 1994; Biere et al., 1996; Matsubara et al., 1999). However, the mechanism underlying the formation of these Aβ-lipid complexes is poorly understood, and their significance in cholesterol metabolism in the CNS or in the pathogenesis of AD remains to be elucidated.
We have recently shown that apoE modulates cellular cholesterol metabolism in an isoform-specific manner (Michikawa et al., 2000), and tau phosphorylation is enhanced in cholesterol-deficient neurons (Fan et al., 2001) and in the brains of Niemann-Pick disease type C mice (Sawamura et al., 2001), suggesting that cholesterol is the key molecule in the pathogenesis of tauopathy. These lines of evidence have led us to the question of whether Aβ affects cholesterol homeostasis in neurons, leading to the abnormal phosphorylation of tau. In this study, we conducted experiments to examine the effect of Aβ on cholesterol metabolism in cultured neurons.
MATERIALS AND METHODS
Cell culture. All experiments were performed in compliance with existing laws and institutional guidelines. Neuron-rich cultures were prepared from cerebral cortices as described previously (Michikawa and Yanagisawa, 1998), with some modifications. In brief, uteri of gravid rats at embryonic days (E) 17–18 were removed under anesthesia. Cerebral cortices from fetal rat brains were dissected, freed of meninges, and diced into small pieces; the cortical fragments were incubated in 0.25% trypsin and 20 mg/ml DNase I in PBS (8.1 mmNa2HPO4, 1.5 mmKH2PO4, 137 mm NaCl, and 2.7 mm KCl, pH 7.4) at 37°C for 20 min. The fragments were then dissociated into single cells by pipetting. The cells were suspended in the feeding medium and plated onto poly-d-lysine-coated 24-well plates at a cell density of 2 × 104/cm2. The feeding medium consisted of DMEM nutrient mixture (DMEM/F12, 50:50) and N2 supplements. More than 99% of the cultured cells were identified as neurons by immunocytochemical analysis using monoclonal antibody against microtubule-associated protein 2, a neuron-specific marker, at 3 d in culture. For astrocyte-rich cultures, mixed glial cells were prepared according to a previously described method (Isobe et al., 1999). In brief, dissociated cells were prepared from E17–18 rat cerebral cortices as described above and seeded in 75 cm2 flasks at a cell density of 1 × 107 in DMEM containing 10% FBS. After 2 weeks of incubation in vitro, the cells in the astrocytic monolayer were removed by vigorously shaking the flasks. The medium with floating cells was removed, and the remaining monolayer cells were trypsinized (0.1%) and reseeded onto 12-well plates.
Preparation of oligomeric Aβ. Synthetic Aβ1–40 (TFA salt) was purchased from Peptide Institute (Osaka, Japan; lot numbers 490703, 491131, 500324, 500520, 500701, and 501001), Bachem (Bubendorf, Switzerland; lot numbers 518765 and 519600), and Sigma (St. Louis, MO; lot number 38H49581). Aβ was dissolved in dimethyl sulfoxide (DMSO) at 13.3 m and diluted with PBS to obtain a 350 μm stock solution. Aβ solution was then incubated for 24 hr at 37°C (iAβ). After incubation, Aβ solution was filtered with a 0.45 μm Millipore filter (Millipore, Bedford, MA). Peptide concentrations of both nonfiltered and filtered Aβ were determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). The aggregation state of Aβ in both solutions was monitored by measuring the intensity of thioflavin-T fluorescence. Fresh Aβ1–40 was dissolved in the same manner to obtain 350 μm solution and used for experiments immediately after determination of its peptide concentration. For electron microscopic analysis, each sample was centrifuged in PBS in a SW 41-Ti swing rotor (at 4°C) for 48 hr at 34,200 rpm using a Beckman TL-70. After centrifugation, electron microscopic analysis of the lower part of each solution containing resuspended pellet, if any, was performed.
Quantification of released and intracellular cholesterol and phosphatidylcholine. Neurons in 6- or 12-well plates were labeled in DMEM supplemented with N2 supplements containing 37 Bq/ml of [14C]acetate (DuPont NEN) for 48 hr, the time period necessary to achieve isotopic steady state in these cells, as has been described previously (Michikawa et al., 2000). Astrocytes in 12-well plates were labeled in DMEM with 5% FBS containing 37 Bq/ml of [14C]acetate (DuPont NEN) for 48 hr. The labeled neurons and astrocytes were rinsed three times with fresh DMEM and treated with the reagents that were examined. Aliquots of 1.0 ml of each conditioned medium were filtered with a 0.45 μm Millipore filter and then transferred into clean glass tubes containing 4.0 ml of chloroform/methanol (2:1 v/v). The organic phase was separated from the aqueous phase, washed twice by vigorous shaking with 3 ml of chloroform/water (1:1 v/v), separated from the aqueous phase by centrifugation, and dried under N2 gas. For extraction of intracellular lipids, dried cells were incubated in hexane/isopropanol (3:2 v/v) for 1 hr at room temperature. The solvent from each plate was removed and dried under N2gas. The organic phases were redissolved in 50 μl of chloroform, and 10 μl of each sample was spotted on activated silica gel high-performance thin-layer chromatography (HPTLC) plates (Merck, Darmstadt, Germany). The lipids were separated by sequential one-dimensional chromatography using the chloroform/methanol/acetic acid/water (25:15:4:2, v/v/v/v) solvent system, followed by another run in hexane/diethyl ether/acetic acid (80:30:1). [14C]cholesterol, [14C]sphingomyelin, and [14C]phosphatidylcholine were used as standards. The chromatography plates were exposed to radiosensitive films, and each lipid was visualized and quantified with BAS2500 (Fuji Film, Tokyo, Japan). The amount of lipid release was calculated as the percentage of released lipid relative to the total lipid content (released plus intracellular lipid).
Density gradient ultracentrifugation. After incubation with iAβ at 8 μg/ml for 24 or 48 hr, the neuronal or astrocyte culture medium was filtered with a 0.45 μm Millipore filter. A discontinuous KBr gradient was prepared in a 14 × 89 mm ultracentrifuge tube (Ultraclear, Beckman) from the bottom to the top with 2 ml of sample at a density of 1.30 gm/ml, 3 ml at 1.21 gm/ml, 2 ml at 1.063 gm/ml, 2 ml at 1.19 gm/ml, and 4 ml at 1.006 gm/ml KBr solution (all salt solutions contained 0.1% disodium-EDTA and 0.002% sodium azide, pH 7.4). The sample in the KBr gradient was centrifuged using a Beckman TL-70 ultracentrifuge in a SW 41-Ti swing rotor (at 4°C) for 48 hr at 34,200 rpm. After density gradient centrifugation, 12 fractions (1.0 ml each) were collected from the top of the gradient using a micropipette. The densities of the fractions were determined by measuring the weight of each 100 μl fraction collected. The last fraction was stirred to dissolve the pellet. The cholesterol and phospholipid contents were determined in each fraction. Five milliliters of chloroform/methanol (2:1 v/v) solvent were added to 1 ml of each fraction, and the mixture was stirred vigorously. After centrifugation, the organic phase was removed from each fraction and dried under N2 gas. The organic residue was dissolved in a small volume of chloroform, and the total cholesterol and phospholipid contents were determined using cholesterol and phospholipid determination kits (Kyowa Medix, Tokyo, Japan and Wako, Osaka, Japan, respectively).
Viability assay. The release of the cytoplasmic enzyme, lactate dehydrogenase (LDH), into culture medium was determined for the quantification of cell death. Fifty microliters of culture medium were transferred to a fresh 96-well flat-bottomed plate and a colorimetric LDH-release assay was performed according to the instructions of the manufacturer (Promega, Madison, WI); absorbances were read at 490 nm immediately thereafter. For determination of total LDH, the neuronal cultures were incubated with 100 mmH2O2 for 10 min at room temperature and released LDH was determined, and the percentage of released LDH per total LDH in each culture was calculated.
