

Abstract
α-Synuclein mutations have been identified in certain families with Parkinson's disease (PD), and α-synuclein is a major component of Lewy bodies. Other genetic data indicate that the ubiquitin-dependent proteolytic system is involved in PD pathogenesis. We have generated stable PC12 cell lines expressing wild-type or A53T mutant human α-synuclein. Lines expressing mutant but not wild-type α-synuclein show: (1) disruption of the ubiquitin-dependent proteolytic system, manifested by small cytoplasmic ubiquitinated aggregates and by an increase in polyubiquitinated proteins; (2) enhanced baseline nonapoptotic death; (3) marked accumulation of autophagic–vesicular structures; (4) impairment of lysosomal hydrolysis and proteasomal function; and (5) loss of catecholamine-secreting dense core granules and an absence of depolarization-induced dopamine release. Such findings raise the possibility that the primary abnormality in these cells may involve one or more deficits in the lysosomal and/or proteasomal degradation pathways, which in turn lead to loss of dopaminergic capacity and, ultimately, to death. These cells may serve as a model to study the effects of aberrant α-synuclein on dopaminergic cell function and survival.
Keywords: Parkinson's disease, Lewy body, ubiquitin, autophagy, proteasome, lysosome, dopamine
Parkinson's disease (PD) is a neurodegenerative disorder characterized by resting tremor, rigidity, hypokinesia, and postural instability. Pathological examination shows loss of pigmented dopaminergic neurons and accumulation of eosinophilic inclusions termed Lewy bodies in the substantia nigra pars compacta and other brainstem nuclei (Fahn and Przedborski, 2000). The cause of PD is unknown, but recent studies indicate that two separate α-synuclein mutations, A53T and A30P, are responsible for certain rare familial forms of the disease (Polymeropoulos et al., 1997; Kruger et al., 1998). α-Synuclein is a protein of unknown function that localizes within presynaptic terminals in the CNS (Clayton and George, 1998,1999) and, together with ubiquitin, is among the major components of Lewy bodies (Spillantini et al., 1997, 1998), suggestive again of an association with PD pathogenesis.
Two other genetic causes of PD have been identified. Mutations in the gene encoding Parkin are identified in cases with autosomal recessive PD (Kitada et al., 1998), and a mutation in the gene encoding for ubiquitin C-terminal hydrolase L-1 (UCHL-1) is associated with PD in one family (Leroy et al., 1998). Both of these proteins are involved in the ubiquitin-dependent degradation of intracellular proteins (Ciechanover, 1998). Proteins that are degraded through this system are tagged with polyubiquitin chains through a series of enzymatic reactions and then degraded by the proteasome, a multicatalytic complex. Some ubiquitinated proteins may also be degraded by the lysosomal system (Ciechanover, 1998). Parkin is an E3 ligase, responsible for the attachment of ubiquitin to substrates such as CDCrel-1, Pael receptor, and, possibly, a glycosylated form of α-synuclein (Shimura et al., 2000; Zhang et al., 2000; Imai et al., 2001; Shimura et al., 2001), and UCHL-1 is a member of the family of deubiquitinating enzymes, which remove polyubiquitin chains once the substrate has been attached to the proteasome (Ciechanover, 1998).
Induced expression of wild-type or mutant α-synuclein has been achieved in a number of cellular systems. The results have been variable, with some studies reporting no effects with overexpression alone (Ko et al., 2000), others reporting adverse effects with wild-type or mutant α-synuclein overexpression (Ostrerova et al., 1999; Saha et al., 2000), and others reporting adverse effects only with mutant overexpression (Zhou et al., 2000; Lee et al., 2001). Many studies have also reported increased toxicity of wild-type α-synuclein overexpression and, more consistently, mutant α-synuclein overexpression in cells only after challenge with a variety of agents, particularly those that induce oxidative stress (Kanda et al., 2000; Ko et al., 2000; Ostrerova-Golts et al., 2000;Tabrizi et al., 2000).
To investigate the effects of wild-type or mutant α-synuclein in a dopaminergic cell system, we have generated stable rat pheochromocytoma PC12 cell lines (Greene and Tischler, 1976) expressing the wild-type and A53T mutant forms of the human protein. PC12 cells were selected because they are dopaminergic and have been extensively studied as models of neuronal degeneration (Greene, 1978; Batistatou and Greene, 1991; Rukenstein et al., 1991; Mesner et al., 1992; Stefanis et al., 1996). We have placed particular emphasis on the effects of α-synuclein on the cellular degradation machinery, because of the genetic and pathological evidence suggesting dysfunction of this system in PD (Kitada et al., 1998; Leroy et al., 1998; Shimura et al., 2000; McNaught and Jenner, 2001; McNaught et al., 2001), and on the dopaminergic phenotype, because this seems to be affected out of proportion to the degree of dopaminergic cell loss in PD (Hornykiewicz, 1996). We find that expression of mutant α-synuclein enhances baseline levels of death, induces accumulation of autophagic–vesicular structures, and results in a loss of catecholamine storage granules and the capacity for depolarization-induced dopamine release. These effects are accompanied by, and may be consequences of, defects in the lysosomal and proteasomal degradation systems.
MATERIALS AND METHODS
Cell culture
PC12 cells were grown as described previously (Greene and Tischler, 1976; Rukenstein et al., 1991) on rat-tail collagen-coated dishes in RPMI 1640 medium containing 5% fetal bovine serum and 10% heat-inactivated horse serum (complete medium).
Generation of constructs and transfection of PC12 cells
Wild-type α-synuclein in TA vector (Invitrogen, Carlsbad, CA) was generated as described previously (Stefanis et al., 2001). The A53T mutation was induced by PCR-based site-directed mutagenesis. Both inserts, wild type and mutant, were subcloned in theHindIII–XhoI sites of a PCDNA3 expression vector (Invitrogen), downstream of a cytomegalovirus promoter. PC12 cells were transfected by electroporation with empty vector, wild-type α-synuclein, or mutant α-synuclein; individual colonies were subsequently selected in the presence of neomycin (Greene et al., 1998;Stefanis et al., 2001).
Preparation of cell lysates for Western blotting
Cells were rinsed in cold PBS and then collected in a buffer of 25 mm HEPES, pH 7.5, 5 mm EDTA, 1 mm EGTA, 5 mm MgCl2, 0.5% Triton X-100, 2 mm DTT, 10 μg/ml each of pepstatin and leupeptin, and 1 mm PMSF. The cellular material was left for 20 min on ice. The lysate was then centrifuged for 20 min at 160,000 × g, and the supernatant was collected. The detergent-insoluble pellet was solubilized in SDS–sample buffer and sonicated. Protein concentrations were measured using the Bradford assay (Bio-Rad, Richmond, CA).
Western immunoblotting
Equal volumes of 2× sample buffer were added to Triton-soluble lysates (200 μg of protein). Alternatively, Triton-insoluble material solubilized in sample buffer (200 μg of protein) was used. The samples were boiled for 5 min, resolved by SDS-PAGE electrophoresis, transferred to nitrocellulose membranes, and immunoblotted with mouse anti-synuclein-1 (1:1000; Transduction Laboratories, Lexington, KY), rabbit anti-ubiquitin (1:1000; Dako, Glostrup, Denmark), or mouse anti-actin (1:5000, Sigma, St. Louis, MO) antibodies according to previously described procedures (Stefanis et al., 1998).
Assessment of cell death
Trypan blue assay. The day before the assay, subconfluent cultures were rinsed twice with complete medium to remove cells at late stages of degeneration. The following day the cells were triturated off the dish and five drops of the cellular suspension were added in a tube together with five drops of Trypan blue solution (0.4%; Sigma). After light mixing, the cells were placed on a glass slide, coverslipped, and visualized under 20× magnification. For each line, three fields of 100 cells each were assessed for the percentage of Trypan blue-positive cells. The results reported are the mean ± SEM (n = 3).
Nuclear staining. Cells were plated in 35 mm collagen-coated dishes and then fixed and stained on the following day with the nuclear dye Hoechst 33342 (1 μg/ml; Sigma).
Immunocytochemistry
PC12 cells from the various lines were fixed and immunostained with the mouse anti-synuclein-1 antibody (1:50) in combination with the rabbit anti-ubiquitin antibody (1:100) or a rabbit polyclonal antibody to cathepsin D (a generous gift from Dr. Eiki Kominami, Juntendo University, Tokyo, Japan; used at 1:200), using previously described procedures (Stefanis et al., 1999, 2001). For indirect immunofluorescence studies, cells were plated on 35 mm dishes; for colocalization studies with confocal microscopy, cells were plated on glass coverslips coated with poly-d-lysine. For confocal microscopy, we used a Zeiss (Thornwood, NY) LSM 410 scanning laser confocal attachment mounted on a Zeiss Axiovert 100 TV inverted fluorescence microscope.
