

Abstract
We studied paired-pulse depression (PPD) of GABAAergic IPSCs under conditions of reduced transmitter release (caused by Cd2+, baclofen, or reduced stimulus intensity) with whole-cell voltage clamp in CA1 pyramidal cells in vitro. The use-dependent model of paired-pulse responsiveness holds that a decrease in the probability of neurotransmitter release during the first stimulus will cause predictable changes in the paired-pulse ratio (PPR, the amplitude of the second IPSC divided by that of the first). However, the applicability of the use-dependent model to inhibitory synapses is controversial. Our results are inconsistent with this model, but are consistent with the hypothesis that random fluctuations in response size significantly influence PPR. PPR was sensitive to the extracellular stimulus intensity in all conditions. Changes in PPR were not correlated with changes in the first IPSC, but were correlated with changes in variability of the PPRs of individual traces. We show that spurious paired-pulse facilitation (PPF) can result from averaging randomly fluctuating PPRs because the method of calculating PPR as the mean of individual PPRs is biased in favor of high values of PPR. Spurious PPF can mask the intrinsic paired-pulse property of the synapses. Calculating PPR as the mean of the second response divided by the mean of the first avoids the error. We discuss a simple model that shows that spurious PPF depends on both the number of synapses recruited for release and the probability of release at each release site. The random factor can reconcile some conflicting published conclusions.
Keywords: short-term plasticity, neurotransmitter release, use dependency, release probability, hippocampus, GABAergic IPSC
The small size and inaccessibility of most nerve terminals in the CNS make direct functional studies of synapses difficult or impossible, and indirect methods are used to study the control of neurotransmitter release. One method involves paired-pulse stimulation of presynaptic cells (for review, see Zucker, 1989; Thomson, 2000). Two stimuli are delivered at a short interval (<5 sec), and a difference between the amplitudes of the first (A1) and second (A2) responses, expressed as the paired-pulse ratio (PPR), is taken to reflect a transient change in the probability of release. Often there is an inverse relationship: the larger the A1, the smaller the A2 and vice versa. This is the basis of the use-dependent model of PPR. Neuromodulators or physiological processes that affect transmitter release probability (Pr) affect PPR. Conversely, changes in PPR often are assumed to reflect changes in the probability of neurotransmitter release. Use dependence describes paired-pulse responsiveness at many excitatory synapses (e.g., in dissociated hippocampal cell culture) [Dobrunz and Stevens (1997), although cf. Brody and Yue (2000)].
When inhibitory synapses are activated by paired stimulation, A2 is normally smaller than A1 [i.e., paired-pulse depression (PPD) occurs]. Less transmitter is released by the second stimulus because of activation of GABAB autoreceptors in the CA1 (Davies et al., 1990) and dentate gyrus regions in the hippocampal slice (Mott and Lewis, 1991) or because of transmitter depletion resulting from the first release, as in CA3 cells in the slice preparation (Lambert and Wilson, 1993, 1994) and at inhibitory synapses in tissue culture (Wilcox and Dichter, 1994). Both are use-dependent processes. Nevertheless, the applicability of the use-dependent model to changes in PPD at inhibitory synapses is not clear: decreasing the probability of release by the first stimulus (e.g., with low [Ca2+]o) may not increase PPR, as in synaptically coupled dentate granule cell–interneuron pairs (Kraushaar and Jonas, 2000). Within pairs, A2 and A1 may be uncorrelated, as also found by Kraushaar and Jonas (2000) and by Waldeck et al. (2000) at the axo–axonic connections between Mauthner cells and cranial relay interneuron pairs. GABAAergic inhibition plays vital roles in the regulation of CNS excitability and is affected by numerous presynaptic modulatory effects (for review, see Thompson, 1994; Alger and Le Beau, 2001); hence it is very important to understand the plasticity of inhibitory synaptic transmission. We have reexamined the PPR of monosynaptic GABAAergic IPSCs in the hippocampal slice to resolve the apparent discrepancies between the use-dependent model of PPR and the findings referred to. The use-dependent model makes several predictions concerning the effects of an experimental treatment on PPR. (1) There should be a direct relationship between the change in A1 and the change in PPR. (2) Decreasing the probability of release should cause a consistent increase in PPR. (3) The change in PPR should be independent of the number of synapses that have been activated. We tested these predictions by analyzing the changes in PPR that occur when IPSCs are decreased by Cd2+, baclofen, or reduction of the stimulus intensity. Our results are inconsistent with the use-dependent model and imply instead that random response variation can play a significant role in apparent shifts from PPD to paired-pulse facilitation (PPF) when “mean PPR” is calculated as the mean of the individual PPRs. Unless otherwise stated, mean PPR refers to this method of calculation. The findings have practical and theoretical importance and raise questions concerning some previous conclusions based on PPR.
MATERIALS AND METHODS
Preparation of slices. Hippocampal slices were obtained, using conventional techniques, primarily from 4- to 6-week-old male Sprague Dawley rats, although ∼20% of the experiments were done on 16- to 20-d-old rats. There was no obvious difference between the results with animals from these different age groups, and all of the results have been combined. All experiments were performed in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. After the animals were anesthetized deeply with halothane and decapitated, the hippocampi were removed and sectioned into slices 400 μm thick in ice-cold saline, using aVibratome (Technical Products International, St. Louis, MO). The slices were maintained at room temperature in an interface holding chamber in a humidified atmosphere saturated with 95% O2/5% CO2. The slices were used at least 1 hr after sectioning. The recording chamber warmed the submerged slice, and experiments were performed at 30 ± 1°C (Nicoll and Alger, 1981).
Electrophysiology. Whole-cell voltage-clamp recordings of CA1 pyramidal cells were performed via the “blind” patch method (Blanton et al., 1989) with patch electrodes (2–5 MΩ in the bath). Recordings with series resistance <30 MΩ were accepted. During the experiments series resistance was checked by −1 mV hyperpolarizing voltage steps, and data associated with obvious changes of series resistance or unstable current baseline were discarded. Holding potential was −70 mV in all experiments. Monosynaptic IPSCs were elicited by 100 μsec extracellular stimuli delivered with concentric bipolar stimulating electrodes (David Kopf Instruments, Tujunga, CA) placed in stratum pyramidale 0.5–1 mm apart from the recording site, with the tip of the electrode lowered ∼250 μm into the slice. Evoked IPSC data were collected with an Axopatch 1C or Axoclamp 2B amplifier (Axon Instruments, Union City, CA), filtered at 2 kHz, and digitized at 5 kHz with a Digidata 1200 and Clampex7 software (Axon Instruments).
The intracellular recording solution contained (in mm): 90 or 85 CsCH3SO3, 50 CsCl2, 0.2 CaCl2, 1 MgCl2, 2 Mg-ATP, 2 Cs4-BAPTA, 10 HEPES, and 5 QX-314, pH 7.20 with CsOH (295 mOsm). The extracellular solution included (in mm): 120 NaCl, 3 KCl, 25 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 2 MgSO4, and 15 glucose (300 mOsm). The extracellular solution was oxygenated with 95% O2/5% CO2 gas and flowed continuously through the recording chamber at a rate of ∼1 ml/min.