Measurement of thioflavin-T binding to aggregated Aβ. Determination of the aggregated Aβ state in solution was performed on the basis of a previously established method (LeVine, 1995; LeVine, 1999). A 350 μm stock solution of Aβ1–40 was prepared as described above and incubated for 24 hr at 37°C. The solution was then diluted to threefold with PBS. One-half the amount of the solution was filtered with a 0.45 μm pore-sized Millipore filter. The protein concentration of nonfiltered and filtered solution was determined using a bicinchoninic acid protein assay kit. Freshly dissolved Aβ was prepared as described above and diluted to threefold with PBS, and its protein concentration was determined. The concentration of each solution was then adjusted to 50 μm with PBS. Steady-state fluorescence measurements for Aβ were performed with a multiplate reader (Fluoroskan Ascent, Labsystems Inc., Franklin, MA) (excitation 446 nm, emission 490 nm) in 48-well plates. Each well contained 10 μl of 50 μm Aβ in 1000 μl/well of 5 μm thioflavin-T in 50 mm glycine-NaOH, pH 8.5.
Immunoblot analysis of Aβ, apoE, apoJ, and GM1 ganglioside. For immunoblot analysis of Aβ, samples of each fraction isolated by density gradient ultracentrifugation were dissolved in sample solubilizing buffer consisting of 50 mm Tris-HCl, pH 6.8, 10% glycerol, 4% SDS, 10% mercaptoethanol, 8 m urea, and 0.01% bromophenol blue. For analysis of apoE and GM1 ganglioside, samples of each fraction were dissolved in equal volumes of Laemmli buffer. They were then subjected to 4–20% gradient Tris/tricine SDS-PAGE (Dai-ichi Pure Chemical Co., Tokyo, Japan). The separated proteins were transferred onto an immobilon or polyvinylidene difluoride membrane (Millipore) with a semidry electrophoretic transfer apparatus (Nihon Eido, Tokyo, Japan) using a transfer buffer (0.1m Tris, 0.192 m glycine, and 20% methanol). The blots were blocked with 100% Block Ace (Dainippon Pharmaceutical Co., Osaka, Japan) for 1 hr and incubated with primary antibodies overnight at 4°C. The primary antibodies used were monoclonal antibodies specific for the Aβ1–40 ending site, BA27 (Suzuki et al., 1994) (at a final concentration of 3.1 μg/ml), specific for human Aβ1–17, 6E10 (Kim et al., 1990) (Senetek, St. Louis, MO) (at a final concentration of 1 μg/ml), polyclonal anti-apoE antibody, AB947 (Chemicon, Temecula, CA) (1:1,000), and anti-apoJ antibody (Rockland, Gilbertsville, PA) (at a final concentration of 0.2 μg/ml). The blots were washed four times with PBS-T (PBS containing 0.05% Tween 20) within a period of 60 min and then incubated with secondary antibodies (horseradish peroxidase-conjugated anti-goat or anti-mouse antibodies, used at a final concentration of 0.4 μg/ml) for 1 hr. For the detection of GM1 ganglioside, the membrane was probed with horseradish peroxidase-conjugated cholera toxin B (Sigma) (final concentration at 42 ng/ml) overnight at 4°C. In between steps, the blots were washed four times with PBS-T for 15 min. Bound antibodies or cholera toxin was detected using ECL (Amersham Pharmacia Biotechnology).
Immunoprecipitation of lipids in association with Aβand apoE. The neurons and astrocytes were labeled with 37 Bq/ml of [14C]acetate (DuPont NEN) for 48 hr, followed by three washes in DMEM, and treated with 8 μm iAβ. The conditioned media, in which neurons and astrocytes were cultured in the presence of iAβ for 24–48 hr, were filtered. The filtered conditioned media were then incubated with 1 μl of mouse monoclonal antibody, 6E10, or goat polyclonal antibodies, AB947, and anti-apoJ antibody and mouse normal mouse IgG, together with 100 μl of 20% protein G-Sepharose (Amersham Pharmacia Biotechnology) slurry under rotation at 4°C overnight. The immunoprecipitated lipids associated with protein G-Sepharose were washed in PBS-T three times and solubilized in a solution of chloroform/methanol (2:1 v/v), and the solution was evaporated by N2 gas. The organic phases were redissolved in 20 μl of chloroform, and all samples were spotted on activated silica gel HPTLC plates; the lipids were separated by sequential one-dimensional chromatography using the chloroform/methanol/acetic acid/water (25:15:4:2) solvent system, followed by another run in hexane/diethyl ether/acetic acid (80:30:1). The chromatography plates were exposed to radiosensitive films, and each lipid was visualized and quantified with BAS2500. The amount of immunoprecipitated lipid was calculated as the percentage of lipid relative to the total lipid in the media.
Immunoelectron microscopy. The solutions containing Aβ-lipid particles were obtained by density gradient ultracentrifugation, diluted with H2O to a density of 1.006, and centrifuged again at 46,000 rpm in a SW 50.1-Ti rotor for 24 hr at 4°C. The 400 μl portion at the bottom was used for electron microscopic study. The solutions were placed on a carbon-coated electron microscopy grid. Nonspecific binding was blocked by incubation in PBS with 1% bovine serum albumin (BSA) for 10 min. The grids were then placed on a droplet of either the Aβ-specific antibody, 6E10 (at final concentration of 5 μg/ml), or normal mouse IgG for 60 min (both diluted in PBS, 0.1% BSA), and passed over seven droplets of washing solution (PBS) for 1 min each. The grids were placed on a droplet of anti-mouse IgG conjugated to 5 nm colloidal gold particles for 60 min (Sigma; diluted 1:20 in PBS, 0.1% BSA), passed over seven droplets of washing solution (PBS), and passed over another seven droplets of distilled water. The specimens were then negatively stained with 2% sodium phosphotungstate.
Statistical analysis. Statistical analysis was performed using StatView computer software (Macintosh), and multiple pairwise comparisons among the sets of data were performed using ANOVA and the Bonferroni t test.
RESULTS
Characterization of Aβ used in this study
We studied the effect of Aβ1–40 on lipid metabolism from the point of view of lipid release from neurons and astrocytes in culture. Because Aβ1–42 is known to be highly amyloidgenic and assumed to play a critical role in the pathogenesis of AD, the effect of Aβ1–42 on cellular lipid metabolism is also an important issue that needs to be addressed. However, the fact that synthetic Aβ1–42 is very difficult to handle and that oligomerized Aβ1–40 as well as Aβ1–42 can be associated with lipids led us to use Aβ1–40 in the present study. To characterize Aβ used in this study, Aβ1–40 incubated for 24 hr at 37°C at 350 μm(iAβ-nonfiltered), Aβ1–40 incubated in the same way followed by filtration through a 0.45 μm Millipore filter (iAβ-filtered), and freshly dissolved Aβ (fresh Aβ) were subjected to thioflavin-T assay, Western blot analysis, and electron microscopy. Determination of Aβ peptide concentration in each sample was performed using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). The concentration of Aβ in each solution was then adjusted to 100 μm using PBS, and the solutions were used for the experiments. As we reported previously (Isobe et al., 2000), the intensity curve of thioflavin-T reaction with Aβ, which was incubated at 350 μm at 37°C, was saturated at 24 hr of incubation. The fluorescence intensity of iAβ-filtered was similar to that of Aβ-nonfiltered, whereas that of fresh Aβ was as low as background levels of PBS (Fig.1a). Immunoblot analysis showed that tetrameric, trimeric, dimeric, and monomeric Aβ were found in the samples of iAβ-nonfiltered and iAβ-filtered, whereas only dimeric and monomeric Aβ were found in fresh Aβ (Fig.1b). Electron microscopy showed that fibrils were formed in the samples from iAβ-nonfiltered, 7.9 ± 0.5 nm in diameter and 118 ± 14 nm in length for measurable fibrils (Fig.1c), although the length was difficult to determine because of the twisted configuration. The morphological characteristics of these fibrils are identical to those of protofibrils with curvilinear structures of 4–11 nm in diameter and <200 nm in length as has been reported previously (Walsh et al., 1997). In contrast, electron microscopy of iAβ-filtered and fresh Aβ did not reveal any structures such as protofibrils or Aβ-derived diffusible ligands (ADDLs) (Fig. 1d,e).
Fig. 1.

