

Abstract
We have explored the processes regulating presynaptic calcium concentration ([Ca2+]i) in the generation of post-tetanic potentiation (PTP) at crayfish neuromuscular junctions, using spectrophotometric dyes to measure changes in [Ca2+]i and [Na+]i and effects of inhibitors of Ca2+-transport processes. The mitochondrial Na+/Ca2+ exchange inhibitor CGP 37157 was without effect, whereas the reverse mode plasmalemmal Na+/Ca2+ exchange inhibitor KB R7943 reduced PTP and Ca2+ accumulation caused by increased [Na+]i. Exchange inhibitory peptide and C28R2 had opposite effects, consistent with their block of the plasma membrane Ca2+ ATPase. All drugs except CGP 37157 reduced Ca2+ accumulation caused by Na+ accumulation, which occurred on block of the Na+/K+ pump, acting in proportion to their effects on plasmalemmal Na+/Ca2+ exchange. We find no role for mitochondrial Na+/Ca2+exchange in presynaptic Ca2+ regulation. The plasma membrane Na+/Ca2+ exchanger acts in reverse mode to admit Ca2+ into nerve terminals during and for some minutes after tetanic stimulation, while at the same time the plasma membrane Ca2+ ATPase operates as an important Ca2+ removal process. The interplay of these two Ca2+ transport processes with Na+-independent mitochondrial Ca2+ fluxes and the plasmalemma Na+/K+ pump determines the magnitude of tetanic [Ca2+]iaccumulation and potentiation of excitatory transmission, and the post-tetanic time courses of decay of elevated [Ca2+]i and PTP.
Keywords: post-tetanic potentiation, Na+/Ca2+ exchange, Ca2+ ATPase, mitochondria, transmitter release, crayfish, neuromuscular junction, synaptic transmission
When presynaptic activity persists at high levels for several minutes, many synapses display a gradual potentiation of synaptic transmission. After activity subsides, individual action potentials continue to evoke potentiated postsynaptic potentials for several minutes, a process called post-tetanic potentiation (PTP) (Zucker, 1989). At crayfish neuromuscular junctions, PTP reflects an increase in the number of quanta released by action potentials (Wojtowicz and Atwood, 1986) that is caused by a persistent increase in presynaptic calcium concentration ([Ca2+]i) (Delaney et al., 1989). This so-called “residual Ca2+” remaining in nerve terminals after bouts of activity arises primarily from efflux of Ca2+ from mitochondria that become Ca2+ loaded during the conditioning activity (Tang and Zucker, 1997). It appears to induce PTP by acting on a presynaptic Ca2+ target distinct from those involved in exocytosis and short-term synaptic facilitation (Kamiya and Zucker, 1994). Excessive Na+loading of presynaptic terminals also enhances and prolongs PTP and the persistence of residual Ca2+ (Mulkey and Zucker, 1992), suggesting that accumulation of Na+ retards Ca2+ extrusion by Na+/Ca2+exchange, contributing to the genesis of PTP.
There are two Na+/Ca2+exchangers that could mediate an influence of Na+ on [Ca2+]i: plasmalemmal Na+/Ca2+exchange (Blaustein and Lederer, 1999) and mitochondrial Na+/Ca2+exchange (Friel, 2000; Gunter et al., 2000). In the first case, tetanic [Na+]i elevation could retard or even reverse the direction of Ca2+ flux through the plasmalemmal Na+/Ca2+exchanger, resulting in Ca2+ influx into cytoplasm from the external medium. In the second case, [Na+]i elevation could activate mitochondrial Ca2+ efflux into cytoplasm. Both plasmalemmal Na+/Ca2+exchange (Luther et al., 1992; Gleason et al., 1994; Kobayashi and Tachibana, 1995; Regehr, 1997; Scotti et al., 1999) and mitochondrial Ca2+ fluxes (Alnaes and Rahamimoff, 1975;David et al., 1998; Peng, 1998; Brodin et al., 1999) are known to be involved in Ca2+ regulation at nerve terminals.
Two other important Ca2+ regulatory processes are the plasmalemma Ca2+ ATPase (Garcia and Strehler, 1999) and Ca2+channels in endoplasmic reticulum (Pozzan et al., 1994; Simpson et al., 1995). Both have been implicated in controlling [Ca2+]i at nerve terminals (Fossier et al., 1994; Kobayashi and Tachibana, 1995; Tucker and Fettiplace, 1995; Peng, 1996; Smith and Cunnane, 1996; Morgans et al., 1998; Juhaszova et al., 2000; Zenisek and Matthews, 2000).