The local anesthetic QX-314 (Sigma, St. Louis, MO) was included in the recording pipette to block sodium-dependent action potentials (Connors and Prince, 1982) as well as postsynaptic GABABresponses in the pyramidal cell (Nathan et al., 1990; Andrade, 1991). To isolate monosynaptic IPSCs, we included ionotropic glutamate receptor blockers 10 μm 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX; Sigma) and 50 μmdl-2-amino-5-phosphonopentanoic acid (dl-AP-5; Sigma) (Davies et al., 1990) in the bath solution throughout the experiments. Water-based stock solutions of CdCl2(Sigma) or (±)baclofen (Research Biochemicals, Natick, MA or Sigma) were added to the bath solution and perfused into the recording chamber when needed.
Data analysis. Except where discussed in the text, PPR was calculated by dividing A2 by A1. A1 and A2 were determined by measuring the difference between baseline amplitude immediately before the first or the second stimulus and peak amplitude. To calculate a mean PPR in a given condition, we recorded 20–50 individual traces, calculated PPRs of individual traces, and averaged them. We called these 20–50 traces in a given condition a “trace group.” The stimulus intensity was adjusted so that failures of evoked IPSC almost never occurred. Only a few trace groups contained any failures, and when a failure did occur, it was excluded from the trace group.
Data analysis was done with Clampfit 6.0.5 (Axon Instruments) and Excel 97 (Microsoft, Redmund, WA), and graphs were drawn in SigmaPlot 2000 (SPSS, Chicago, IL). Statistical tests were done in Excel 97 and SigmaStat10 (SPSS), and the regression tests were performed in SigmaPlot 2000 and Origin 6.0 (Microcal Software, Northampton, MA). Allt tests were two-tailed tests; the p value for significance was < 0.05. Random number generation in Figure 1 was done with Microsoft Excel 97 or SigmaPlot 2000. The neuronal network stimulation program, Stella Research Software 5.1.1 (High Performance Systems, Hanover, NH), was used for the simulations in Figures 1 and7.
Fig. 1.

Simulation of paired-pulse ratios (PPRs) by means of randomly generated numbers. Simulated PPRs were generated assuming that the distribution of Pr across the simulated population is either uniform (A,B) or nonuniform (C, D).A, To test whether spurious PPF can be obtained by the simple occurrence of random numbers, we generated 50 pairs of random numbers between 10 and 100 and took the ratio (simulated PPR) of the second (A2) to the first (A1). When PPR is plotted against A1, the result resembles the output of a use-dependent process. The mean of the PPRs of this trace group is 1.51. B, Spurious PPF is related to the CV of the distribution of sampled random numbers. We varied the CV by restricting the range of random numbers that were sampled and plotted the means of trace groups generated as inA; one dot represents the mean PPR of one trace group. The least variable numbers were generated between 90 and 100, and the most variable numbers were generated between 5 and 100. Spurious PPF increased monotonically as the CV of the population of sampled numbers increased. In all figures CV is given as its absolute value and is plotted as descending to the right to emphasize that large values of CV are associated with small response sizes. C, D, The analysis is the same as in A and B except that a nonuniformPr distribution of a population of simulated synapses was modeled with a Γ density function (see Materials and Methods). Simulation of probabilistic release was accomplished by comparing an assigned Pr at a synapse with a randomly generated number between 0 and 1; ifPr was greater than a random number, then a release was counted. C, Fifty pairs of random responses were generated from a Γ distribution ofPr, with a mean of 0.033 (to simulate low Pr conditions) and individualPr values distributed across 200 synapses. Note again the inverse relationship between simulated PPR and A1. The mean PPR of this simulated trace group is 1.32. D, CVs of the nonuniform Pr distributions were varied by varying the mean Pr of the Γ distributions (from 0.013 to 0.645). Again, spurious PPF increases with increasing the CV of simulated responses.
Fig. 7.

Theoretical model of dependence of spurious PPF onPr and stimulus strength when mean(A2/A1) is used. Random PPRs were simulated by a nonuniformPr distribution (Γ density function) as in Figure 1, C and D. The total number of activated synapses (n) was varied from 10 to 4000. [The bin size for the Pr distribution (see Materials and Methods) was 0.05 except when n was <100 and Pr = 0.645; then it was 0.1.] This simulation was done with three meanPr values of the Γ functions (indicated by different symbols). Each symbolrepresents the mean of 6–15 trace groups ± SEM, and each trace group comprised 50 individual PPRs. Note that PPR is dependent both on the mean Pr of the population and on the number of activated synapses. Moving along the y-axis at a particular x point simulates the addition of Cd2+ or baclofen application (i.e., decreasingPr) at a fixed stimulus intensity. AsPr decreases, spurious PPF increases. Moving along the x-axis simulates increasing the stimulus intensity (increasing the number of activated synapses). For every value of Pr, increasing the number of activated synapses decreases spurious PPF; decreasing this number increases PPF. Therefore, spurious PPF will increase as stimulus intensity is decreased.
Simulation. Pairs of random integers were generated initially by the random number generators in Excel 97 by using “randbetween” or in SigmaPlot 2000 by using the “rand” commands. The ratio of the second number divided by the first simulated a PPR, and one trace group contained 50 PPRs. The results obtained with both programs were indistinguishable, demonstrating that our conclusions do not depend on the use of one particular random number generator. The Excel random number generator was used for the experimental results that are shown. Because each number has an equal chance of occurrence, this type of model simulates a uniform spatial distribution of Pr. We obtained virtually identical results (data not shown) by using the Gaussian random number generator in SigmaPlot 2000, suggesting that a uniformPr distribution is also inessential to the main conclusions.
To simulate physiological random PPRs more realistically, we generated pairs of random numbers by assuming that Pr is distributed in a spatially nonuniform manner and can be modeled by nonuniform Pr distributions by Γ density functions (Martin, 1973) of the form:
| Equation 1 |
where
Γ Distributions have been used to describe thePr distributions of synapses in tissue-cultured hippocampal neurons (Murthy et al., 1997). pand f(p;α,β) in Equation 1represent Pr and the number of synapses that have the release probability of p, respectively. Two methods were used to evaluate the value of Equation1. First, the integral value of Γ(α) was calculated in SigmaPlot 2000 in the range from 0 to 5000 with increments of 0.05. Then the integral value was plugged into Equation 1. With the second method the value of Equation 1 was calculated with the built-in function of Excel 97, “gammadist.” In the α range of 0.5–2, the difference in Equation 1 values between the two methods was from 24 to 0.02%, respectively, whereas the difference between them was <10−5% when α was >2. When the values of Equation 1 were multiplied by scaling factors consistent with the total number of synapses being 200 (see below) or other numbers as appropriate (see Fig. 7), the distributions were identical. We fixed β at 1/11 (cf. Murthy et al., 1997) and varied α to obtainPr distributions with various meanPr values. When the meanPr is low, the shape of thePr distributions is skewed negatively, and when the mean Pr is high, the distributions are skewed positively. These Γ distributions closely resemble β density functions used in modeling climbing fiber–Purkinje cell synapses (Silver et al., 1998).