Assay for chymotrypsin-like activity of the proteasome
Cells from the different lines were triturated off the dish, centrifuged, and washed in PBS. The resulting pellets were resuspended in 200–500 μl of lysis buffer (10 mm Tris-HCl, pH 7.8, with 1 mm ATP, and 10% glycerol) (Figueiredo-Pereira et al., 1994). The cells were left for 20 min on ice and then lysed with 30 strokes of a Dounce homogenizer. The lysates were then centrifuged at 10,000 × g for 10 min. Fifty micrograms of protein of the resulting supernatants was included in an assay buffer of 100 mm Tris-HCl, pH 8.0, 2 mm CaCl2, 1 mm ATP, and a 100 μmconcentration of the fluorogenic substrate LLVY-7-amino-4-trifluoromethyl coumarin (AFC) (Enzyme Systems Products, Livermore, CA) in a total final volume of 1 ml. The assay was performed at 37°C for 10 min (Figueiredo-Pereira et al., 1994). The activity was then measured in a SLM 8000 fluorometer, with assay buffer without lysate as blank. The activity was linear with respect to the amount of protein (in the range of 25–200 μg of protein). In addition, lysates of cells treated overnight with the proteasomal inhibitor MG132 (0.5 μm;Biomol, Plymouth Meeting, PA) showed <5% of the activity, indicating that this activity was specific to the proteasome.
Electron microscopy
Cells from the various lines were plated on poly-d-lysine and laminin-coated aclar in 35 mm dishes with punch holes. At 1–2 d after plating, the cells were rinsed with PBS and then fixed for 60 min at 4°C with 2% glutaraldehyde in 2 mm CaCl2 and 100 mmsodium cacodylate, pH 7.4. The fixed cultures were maintained in 100 mm sodium cacodylate and then processed for electron microscopy (EM) using standard methods (Tennyson et al., 1993).
Dopamine release
Depolarization-induced dopamine release was quantified as described previously (Pothos et al., 2000) by HPLC coupled with electrochemical detection on an ESA (Bedford, MA) Coulochem II HPLC equipped with a model 5011 analytical cell with an applied potential of 400 mV and a Velosep RP-18 column (Applied Biosystems, Foster City, CA). The mobile phase contained 6.8 gm/l sodium acetate, 18.6 mg/l EDTA, 142 mg/l heptanesulfonic acid, and 10% methanol (adjusted to a pH of 4.6 with acetic acid). Briefly, cells from the various cell lines were plated in 24-well dishes. The cultures were rinsed once in PBS and then exposed to normal incubation medium (2 mm KCl) or to medium containing high potassium (80 mm KCl) for 1 min (Pothos et al., 2000). The medium was then harvested in ice-cold 0.1N perchloric acid and analyzed for dopamine content. Depolarization-induced dopamine release was quantified as the difference in extracellular dopamine between cultures exposed to high potassium compared with normal incubation medium. To measure intracellular monoamine levels, medium was removed and the cells were rapidly solubilized in 100 μl of 0.3 mperchloric acid. The samples were centrifuged at 15,000 rpm for 15 min at 4°C and stored at −80°C until HPLC analysis.
Labeling of living cells: labeling of functional lysosomes
Cells from the various lines were plated in 24-well dishes and incubated for 20 min at 37°C with the cell-permeable dye Lysotracker red (50 nm; Molecular Probes, Eugene, OR), which labels acidic organelles. Cells were then washed twice in PBS and visualized at 40× magnification under an epifluorescent microscope. In some experiments, for superior resolution, cells were plated on glass coverslips and visualized with oil immersion at 60× magnification in an inverted microscope.
To further investigate lysosomal function, we labeled the cells with Lysosensor Yellow/Blue Dextran (Molecular Probes). This lysosensor-tagged dextran is endocytosed by the cells and degraded through the lysosomal system. It emits in the UV spectrum at a neutral pH and emits in the rhodamine–fluorescein spectrum at an acidic pH. We plated the cells from the different cell lines in 96 well plates and incubated the cultures with 2 mg/ml Lysosensor Yellow/Blue Dextran for various periods of time (8–20 hr). We then rinsed the cultures three to four times with complete medium, plated the cells on a glass slide, placed a glass coverslip on top, and visualized the cells with oil immersion at 100× magnification in an upright microscope. Images were obtained using the UV and the rhodamine filters. Representative pictures were obtained using identical exposure times across the different lines.
RESULTS
Generation of stable PC12 cell lines expressing various levels of the wild-type and A53T mutant form of human α-synuclein
After the generation of various clonal lines transfected with empty vector, wild-type α-synuclein, and A53T mutant α-synuclein, Triton-soluble lysates were assessed by Western immunoblotting for the level of expression of α-synuclein. Only a small fraction of the detectable synuclein protein was Triton-insoluble. As we have reported previously (Stefanis et al., 2001) in empty-vector controls (designated P1-P5) as well as in nontransfected PC12 cells, only a 45 kDa band was detected using a mouse monoclonal anti-synuclein antibody (Fig.1). The 45 kDa band may represent endogenous post-translationally modified rat synuclein-1 or a cross-reacting protein (Stefanis et al., 2001). Lysates of clonal lines transfected with wild-type α-synuclein (S4, S10, and S12) or mutant α-synuclein (M1, M13, M15, and M18) contained a broad band of ∼18 kDa that comigrated with the dominant band of α-synuclein detected in rat cortical lysates. Various levels of expression were achieved in different clonal lines. M1, M15, and M18, three lines expressing the highest levels of mutant α-synuclein, were comparable in terms of level of expression to S12, which expressed the wild-type form. Lines M13, S4, and S10 had considerably lower levels of expression. The levels of α-synuclein in the highest expressing lines were comparatively less than in rat cortex and therefore were well within the physiological range (Fig. 1). As we have reported, levels of the 18 kDa α-synuclein band are induced in PC12 cells with NGF treatment (Stefanis et al., 2001). The levels of α-synuclein achieved in these lines were within the range of such induction.
Fig. 1.

Various levels of expression of wild-type and A53T mutant α-synuclein in clonal PC12 cell lines. Various clonal PC12 cell lines were generated expressing empty vector (P1 and P2), wild-type α-synuclein (S4, S10, and S12), or A53T mutant α-synuclein (M1, M13, M15, and M18). Triton-soluble lysates (200 μg of protein) were generated, resolved by 13% SDS-PAGE, and immunoblotted with a monoclonal synuclein antibody (1:1000; Transduction Laboratories). Two separate blots are presented. In the blot on the left, a control lysate from rat cortex (30 μg) was used in the first lane. Theasterisk indicates the broad 18 kDa band seen in the control lysate and in the lines expressing α-synuclein. Note the 45 kDa band (arrow) present in the control lysate, the empty-vector controls, and the overexpressing lines. In the blot on theright, lysates of the M15 and M18 lines were similarly generated and processed. Three more empty vector lines (P3, P4, and P5) were generated and showed a pattern that was similar to that seen for P1 and P2 on immunoblotting (Stefanis et al., 2001).
Clonal lines expressing A53T α-synuclein show altered morphological characteristics
Cell lines expressing wild-type α-synuclein were indistinguishable from empty-vector control-transfected lines as well as nontransfected parental cells in terms of their appearance (Fig.2) (Stefanis et al., 2001). In contrast, M1 and M15 cells, which expressed high levels of mutant A53T α-synuclein, displayed a number of morphological abnormalities, including increased size, occasional stellate appearance, increased tendency to extend processes in the absence of NGF treatment, and increased cellular degeneration. Degenerating cells had a vacuolar–granular appearance (Fig. 2A). A similar phenotype was seen in the M18 line. Line M13, which expressed less of the exogenous mutant α-synuclein, displayed an intermediate appearance with no significant overall increase in cell size but the occasional presence of very large cells and some granular degenerating cells (data not shown).
Fig. 2.