Characterization of Aβ1–40. Aβ1–40 was prepared as described in Materials and Methods. a, The aliquots of iAβ-nonfiltered,iAβ-filtered, fresh Aβ, and PBS were subjected to thioflavin-T assays as described in Materials and Methods. Three independent experiments were performed, and similar results were obtained.b, The equal volume of 2× sample solubilizing buffer was added to each Aβ solution, of which the concentration was normalized with PBS. The samples were then subjected to 4–20% Tris/tricine SDS-PAGE, followed by Western blot analysis.c, Electron micrograph of each sample is shown. The samples were centrifuged at 34,500 rpm for 48 hr using a SW 41-Ti rotor. Electron microscopic analysis of the lower part of each solution containing the resuspended pellet was performed. Results of negative staining show that fibrillar structures are found in the sample ofiAβ-nonfiltered(c); however, no fibril is detected in the samples of iAβ-filtered(d) or fresh Aβ (e). Scale bar, 50 nm.
Oligomeric Aβ promotes lipids release from neurons
When we analyzed lipids released from the cells, all the conditioned media of the cultures treated with iAβ were examined after filtration with a 0.45 μm Millipore filter. Electron microscopic study did not reveal any structures such as fibrils, protofibrils, or ADDLs in the conditioned media of the neuronal cultures incubated with iAβ-nonfiltered or iAβ-filtered for 24 hr (data not shown). The dose–response curves for the release of cholesterol and phosphatidylcholine from neurons at 4 hr of incubation with iAβ-filtered are shown in Figure2a. Incubation with iAβ promoted the release of cholesterol (essentially all unesterified) and phosphatidylcholine from neurons in a dose-dependent manner. Saturation of Aβ-mediated lipid release was observed at Aβ concentrations higher than 1 μm, which can be expected to be present in local extracellular spaces in vivo. The time course of cholesterol and phosphatidylcholine release from cultured neurons into the medium in the presence of 8 μmAβ is shown in Figure 2b. The cholesterol and phosphatidylcholine release mediated by Aβ increased in a time-dependent manner. We used six batches of Aβ1–40 obtained from Peptide Institute, one batch of Aβ1–40 from Sigma, and two batches of Aβ1–40 from Bachem. The iAβ prepared from these batches promoted lipid release from neurons. Because among these Aβ peptides tested, Aβ peptide from Peptide Institute has the strongest ability to promote lipid release (data not shown), we used Aβ peptide obtained from Peptide Institute.
Fig. 2.
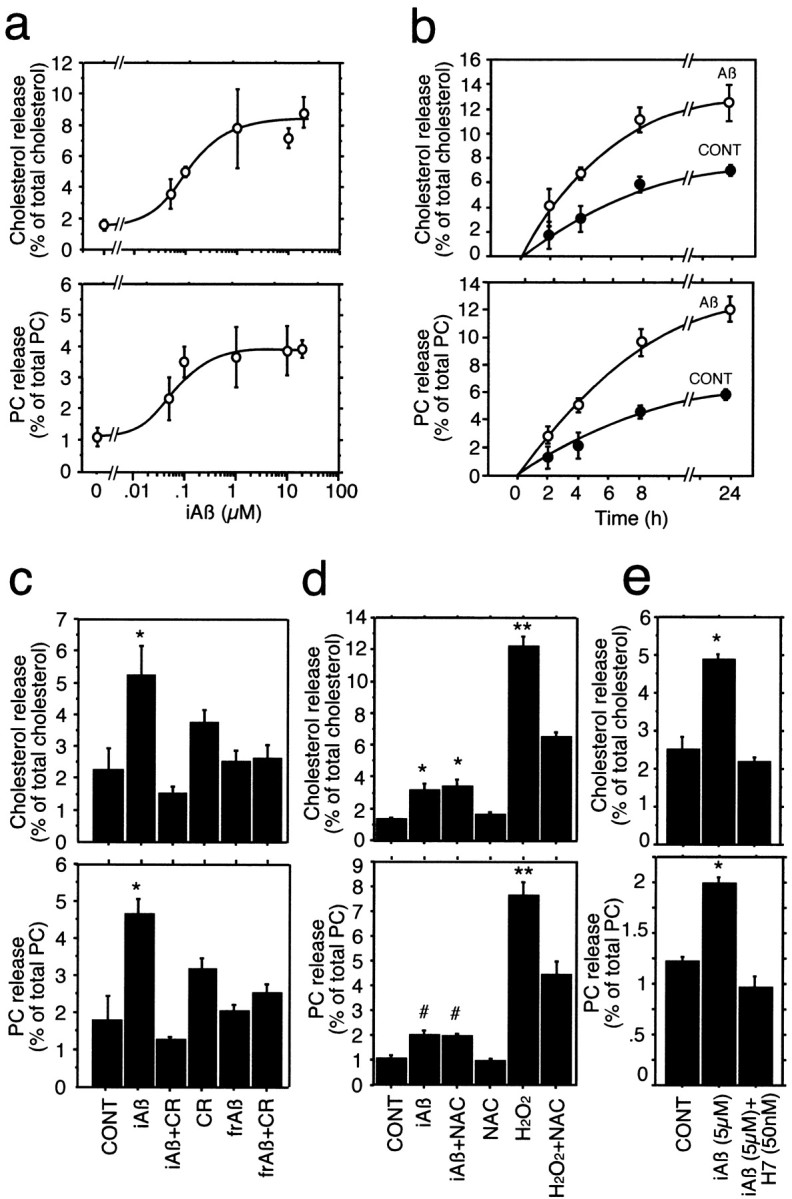
Effect of iAβ on cholesterol and phosphatidylcholine release from neurons in culture. Neuron-rich cultures were labeled with [14C]acetate for 48 hr as described in Materials and Methods. Cells were then washed three times with DMEM and incubated with iAβ at various concentrations for 4 hr. Synthetic Aβ1–40 dissolved at high concentration and incubated at 37°C for 24 hr, followed by filtration, was used. The released lipids in the media and the cellular lipids were extracted and analyzed as described in Materials and Methods. The iAβ-mediated release of cholesterol and phosphatidylcholine (PC) (a) was significantly increased in a dose-dependent manner. Each data point represents mean ± SE for three samples. For the time course study of iAβ-mediated lipid release from neurons, cultured neurons were labeled with [14C]acetate for 48 hr and then washed three times with DMEM and incubated with iAβ at a final concentration of 8 μm. The iAβ-mediated release of cholesterol and PC (b) increased with incubation time. Each data point represents mean ± SE for three samples. Effect of Congo red on iAβ-mediated lipid release and effect of fresh Aβ on lipid release from neurons were investigated using labeled neurons with [14C]acetate for 48 hr. c, Cells were washed three times with DMEM and then incubated with iAβ (10 μm), iAβ (10 μm) with Congo red (CR) (10 μm), CR alone (10 μm), freshly dissolved Aβ (frAβ) (10 μm), and frAβ plus CR (10 μm) in serum-free N2 medium for 24 hr. The release of cholesterol and PC in iAβ-treated culture medium was abolished by concurrent treatment with Congo red. Freshly dissolved Aβ1–40 did not promote lipid release from these cells. Each data point represents mean ± SE for four samples. *p < 0.005 versus CONT, iAβ + CR, frAβ, and frAβ + CR. CONT, Control cultures;iAβ, incubated Aβ1–40; CR, Congo red; frAβ, fresh Aβ1–40. d, Cells were washed three times with DMEM and then incubated with none (CONT), iAβ (5 μm), iAβ (5 μm) + NAC (1 mm), NAC (1 mm), H2O2 (2 mm), and H2O2 (2 mm) + NAC (1 mm). *p < 0.001 versus CONT and NAC; **p < 0.0001 versus H2O2 + NAC; #p < 0.06 versus CONT and NAC.NAC, N-acetyl-l-cysteine.e, The cultures were washed three times with DMEM and then incubated with none (CONT), iAβ (5 μm), and iAβ (5 μm) + H7 (30 nm) for 16 hr at 37°C, and the lipids in the medium and the cells were quantified as described in Materials and Methods. *p < 0.004 versus CONT and iAβ + H7.