Pr was binned by 0.05 from 0 to 1. For each Pr the number of synapses was calculated with Equation 1. Equation 1 was multiplied by a scale factor so that the total number of synapses for a distribution was 200, except for Figure 7, in which the total number of synapses was varied. We chose to model 200 synapses because this approximated the number expected to generate a typical IPSC amplitude in our experiments (∼1 nA) assuming a quantal IPSC amplitude of ∼20 pA (Morishita and Alger, 1997), the release of a single quantum per action potential per synapse, and the estimatedPr of individual release sites ∼0.4 (based on data in Miles and Wong, 1984; Buhl et al., 1995; Vida et al., 1998). If the true Pr differs from 0.4, then the number of synapses could be too high or too low, but in general these numbers represent reasonable estimates of the actual physiological conditions. To simulate a probabilistic release event from this nonuniform distribution, we compared thePr at each synapse with a random number generated between 0 and 1; ifPr was greater than the random number, the synapse was considered to have released a quantum, and ifPr was less than the random number, a failure of release was counted. The response size (i.e., amplitude) was taken as the sum of releases across the population of synapses for a given trial, with each response of a pair being simulated by generating a new random number distribution. Simulated PPR was obtained from the ratio of two response sizes simulated in this way. A trace group consisted of 50 simulated PPRs.
RESULTS
Computer simulations
A widely used method of calculating the PPR is to take the mean of the ratios of responses to pairs of stimuli, A2/A1 [i.e., mean(A2/A1)]. If PPR > 1, an increase in the probability of transmitter release is said to have occurred. However, the mean(A2/A1) method of calculating PPR is biased systematically in favor of detecting PPF. Consider two numbers (representing IPSC amplitudes)a and b (where a, b > 0 and a ≠ b); if a/bis >1.0, then b/a will be <1. Moreover, becausea/b can be arbitrarily large (between 1 and infinity) but b/a must be between 0 and 1, then ‖a/b − 1‖ > ‖b/a − 1‖. The mean of a set of sucha, b ratios will be >1 (i.e., it will appear that PPF has occurred) even if the values a and boccur randomly and are equally likely to occur as A2 or A1.
To show this graphically, we first simulated a paired-pulse experiment by using a random number generator and assuming a uniform distribution of random numbers (see Materials and Methods). We took pairs of random numbers (ranging between 10 and 100 to simulate the process of normalizing across experiments) and calculated the ratios of the pairs, simulated PPRs, with 50 PPRs in each experimental trace group. We then plotted the PPR against the corresponding value of simulated A1. An inverse relationship between PPR and A1 has been considered to reflect a use-dependent mechanism, because a small A1 should cause a large increase in PPR. Nevertheless, Figure1A shows that, when individual PPRs of a simulated trace group are plotted against A1, an inverse relationship is generated. In Figure 1A, the largest values of simulated PPR were associated with the smallest values of A1 simply because a small value of A1 is statistically likely to be followed by a larger value for A2 (PPF); conversely, a larger first number is likely to be followed by a smaller second number (PPD). Thus, the inverse relationship does not imply a physiological, use-dependent mechanism. The mean PPR of all of the simulated data in Figure 1A is 1.51 (i.e., it shows the bias toward PPF introduced by random variability).
The important factor leading to spurious PPF is not the small size of A1 per se, but rather the coefficient of variation (CV) of the populations of A1 and A2. This can be seen in Figure1B, in which each dot represents the mean PPR of an entire simulated trace group, such as the one seen in Figure1A. To vary CV systematically, we varied the ranges of the populations from which A1 and A2 were drawn randomly. Groups with the lowest CV had random numbers between 90 and 100, and groups with the highest CV ranged between 5 and 100. The mean PPR of randomly generated pairs calculated with the mean(A2/A1) method was plotted against the CV (Fig. 1B). The plot shows that PPF is largest when CV is largest. Note that the values of PPR approach 1.0 as the CV decreases toward 0. The CV for the simulated trace group in Figure 1A is 0.48.
In these initial simulations we used simple random models to demonstrate the generality of the result. Implicitly, these models resemble the physiological case in which uniform distributions of release probabilities exist for populations of synapses. Yet uniform distributions of Pr do not characterize populations of synapses in the brain. Nevertheless, spurious PPR can occur if we assume nonuniformPr distributions. To show this, we used the program Stella and made a model based on data from the population of glutamatergic synapses in tissue culture, which shows a nonuniform Pr distribution that is well described by the Γ distribution (Murthy et al., 1997). This is a realistic and convenient model that bears a close resemblance to the β distribution that also has been used to characterize possible nonuniform Pr distributions (Silver et al., 1998). As can be seen in Figure 1C, the resulting plot of PPR versus A1 for this simulated trace group resembles the simulation in Figure 1A. To test the prediction that spurious PPF would be correlated with the CV of the response amplitudes, we produced six different Γ distributions (by fixing β and varying α) that had mean Prvalues that varied from 0.013 to 0.645. ThePr distributions determined from physiological experiments that characterize single release sites at GABAergic synapses are generally thought to lie within this range (see Materials and Methods). In Figure 1D each dot represents the mean of a simulated trace group of 50 PPRs generated randomly from these distributions, plotted against the CV of the trace group. As in Figure 1B, the magnitude of spurious PPF increases smoothly with increases in CV. Thus the conclusion that spurious PPF can be produced readily by the mean(A2/A1) method does not depend on the assumption of a uniformPr distribution.
These considerations do not prove that PPF calculated by the mean(A2/A1) method is spurious (genuine, use-dependent PPF would be detected with this method as well), but they do raise the concern that PPF calculated in this way might be spurious. To determine whether the presence of spurious PPF contributes to experimental paired-pulse response data as well as to these computer simulations, we tested several predictions of the use-dependent model.
Experimental tests of the use-dependent model of PPF
Evoked monosynaptic GABAA receptor-mediated IPSCs were recorded from pyramidal cells in the rat hippocampal CA1 region by stimulating extracellularly in stratum pyramidale in the presence of 50 μmdl-AP-5 and 10 μm NBQX (Davies et al., 1990). The paired-pulse interval was 100 msec, and the interval between consecutive pairs of stimuli was usually 5 sec, although in different cells it was between 4 and 9 sec to permit full recovery of the IPSCs between pairs. To examine the effects of decreasing the initial IPSC amplitude on PPR, we treated the slices with 10– 60 μmCd2+ or 2–3 μm baclofen or reduced the stimulus intensity. The first two manipulations should change Pr, and hence PPR, under the use-dependent model. The last will change the number of terminals activated with less, if any, effect onPr. In normal saline, pairs of IPSCs were recorded at a few different stimulus intensities and then recorded again in the presence of Cd2+ or baclofen at the same intensities. Data were recorded from a total of 36 cells; of these 14 were treated with Cd2+ and 13 with baclofen. For the remaining nine cells the data were recorded only in normal saline at different stimulus intensities.