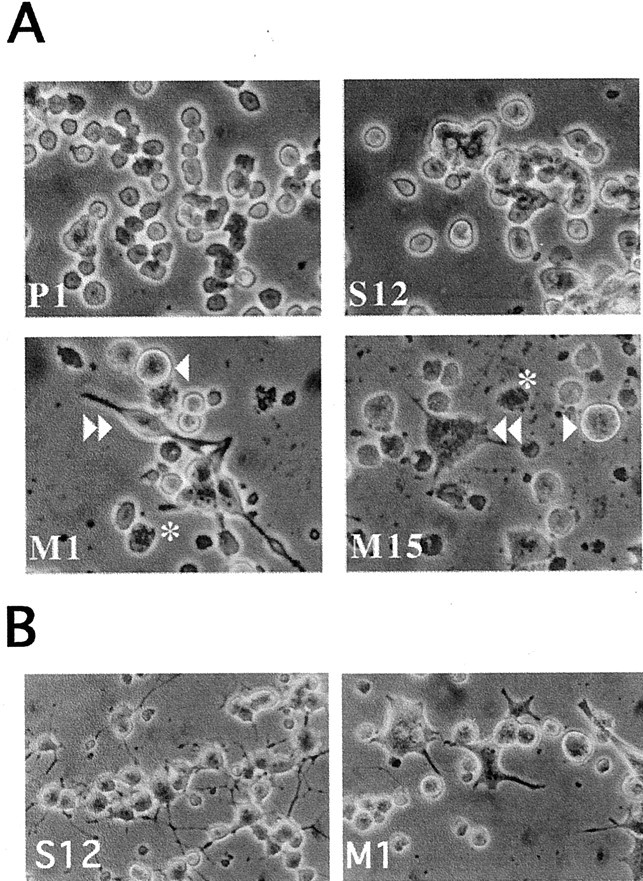
Morphological alterations in PC12 cells expressing A53T α-synuclein. A, Photomicrographs of naive PC12 cells expressing empty vector (P1), wild-type α-synuclein (S12), or A53T mutant α-synuclein (M1 and M15). Theasterisks indicate granular degenerating cells. Thesingle arrowhead indicates very large cells. Thedouble arrowhead indicates a neuritic-like extension in an M1 cell and a stellate appearance in a M15 cell. B, Photomicrographs of PC12 cells expressing wild-type α-synuclein (S12) or A53T mutant α-synuclein (M1) and treated for 9 d with NGF. Note the neuritic network present in the S12 cultures but not in the M1 cultures. Also note the large number of degenerating cells in the cultures derived from the M1 line.
PC12 cells treated with NGF assume a neuronal-like phenotype (Greene and Tischler, 1976). Wild-type α-synuclein-expressing cells responded to treatment with 100 ng/ml NGF in a manner similar to that seen for empty-vector controls, establishing a neuritic network after 9 d of NGF treatment (Fig. 2B) (Stefanis et al., 2001). In contrast, cells expressing mutant A53T α-synuclein showed a limited response to NGF, extending only short, stumpy processes. Many of these cells were very large and had bizarre shapes. As in the naive state, many granular–vacuolar degenerating cells were noted in these cultures (Fig. 2B).
Clonal lines expressing A53T α-synuclein show enhanced cellular degeneration
To quantify the impression of increased cellular degeneration in lines expressing mutant α-synuclein, we assessed the percentage of Trypan blue-positive cells in each line (Fig.3A). There was a markedly higher percentage of these degenerating cells in lines M1 and M15 compared with control cell lines or with lines expressing wild-type α-synuclein. Line M13 showed a more modest increase in degenerating cells (Fig. 3A).
Fig. 3.

Increased nonapoptotic death in PC12 cells expressing A53T α-synuclein. A, PC12 cells from various clonal lines were tested for Trypan blue uptake. The percentage of Trypan blue-positive cells is reported as the mean ± SEM (n = 3). M1 and M15lines (p < 0.001, determined by one-way ANOVA with Neuman–Keuls post hoc tests) and, to a lesser extent, M13 (p < 0.05) showed a higher percentage of Trypan-blue-positive cells compared with empty vector or wild-type synuclein overexpressors.B, PC12 cells from various clonal lines were fixed and stained with Hoechst 33342 and nuclear morphology was evaluated. The percentage of apoptotic nuclei for each line is reported as the mean ± SEM (n = 3). As a positive control, cells from the P1 line were deprived of serum overnight and subsequently fixed and stained with the Hoechst dye (SD).
To evaluate the morphological features of this cell death, we used the nuclear dye Hoechst 33342 and counted the percentage of apoptotic nuclei in each line. The proportion of apoptotic cells was low (<5%), and there was no significant difference in the relative percentage of apoptotic nuclei across cell lines. In contrast, a positive control of P1 cells undergoing death caused by serum deprivation showed a large proportion of apoptotic nuclei (Fig. 3B).
To test for potential involvement of caspases in cellular degeneration associated with expression of A53T α-synuclein, we examined control, wild-type, and mutant lines for basal caspase-3-like activity, as measured by cleavage of the fluorogenic substrate DEVD-AFC (15 μm; Enzyme Systems Products). We found no difference in basal activity among cell lines (data not shown). Positive controls from serum-deprived cultures showed the expected caspase-3-like activity (Stefanis et al., 1996). In addition, the pan-caspase inhibitor Boc-aspartate-fluoromethyl ketone (50 μm; Enzyme Systems Products) did not affect the percentage of Trypan blue-positive cells in the mutant lines (data not shown)
We conclude that PC12 cell lines expressing A53T mutant α-synuclein display enhanced cellular degeneration that appears nonapoptotic by morphological and biochemical criteria.
Clonal lines expressing A53T α-synuclein show cytoplasmic ubiquitinated aggregates
To evaluate further the nature of the different phenotypes in the lines expressing A53T α-synuclein, we immunostained the various lines with anti-synuclein. Empty-vector controls showed a low level of cytoplasmic staining (Stefanis et al., 2001). A higher level of staining in the same distribution was seen in lines S4, S10, and M13 expressing low levels of α-synuclein. In contrast, lines S12, M1, and M15, which expressed higher α-synuclein levels, showed occasional nuclear staining in addition to cytoplasmic staining (Fig.4A,B). Overall, there was no detectable difference in the pattern of staining between lines expressing the wild-type or mutant α-synuclein.
Fig. 4.
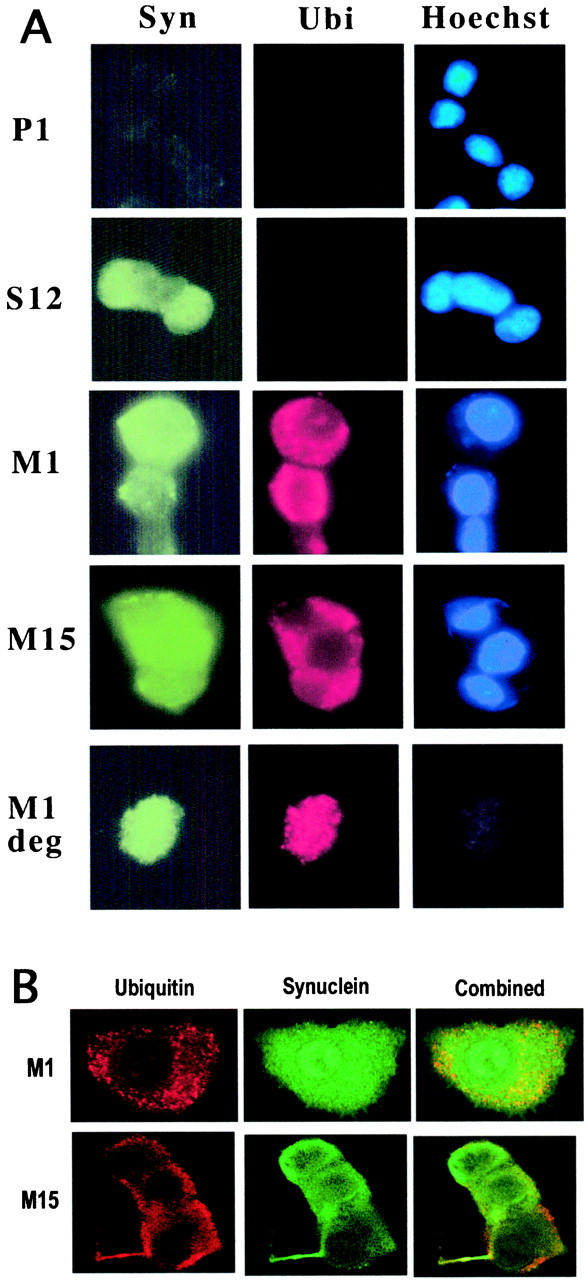
Ubiquitinated aggregates in PC12 cells expressing A53T α-synuclein. A, PC12 cells from the various lines were fixed and stained with a monoclonal synuclein antibody (1:50; Transduction Laboratories) (left column;Syn), a polyclonal ubiquitin antibody (1:100; Dako) (middle column; Ubi), and the Hoechst dye 33342 (1 μg/ml; Sigma) (right column;Hoechst). Ubiquitin staining in control and wild-type α-synuclein expressors was diffuse and low level and was not evident with this exposure. In contrast, with identical exposure time, note the strong staining for ubiquitin in the cytoplasm of M1 andM15 cells. In the last row, a degenerating (deg) M1 cell that has lost nuclear staining shows even more discrete punctate ubiquitin staining.B, Confocal microscopy of M1 or M15 cells shows ubiquitin (left column), synuclein (middle column), and combined (right column) immunostaining. The intense, punctate nature of ubiquitin immunostaining is apparent. There is little synuclein immunoreactivity within these aggregates. Nuclear synuclein immunostaining is evident in the cell shown from the M1 line.