Because Congo red is known to inhibit oligomerization of Aβ by stabilizing Aβ monomer (Podlisny et al., 1995, 1998), we next examined whether Aβ-mediated lipid release is inhibited after concurrent treatment with Congo red. Aβ was incubated at high concentration for 24 hr at 37°C, filtered, and added into neuronal cultures. As shown in Figure 2c, iAβ promoted lipid release from neurons, whereas Aβ incubated with 10 μm Congo red for 24 hr at 37°C lost its function as a lipid acceptor. In addition, freshly dissolved Aβ did not induce lipid release from neurons (Fig. 2c), suggesting that the aggregated form of Aβ is necessary for acquiring a lipid acceptor function.
Because the concentrations of Aβ used in this study were high, they may have induced oxidation of cell membranes and thereby could be cytotoxic (Schubert et al., 1995; Mark et al., 1996), leading to lipid leakage from cells damaged by Aβ. Therefore, we next performed experiments to determine whether free radicals are involved in iAβ-mediated lipid release in the neuronal cultures. The neuronal cultures were subjected to the following treatments: none, iAβ, iAβ + N-acetyl-l-cysteine (NAC), NAC, H2O2, and H2O2 + NAC for 8 hr. The amount of lipids released into the culture media in each culture was determined. NAC at a concentration of 1 mm had no effect on iAβ-mediated lipid release, whereas lipid leakage caused by H2O2 was significantly inhibited by 1 mm NAC (Fig. 2d).
It is known that lipid release is an active cellular process and that intracellular signaling molecules such as PKC are involved in cellular cholesterol release (Theret et al., 1990; Mendez et al., 1991; Li and Yokoyama, 1995; Mendez, 1997). To confirm that the iAβ-mediated lipid release is an active cellular process and not a nonspecific physicochemical phenomenon, we next examined the effect of a PKC inhibitor, H7, on the iAβ-mediated lipid release from neurons. As shown in the Figure 2e, H7 completely inhibited lipid release mediated by iAβ.
Density gradient analysis of lipid particles produced by neurons in the presence of iAβ
The characteristics of the released cholesterol and phospholipids and the localization of iAβ added exogenously to the serum-free media in cultured neurons were examined. The conditioned media of the neuronal cultures treated with iAβ (10 μm) were filtered with a 0.45 μm Millipore filter and centrifuged at 34,500 rpm for 48 hr at 4°C in a tube using a Beckman SW 41-Ti rotor. Figure 3, a andb, show the results of density gradient ultracentrifugation of the conditioned medium containing iAβ. They show that most of the cholesterol and phospholipids are distributed similarly across the gradient, with both having densities of ∼1.08–1.18 gm/ml (fractions 5, 6, 7, and 8), which correspond to the densities of HDL (Fig.3a). The cholesterol to phospholipid ratio (w/w) at peak density was 0.35. Western blotting analysis using anti-Aβ antibody, BA27, and anti-apoE antibody, AB947, was performed with the samples from the fractions. Exogenously added Aβ was recovered mainly from fractions 4–8 (Fig. 3b). Aβ in these fractions was detected as monomers and dimers under denatured conditions. Endogenous GM1 ganglioside was identified in fractions 5–8 (Fig. 3b). However, apoE was not detected in these fractions, and apoJ, which is secreted by neurons, was localized in fractions 10–12 (Fig.3b). GM1 is known to have a strong positive curvature-imposing potency, which is required for the formation of small particles such as HDL (Epand, 1998), suggesting that GM1 may contribute to the formation of Aβ-lipid particles by bending of lipid membranes in a convex manner.
Fig. 3.

Density gradient ultracentrifugation analysis of the culture medium of neurons in the presence of iAβ or H2O2. Neuronal cultures plated in six-well plastic plates were incubated with Aβ1–40 (10 μm) in serum-free N2 medium for 24 hr. The culture medium was collected, filtered through a 0.45 μm filter, and subjected to an initial discontinuous density gradient prepared using KBr solution as described in Materials and Methods. a, After ultracentrifugation, fractions were obtained, and the density, cholesterol, and phospholipids content in each fraction were determined. b, Aliquots of 10 μl from each fraction were mixed with the same volume of SDS buffer, subjected to SDS gel electrophoresis, and immunoblotted with antibodies against Aβ (BA27), apoE (AB947), and apoJ. GM1 ganglioside in each fraction was detected with HRP-conjugated chorea toxin-B. c, Forty-eight hours after plating in serum-free N2 medium, the neuronal cultures plated in six-well plastic plates were washed in DMEM and incubated with Aβ1–40 (10 μm) or 5 mmH2O2 in serum-free N2 medium for indicated periods. The percentage of LDH released from the cultures was determined as described in Materials and Methods. The data are mean ± SE of triplicate. d, Forty-eight hours after plating in serum-free N2 medium, the neuronal cultures plated in six-well plastic plates were washed in DMEM and incubated with 5 mm H2O2 in serum-free N2 medium for 24 hr. The culture medium was collected, filtered through a 0.45 μm filter, and subjected to an initial discontinuous density gradient prepared using KBr solution as described in Materials and Methods. After ultracentrifugation, fractions were obtained, and the density and concentrations of cholesterol and phospholipids were determined.
These results indicate that iAβ-mediated lipid release results in the formation of Aβ-lipid particles. To confirm that iAβ-mediated lipid release is not the nonspecific lipid leakage from neuronal cultures by free radicals generated by iAβ, we determined whether HDL-like particles were generated in the conditioned medium of the neuronal cultures damaged by H2O2. As shown in Figure3c, iAβ at 20 μm has no toxic effect on neuronal cultures until 144 hr of treatment, whereas H2O2 at 5 mm exhibits a toxic effect on neuronal cultures at 24 hr of treatment assayed by LDH release (Fig. 3c). The conditioned media of these cultures were collected, and lipids, the release of which from neurons was caused by H2O2, were analyzed by density-gradient ultracentrifugation. As shown in Figure 3d, the distribution pattern of lipids shows no peak of HDL and was quite different from that mediated by iAβ (Fig. 3a). These lines of evidence together with the finding shown in Figure 2dclearly indicate that lipid release mediated by iAβ is not nonspecific lipid leakage from damaged cells by a cytotoxic effect of iAβ.
Oligomeric Aβ promotes lipid release from astrocytes
We further examined the effects of iAβ on the release of cholesterol and phosphatidylcholine from cultured astrocytes. The astrocyte cultures were labeled with [14C]acetate for 48 hr, washed in DMEM, and incubated with iAβ at various concentrations for 4 hr. Lipid concentration released into the media and cellular lipid content were determined. The dose–response curves for the release of cholesterol and phosphatidylcholine at 4 hr of incubation with iAβ are shown in Figure 4, a and b, respectively. Incubation with iAβ promoted the release of cholesterol and phosphatidylcholine from astrocytes in a dose-dependent manner. Saturation of iAβ-mediated release of these lipids was observed at iAβ concentrations higher than 1 μm. In contrast to iAβ, freshly solubilized Aβ at 10 and 30 μm did not promote lipid release from astrocytes (Fig. 4, a and b, respectively).
Fig. 4.

iAβ promotes lipid release from astrocytes in culture. Astrocyte-rich cultures were labeled with [14C]acetate for 48 hr as described in Materials and Methods. Cells were then washed three times with DMEM and incubated with iAβ1–40 or fresh Aβ at various concentrations for 4 hr. The conditioned media were collected and then filtered. The lipids that were released into the medium and the intracellular lipids were extracted and analyzed as described in Materials and Methods. iAβ (○) promoted the release of cholesterol (a) and phosphatidylcholine (PC) (b) in a dose-dependent manner; fresh Aβ (●) did not. Data are mean ± SE for four samples. The scale bars are smaller than the symbol size at 0 μm (a and b). Density gradient ultracentrifugation analysis was performed with the conditioned medium of astrocytes in the presence of iAβ. Astrocytes plated in six-well plastic plates were incubated with Aβ1–40 (10 μm) in DMEM for 24 hr. The culture medium was collected, filtered through a 0.45 μm filter, and subjected to an initial discontinuous density gradient prepared using KBr solution as described in Materials and Methods. After ultracentrifugation, the solution was fractionated. The density and cholesterol and phospholipid content in each fraction were determined (c). Aliquots of 10 μl from each fraction were mixed with the same volume of SDS buffer, subjected to SDS gel electrophoresis, and immunoblotted with antibodies against Aβ (BA27), apoE (AB947), and apoJ. GM1 ganglioside in each fraction was detected with HRP-conjugated chorea toxin-B (d).