Inconsistent change of PPR and deviation from the use-dependent model
The use-dependent model suggests that PPR should increase consistently when the probability of release is reduced (i.e., less release on the first response will lead to greater release during the second). However, we did not observe a consistent change in PPR in any of the treatments, although they all produced significant decreases in A1. Rather, the effects were mixed: PPR was increased or did not change when Cd2+ or baclofen was applied or when stimulus intensity was reduced (Fig.2A,B). It was surprising that both kinds of effects (increase or no change) could be seen even in a single cell (Fig. 2A,C). In general, Cd2+ or baclofen had no effect on PPR of IPSCs evoked by higher stimulus intensities, but increased PPR when lower intensities were used. This observation is shown graphically in Figure 2C, in which we plotted the change in PPR, expressed as a percentage of control PPR, for a given cell and treatment condition obtained at two levels of stimulus intensity: weak and strong. To compare the effects of reduced stimulation at two different stimulus intensities (Fig. 2C, circles), we recorded PPR at three intensities from a cell: strong, medium, and weak. The variability in recording conditions from slice to slice precludes general, precise definitions of these terms. A strong stimulus was in the range 140–600 μA, and the responses that were produced were very stable from trial to trial; the weak stimulus was in the range of 50–130 μA, and the responses varied considerably, but there were no, or few, response failures. The medium stimulus intensity was between the other two. The PPR change from strong to medium stimulus intensities is considered an effect at stronger intensity (y-axis in Fig. 2C), and the PPR change from medium to weak is an effect at weaker intensity (x-axis in Fig. 2C). If the same degree of PPR change had been produced at both stimulus intensities, the data points would fall on the dotted line. In fact, they mostly fall to the right of the line, meaning that there was a greater tendency toward PPF, or PPR increase, when weak stimulation was used than when strong stimulation was used, although both levels of stimulation were given to the same cell in the same treatment condition. GABAB-dependent PPD increases with increasing stimulus intensity (Davies et al., 1990;Lambert and Wilson, 1994); therefore, differences in GABAB-mediated autoinhibition cannot account for this result. Because increases in stimulus intensity should increase the numbers of fibers and synapses that have been activated without changing the Pr, these data are not predicted by a use-dependent model.
Fig. 2.

Effects of experimental treatments on measured PPR. Cd2+ (10–60 μm), baclofen (2–3 μm), or reduced stimulus intensity had inconsistent effects on PPR change. A, Whole-cell voltage-clamp recordings of IPSCs evoked by paired-pulse stimuli at a holding potential of −70 mV. Stimulus artifacts were removed graphically. The PPR in control solution with strong stimulation was 0.75; with weak stimulation it was 0.63 in the same cell. Baclofen (2 μm) did not change the PPR significantly when the strong (140 μA) stimulus was used (PPR was 0.83; i.e., 110% of control PPR) but increased it in the same cell when weak (103 μA) stimulation was used (PPR was 0.90; i.e., 143% of control PPR). Although the mean PPR was averaged from 30 individual PPR traces, only five traces per condition are shown for clarity. PPR was not altered by reduced stimulus intensity in the control saline before adding baclofen in this example.B, Histogram of PPR change after Cd2+(n = 14 cells; 29 trace groups), baclofen (n = 13 cells; 34 trace groups), or reduced stimulus intensity (n = 15 cells; 30 trace groups) compared with control PPR. Note the skew of distribution toward higher values of PPR. The distributions of PPR change for individual treatment are shown as a horizontal dot plot on the top of the histogram; each dot represents mean PPR from one trace group. The patterns of PPR change are similar in all treatments.C, When data were obtained from a given cell at two or more stimulus intensities, PPR changes at two intensities were analyzed. One dot represents PPR changes in one cell at two different stimulus intensities. Deviation of asymbol from the dotted line means a different change of PPR was recorded in the cell at different stimulus intensities. In general, PPR increases at the lower intensity and does not change at the higher intensity. D, The degree of PPR change was not correlated with the degree of IPSC amplitude change. Onedot represents one trace group; PPR changes vary independently of IPSC amplitude changes. The same conclusion can be reached if the Cd2+, baclofen, and stimulus intensity groups are plotted separately, as can be seen from inspection of the individual symbols.
According to the use-dependent model, decreases inPr should cause increases in PPR, and there should be an inverse relationship between the degree of depression of A1 and the change in PPR. The cell-to-cell variation of IPSC reduction induced by the experimental treatments was very large and ranged from 5 to 90% of control amplitude. However, there was no correlation between the PPR change and IPSC amplitude changes that were observed (Fig. 2D). We analyzed the Cd2+, baclofen, and stimulus intensity groups separately and found no correlation between PPR and degree of depression of A1 within these groups (data not shown); therefore, the data were combined in Figure 2D.
Another commonly used method of assessing use dependence is to plot A2 versus A1 for each pair of responses (Kraushaar and Jonas, 2000; Waldeck et al., 2000); the use-dependent model predicts an inverse relationship between them. Accordingly, we plotted A2 against A1 for every trace group from 25 cells [57 trace groups in normal saline, 20 trace groups in Cd2+, and 22 trace groups in baclofen (data not shown)]. Of 99 trace groups the great majority (92 of 99) had anr2 value of <0.1 (i.e., no correlation), and only seven showed a significant negative relationship (p < 0.05;r2 = 0.33; slope, −0.53) between A2 and A1. Therefore, in agreement with previous reports (Kraushaar and Jonas, 2000; Waldeck et al., 2000), we conclude that use dependence may not be the main underlying mechanism of PPR of IPSCs.
Both larger and smaller values of PPR are associated with increases in mean PPR
We find that the variability of individual PPRs increases along with the increase in the mean PPR of the trace group, as might be expected from the increased variance of individual responses predicted by the variance–mean analysis of transmission (Silver et al., 1998). However, there is also a greatly increased occurrence of individual PPRs that are smaller than the PPRs in control, which would not be expected in a simple use-dependent model. Examples of traces from a single cell are shown in Figure3A. Both the first and second IPSCs in the pairs were relatively constant over several trials in control solution. After Cd2+ (30 μm for this cell) was added to the bathing solution, both responses (A1 and A2) became much more variable, with some individual PPRs being higher and many others lower than those in control. The complete set of data from this cell is shown in the graph below the traces.
Fig. 3.