We also stained the various cell lines with anti-ubiquitin in parallel. We found that lines M1 and M15 showed a marked increase in ubiquitin staining compared with lines S12, S4, and S10 and empty-vector controls (Fig. 4A). Instead of the low-level diffuse staining seen in the other lines, the majority of M1 and M15 cells showed an intense punctate cytoplasmic staining. Cells of the M18 line showed a similar pattern, whereas M13 cells had an intermediate phenotype, with only occasional intense cytoplasmic ubiquitin staining (data not shown). To ensure that the increased level of ubiquitin staining was not simply a nonspecific response to stress, we also used anti-ubiquitin antibody to stain serum-deprived cells from the P1 clonal line. The intense cytoplasmic punctate staining was not seen under these circumstances (data not shown).
To characterize better the ubiquitin and synuclein immunostaining in the mutant lines, we performed confocal microscopy in these cell lines after staining them for ubiquitin and α-synuclein. Representative cells are shown from lines M1 and M15 (Fig. 4B). Synuclein staining was diffuse, occasionally including the nucleus. There was no evidence of synuclein aggregation. Immunostaining with a polyclonal synuclein antibody (Chemicon, Temecula, CA) also failed to show synuclein aggregation (data not shown). In contrast, ubiquitin immunostaining was primarily in the form of small discrete punctate accumulations of staining, which represent aggregates. Only a very limited proportion of these ubiquitinated aggregates stained for synuclein (Fig. 4B). We did not detect large ubiquitinated inclusions resembling Lewy bodies in these cells.
Cytoplasmic ubiquitinated aggregates were not seen in any of >10 wild-type α-synuclein-expressing lines, 5 empty-vector control lines, or multiple batches of untransfected PC12 cells. Furthermore, when some cultures derived from the two mutant lines, M1 and M15, eventually lost synuclein expression (when maintained in the absence of the selecting agent G418), they also lost the pattern of aggregated ubiquitin immunostaining (data not shown). This suggests that mutant α-synuclein expression is causally related to the accumulation of ubiquitinated aggregates.
In summary, PC12 cells expressing A53T α-synuclein show cytoplasmic ubiquitinated aggregates. These appear to be different from Lewy bodies in a number of respects, including their small size and the paucity of colocalization with α-synuclein.
A number of proteins, but not α-synuclein itself, are preferentially ubiquitinated in the mutant α-synuclein cell lines
The antibody that we used for ubiquitin immunostaining could potentially identify free ubiquitin, mono-ubiquitinated, or polyubiquitinated proteins. To verify the immunostaining results and to assess whether the increased levels of ubiquitination in the mutant α-synuclein lines were attributable to polyubiquitinated proteins marked for degradation, we performed Western immunoblotting with the ubiquitin antibody. We divided cell lysates into Triton-soluble and -insoluble fractions, and the latter was solubilized in SDS–sample buffer to assess the relative solubility of proteins. In Triton-soluble extracts there was little apparent difference in the high molecular mass pattern of ubiquitination among cell lines. A doublet at ∼52–54 kDa was more prominent in lines M1 and M15 compared with the other lines (Fig.5A). The Triton-insoluble fractions of lines M15 and M1 showed a considerable increase in high molecular mass (>80 kDa) polyubiquitinated proteins, which are signaled for degradation, compared with controls or wild-type α-synuclein expressors (Fig. 5B, bracket). This increase was also apparent in the stacking gel, implying an increase of insoluble polyubiquitinated proteins. A few discrete bands, including bands migrating at 52–54 kDa, were more abundant in the mutant lines (Fig. 5B, arrows).
Fig. 5.
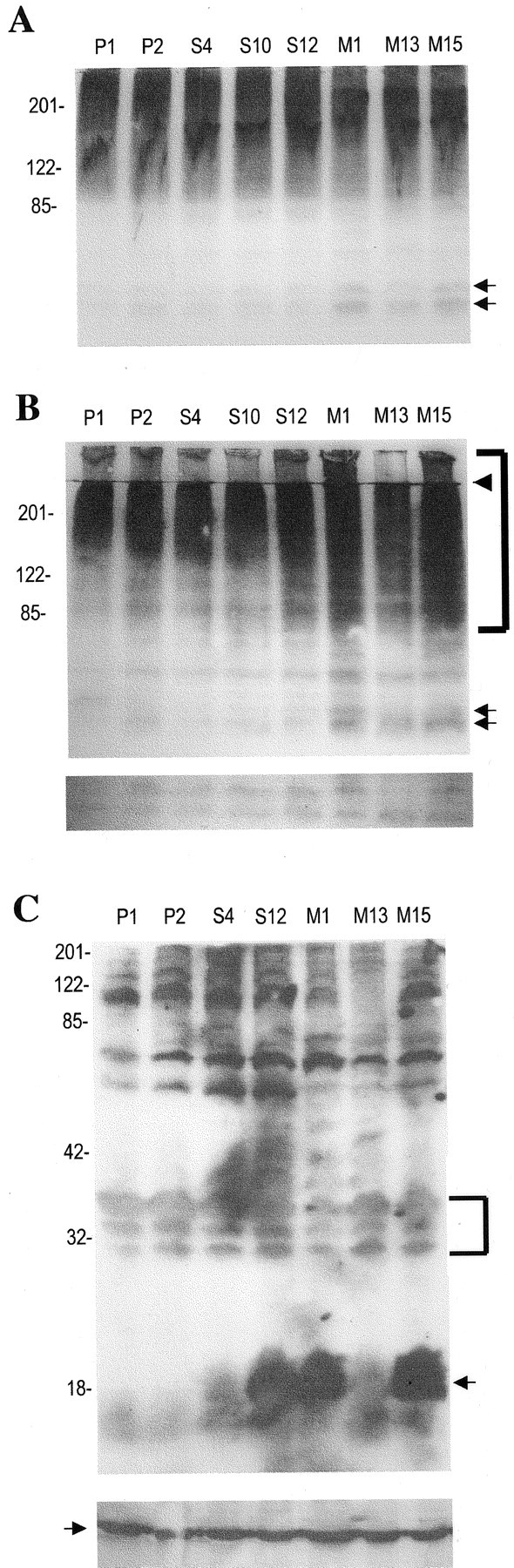
Increase of multi-ubiquitinated proteins in Triton-insoluble lysates of PC12 cells expressing A53T α-synuclein.A, Cell lysates (200 μg of protein) from various clonal lines were resolved on an 8% SDS-PAGE gel and immunoblotted with an anti-ubiquitin polyclonal antibody (1:1000; Dako). Thearrows indicate a doublet at 52–54 kDa that was selectively increased in M1 and M15 cell lysates. This blot represents one of two independent experiments, which yielded similar results.B, Triton-insoluble, sample buffer-soluble lysates (200 μg of protein) were resolved on an 8% SDS-PAGE gel and immunoblotted with an anti-ubiquitin polyclonal antibody (1:1000; Dako). Thearrows indicate bands at 52–54 that were more prominent in the M1 and M15 lines. Thebracket indicates higher molecular mass proteins, which were more prominent in the M1 and M15 lines. The arrowheadindicates the end of the stacking gel. The bottom panel is from a longer exposure (3 min vs 1 min) of the same blot, at the level of 30–35 kDa. The bands seen are presumably background bands, with similar intensity across the lanes. Equal protein loading was also verified by Ponceau S staining. This blot represents one of three independent experiments, which yielded similar results, except for the fact that an increase in polyubiquitinated proteins was inconsistently found for M13. C, Triton-insoluble, sample buffer-soluble lysates (200 μg of protein) from various clonal lines were resolved on a 12% SDS-PAGE gel and immunoblotted with anti-synuclein. The arrow indicates the 18 kDa α-synuclein band. Note that the exposure for this blot was at least 10 times longer than for the blots used to obtain similar band intensities from Triton-soluble lysates. The blot was intentionally overexposed to detect low abundance α-synuclein-specific bands in the upper portion of the blot. Such bands were not seen. The bands detected at the top portion of the blot are presumably nonspecific background bands, because they are also seen in the empty-vector control lysates. Thebracket indicates background bands at 30–35 kDa that are of similar intensity across the various samples. To ensure equal protein loading, the same samples were loaded on another gel and immunoblotted with an anti-actin monoclonal antibody (1:5000; Sigma) (bottom panel). The arrow in thebottom panel indicates the 44 kDa actin band.