Here we explore roles for these processes in controlling the residual Ca2+ responsible for the induction of PTP at crayfish motor nerve terminals. We report effects of drugs that target the plasmalemmal or mitochondrial Na+/Ca2+exchanger or the Ca2+ ATPase. Although these drugs were developed in mammalian preparations, the high genetic and immunological conservation of these molecules across vertebrates and invertebrates, particularly for the plasmalemmal Na+/Ca2+exchanger (Blaustein and Lederer, 1999), encouraged us to apply them to this preparation. We observe important effects of plasmalemmal Na+/Ca2+exchange and the Ca2+ ATPase, whereas we could detect no role for mitochondrial Ca2+ efflux via Na+/Ca2+exchange.
MATERIALS AND METHODS
Animals, solutions, and drugs. Experiments used isolated opener muscles of the first walking leg of crayfish (Procambarus clarkii, 2–2.5 inches) obtained from KLM Bioscientific (San Diego, CA) and Niles Biological (Sacramento, CA). Autotomized first walking legs were pinned in a Sylgard-lined chamber continuously perfused with a solution containing (in mm): 195 NaCl, 13.5 CaCl2, 5.4 KCl, 2.6 MgCl2, and 10 Na-HEPES, pH 7.4, at 15–17°C. Opener muscles and exciter axons were exposed as described previously (Delaney and Tank, 1991; Landò and Zucker, 1994). Chemicals were obtained from the following suppliers: 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea (KB R7943) and 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP 37157) from Tocris (Ballwin, MO); exchange inhibitory peptide (XIP) (RRLLFYKYVYKRYRAGKQRG) and C28R2 (Ca2+ ATPase inhibitor, LRRGQILWFRGLNRIQTQIRVVKAFRSS) from Alpha Diagnostic International (San Antonio, TX); ouabain and palytoxin from Sigma (St. Louis, MO); fura-2 pentapotassium salt from Molecular Probes (Eugene, OR); sodium-binding benzofuran isophthalate (SBFI) from TefLabs (Austin, TX); and gramicidin D and monensin from Calbiochem (San Diego, CA). Stock solutions of KB R7943 (100 mm) and CGP 37157 (25 mm) were made in dimethyl sulfoxide and diluted to the desired concentration in crayfish saline before experiments. The final concentration of solvent did not exceed 0.1%. Ouabain was freshly prepared in crayfish saline before each experiment.
Electrophysiology. The exciter motor neuron to the opener muscle was stimulated with a suction electrode on the leg nerve exposed in the meropodite while excitatory junction potentials (EJPs) were recorded via microelectrodes (12–25 MΩ) filled with 3m KCl impaled in central–proximal or proximal muscle fibers. Electrical signals were amplified and filtered at 2 kHz (Neuroprobe 1600, A-M Systems, Everett, WA) and digitized at 5 kHz (DigiData 1200A, Axon Instruments, Foster City, CA). In some experiments, axon action potentials were recorded using a beveled microelectrode (25–45 MΩ) penetrating a primary or secondary branch of the exciter axon and amplified (Getting Microelectrode Amplifier Model 5, Iowa City, IA) before digitization. After penetrating a nerve terminal, a second microelectrode was placed into an adjacent postsynaptic muscle fiber within 100–200 μm of the presynaptic impalement site to measure EJPs. Intracellular recordings from synaptic terminals and muscle fibers were stable for several hours. Recordings from the Y branch or a secondary branch were within 0.3–0.5 space constants of imaged sites of excitatory transmitter release (Baxter and Bittner, 1981, 1991). The average resting membrane potential was −71 ± 3 mV (n = 4). EJPs and action potentials were stored on a personal computer using pClamp7 software (Axon Instruments). EJP amplitudes were analyzed off-line (Clampfit 6.05,Axon Instruments).
Presynaptic peptide injection. The primary or secondary branch of the excitatory axon was penetrated with a beveled electrode containing XIP (0.33 mm) or C28R2 (0.23 mm) in a dye-marked carrier solution (6 mm fura-2, 200 mm KCl, 10 mm HEPES, pH 7.4). Pressure injection used trains of pressure pulses (30–40 psi, 400 msec duration, 0.33 Hz) for 1 hr. From the fluorescence intensity of fura-2, the dye concentration was estimated as described previously (Mulkey and Zucker, 1992) and used to estimate peptide concentrations in presynaptic terminals. For EJP measurements, responses to a control tetanus were obtained before peptide injection and then after injection responses to a second tetanus were recorded. [Ca2+]imeasurements were obtained from another group of animals in which peptide with fura-2 was injected and effects of tetanic stimulation were recorded. Controls consisted of identical experiments performed in different cells injected with fura-2 but no peptide. In these experiments, fura-2 (17 mm in 200 mm KCl) was iontophoresed into the axon using 10–15 nA of continuous hyperpolarizing current for ∼30 min. The final concentration of fura-2 was ∼150 μm.