When variability of individual PPRs increases, mean PPRs also increase. A, The individual traces at thetop are representative samples from a single cell before and after Cd2+ (30 μm) application. The mean PPR increased from 0.78 to 1.16 (dotted lines) as in the graph of the complete set of data from this cell. Onedot represents one individual PPR. Individual PPRs range from very low to very high. B, Trace groups with larger mean PPRs have higher variability of individual PPRs. SD values of PPR (SDPPR) within a trace group were plotted against the mean PPR of the trace group. One dot represents one trace group. The variability is fairly constant below a PPR of ∼0.85 in contrast to the variability when the PPR > 1.0 (the PPF region), in which variability increases proportionally with PPR. In the PPR region between 0.85 and 1.0 there is an intermediate pattern of variability. C, When mean PPR increased, the largest individual PPRs increased, and the smallest PPRs decreased. The means of the three largest PPRs (open circles) and of the three smallest PPRs (filled circles) in a trace group were calculated from trace groups that showed a mean PPR increase after the treatments. Data were divided into three groups according to the PPR after treatment (PPRtreat). In the group of PPRtreat < 0.85 (n = 15 trace groups), the largest and the smallest PPRs did not change (pairedt tests, p > 0.1). In the other two groups (n = 23 trace groups each), however, the largest PPRs increased and the smallest PPRs decreased when mean PPR increased. Data from all treatments were pooled because they showed similar patterns individually. The left dot in each pair represents the control value, and the right dotrepresents the value after treatment. The error bars are SEM. The numbers of trace groups treated by reduced stimulation, Cd2+, or baclofen, respectively, included the following: for PPR < 0.85: 3, 4, and 8; for PPR between 0.85 and 1: 6, 9, and 8; for PPR > 1: 11, 8, and 4. *Pairedt test, p < 0.01. D, The largest and smallest PPRs are related inversely in groups showing mean PPF. Each symbol represents the mean of the three largest PPRs of a trace group plotted against the mean of three smallest PPRs of the given trace group. Data are from trace groups for which the mean PPRs were >1.0. The solid lines inD–F are the y = 1/xcurves, which describe a perfect inverse relationship.E, The same plot as in D made for groups with mean PPRs below 0.85. F, The same plot as inD and E for simulated data. Filled circles are trace groups with a CV of A1 > 0.3;open circles are trace groups with a CV of A1 < 0.3.
In all treatments we found that the trace groups with higher mean PPRs had higher variability of individual PPRs than those groups with lower mean PPRs. This relationship is shown in Figure 3B, in which we plotted the variability (SD) of PPR within a group against the mean PPR of that group in all conditions. Data in Figure 3B were from all 36 cells (178 trace groups). The relative variability was fairly low and constant when PPR was below 1 (i.e., PPD was present) but then began to rise and was very large in the region when PPR was >1 (PPF).
To determine whether it is generally true that when individual PPRs became variable the mean PPR increases, we took the largest three individual values of PPR for a given trace group and the smallest three values of that group as a measure of the range of values in the group. We then compared these values for control and experimental conditions in cases in which the mean PPR increased in the experimental treatment, because this was the direction of change predicted by the use-dependent model. Because the changes in variability of PPR were clearly dependent on the value of the mean PPR in a given condition (Fig. 3B), we analyzed the data in three groups: final value of PPR below 0.85, PPR between 0.85 and 1.0, and PPR >1.0. These data are summarized in Figure 3C. When the final PPR was low, there was hardly any change in the distribution of values of the PPR (paired ttest between control group and treatment group, p > 0.1). In the other two groups the lowest values of PPR were significantly lower than control, and the highest values were significantly higher than control (paired t test,p < 0.01).
The changes in variability of PPR are consistent with the random fluctuation model, but may not exclude use-dependent models, because increases in absolute response variance associated with decreases inPr can be seen in variance–mean plots (Silver et al., 1998). To determine whether the variability in PPR that we recorded actually reflected random response fluctuations, we applied several tests.
Highly variable PPRs might arise from random A1 and A2
One prediction of the random model is that there will be an inverse relationship between the largest PPR and the smallest PPR within a trace group. Consider two of the possible values for A1 and A2, say a and b. If a and boccur randomly within an experiment, then the value of PPRa/b should occur as frequently as the valueb/a. The occurrence of a/band b/a will be related inversely (i.e., within a given trace group the largest values of PPR will be related inversely to the smallest values of PPR). To test this prediction, in Figure3D we plotted the mean of the three largest PPRs,y, in a trace group against the mean of the three smallest PPRs, x, of the same trace group for the Cd2+, baclofen, and low-stimulus-intensity data. When the mean of the PPRs was >1.0, the relationship between the largest and smallest PPRs within the group was close to the inverse relationship, y = 1/x, predicted by the random model (Fig. 3D, solid line). However, such a relationship was not found when PPR was <0.85 (Fig. 3E) or when 0.85 < PPR < 1.0 (data not shown). Finally, Figure3F shows the fit of y = 1/x to simulated PPR data generated by the random model, using either Γ functions having different CVs (which was necessary to obtain the full range of PPR ratios to make the comparison) or a uniformPr model.
Additional tests of the random model
If the pattern of individual PPRs reflects random variability, it would be the same with long interstimulus intervals at which there is no short-term plasticity as with the 100 msec interval. If the pattern of individual PPRs at the 100 msec interstimulus interval reflects a genuine physiological property, it should change with long interstimulus intervals because of recovery from short-term plasticity at these intervals. In these experiments the long interstimulus intervals were between 4 and 9 sec for different trace groups, although within a trace group the interval was fixed. To calculate PPRs with 4–9 sec interstimulus intervals, we divided a given A1 by the previous A1 within the same trace group (Fig. 4). Although A2 was evoked between A1 and the next A1, the interval between A2 and the next A1 was long enough to eliminate any use dependence.
Fig. 4.

Comparison between PPR at short and long interstimulus intervals. The short interval was 100 msec; the long interval varied between different cells between 4 and 9 sec (but was constant for a given cell). PPR at long intervals was calculated by dividing A1 from one pair by the A1 of the immediately preceding pair. Because the interpair interval was 4-9 sec, “A1/previous A1” is a good approximation to long intervals.A1, Representative cell with PPR < 0.85. At the 100 msec interval this cell showed PPD (filled circles), but the mean PPR changed to ∼1.0 at the long interval (open circles). Onedot is one PPR. A2, Group data (n = 87 trace groups) in which PPR < 0.85 shows significant changes in mean PPR and in the distribution of individual PPRs. The histogram compares the mean PPRs, the three largest PPRs, the three smallest PPRs, and the SDPPR of the trace groups at the 100 msec (filled bar) and long (open bar) intervals. The asterisksindicate significant differences (paired t tests,p < 0.01). B1, Representative trace groups from a cell with PPR > 1.0 both at the 100 msec and at the long intervals.B2, Group data for 40 groups that showed PPF at the 100 msec and at the 4–9 sec intervals. The histogram shows that the mean PPR and the PPR distribution patterns at these two intervals did not differ (paired t test,p > 0.05).
In the trace groups with mean PPR < 0.85 (i.e., PPD), the mean PPR increased to ∼1 at the 4–9 sec interstimulus intervals (Fig.4A). Of 178 trace groups gathered from all cells, 87 had mean PPR < 0.85, and these changed from 0.74 ± 0.010 to 1.01 ± 0.0035 (paired t test, p < 0.01; Fig. 4A2) with the change of interval from 100 msec to 4–9 sec. This suggests that PPD was a genuine property of the responses. In contrast, when the mean of the PPRs was >1.0 (i.e., PPF) at 100 msec (40 of 178 trace groups), then PPR did not change with the interval change from 100 msec to 4–9 sec (Fig. 4B); the mean PPR was 1.23 ± 0.031 at the 100 msec interval and was 1.29 ± 0.052 at the 4–9 sec interval (paired t test, p > 0.05; Fig.4B2). We again compared the three largest PPRs, the three smallest PPRs, and SD of PPRs within a trace group. In addition to the lack of change in the mean PPR, the pattern or distribution of individual PPRs within a trace group was not significantly different at the two intervals (pairedt test, p > 0.05; Fig.4B2). The lack of change in PPR between the 100 msec and the 4–9 sec PPR intervals strongly implied that apparent PPF was caused by random fluctuations of A1 and A2.