Density gradient analysis of lipid particles produced by astrocytes in the presence of iAβ
The conditioned media of the astrocyte cultures treated with iAβ (10 μm) were filtered with a 0.45 μmMillipore filter and centrifuged at 34,500 rpm for 48 hr at 4°C in a tube using a Beckman SW 41-Ti rotor. Density gradient analysis of lipid released from astrocytes showed that a major part of cholesterol and phospholipids distributed similarly across the gradient, with both having densities at ∼1.08–1.17 gm/ml (fractions 5, 6, 7, and 8), which corresponded to the densities of HDL (Fig. 4c). The cholesterol to phospholipids ratio (w/w) at peak density was 2.22. Western blotting analysis using anti-Aβ antibody, BA27, and anti-apoE antibody, AB947, was performed with the samples from the fractions. Exogenously added Aβ was recovered mainly in the fractions between 4 and 8 (Fig. 4d). Aβ was detected as monomer and dimer under denaturing conditions. Endogenous GM1 ganglioside and apoE were identified in the fractions between 5 and 8 (Fig. 4d). However, apoJ, which is secreted from neurons, was localized in fractions 10, 11, and 12 (Fig. 4d).
Morphological and biochemical analysis of lipid particles associated with Aβ
To confirm the conformation of Aβ-lipid particles isolated by density gradient analysis of filtered culture medium of neurons in the presence of iAβ, fractions containing Aβ-lipid particles were diluted with distilled water to a density of 1.006 gm/ml and centrifuged at 34,500 rpm for 48 hr at 4°C in a tube using a Beckman SW 41-Ti rotor. The conditioned media of the neuronal cultures in the presence of 0.25 μm of apolipoprotein E3 (apoE3) were also analyzed. The lower part of each solution was subjected to immunoelectron microscopic study. Analysis of samples from neuronal conditioned media in the presence of iAβ revealed that lipoprotein particles were spherical, with a mean diameter of 29.4 ± 1.1 nm, similar to the appearance of, but larger than, HDL-like particles formed via the apoE-mediated manner, the mean diameter of which is 11.4 ± 0.5 nm (Fig.5a,b). In addition, Aβ was found to be associated with lipid particles as demonstrated by the immunoreactivity of lipid particles against the anti-Aβ antibody, 6E10, using electron microscopy (Fig. 5c). In contrast, Aβ immunoreactive lipid particles were not detected in negative control samples (Fig. 5d), suggesting that iAβ is directly associated with lipid particles.
Fig. 5.
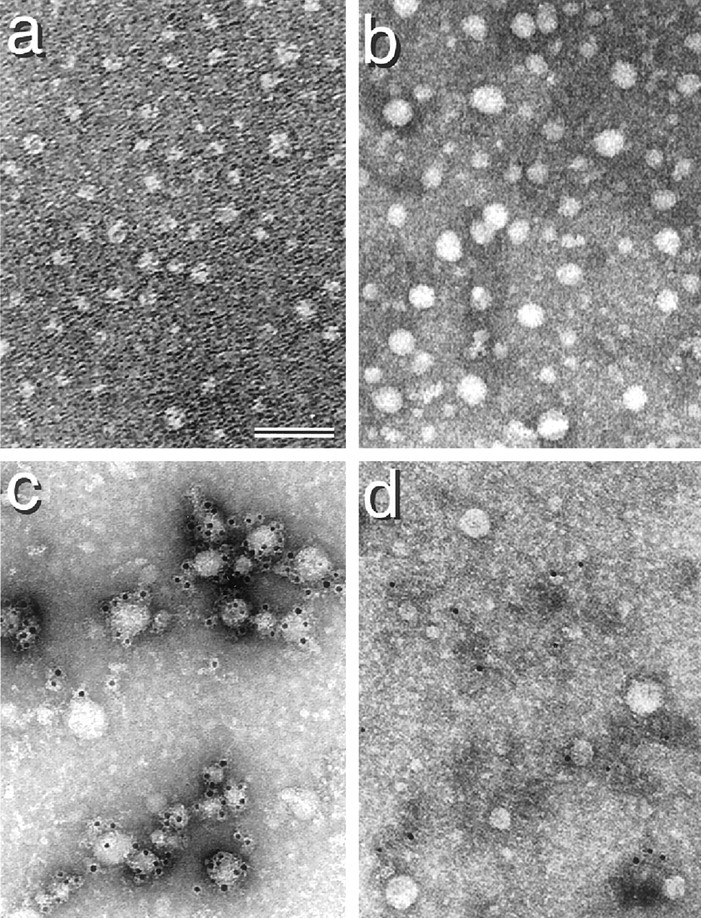
Electron micrographs of lipid particles associated with iAβ and apoE3. Neuronal cultures were incubated with 10 μm iAβ or 0.25 μm apoE3 for 24 hr. The conditioned media of these cultures were collected, filtered with a 0.45 μm Millipore filter, subjected to an initial discontinuous density gradient prepared using KBr solution, and centrifuged at 34,500 rpm for 48 hr using a SW 41-Ti rotor. After centrifugation, 12 fractions were isolated, and lipid concentration and the density in each fraction were determined. HDL fractions were then diluted with 10-fold volumes of distilled water, followed by centrifugation at 34,500 rpm for 48 hr using a SW 41-Ti rotor. Electron microscopic examination of the lower part of each solution was performed. Negative staining of electron micrographs of lipid particles in the presence of apoE and iAβ is shown (a and b, respectively). Results of immunoelectron microscopy show that exogenously added iAβ1–40 forms complexes with lipid particles as demonstrated using the antibody directed against human Aβ1–17, 6E10 (Kim et al., 1990). c, Gold labeling is considered to be associated with lipid particles. d, In contrast, lipid particles were not labeled with gold without 6E10. Scale bar, 50 nm.
We further examined whether Aβ-lipid particles contain other acceptors such as apoE and apoJ. The filtered conditioned medium of neuronal cultures was subjected to immunoprecipitation using antibodies including anti-apoE antibody, AB947, anti-Aβ antibodies, 6E10, anti-apoJ antibody, or normal mouse IgG as a negative control. The ratio of cholesterol and phospholipids associated with protein G-Sepharose to those in the conditioned medium is shown in Figure6. Approximately 12% of total cholesterol and phospholipids in the conditioned medium was immunoprecipitated with 6E10, whereas the percentages of both lipids immunoprecipitated with AB947, anti-apoJ antibody, and normal mouse IgG were significantly lower than those with 6E10 in neuronal culture (Fig.6Aa,b). In the conditioned medium of astrocytes incubated with 10 μm iAβ, the percentages of cholesterol and phospholipids immunoprecipitated with 6E10 were 4.7 and 3.1%, respectively, of those in the conditioned medium, and those immunoprecipitated with AB947 were 4.7 and 3.2% of those in the conditioned medium, respectively (Fig.6Ba,b). However, as seen in neuronal cultures, the percentages of either lipid immunoprecipitated with anti-apoJ antibody and normal mouse IgG were significantly lower than those with 6E10 or AB947 in astrocyte cultures.
Fig. 6.

Binding of Aβ by immobilized anti-Aβ antibody, 6E10, results in capture of lipids. Filtered conditioned medium of neuronal and astrocyte cultures incubated with iAβ for 48 hr was subjected to immunoprecipitation using anti-Aβ antibody (6E10), anti-apoE antibody (AB947), anti-apoJ antibody, and normal mouse IgG.A, The protein-G-Sepharose-associated lipids were determined using the kits described in Materials and Methods. The quantity of cholesterol (a) and phospholipids (b) immunoprecipitated with 6E10 from the conditioned medium of neuronal culture incubated with iAβ was ∼12% of the total cholesterol and phospholipids in the initial conditioned medium. However, those immunoprecipitated with anti-apoE antibody, anti-apoJ antibody, or normal mouse IgG were significantly low.B, In astrocyte culture medium, the quantity of cholesterol (a) and phospholipids (b) immunoprecipitated with 6E10 and AB947 from the conditioned medium incubated with iAβ was significantly higher than those with anti-apoJ antibody or normal mouse IgG. Western blot analysis using anti-apoE antibody (AB947) and anti-Aβ antibody (BA27) was performed with the immunoprecipitates. The bands corresponding to Aβ monomers and dimers is labeled only in immunoprecipitates by 6E10, whereas no band or a faint band was detected in those by AB947, anti-apoJ antibody, and normal mouse IgG in neurons (Ac). The bands corresponding to Aβ monomers and dimers and apoE are labeled in both immunoprecipitates by 6E10 and AB947, respectively, whereas no band or a faint band was detected in those by anti-apoJ and normal mouse IgG for astrocytes (Bc). Data are mean ± SE for four samples. *p < 0.01 versus 6E10, anti-apoJ, and normal IgG (Ac); *p < 0.01 versus anti-apoJ and normal IgG (Bc).