[Ca2+]i measurement. Fura-2 fluorescence was detected with a silicon-intensified target (SIT) camera (Dage MTI, model 66), via a 40× 0.7 numerical aperture water immersion objective (Olympus, Lake Success, NY). Fluorescence was alternately excited through 350 ± 10 and 382 ± 5 nm filters (Omega Optical, Battleboro, VT). A dichroic mirror (455 nm; Nikon, Tokyo, Japan) separated excitation and emission wavelengths, and a barrier filter (530 ± 20 nm) (Omega Optical) limited interference by autofluorescence. An area near the imaged bouton with uniform intensity similar to that around the bouton was chosen for obtaining tissue background. Background subtraction and shading correction were performed automatically in an image processor (FD5000; Gould Inc., Fremont, CA). Shading correction removes errors attributable to changes in the color of the excitation illumination with age of the bulb or other variable optical chromatic distortions.
Averages of 32 sequential images excited at 350 and 385 nm were stored on an optical disk recorder (TQ-2028F, Panasonic, Secaucus, NJ). The imaging processor, optical disk recorder, and filter changer were under the control of a Scientific Microsystems SMS 1000 computer (Mountain View, CA), using software written by Dr. Roger Tsien (Pharmacology Department, University of California, San Diego). Fura-2 images were calibrated by measuring the fluorescence ratio obtained with 50 μm fura-2 in solutions at 280 mm ionic strength, resembling crayfish cytoplasmic solution (250 mmK-gluconate, 15 mm NaCl, 15 mm K-HEPES, pH 7.02) with zero-calcium (10 mmK2EGTA), 5 mmCa2+, or [Ca2+]i buffered to 500 nm with 10 mmK2EGTA and 5 mmCaCl2. Ratios measured in terminals were converted to [Ca2+]i (Grynkiewicz et al., 1985) after application of a viscosity correction corresponding to a 30% reduction in the minimum and maximum 350 nm/385 nm fluorescence ratios (Mulkey and Zucker, 1992). Calibrations used values ofRmax/Rmin= 22.9 and KD = 523 nm.
[Na+]i measurement. The [Na+]imeasurements were done in separate preparations from those used for EJP and [Ca2+]imeasurements. The exciter axon was penetrated with a beveled microelectrode (25–45 MΩ) containing 20 mm SBFI in 200 mm KCl, 10 mm HEPES, pH 8.5. Dye was iontophoresed (−10 nA for 30 min) to a final concentration of ∼0.5 mm. Ratiometric SBFI images were produced in the same manner as that used for fura-2. A 50% neutral density filter reduced excitation light intensity to minimize photobleach.
SBFI was calibrated in situ (Harootunian et al., 1989). Axons injected with SBFI were permeabilized by addition of sodium ionophores palytoxin (0.1 μm), gramicidin D (10 μm), and monensin (10 μm) and subsequently perfused with solutions comprising varying [Na+]. This was achieved by mixing a “high Na+” solution [containing (in mm): 13 NaCl, 244 Na-gluconate, 10 Cs-HEPES, 5 CaCl2, 1 MgCl2, pH 7.00] with a “Na-free” solution [containing (in mm): 13 KCl, 244 K-gluconate, 10 Cs-HEPES, 5 CaCl2, 1 MgCl2, pH 7.00] so that ionic strength remained at 280 and [Na+] could be set from 0–257 mm. Palytoxin also inhibits the Na+/K+ ATPase (Habermann, 1989), permitting [Na+]i to rise to high levels when external [Na+] is high. Ratiometric fluorescence images were obtained at different [Na+].Rmax/Rminwas 1.88 and the KD of Na+ binding to SBFI was determined as 21 ± 2 mm (n = 3) using Equation 5 of Grynkiewicz et al.(1985).
Data analysis. Curve-fitting algorithms in Prism (GraphPad Software, San Diego, CA) were used to determine the decay time constants of PTP and [Ca2+]i. EJP amplitudes normally showed a double exponential decay. We quantified PTP by fitting an exponential function to the slowly decaying component of the post-tetanic decay in EJP amplitude. A two-sided Student's t test on percentage changes from control in each pair was used to estimate statistical significance unless indicated otherwise.