To test for the possibility that synuclein itself was ubiquitinated, we probed Western blots of Triton-insoluble lysates from the various lines with anti-synuclein. We did not identify the characteristic ladder pattern of ubiquitination (Fig. 5C).
These results suggest that a number of proteins, but not α-synuclein itself, are preferentially polyubiquitinated in the mutant α-synuclein cell lines.
PC12 cell lines expressing A53T α-synuclein show reduced proteasomal activity
The phenotype of the lines expressing A53T α-synuclein is reminiscent in some respects of the phenotype of cells exposed to proteasomal inhibitors. These inhibitors can cause neurite sprouting, increased cell size, increased levels of polyubiquitinated proteins, and cell death (Drexler, 1997; Lopes et al., 1997; Ohtani-Kaneko et al., 1998; Obin et al., 1999; Rideout et al., 2001). To evaluate the possibility that the A53T cell lines had diminished proteasomal activity, we measured the proteasomal chymotrypsin-like activity in various cell lysates. We found that M1 and M15 had 25–35% lower chymotrypsin-like activity compared with lines P5 or S12. This difference was statistically significant (Fig.6).
Fig. 6.

Decrease in proteasomal activity in PC12 cell lines expressing mutant A53T α-synuclein. PC12 cell lysates were generated and assayed for chymotrypsin-like proteasomal activity. The results for each line are reported as mean ± SEM (n = 8 for S12 and M15; n = 10 for P5 and M1). Both M1 and M15 showed a statistically significant decrease in proteasomal activity compared with S12 or P5 (Student's nonpaired t test; *p < 0.05; **p < 0.01).
PC12 cell lines expressing A53T mutant α-synuclein show accumulation of vesicular structures suggestive of autophagy
To further investigate the cellular degeneration seen in the A53T expressors, we examined the P1, S12, M1, and M15 cell lines by electron microscopy. There was no evidence of increased apoptotic death in lines M1 or M15 compared with lines P1 and S12. There was however a marked accumulation of vesicular–autophagic structures in the A53T expressors. Many had double membranes. In many cases, these structures appeared to engulf intracellular organelles, in particular mitochondria. Some mitochondria were swollen or showed electron-dense material, consistent with calcification and degeneration. Other structures resembled more mature lysosomal organelles (Fig.7). Another interesting feature of these lines was that there was a complete absence of dense core granules (DCGs), which are the catecholamine-secreting vesicular structures in PC12 cells. DCGs were readily apparent in the P1 and S12 lines (Fig. 7and Table 1).
Fig. 7.
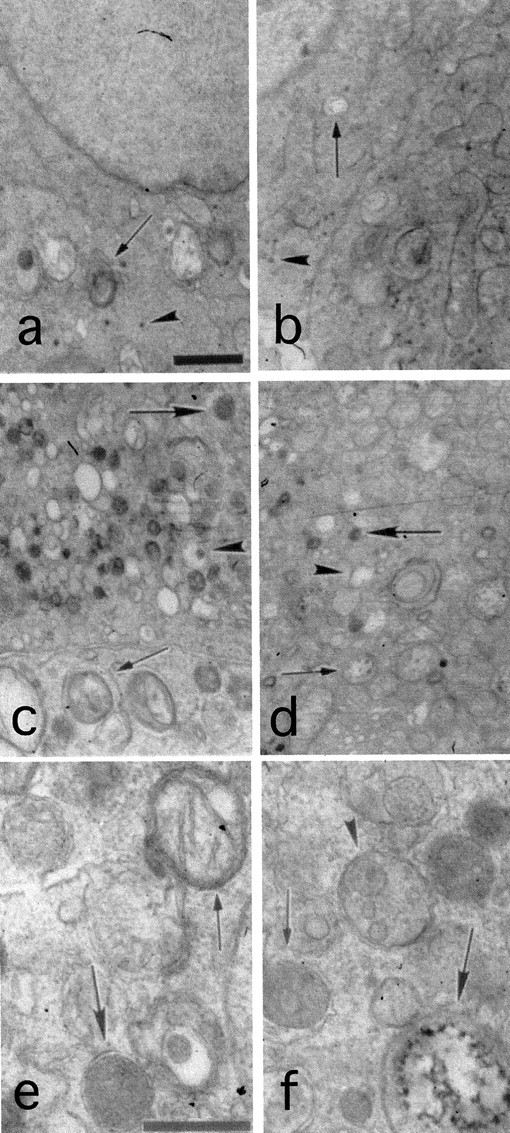
Features of autophagy in the A53T α-synuclein-expressing lines. a–d, Cells from the P1 (a), S12 (b), M1 (c), and M15 (d) clonal lines were processed for EM. Scale bar: a, 1 μm (applied to a–d). Note the normal appearance of the P1 and S12 cells, with only rare lysosomal–vacuolar structures (arrows in a and b). Dense core granules are indicated by arrowheads, and were actually more prominent overall in the S12 cells compared with the P1 cells. Such granules were entirely absent from the mutant lines. Note the granular–vacuolar appearance of cells from the mutant lines inc and d and the dense packaging with lysosomal-like structures that range from electron dense (large arrows) to vacuolar (arrowheads). On many occasions double membrane-bound structures (small arrowsin c and d) were noted to engulf intracellular organelles, and in particular mitochondria. e, f, M1 (e) and M15 (f) cells shown at a higher magnification. Scale bar: e, 0.5 μm. Note again the numerous membrane- bound structures. Some of these are reminiscent of multivesicular bodies (arrowhead in f). Thesmall arrow in f shows one such structure within a vacuole. Note a degenerating mitochondrion (large arrow in f) and a double-membrane structure engulfing a mitochondrion (small arrow ine). The large arrow in edenotes a relatively electron-dense lysosomal-like structure with a double membrane.
Table 1.
Absence of DCGs in PC12 cell lines expressing mutant α-synuclein
| Mean number of DCGs per section | SEM | |
|---|---|---|
| P1 | 54.9 | 21.1 |
| S12 | 113.8 | 34 |
| M1 | 0.8 | 0.6 |
| M15 | 1 | 0.7 |
PC12 cells from the various lines were fixed and processed for electron microscopy. For each line, nine random sections of 10 × 13 μm were scanned and the number of DCGs was assessed.
Together, these EM data suggest that the cellular degeneration seen in the A53T-expressing lines more closely resembled an autophagic form of cell death (Clarke, 1990; Ohsawa et al., 1998). In addition, these lines demonstrate an accumulation of aberrant vesicular structures that appear to be part of the lysosomal degradation system and show an absence of the dopamine-secreting dense core granules.
PC12 cell lines expressing A53T mutant α-synuclein do not release dopamine after depolarization
To investigate whether the absence of dense core granules in the A53T-expressing cells would have functional consequences, we measured depolarization-induced dopamine release from the various lines using HPLC. We found no stimulation-dependent dopamine release from cultures of the M1, M15, or M18 lines (Fig. 8). In contrast, cultures expressing wild-type human α-synuclein retained the capacity for dopamine release, although to a somewhat reduced degree compared with an empty-vector control line (Fig.8A). The mutant lines did contain intracellular dopamine, albeit at lower levels compared with the empty vector or the wild-type synuclein-expressing lines (Fig. 8B). Such lower intracellular levels could be attributable to degradation of nonsequestered dopamine or to mechanisms of feedback inhibition that operate when dopamine is prevented from entering the vesicular compartment (Goldstein and Greene, 1987; Fon et al., 1997).
Fig. 8.

Absence of depolarization-induced dopamine release from PC12 cells expressing A53T α-synuclein. Cells from theP4, S12 (S12a andS12b, from two separate subclones in two different experiments), M1, M15, andM18 lines were plated on 24-well dishes, and depolarization-induced dopamine release (A) and intracellular dopamine (B) were assessed as described in Materials and Methods. The results are reported as mean ± SD (n = 4).
PC12 cells expressing mutant α-synuclein therefore manifest an absence of depolarization-induced dopamine release, consistent with the absence of dense core granules in these cells.