Western blotting analysis using anti-Aβ antibody, BA27, and anti-apoE antibody, AB947, was performed with the immunoprecipitates. The pellet fractions from the immunoprecipitation of neuronal conditioned medium with the anti-Aβ antibody contained exogenously added Aβ, being compatible with lipid content in association with immunoprecipitates (Fig. 6Ac). In contrast, those from the immunoprecipitation of astrocyte-conditioned medium with the anti-Aβ antibody contained both exogenously added Aβ and endogenous apoE (Fig. 6Bc). In addition, those of astrocyte-conditioned medium with the anti-apoE antibody contained both exogenously added Aβ and endogenous apoE (Fig.6Bc).
Binding and internalization of Aβ-lipid particles into neurons
Because iAβ1–40 was found to be an acceptor of lipids from neurons and astrocytes to form Aβ-lipid particles, it was considered appropriate to perform further study on binding and internalization of Aβ-lipid particles produced by iAβ. The conditioned media of [14C]acetate-labeled astrocytes in the presence of iAβ at 10 μm or apoE3 at 0.25 μm in DMEM after 24 hr incubation were collected. Aβ-lipid particles were recovered from HDL fractions by density gradient centrifugation and dialyzed. The fractions containing Aβ-lipid particles were collected and dialyzed, and the radioactivity contained was normalized by a scintillation counter. Neuronal cultures were then incubated with the equal amount of [14C]labeled-Aβ-lipid particles in DMEM for 20 min at 4 or 37°C. The cultures were washed with cold PBS three times and dried under the fresh airflow. The lipids were extracted with hexane/isopropanol (3:2 v/v), separated by HPTLC, and quantified with BAS2500. Figure 7demonstrates the binding activity and internalization efficacy of Aβ-lipid particles and HDL-like particles formed by exogenously added iAβ and apoE3, respectively. The ratio of the labeled-cholesterol activity associated with neurons per total cholesterol activity in the added medium was significantly lower at both 4 and 37°C in the cultures incubated with conditioned medium treated with iAβ1–40 (Fig. 7a). In contrast, it was significantly higher both at 4 and 37°C in the cultures incubated with apoE. Similar results were observed for the ratio of the labeled phosphatidylcholine in association with the cells (Fig. 7b).
Fig. 7.
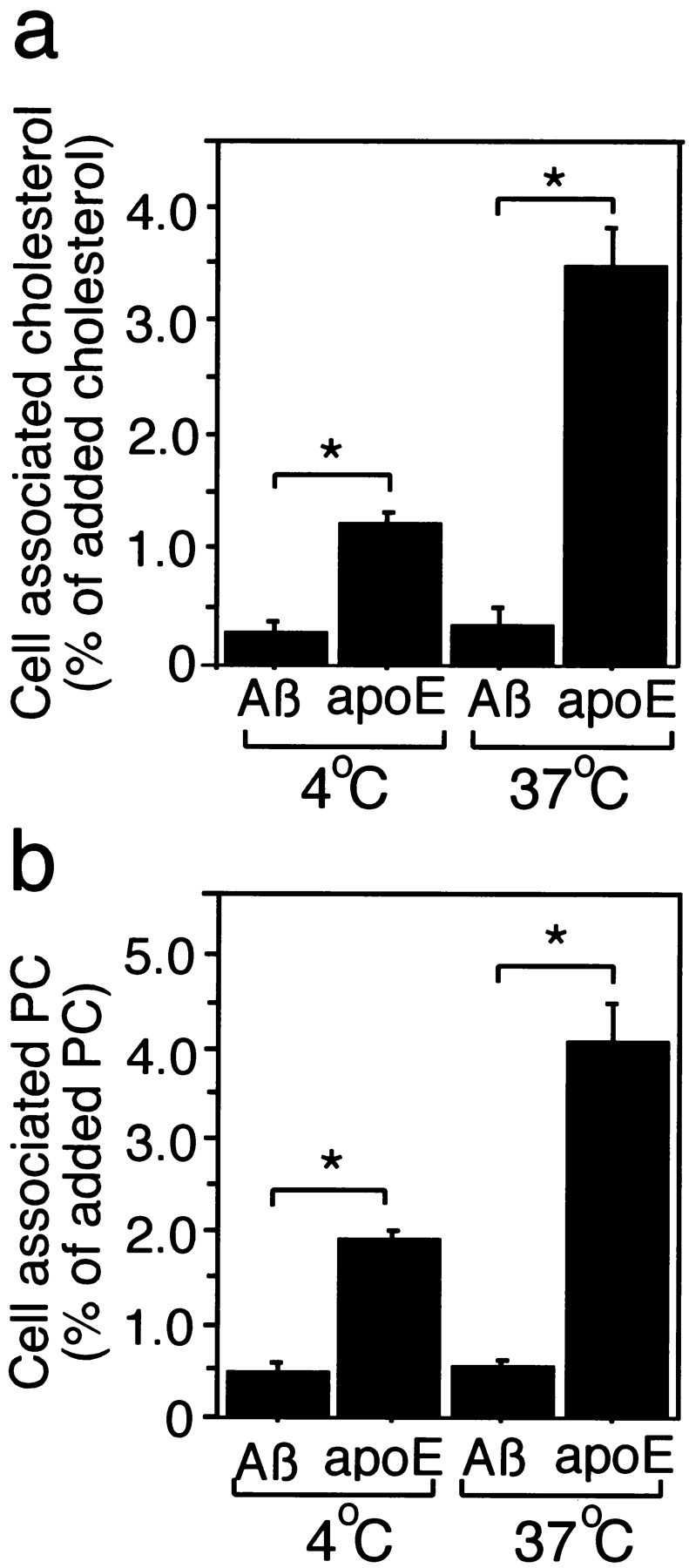
Binding affinity and internalized efficacy of Aβ-lipid particles into neurons. Astrocyte-rich cultures were labeled with [14C]acetate for 48 hr as described in Materials and Methods. Cells were then washed three times with DMEM and incubated with 10 μm iAβ 1–40 or 0.25 μmhuman recombinant apoE3 for 24 hr. The conditioned media were obtained, filtered using a 0.45 μm filter, and subjected to density-gradient ultracentrifugation at 34,000 rpm for 48 hr in a Beckman SW 41-Ti rotor. The HDL fractions were then collected and dialyzed. The radioactivity in each sample was determined by a scintillation counter and normalized with DMEM. The normalized conditioned medium containing iAβ or apoE was added to neuronal cultures at 4 or 37°C. Twenty minutes after the addition, the cultures were washed three times with cold PBS and dried under air flow at room temperature. The lipids in each culture were extracted by incubation with hexane/isopropanol (3:2 v/v) solution for 1 hr. Then the solution was moved into tubes and dried under N2 gas. The extracted lipids were then dissolved in chloroform, developed in HPTLC, and quantified by BAS2500 (Fuji Film, Tokyo, Japan). a, The ratio of the labeled cholesterol associated with neurons was significantly lower at both 4 and 37°C in the cultures incubated with conditioned medium treated with iAβ1–40. However, it was significantly higher at both 4 and 37°C in the cultures incubated with apoE. Similar results were observed for the ratio of the labeled phosphatidylcholine in association with the cells (b). Data are mean ± SE for six samples. *p < 0.003.