Spurious PPF is eliminated by the meanA2/meanA1 method
If A1 and A2 differ simply because of random variation, then within a trace group mean A1 should be the same as mean A2. If PPR were calculated by dividing mean A2 by mean A1, spurious PPF would be eliminated because the random fluctuations that give rise to it would average out. To test this, we recalculated mean PPR for each trace group by the meanA2/meanA1 method and plotted these values against the mean PPR calculated by mean(A2/A1) (Fig.5). PPRs > 1 calculated with mean(A2/A1) changed to ∼1 with the meanA2/meanA1 method, as predicted by the random model. Interestingly, many of the PPRs that were <1 also shifted toward smaller values (Fig. 5B), implying that the effects of random fluctuations also bias the mean PPR in this region, even when overt PPF is not calculated.
Fig. 5.
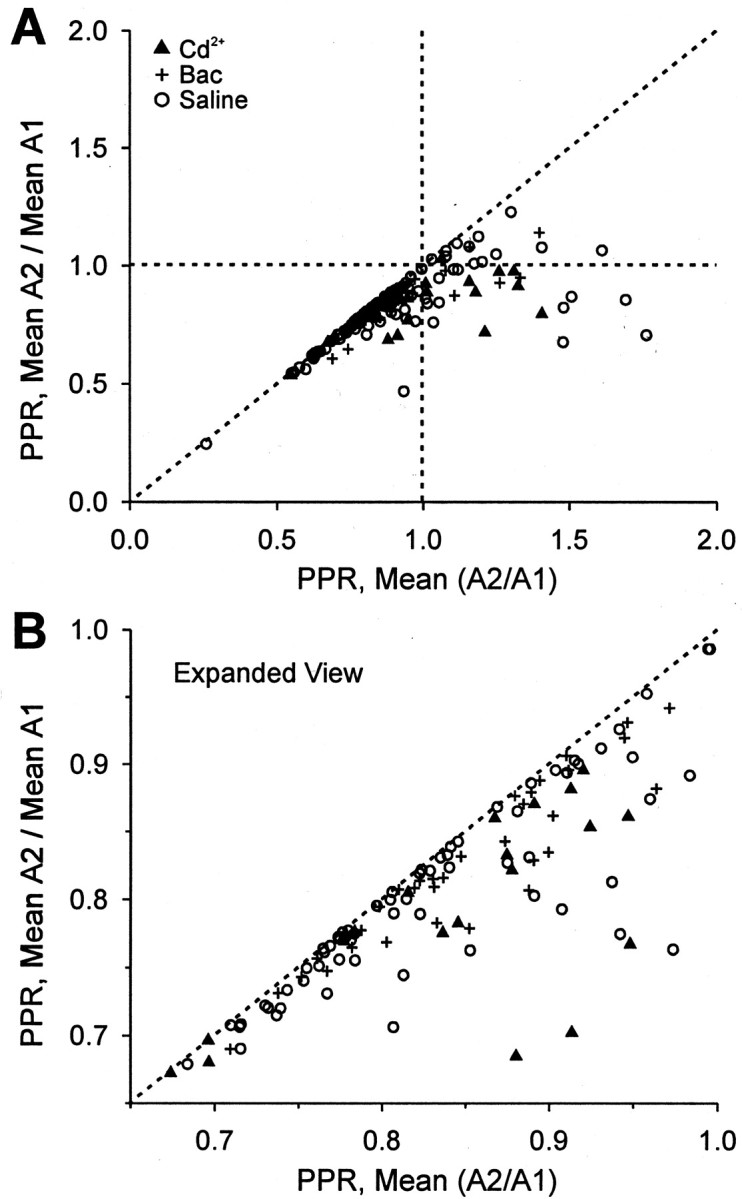
Spurious PPF disappears when PPR is calculated by meanA2/meanA1. A, For each trace group the PPR was calculated by dividing the mean of all of the A2 values for a given group by the mean of the A1 values for that group (meanA2/meanA1); this PPR value was plotted against the PPR calculated by mean(A2/A1). Note that PPF calculated as the mean(A2/A1) (i.e., values >1.0 on thex-axis) disappeared with the meanA2/meanA1 method.B, The region between 0.65 and 1.0 of the plot inA is expanded. Deviation of PPR values from thedotted line also can be seen in the PPD region (values <1.0), indicating that random fluctuations also influence PPD values calculated by mean(A2/A1) even when spurious PPF is not apparent.
Because the spurious PPF is dependent on the CV of the data points as well as on the method of calculation, the random model predicts that spurious PPF in the experimental data should depend on the CV of A1, as was true in the simulated data in Figure 1. We therefore replotted our experimental PPR data, using mean(A2/A1), against the CV of A1 for each experimental trace group. PPF clearly was associated with the higher values of CV (Fig. 6A). If PPF in this case is spurious, then replotting the experimental PPR data by using meanA2/meanA1 against the CV of A1 should remove it. Figure 6B confirms this expectation: virtually all of the points fall below the value 1.0, revealing the consistent occurrence of PPD throughout the range of experiments. Figure6C presents a similar plot of meanA2/meanA1 versus CV of A1 for data simulated by using Γ functions, showing, as expected, that the points scatter about 1.0 (i.e., there is no tendency for PPF or PPD in the random data when meanA2/meanA1 is used).
Fig. 6.
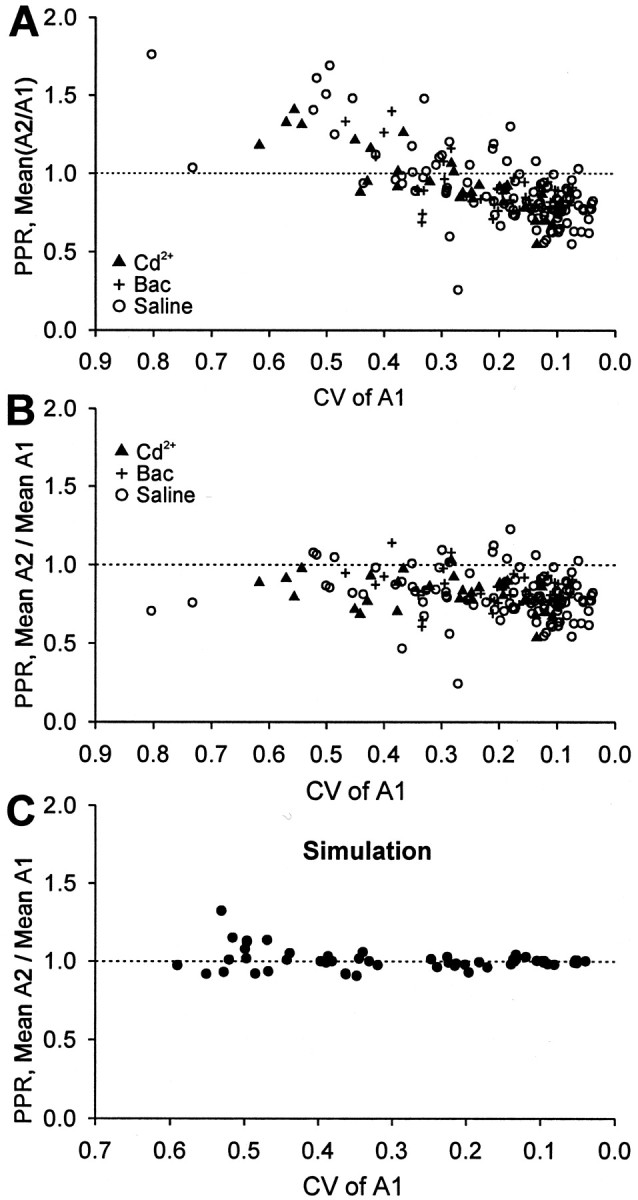
Spurious PPF as a function of CV in experimental and simulated data. A, Experimental data were plotted as the mean PPRs of a trace group versus a CV of the same trace group, as in Figure 1, B and D. PPF increases as CV increases as in the simulated pattern in Figure 1, B andD. B, Recalculation of the data inA by meanA2/meanA1 and plotting it against a CV of A1 removes spurious PPF without altering PPD. C, The simulated PPR data in Figure 1D were recalculated with meanA2/meanA1 and plotted against CV. Spurious PPF in large part disappeared; the symbols vary ∼1.0.