PC12 cell lines expressing A53T mutant α-synuclein show lysosomal dysfunction
To examine whether the apparent increase in lysosomal–autophagic structures reflected an alteration in lysosomal activity in the A53T-expressing lines, we labeled the cells with an ionic dye, Lysotracker red (50 nm; Molecular Probes), which is selectively sequestered in acidic organelles. In control cells and cells expressing wild-type α-synuclein, we observed a fine punctate pattern of labeling within the cytoplasm, consistent with lysosomal staining. There was a widespread, marked decrease of such punctate staining in cells from the M1 and M15 lines. Rare cells from the mutant lines (<2%) showed intense labeling of large cytosolic structures, which resembled vacuoles/inclusions (Fig.9). A similar phenotype was seen in the M18 line (data not shown).
Fig. 9.

Reduced presence of acidic organelles in the A53T α-synuclein-expressing lines. A, Cells from the various lines were labeled with the dye Lysotracker red (50 nm; Molecular Probes) and then visualized under a fluorescent microscope with a 40× objective. Representative pictures are shown. Note the very low level of labeling in linesM1 and M15. The arrowsdenote a rare vacuolar/inclusion-like structure intensely labeled with Lysotracker. Similar results were achieved in five independent experiments. B, For superior resolution, cells labeled as in A were visualized with an oil-immersion 60× objective. Note the discrete punctate labeling of P1 and S12 and the absence of such labeling in M1 and M15. An example of a large accumulation of Lysotracker labeling is shown again for M15.
To further examine lysosomal function in these cell lines and to follow the fate of a lysosomal substrate within the cells, we incubated the cultures with Lysosensor Yellow/Blue Dextran, a substrate that is endocytosed by the cells and degraded in the lysosomes. The wavelength of the fluorescence emitted is pH-dependent. In an acidic environment, there is emission in the yellow spectrum; this was captured in our experiments with a rhodamine filter. In a neutral or basic environment, emission is in the UV spectrum. We found that after a 20 hr incubation with Lysosensor Yellow/Blue Dextran, S12 cells showed intense punctate labeling in the rhodamine spectrum (red), indicative of normal lysosomal acidification, whereas M1 or M15 cells did not (Fig.10, bottom row). All cell types showed punctate labeling with the UV filter (blue), but the A53T-expressing cells were of higher intensity (Fig. 10, top row). P4 cells did not show much fluorescence in either spectrum when incubated for 20 hr, presumably because the substrate had already been degraded; however, with 8 hr of incubation, P4 cells showed a pattern of labeling similar to the S12 cells (data not shown). The difference in incubation time needed to achieve similar levels of fluorescence may be related to different rates of endocytosis between the control and the synuclein-expressing lines. These results indicate that PC12 cells expressing mutant α-synuclein manifest an impairment of the lysosomal degradation system.
Fig. 10.

Reduced degradation of a lysosomal substrate in PC12 cells expressing A53T α-synuclein. PC12 cells from the various cell lines were incubated with 2 mg/ml Lysosensor Yellow/Blue Dextran for 20 hr, rinsed three times in complete medium, and then visualized under a 100× oil-immersion objective. Images were captured in a UV (top) or a rhodamine (bottom) filter. Note the increased fluorescent signal in cells from the M1 and M15 lines in the UV range, indicative of accumulation of the substrate in nonacidic organelles, and the absence of fluorescence in the rhodamine spectrum, indicative of the absence of degradation of the substrate in acidic organelles. Identical exposure times were used across the various lines. The experiment was repeated twice with similar results.
Limited colocalization between α-synuclein and cathepsin D
In view of the lysosomal dysfunction detected in the A53T mutant α-synuclein-expressing lines, we wanted to test whether α-synuclein could directly interact with lysosomes to cause these effects. Cathepsin D is a major lysosomal enzyme, and immunostaining with antibodies directed against cathepsin D has been used as a lysosomal marker. Kegel et al. (2000) recently reported that overexpressed mutant Huntingtin colocalizes with cathepsin D in cytoplasmic vacuoles and stimulates autophagy in cloned striatal neuronal cells. Therefore, we performed double immunostaining for α-synuclein and cathepsin D in wild-type and mutant A53T-expressing cells and examined the cells by confocal microscopy. We did not find specific accumulation of α-synuclein within cathepsin-positive structures. Overall, there was limited colocalization between α-synuclein and cathepsin D, and the extent of colocalization did not differ in cells expressing wild-type or mutant α-synuclein (Fig. 11).
Fig. 11.

Limited colocalization between α-synuclein and cathepsin D. PC12 cells from the S12 orM18 lines were fixed and immunostained with synuclein-1 and cathepsin D antibodies. Immunostaining was then evaluated by confocal microscopy. Representative pictures are shown. Limited colocalization was noted between cathepsin D (left column) and α-synuclein (middle column), and the extent of colocalization did not differ among the two cell lines. Combined images are shown in the right column. A similar immunostaining pattern was seen in M1 and M15 cells (data not shown).
The fact that α-synuclein does not significantly colocalize with cathepsin D suggests that the effects of mutant α-synuclein on lysosomal function may be mediated at an earlier stage of this degradation pathway, at the level of endosomes or early autophagosomes. Alternatively, the effects on lysosomal function may be indirect, caused by, for example, oxidative stress, which has been shown to induce lysosomal dysfunction (Brunk et al., 1995).
DISCUSSION
Expression of A53T α-synuclein alters the cellular phenotype
We show here that expression of mutant A53T α-synuclein in PC12 cells leads to the formation of small ubiquitinated aggregates, to autophagic cellular degeneration, and to an absence of both DCGs and evoked dopamine release. M13, a line that had lower levels of the mutant protein, showed an intermediate phenotype, indicating that the effect is dose-dependent.
The levels of expression achieved were at least an order of magnitude lower compared with endogenous synuclein in rat brain on a per milligram of protein basis. At this expression level, there was a selective deleterious effect of mutant but not wild-type α-synuclein. We have not generated cell lines overexpressing rat synuclein-1, but, as we have reported previously, PC12 cells and sympathetic neurons upregulate levels of endogenous synuclein-1 in response to NGF, and this upregulation has no obvious detrimental effects (Stefanis et al., 2001). Threonine at position 53 is the normal amino acid in rodents, and this has raised doubts about modeling the disease based on expression of this mutant form in rodent cells. However, human A53T α-synuclein is different from rodent synuclein-1 in a number of other amino acids. Our results and those of others (Zhou et al., 2000) suggest that A53T mutant α-synuclein, but not rat synculein-1, induces selective detrimental effects in a rodent background.
We have sought to replicate our findings in a different background of PC12 cells, by generating PC12 cell lines expressing the A53T or the A30P mutant or wild-type α-synuclein using a Tet-on system, with cells supplied by the manufacturer (Clontech, Palo Alto, CA). We achieved high levels of expression of α-synuclein in a number of lines, even without tetracycline, presumably because of the leakiness of the system. Wild-type α-synuclein expressors were again indistinguishable from controls. Cells expressing the A53T or A30P mutant form did not demonstrate the dramatic phenotype seen in our original transfected lines, in that their size and morphological features appeared grossly normal and there was no enhancement of baseline cell death. However, these cells did show, albeit to a lesser degree than the original lines, small cytoplasmic ubiquitinated aggregates that again did not colocalize with α-synuclein (data not shown). The similarities and differences between the two sets of transfected cells underscore the potential importance of the cellular background or other unidentified factors in phenotype penetrance. Phenotypic heterogeneity has also been observed in families with α-synuclein mutations (Papadimitriou et al., 1999) and in transgenic mouse models overexpressing wild-type or mutant α-synuclein (Masliah et al., 2000; Matsuoka et al., 2001; Rathke-Hartlieb et al., 2001). Our cell lines may thus present the opportunity to identify molecules that modulate the risk of degeneration associated with expression of mutant α-synuclein.
Effects of mutant α-synuclein appear independent of self-aggregation: potential role of proteasomal/lysosomal dysfunction
Accumulating evidence suggests that α-synucleins, and in particular the mutant forms, aggregate in vitro (Conway et al., 1998; Giasson et al., 1999), accumulate in Lewy bodies (Spillantini et al., 1997, 1998) and, in an insoluble form, are increased in PD brains (Baba et al., 1998), leading to the idea that α-synuclein forms aggregates that serve as a nidus for the formation of Lewy bodies, disruption of cell homeostasis, and death (Trojanowski et al., 1998). In our system, there was no evidence of α-synuclein aggregation or ubiquitination or of significant colocalization of α-synuclein within the ubiquitinated aggregates. Therefore, it appears that A53T α-synuclein in our system leads to the formation of ubiquitinated aggregates through an indirect mechanism. Similarly, α-synuclein expression enhances the formation of Huntingtin-containing inclusions but does not colocalize in the aggregates (Furlong et al., 2000).