DISCUSSION
In the present study, we found a novel action of Aβ: oligomeric Aβ can promote lipid release from astrocytes and neurons to form Aβ-lipid particles consisting of cholesterol, phospholipids, GM1 ganglioside, and Aβ. Aβ-lipid particles produced by oligomeric Aβ have very low binding affinity to neurons and therefore are not internalized into neurons, suggesting that oligomeric Aβ may affect intracellular lipid metabolism. Because high concentrations of Aβ are known to induce oxidation and can be cytotoxic (Schubert et al., 1995;Mark et al., 1996), we have examined the toxicity of Aβ used in this study and found that iAβ has no cytotoxic effect on neurons until 144 hr of treatment, as demonstrated by LDH assay. We have also found that NAC, a potent antioxidant molecule, has no effect on iAβ-mediated lipid release, and lipids released from the cells after the addition of H2O2 do not form lipid particles, which were recovered in HDL fractions. These lines of evidence clearly indicate that lipid release mediated by iAβ is not nonspecific lipid leakage from damaged cells by cytotoxic effect of iAβ. Because Congo red is a well known dye that not only binds to Aβ fibrils and Aβ oligomers to inhibit fibril formation but also inhibits Aβ oligomerization by stabilizing Aβ monomer (Podlisny et al., 1995, 1998), we examined the effect of Congo red on iAβ-mediated lipid release. We also performed experiments to determine whether freshly dissolved Aβ can serve as a lipid acceptor. We found that neither freshly prepared Aβ nor iAβ incubated with 10 μm Congo red removes lipids from neurons. These findings suggest that oligomerized Aβ is required for acquiring the ability to promote lipid release from the cells. In addition, iAβ-filtered does not contain any fibrils or ADDLs (Fig. 1c). Moreover, all conditioned media of cultures treated with iAβ were examined after filtration, and electron microscopic study did not detect any fibrils in the samples after 24 hr incubation (data not shown). These lines of evidence exclude the possibility that Aβ fibrils, but not Aβ oligomers, act as lipid acceptors. Taken together, oligomerization to dimers, trimers, tetramers, and possibly larger assemblies is indispensable for Aβ to acquire the ability to promote lipid release.
The molecular mechanism by which oligomeric Aβ, but not monomeric Aβ, promotes cholesterol and phospholipid release remains obscure. However, one can explain the mechanism based on the differential binding affinity of Aβ to lipids. It has been reported that the binding efficacy of lipids, including cholesterol, phosphatidylcholine, and free fatty acids, to Aβ is increased when the added Aβ forms polymers (Avdulov et al., 1997). Aggregated Aβ exhibits strong electrostatic interactions with the surface of model membranes, which appear to mediate its neurotoxicity (Hertel et al., 1997). In addition, a synthetic peptide in an amphipathic β-sheet structure was found to associate with lipids, including cholesterol and phosphatidylcholine, and is being proposed as a model for apolipoprotein B (Osterman et al., 1984), suggesting that cholesterol and other lipids bind to hydrophobic areas of amphipathic β-sheet. These lines of evidence suggest that cholesterol and other lipids may bind to hydrophobic areas of aggregated Aβ to form Aβ-lipid particles.
Another interesting point is that Aβ-lipid particles generated in the cultured astrocytes are cholesterol rich compared with HDL-like particles produced by apolipoproteins, such as apoE or apoAI (Ito et al., 1999; Michikawa et al., 2000). In addition, our finding that the average size of Aβ-lipid particles produced by iAβ (29.4 ± 1.1 nm) is larger than that of HDL-like particles produced by apoE (11.4 ± 0.5 nm) in neuronal cultures suggests that there could be two species of lipid particles, one produced by iAβ and the other produced by apoE. The size of HDL-like particles produced by apoE is consistent with that previously reported in astrocyte conditioned medium or in CSF (LaDu et al., 1998; Fagan et al., 1999). The mechanisms underlying these cell type- and acceptor type-specific discrepancies are not clear, and further studies are needed to address these issues.
Interestingly, Aβ-lipid particles produced by iAβ cannot bind to neurons and thus are not internalized into the cells, whereas HDl-like particles produced by apoE can do so. This may be because neurons have no receptors to bind Aβ. It has been shown that under physiological conditions, a significant amount of soluble Aβ is associated with apoE-containing HDL particles in CSF, AD brain, and culture medium (Koudinov et al., 1994, 1996; Biere et al., 1996). In contrast to oligomeric Aβ, when monomeric Aβ forms complexes with such particles, apoE receptors bind and internalize these lipid complexes into the cells, thereby modulating the amount of Aβ in the brain parenchyma through cellular clearance mechanisms (Holtzman et al., 1999). This may be because the protein component of HDL under physiological conditions is predominantly apoE and not Aβ, and thus apoE can function as a ligand to its receptors. These lines of evidence raise the question of whether Aβ directly binds to apoE and not to lipids, or directly binds to lipids to form apoE-lipid-Aβ complexes. It is difficult to answer this question using astrocyte cultures, because astrocytes synthesize and secrete both apoE and Aβ (Figs.4d, 6Bc). However, our data on iAβ-mediated lipid release from neurons can exclude the former possibility, because lipid particles associated with iAβ contained neither apoE nor apoJ (Figs. 3b, 6Ac). These data indicate that iAβ interacts directly with lipid particles. Previous studies have shown that Aβ oligomers (dimers, trimers, tetramers, and possibly larger assemblies) are formed in the conditioned media of certain cell lines that constitutively secrete Aβ and that endogenous and synthetic Aβ can assemble into stable oligomers at physiological concentrations in culture (Podlisny et al., 1995, 1998; Xia et al., 1997). Recently, Aβ oligomers have been identified in CSF of AD patients (Pitschke et al., 1998). These lines of evidence together with our present results may allow us to assume that oligomeric Aβ accumulates extracellularly under pathophysiological conditions such as the AD brain, and this oligomeric Aβ, in turn, may stimulate lipid release from neurons, leading to disruption of cholesterol homeostasis in the CNS.
We have recently found that the deficiency in intracellular cholesterol content causes tau phosphorylation (Fan et al., 2001). We have also demonstrated that tau is hyperphosphorylated in brains of mice with Niemann-Pick disease type C (Sawamura et al., 2001), which is known as a cholesterol storage disease involving late endosomes and lysosomes with defective intracellular sterol trafficking (Scriver et al., 1995). On the basis of these lines of evidence, together with the findings reported here, we hypothesize that oligomeric Aβ promotes lipid release, which in turn may reduce cellular cholesterol levels, thereby promoting tau phosphorylation and neurodegeneration, as observed in AD brain. Our observations in the present study also provide new insight into the central issue concerning the pathogenesis of AD, that is, the relationship between amyloid plaque formation and the development of neurofibrillary tangles in neurons.
Footnotes
We are grateful to K. Matsuzaki and S. Yokoyama for helpful comments and suggestions. This study was supported by a research grant for Longevity Sciences (H11-001), Research on Brain Science from Ministry of Health and Welfare, by CREST (Core Research for Evolutional Sciences and Technology), Japan, and by Ono Research Foundation and Life Science Foundation of Japan.
Correspondence should be addressed to Dr. Makoto Michikawa, National Institute for Longevity Sciences, 36-3 Gengo, Morioka, Obu, Aichi 474-8522, Japan. E-mail: michi@nils.go.jp.