Sources of random fluctuations
For simplicity, we assume that the random variability that dominates experiments such as these arises principally from variability in the numbers of synapses that have been activated and in the probability of transmitter release from each synapse. We manipulated these factors experimentally by changing the stimulus intensity or by the addition of either Cd2+ or baclofen and found that both kinds of manipulations appear to be equivalent in their tendency to produce spurious PPF. To verify that the resulting changes in CV alone were sufficient to cause spurious PPF, we simulated PPR calculated by mean(A2/A1) and varied both the numbers of synapses that had been activated and the meanPr of nonuniform distributions ofPr, as in Figure 1. The results, shown in Figure 7, make two points: decreasingPr and decreasing the number of activated synapses dramatically increase spurious PPF. Conversely, increasing the number of synapses that have been activated (analogous to increasing stimulus intensity) decreases spurious PPF for any givenPr. This latter result explains the observation in Figure 2 that varying the stimulus intensity in a given condition alters the PPR.
DISCUSSION
We conclude that changes in PPR do not necessarily reflect changes in the probability of neurotransmitter release. The unrecognized influence of random fluctuations in response amplitudes can lead to erroneous interpretations of experiments that use the PPR calculated by mean(A2/A1). When A1 and A2 are small and vary randomly, then mean PPR will tend to be high by chance alone, and the intrinsic paired-pulse property of the synapse can be masked. This is a particular problem because the effect of the random error is in the direction of the changes predicted by use-dependent models. In these experiments we used Cd2+ and baclofen to reduce IPSCs by reducing transmitter output at individual synapses, and we used a decrease in stimulus intensity to reduce IPSCs without reducing the amount of transmitter released by an individual synapse. Both GABAB-dependent and GABAB-independent mechanisms for PPD exist (Davies et al., 1990; Mott and Lewis, 1991; Lambert and Wilson, 1993, 1994). Reductions in GABA release by Cd2+ should reduce both forms of PPD at all GABAA synapses, and baclofen will reduce PPD at all GABAA synapses with presynaptic GABAB receptors. Cd2+ and baclofen therefore should have qualitatively similar effects on PPD under the use-dependent model. Reduced stimulus intensity could decrease GABAB-dependent PPD disproportionately (Lambert and Wilson, 1994), but because it accounts for a lesser fraction of total PPD, even the loss of GABAB-dependent PPD could not explain our results. Our finding that the same results on PPD were obtained with each method of reducing IPSCs illustrates the problem in using PPR to probe mechanisms of release from an individual synapse. The dependence of PPR on stimulus intensity might suggest that the different populations of interneurons with different inherent PPR properties were activated by different stimulus intensities (i.e., interneurons may vary in their tendency to show PPF or PPD). Although we cannot rule out a small contribution by this factor, our data cannot be explained by such differences. If, for example, low stimulus intensities selected for interneurons that tended to show real PPF, then PPF should have been apparent in the meanA2/meanA1 plots and should have disappeared at the 4–9 sec interstimulus intervals.
Figure 7 illustrates that spurious PPF can arise when few synapses are activated either because of decreasedPr or because of other factors that prevent transmitter release. Although nonlinearities in the efficacy of the extracellular stimulation that often is used in experiments like ours can lead to spurious PPF, spurious PPF also will arise from simultaneous recordings from synaptically coupled pairs of cells, as implied by Figure 7. This is because the CV of the responses grows as the number of synapses that have been activated decreases, an effect that is made clear by Silver et al. (1998), who used β distributions to model spatial nonuniformity of Prand to demonstrate the monotonic increase in CV even down to quantal responses. Evidence that spurious PPR may be observed in paired-cell recordings can be inferred from an investigation of unitary IPSCs in CA1 pyramidal cells (Ouardouz and Lacaille, 1997). Analysis of unitary IPSC PPR data by mean(A2/A1) suggested that use-dependent PPF occurred, whereas analysis of the same data with meanA2/meanA1 revealed no PPF. A possible interpretation was that the averaging involved in the calculation of meanA2/meanA1 had masked a true use-dependent PPF that was obvious only when A1 was small. Our results would suggest that the PPF was spurious and that there was no use-dependent plasticity at these synapses.
As shown in Figures 5 and 6, spurious PPF can be avoided if PPR is calculated by meanA2/meanA1, because random fluctuations in the two responses are averaged out before PPR is determined. However, even without averaging, the appearance of high values of PPR during an experimental treatment that dramatically suppresses the basal level of transmitter release also has been interpreted to mean that a presynaptic effect on the probability of release occurred (Luscher et al., 1997). We suggest that the influence of the random effect may be suspected when the variability of the PPRs increases and especially when the variability clearly extends to values much smaller, as well as larger, than control values of PPR (Fig. 3). The variance of the PPR has been suggested to be a more sensitive index of the locus of synaptic modification (Saitow et al., 2000) than the PPR itself, but the role of variability in spurious PPF has not been discussed.
An important issue is how much the use of the mean(A2/A1) method has affected previous work. To estimate this, we examined 32 peer-reviewed papers published between 1990 and 2001 that investigated synaptic function with the use of PPR. In 10 of these the mean(A2/A1) method was used, in six the meanA2/meanA1 (or a variant) was used, in four papers both methods were used, and in 12 papers we were unable to determine which method was used. All experiments involved measurements of synaptic responses with whole-cell or intracellular recording techniques, 12 involved recordings from synaptically coupled pairs, and 24 were studies of inhibitory responses. In many cases the response amplitudes after a manipulation were very small and were comparable in amplitude to the variable responses we have observed. Generally, the data that were reported did not permit reanalysis, so we could not test for the influence of random fluctuations in detail. Nevertheless, the widespread use of the mean(A2/A1) method, together with the example discussed above, suggests that the influence of the random effect may be greater than generally appreciated. We note that protocols that use a brief train of pulses instead of just two also are subject to the random error effect, if a ratio [A(n)/A1, where A(n) is the amplitude of the nth response] ultimately is used to characterize the results.