How could the ubiquitinated aggregates be formed then, if not by α-synuclein aggregation? Our results indicate that the cells in the mutant lines have defects in the two major systems for the degradation of ubiquitinated proteins: lysosomes and the proteasome (Laszlo et al., 1990; Mayer et al., 1992; Ciechanover, 1998). It is likely that such defects would lead to the accumulation of ubiquitinated aggregates: Inhibition of lysosomal cysteine proteases leads to the formation of ubiquitinated aggregates in cell culture and in vivo (Laszlo et al., 1990; Cavanagh et al., 1993). Proteasomal inhibition leads to the accumulation of polyubiquitinated proteins (Ohtani-Kaneko et al., 1998). It should be noted however that the cellular phenotype that we have observed after acute pharmacological proteasomal inhibition of PC12 cells (Rideout et al., 2001), although similar, differs in some respects from the one described here induced by mutant synuclein overexpression. With acute proteasomal inhibition there is cell enlargement, neuritic extension, and induction of polyubiquitinated proteins and cytoplasmic ubiquitin immunostaining. However, we also detect apoptotic death and large single cytoplasmic inclusions that occasionally contain α-synuclein (Rideout et al., 2001). It is possible that more chronic, low-level regimens of pharmacological proteasomal inhibition would lead to a phenotype similar to the one observed here, or that additional factors, apart from proteasomal inhibition, may play a role in the generation of the mutant synuclein phenotype.
A recent study found inhibition of proteasomal activity in PC12 cells expressing A30P α-synuclein (Tanaka et al., 2001). The functional consequences of such inhibition were not examined. Nevertheless, these results, together with our own, achieved with the other α-synuclein mutant, suggest a link between mutant α-synuclein expression and proteasomal inhibition. There is also evidence for increased sensitivity to proteasomal inhibition-induced death with expression of mutant or wild-type α-synuclein (Lee et al., 2001; Tanaka et al., 2001). A molecular basis for an interaction between α-synuclein and the proteasomal system is suggested by the association of α-synuclein with tat binding protein 1, a component of the proteasome (Ghee et al., 2000). Whether α-synuclein itself is degraded by the proteasome is controversial (Bennett et al., 1999; Ancolio et al., 2000; Imai et al., 2000; Rideout et al., 2001).
Death associated with A53T α-synuclein expression is autophagic, not apoptotic
Our findings indicate that the increased cellular degeneration seen in the A53T lines is not attributable to apoptosis. Therefore, our results diverge from those finding induction of apoptotic death with mutant α-synuclein overexpression (Zhou et al., 2000; Lee et al., 2001). This may be related to the more chronic nature of mutant α-synuclein overexpression in our model or to cell-specific differences. Induction of nonapoptotic death with mutant α-synuclein overexpression has been reported previously, but the morphological features were not characterized further (Ostrerova et al., 1999). Our EM data are suggestive of an autophagic mode of cell death (Clarke, 1990; Ohsawa et al., 1998) in which membrane-bound structures engulf intracellular organelles and participate in the destruction of the cell. Features of autophagy, but not death, were also reported in a neuronal cell line expressing murine α-synuclein (Hsu et al., 2000).
Effects on catecholaminergic activity
One reason for using PC12 cells for the current studies was their dopaminergic phenotype. We found a striking deficiency of DCGs in cell lines expressing A53T α-synuclein. Because DCGs are the major structures for storage and release of catecholamines (Sulzer and Pothos, 2000), we tested the mutant lines for evoked dopamine release and found that this was totally absent. As noted below, such observations are novel and are potentially relevant to the dopaminergic deficits of PD.
Suggested model of cellular dysfunction induced by mutant α-synuclein
The multitude of abnormalities observed in the A53T α-synuclein-expressing cells makes it difficult to propose a causal sequence of events leading to cellular dysfunction and death. As mentioned above, it is likely that proteasomal and lysosomal dysfunction lead to ubiquitinated aggregate formation. Such dysfunction could also lead to autophagy, as described previously (Seglen et al., 1996; Wojcik et al., 1996). It is tempting to speculate that the loss of DCGs and consequently of evoked dopamine release is attributable to DCG degradation within autophagic granules, as occurs with other intracellular organelles, such as mitochondria. Death could be attributable to protein aggregation or to the process of autophagy or could be directly related to lysosomal and proteasomal dysfunction or to a combination thereof (Fig. 12).
Fig. 12.

Suggested model of cellular dysfunction and death induced by mutant α-synuclein.
Insights and potential relevance to PD
Our findings provide a number of novel insights into the function of mutant α-synuclein: We have extended the finding of the association of A30P α-synuclein with proteasomal dysfunction to the A53T mutant and have provided evidence of accumulation of polyubiquitinated proteins in the form of small cytoplasmic aggregates. We have observed lysosomal dysfunction, which has not been reported previously and which may play a role in protein aggregation and cellular, and particularly dopaminergic, dysfunction. This is also the first report of nonapoptotic, autophagic cell death induced by an α-synuclein mutant. This finding may have important implications, because the molecular pathways underlying these two different forms of death are distinct, although not mutually exclusive (Xue et al., 1999;Bursch et al., 2000). The issue of the relationship between α-synuclein aggregation and death is controversial. Our findings suggest that A53T α-synuclein can cause toxicity independently of its propensity to aggregate.
There are parallels between our observations and PD. Our findings linking mutant α-synuclein to the ubiquitin-dependent degradation system are especially important, in view of the genetic and pathological data linking defects in this system with PD (Kitada et al., 1998; Leroy et al., 1998; Shimura et al., 2000; McNaught and Jenner, 2001, McNaught et al., 2001). The dot-like cytoplasmic ubiquitinated aggregates are distinct from Lewy bodies but may be the nidus for the eventual formation of such larger inclusions in vivo. The issue of apoptosis in PD remains controversial (Burke, 1998), whereas a recent EM study reported elements of autophagy in degenerating neurons in PD (Anglade et al., 1997). Notably, ultrastructural studies of Lewy bodies formed in sympathetic ganglion neurons of PD patients indicate that these bodies show more vesicular than filamentous features (Forno and Norville, 1976) and may share similarities with the vesicular–autophagic structures we have observed. Moreover, the novel effects we observe on the dopaminergic release system may be related to the decreased availability of dopamine in nigrostriatal terminals out of proportion to the degree of nigral neuron loss in patients with PD (Hornykiewicz, 1996). Mutant α-synuclein-expressing PC12 cells may serve as a useful model to study the molecular mechanisms through which aberrant α-synuclein leads to dopaminergic neuron dysfunction and degeneration in PD.
Footnotes
This work was supported by a Wellcome Burroughs Career Award in Biomedical Sciences to L.S. Additional support was provided by the Parkinson's Disease Foundation, by the Matheson and Lowenstein Foundations, by the National Institute of Neurological Disorders and Stroke (NINDS) (L.S., L.A.G.), and by a Udall Parkinson's Center of Excellence Award (D.S., L.A.G.). We thank Drs. Robert Burke, James Goldman, and Kim Kegel for helpful discussions, Dr. Kominami for his generous gift of the cathepsin D antibody, and Mary Schoenebeck for her invaluable technical assistance with the electron microscopy studies.
Correspondence should be addressed to Leonidas Stefanis at the above address. E-mail: ls76@columbia.edu.