REFERENCES
- 1.Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem. 1997;69:1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- 2.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 3.Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK. Acceleration of amyloid fibril formation by specific binding of Aβ(1–40) peptide to ganglioside-containing membrane vesicles. J Biol Chem. 1997;272:22987–22990. doi: 10.1074/jbc.272.37.22987. [DOI] [PubMed] [Google Scholar]
- 4.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 6.Epand RM. Lipid polymorphism and protein-lipid interactions. Biochim Biophys Acta. 1998;1376:353–368. doi: 10.1016/s0304-4157(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 7.Esiri M, Hyman B, Beyreuther K, Masters C. Aging and dementia. In: Graham D, Lantos P, editors. Greenfield's neuropathology. Arnold; London: 1997. pp. 153–233. [Google Scholar]
- 8.Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA, LaDu MJ. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem. 1999;274:30001–30007. doi: 10.1074/jbc.274.42.30001. [DOI] [PubMed] [Google Scholar]
- 9.Fagan AM, Younkin LH, Morris JC, Fryer JD, Cole TG, Younkin SG, Holtzman DM. Differences in the Abeta40/Abeta42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann Neurol. 2000;48:201–210. [PubMed] [Google Scholar]
- 10.Fan QW, Wei Y, Senda T, Yanagisawa K, Michikawa M. Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J Neurochem. 2001;76:391–400. doi: 10.1046/j.1471-4159.2001.00063.x. [DOI] [PubMed] [Google Scholar]
- 11.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hertel C, Terzi E, Hauser N, Jakob-Rotne R, Seelig J, Kemp JA. Inhibition of the electrostatic interaction between β-amyloid peptide and membranes prevents β-amyloid-induced toxicity. Proc Natl Acad Sci USA. 1997;94:9412–9216. doi: 10.1073/pnas.94.17.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isobe I, Michikawa M, Yanagisawa K. Enhancement of MTT, a tetrazolium salt, exocytosis by amyloid β-protein and chloroquine in cultured rat astrocytes. Neurosci Lett. 1999;266:129–132. doi: 10.1016/s0304-3940(99)00282-7. [DOI] [PubMed] [Google Scholar]
- 16.Isobe I, Yanagisawa K, Michikawa M. A possible model of senile plaques using synthetic amyloid β-protein and rat glial culture. Exp Neurol. 2000;162:51–60. doi: 10.1006/exnr.2000.7316. [DOI] [PubMed] [Google Scholar]
- 17.Ito J, Zhang LY, Asai M, Yokoyama S. Differential generation of high-density lipoprotein by endogenous and exogenous apolipoproteins in cultured fetal rat astrocytes. J Neurochem. 1999;72:2362–2369. doi: 10.1046/j.1471-4159.1999.0722362.x. [DOI] [PubMed] [Google Scholar]
- 18.Kim KS, Wen GY, Banker C, Chen CMJ, Spienza VJ, Hon H, Wisniewski HM. Detection and quantitation of amyloid β-peptide with 2 monoclonal antibodies. Neurosci Res Commun. 1990;7:113–122. [Google Scholar]
- 19.Koudinov A, Matsubara E, Frangione B, Ghiso J. The soluble form of Alzheimer's amyloid beta protein is complexed to high density lipoprotein 3 and very high density lipoprotein in normal human plasma. Biochem Biophys Res Commun. 1994;205:1164–1171. doi: 10.1006/bbrc.1994.2788. [DOI] [PubMed] [Google Scholar]
- 20.Koudinov AR, Koudinova NV, Kumar A, Beavis RC, Ghiso J. Biochemical characterization of Alzheimer's soluble amyloid beta protein in human cerebrospinal fluid: association with high density lipoproteins. Biochem Biophys Res Commun. 1996;223:592–597. doi: 10.1006/bbrc.1996.0940. [DOI] [PubMed] [Google Scholar]
- 21.LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman DM. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 1998;70:2070–2081. doi: 10.1046/j.1471-4159.1998.70052070.x. [DOI] [PubMed] [Google Scholar]
- 22.LeVine H., III Soluble multimeric Alzheimer beta(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- 23.LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Yokoyama S. Independent regulation of cholesterol incorporation into free apolipoprotein-mediated cellular lipid efflux in rat vascular smooth muscle cells. J Biol Chem. 1995;270:26216–26223. doi: 10.1074/jbc.270.44.26216. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Peterson DA, Schubert D. Amyloid β peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proc Natl Acad Sci USA. 1998;95:13266–13271. doi: 10.1073/pnas.95.22.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mark RJ, Blanc EM, Mattson MP. Amyloid beta-peptide and oxidative cellular injury in Alzheimer's disease. Mol Neurobiol. 1996;12:211–224. doi: 10.1007/BF02755589. [DOI] [PubMed] [Google Scholar]
- 28.Mason RP, Estermyer JD, Kelly JF, Mason PE. Alzheimer's disease amyloid β-peptide 25–35 is localized in the membrane hydrocarbon core: x-ray diffraction analysis. Biochem Biophys Res Commun. 1996;222:78–82. doi: 10.1006/bbrc.1996.0699. [DOI] [PubMed] [Google Scholar]
- 29.Mason RP, Jacob RF, Walter MF, Mason PE, Avdulov NA, Chochina SV, Igbavboa U, Wood WG. Distribution and fluidizing action of soluble and aggregated amyloid β-peptide in rat synaptic plasma membranes. J Biol Chem. 1999;274:18801–18807. doi: 10.1074/jbc.274.26.18801. [DOI] [PubMed] [Google Scholar]
- 30.Matsubara E, Ghiso J, Frangione B, Amari M, Tomidokoro Y, Ikeda Y, Harigaya Y, Okamoto K, Shoji M. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol. 1999;45:537–541. [PubMed] [Google Scholar]
- 31.Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated β-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 32.McLaurin J, Chakrabartty A. Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J Biol Chem. 1996;271:26482–26489. doi: 10.1074/jbc.271.43.26482. [DOI] [PubMed] [Google Scholar]
- 33.Mendez AJ. Cholesterol efflux mediated by apolipoproteins is an active cellular process distinct from efflux mediated by passive diffusion. J Lipid Res. 1997;38:1807–1821. [PubMed] [Google Scholar]
- 34.Mendez AJ, Oram JF, Bierman EL. Protein kinase C as a mediator of high density lipoprotein receptor-dependent efflux of intracellular cholesterol. J Biol Chem. 1991;266:10104–10111. [PubMed] [Google Scholar]
- 35.Michikawa M, Yanagisawa K. Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J Neurosci Res. 1998;54:58–67. doi: 10.1002/(SICI)1097-4547(19981001)54:1<58::AID-JNR7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Michikawa M, Fan QW, Isobe I, Yanagisawa K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74:1008–1016. doi: 10.1046/j.1471-4159.2000.0741008.x. [DOI] [PubMed] [Google Scholar]
- 37.Muller WE, Koch S, Eckert A, Hartmann H, Scheuer K. β-Amyloid peptide decreases membrane fluidity. Brain Res. 1995;674:133–136. doi: 10.1016/0006-8993(94)01463-r. [DOI] [PubMed] [Google Scholar]
- 38.Osterman D, Mora R, Kezdy FJ, Kaiser ET, Meredith SC. A synthetic amphiphilic β-strand tridecapeptide: a model for apolipoprotein B. J Am Chem Soc. 1984;106:6845–6847. [Google Scholar]
- 39.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pitschke M, Prior R, Haupt M, Riesner D. Detection of single amyloid β-protein aggregates in the cerebrospinal fluid of Alzheimer's patients by fluorescence correlation spectroscopy. Nat Med. 1998;4:832–834. doi: 10.1038/nm0798-832. [DOI] [PubMed] [Google Scholar]
- 41.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 42.Podlisny MB, Walsh DM, Amarante P, Ostaszewski BL, Stimson ER, Maggio JE, Teplow DB, Selkoe DJ. Oligomerization of endogenous and synthetic amyloid beta-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 43.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 44.Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, Ohno K, Yanagisawa K, Michikawa M. Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice. J Biol Chem. 2001;276:10314–10319. doi: 10.1074/jbc.M009733200. [DOI] [PubMed] [Google Scholar]
- 45.Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA. 1995;92:1989–1993. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scriver CR, Stanbury JB, Wyngaarden JB, Frederickson DS. Neimann-Pick disease type C: a cellular cholesterol lipidosis. McGraw-Hill; New York: 1995. [Google Scholar]
- 47.Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 48.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y. High tissue content of soluble beta 1–40 is linked to cerebral amyloid angiopathy. Am J Pathol. 1994;145:452–460. [PMC free article] [PubMed] [Google Scholar]
- 50.Terzi E, Holzemann G, Seelig J. Self-association of β-amyloid peptide (1–40) in solution and binding to lipid membranes. J Mol Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 51.Theret N, Delbart C, Aguie G, Fruchart JC, Vassaux G, Ailhaud G. Cholesterol efflux from adipose cells is coupled to diacylglycerol production and protein kinase C activation. Biochem Biophys Res Commun. 1990;173:1361–1368. doi: 10.1016/s0006-291x(05)80938-6. [DOI] [PubMed] [Google Scholar]
- 52.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 53.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 54.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]