Because spurious PPF is nonphysiological, it is independent of the intrinsic plasticity properties of a synapse. We suggest that the inhibitory synapses that we have studied are characterized primarily by PPD but that when the IPSCs are small and variable the spurious PPF masks the PPD. The similarity of our experimental data in Figure 6 with the simulated data in Figure 1 supports this conclusion. When variability was low, PPD was prominent in the experimental data; when variability increased, so did the occurrence of spurious PPF. Other evidence for the masking of PPD by random fluctuations is found in Figure 5. Data points in the PPF region fall into the PPD region when recalculated by meanA2/meanA1. In addition, in the region of PPR < 1 many data points are below the dotted line; that is, the PPRs calculated by meanA2/meanA1 are significantly less than the PPRs by mean(A2/A1) (paired t test, p < 0.01). Thus random fluctuations contributed to the mean PPR in this region, and to some extent the degree of PPD was masked by spurious PPF. Of course, we do not suggest that genuine, physiological PPF does not exist at many synapses; it clearly does (for review, see Thomson, 2000). The possibility of spurious PPF is an insidious and underappreciated factor in the determination of PPR by the mean(A2/A1) method, however.
The PPR traditionally has been used for two main reasons: to determine whether a presynaptic or a postsynaptic effect occurred or to investigate the mechanism of a particular presynaptic effect. Our experiments show that spurious PPF can be produced by manipulations that affect presynaptic function. However, a nonuniform postsynaptic effect, one that altered the synaptic strength of a subset of synapses by affecting the probability of activation of receptor patches, for example, also could alter PPR by random effect. Thus changes in PPR probably cannot be used reliably to distinguish presynaptic from postsynaptic factors in cases in which CV is large and the mean(A2/A1) method is used. Finally, although we have focused on the spurious PPF that can occur when synaptic depression occurs, errors of interpretation in the opposite direction could occur as well. Consider an initial small response pair in which spurious PPF contributes to measured PPR. If an experimental treatment leads to larger initial responses and a decrease in relative response variability (CV), then a decrease in PPR will occur because of a decrease in spurious PPF. Because a decrease in PPR also is predicted by a use-dependent model of synapse strengthening, an erroneous conclusion concerning the mechanism of the effect could be drawn.
Footnotes
This work was supported by National Institutes of Health Grants RO1 NS36612 and RO1 NS30219 to B.E.A. J.K. was supported by the Training Program in Neuroscience T32 DE1474. We thank Scott Thompson and Darrin Brager for help with Stella in the modeling studies and for their comments on this manuscript. We also thank Greg Carlson and Namita Varma for reading and commenting on this manuscript.
Correspondence should be addressed to Dr. Bradley E. Alger, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: balger@umaryland.edu.
REFERENCES
- 1.Alger BE, Le Beau FEN. Physiology of the GABA and glycine systems. In: Mohler H, editor. Pharmacology of GABA and glycine neurotransmission. Springer; Berlin: 2001. pp. 3–76. [Google Scholar]
- 2.Andrade R. Blockade of neurotransmitter-activated K+ conductance by QX-314 in the rat hippocampus. Eur J Pharmacol. 1991;199:259–262. doi: 10.1016/0014-2999(91)90467-5. [DOI] [PubMed] [Google Scholar]
- 3.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole-cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 4.Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhl E, Cobb SR, Halasy K, Somogyi P. Properties of unitary IPSPs evoked by anatomically identified basket cells in the rat hippocampus. Eur J Neurosci. 1995;7:1989–2004. doi: 10.1111/j.1460-9568.1995.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 6.Connors BW, Prince DA. Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. J Pharmacol Exp Ther. 1982;220:476–481. [PubMed] [Google Scholar]
- 7. Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 424 1990. 513 531 facilitation, and depletion at central synapses. Neuron 18:995–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 9.Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron–principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert NA, Wilson WA. Heterogeneity in presynaptic regulation of GABA release from hippocampal inhibitory neurons. Neuron. 1993;11:1057–1067. doi: 10.1016/0896-6273(93)90219-h. [DOI] [PubMed] [Google Scholar]
- 11.Lambert NA, Wilson WA. Temporally distinct mechanisms of use-dependent depression at inhibitory synapses in the rat hippocampus in vitro. J Neurophysiol. 1994;72:121–130. doi: 10.1152/jn.1994.72.1.121. [DOI] [PubMed] [Google Scholar]
- 12.Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G-protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 13.Martin LJ. Distributions and transformations. In: Delaunois AL, editor. Biostatistics in pharmacology, Vol I. Pergamon; Oxford: 1973. pp. 524–544. [Google Scholar]
- 14.Miles R, Wong RKS. Unitary inhibitory synaptic potentials in the guinea-pig hippocampus in vitro. J Physiol (Lond) 1984;356:97–113. doi: 10.1113/jphysiol.1984.sp015455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morishita W, Alger BE. Sr2+ supports depolarization-induced suppression of inhibition and provides new evidence for a presynaptic expression mechanism in rat hippocampal slices. J Physiol (Lond) 1997;505:307–317. doi: 10.1111/j.1469-7793.1997.307bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- 17.Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 18.Nathan T, Jensen MS, Lambert JDC. The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular injection of QX-314. Neurosci Lett. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- 19.Nicoll RA, Alger BE. A simple chamber for recording from submerged brain slices. J Neurosci Methods. 1981;4:153–156. doi: 10.1016/0165-0270(81)90049-2. [DOI] [PubMed] [Google Scholar]
- 20.Ouardouz M, Lacaille J-C. Properties of unitary IPSCs in hippocampal pyramidal cells originating from different types of interneurons in young rats. J Neurophysiol. 1997;77:1939–1949. doi: 10.1152/jn.1997.77.4.1939. [DOI] [PubMed] [Google Scholar]
- 21.Saitow F, Satake S, Yamada J, Konishi S. β-Adrenergic receptor-mediated presynaptic facilitation of inhibitory GABAergic transmission at cerebellar interneuron–Purkinje cell synapses. J Neurophysiol. 2000;84:2016–2025. doi: 10.1152/jn.2000.84.4.2016. [DOI] [PubMed] [Google Scholar]
- 22.Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre–Purkinje cell synapses. J Physiol (Lond) 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson SM. Modulation of inhibitory synaptic transmission in the hippocampus. Prog Neurobiol. 1994;42:575–609. doi: 10.1016/0301-0082(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 24.Thomson AM. Facilitation, augmentation, and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- 25.Vida I, Halasy K, Szinyei C, Somogyi P, Buhl E. Unitary IPSPs evoked by interneurons at the stratum radiatum–stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol (Lond) 1998;506:755–773. doi: 10.1111/j.1469-7793.1998.755bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilcox KS, Dichter MA. Paired-pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci. 1994;14:1775–1788. doi: 10.1523/JNEUROSCI.14-03-01775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]