REFERENCES
- 1.Ancolio K, Alves da Costa C, Ueda K, Checler F. α-Synuclein and the Parkinson's disease-related mutant Ala53Thr-α-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett. 2000;285:79–82. doi: 10.1016/s0304-3940(00)01049-1. [DOI] [PubMed] [Google Scholar]
- 2.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 3.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 4.Batistatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of α-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 6.Brunk UT, Zhang H, Dalen H, Ollinger K. Exposure of cells to nonlethal concentrations of hydrogen peroxide induces degeneration-repair mechanisms involving lysosomal destabilization. Free Radic Biol Med. 1995;19:813–822. doi: 10.1016/0891-5849(95)02001-q. [DOI] [PubMed] [Google Scholar]
- 7.Burke RE. Apoptosis in degenerative diseases of the basal ganglia. The Neuroscientist. 1998;4:301–311. [Google Scholar]
- 8.Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann NY Acad Sci. 2000;926:1–12. doi: 10.1111/j.1749-6632.2000.tb05594.x. [DOI] [PubMed] [Google Scholar]
- 9.Cavanagh JB, Nolan CC, Seville MP, Anderson VE, Leigh PN. Routes of excretion of neuronal lysosomal dense bodies after ventricular infusion of leupeptin in the rat: a study using ubiquitin and PGP 9.5 immunocytochemistry. J Neurocytol. 1993;22:779–791. doi: 10.1007/BF01181323. [DOI] [PubMed] [Google Scholar]
- 10.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 12.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration, and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 13.Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–129. [PubMed] [Google Scholar]
- 14.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;11:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 15.Drexler HC. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci USA. 1997;94:855–860. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahn S, Przedborski S. Parkinsonism. In: Rowland LP, editor. Merritt's textbook of neurology, Ed 10. Williams & Wilkins; Philadelphia: 2000. pp. 679–693. [Google Scholar]
- 17.Figueiredo-Pereira ME, Berg KA, Wilk S. A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem. 1994;63:1578–1581. doi: 10.1046/j.1471-4159.1994.63041578.x. [DOI] [PubMed] [Google Scholar]
- 18.Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 19.Forno LS, Norville RL. Ultrastructure of Lewy bodies in the stellate ganglion. Acta Neuropathol (Berl) 1976;34:183–197. doi: 10.1007/BF00688674. [DOI] [PubMed] [Google Scholar]
- 20.Furlong RA, Narain Y, Rankin J, Wyttenbach A, Rubinsztein DC. α-Synuclein overexpression promotes aggregation of mutant huntingtin. Biochem J. 2000;15:577–581. [PMC free article] [PubMed] [Google Scholar]
- 21.Ghee M, Fournier A, Mallet J. Rat α-synuclein interacts with tat binding protein 1, a component of the 26S proteasomal complex. J Neurochem. 2000;75:2221–2224. doi: 10.1046/j.1471-4159.2000.0752221.x. [DOI] [PubMed] [Google Scholar]
- 22.Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein M, Greene LA. Activation of tyrosine hydroxylase by phosphorylation. In: Meltzer H, editor. Psychopharmacology: a third generation of progress. Raven; New York: 1987. pp. 75–80. [Google Scholar]
- 24.Greene LA. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J Cell Biol. 1978;78:747–755. doi: 10.1083/jcb.78.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene LA, Cunningham ME, Farinelli SE, Park DS. Culture and experimental use of the PC12 rat pheochromocytoma cell line. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT Press; Cambridge, MA: 1998. pp. 161–189. [Google Scholar]
- 27.Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1996;18:925–965. [PubMed] [Google Scholar]
- 28.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. α-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;276:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 30.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 31.Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM. Enhanced vulnerability to oxidative stress by α-synuclein mutations and C-terminal truncation. Neuroscience. 2000;97:279–284. doi: 10.1016/s0306-4522(00)00077-4. [DOI] [PubMed] [Google Scholar]
- 32.Kegel KB, Kim M, Sapp E, McIntyre C, Castaño JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 34.Ko L, Mehta ND, Farrer M, Easson C, Hussey J, Yen S, Hardy J, Yen SH. Sensitization of neuronal cells to oxidative stress with mutated human α-synuclein. J Neurochem. 2000;75:2546–2554. doi: 10.1046/j.1471-4159.2000.0752546.x. [DOI] [PubMed] [Google Scholar]
- 35.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 36.Laszlo L, Doherty FJ, Osborn NU, Mayer RJ. Ubiquitinated protein conjugates are specifically enriched in the lysosomal system of fibroblasts. FEBS Lett. 1990;261:365–368. doi: 10.1016/0014-5793(90)80593-8. [DOI] [PubMed] [Google Scholar]
- 37.Lee M, Hyun D, Halliwell B, Jenner P. Effect of the overexpression of wild-type or mutant α-synuclein on cell susceptibility to insult. J Neurochem. 2001;76:998–1009. doi: 10.1046/j.1471-4159.2001.00149.x. [DOI] [PubMed] [Google Scholar]
- 38.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 39.Lopes UG, Erhardt P, Yao R, Cooper GM. p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272:12893–12896. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 41.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, DiMonte DA. Lack of nigral pathology in transgenic mice expressing human α-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- 42.Mayer RJ, Laszlo L, Landon M, Hope J, Lowe J. Ubiquitin, lysosomes, and neurodegenerative diseases. Ann NY Acad Sci. 1992;674:149–160. doi: 10.1111/j.1749-6632.1992.tb27484.x. [DOI] [PubMed] [Google Scholar]
- 43.McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- 44.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 45.Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obin M, Mesco E, Gong X, Haas AL, Joseph J, Taylor A. Neurite outgrowth in PC12 cells: distinguishing the roles of ubiquitylation and ubiquitin-dependent proteolysis. J Biol Chem. 1999;274:11789–11795. doi: 10.1074/jbc.274.17.11789. [DOI] [PubMed] [Google Scholar]
- 47.Ohsawa Y, Isahara K, Kanamori S, Shibata M, Kametaka S, Gotow T, Watanabe T, Kominami E, Uchiyama Y. An ultrastructural and immunohistochemical study of PC12 cells during apoptosis induced by serum deprivation with special reference to autophagy and lysosomal cathepsins. Arch Histol Cytol. 1998;61:395–403. doi: 10.1679/aohc.61.395. [DOI] [PubMed] [Google Scholar]
- 48.Ohtani-Kaneko R, Takada K, Iigo M, Hara M, Yokosawa H, Kawashima S, Ohkawa K, Hirata K. Proteasome inhibitors which induce neurite outgrowth from PC12h cells cause different subcellular accumulations of multi-ubiquitin chains. Neurochem Res. 1998;23:1435–1443. doi: 10.1023/a:1020763009488. [DOI] [PubMed] [Google Scholar]
- 49.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. α-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papadimitriou A, Veletza V, Hadjigeorgiou GM, Patrikiou A, Hirano M, Anastasopoulos I. Mutated α-synuclein gene in two Greek kindreds with familial PD: incomplete penetrance? Neurology. 1999;52:651–654. doi: 10.1212/wnl.52.3.651. [DOI] [PubMed] [Google Scholar]
- 52.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 53.Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haass C, Schulz JB. Sensitivity to MPTP is not increased in Parkinson's disease-associated mutant-synuclein transgenic mice. J Neurochem. 2001;77:1181–1184. doi: 10.1046/j.1471-4159.2001.00366.x. [DOI] [PubMed] [Google Scholar]
- 55.Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78:899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- 56.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saha AR, Ninkina NN, Hanger DP, Anderton BH, Davies AM, Buchman VL. Induction of neuronal death by α-synuclein. Eur J Neurosci. 2000;12:3073–3077. doi: 10.1046/j.1460-9568.2000.00210.x. [DOI] [PubMed] [Google Scholar]
- 58.Seglen PO, Berg TO, Blankson H, Fengsrud M, Holen I, Stromhaug PE. Structural aspects of autophagy. Adv Exp Med Biol. 1996;389:103–111. doi: 10.1007/978-1-4613-0335-0_12. [DOI] [PubMed] [Google Scholar]
- 59.Shimura H, Hattori N, Kubo SI, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 60.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 61.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 62.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stefanis L, Park DS, Yan CYI, Farinelli SE, Troy CM, Shelanski ML, Greene LA. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support. J Biol Chem. 1996;271:30663–30671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- 64.Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA. Caspase-2 (Nedd2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J Neurosci. 1998;18:9204–9215. doi: 10.1523/JNEUROSCI.18-22-09204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stefanis L, Kholodilov N, Rideout HJ, Burke RE, Greene LA. Synuclein 1 is selectively upregulated in response to NGF treatment in PC12 cells. J Neurochem. 2001;76:1165–1176. doi: 10.1046/j.1471-4159.2001.00114.x. [DOI] [PubMed] [Google Scholar]
- 67.Sulzer D, Pothos EN. Presynaptic mechanisms that regulate quantal size. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- 68.Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH. Expression of mutant α-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet. 2000;9:2683–2689. doi: 10.1093/hmg/9.18.2683. [DOI] [PubMed] [Google Scholar]
- 69.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa AL, Dawson V, Dawson TM, Ross CA. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 70.Tennyson VM, Gershon MD, Sherman DL, Behringer RR, Raz R, Crotty DA, Wolgemuth DJ. Structural abnormalities associated with congenital megacolon in transgenic mice that overexpress the Hoxa-4 gene. Dev Dyn. 1993;98:128–153. doi: 10.1002/aja.1001980105. [DOI] [PubMed] [Google Scholar]
- 71.Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM. Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death Differ. 1998;5:832–837. doi: 10.1038/sj.cdd.4400432. [DOI] [PubMed] [Google Scholar]
- 72.Wojcik C, Schroeter D, Wilk S, Lamprecht J, Paweletz N. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71:311–318. [PubMed] [Google Scholar]
- 73.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signaling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human α-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]