RESULTS
To investigate roles played by Ca2+removal processes in PTP, we normally compared the effects of PTP-inducing tetani on presynaptic [Ca2+]iaccumulation and removal, and the induction of PTP, in the same preparation before and after administration of specific blocking agents. [Na+]i was determined in some preparations. We first needed to show that repetition of PTP-inducing tetani produced repeatable effects on [Ca2+]i, [Na+]i, and transmission when there was no drug present for either tetanus. Figure1 illustrates two control experiments, and the results of eight experiments are tabulated in Table1. PTP was induced by stimulation of the exciter motor neuron at 20 Hz for 10 min, and EJPs were sampled at 2 Hz before and after the tetanus, and one ratiometric fura-2 image of [Ca2+]i or SBFI image of [Na+]iwas produced every minute. The second tetanus followed the first by at least 90 min, to allow for full recovery. Pretetanic, tetanic, and post-tetanic EJP amplitudes, and the decay of PTP, remained constant on repetition of PTP induction. [Ca2+]i was measured in four experiments and [Na+]i in one, and pretetanic, tetanic, and post-tetanic [Ca2+]i and [Na+]i levels and their rate of decay also were unchanged.
Fig. 1.
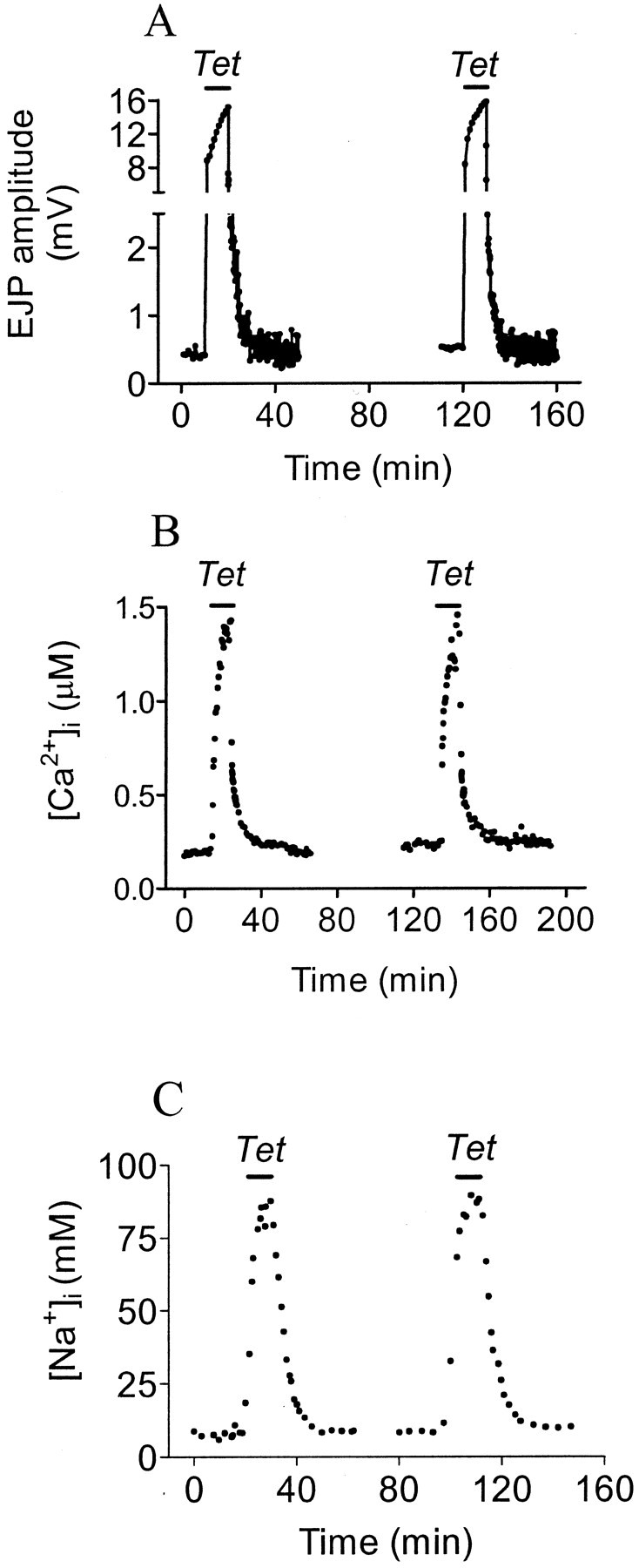
EJP amplitude (A), [Ca2+]i (B), and [Na+]i (C) measured before, during, and after 10 min, 20 Hz tetanic stimulation (Tet). The [Na+]imeasurements were from a separate experiment.
Table 1.
Effects of KB R7943 and CGP 37157 on presynaptic [Ca2+]i and short-term synaptic enhancement
| Measurement | Control | KB R7943 (20 μm) | CGP 37157 (25 μm) | |||
|---|---|---|---|---|---|---|
| First | Second | Before drug | After drug | Before drug | After drug | |
| Pretetanic EJP amplitude (mV) | 0.22 ± 0.02 | 0.23 ± 0.02 (8) | 0.17 ± 0.03 | 0.17 ± 0.02 (8) | 0.19 ± 0.02 | 0.20 ± 0.02 (6) |
| Peak EJP amplitude during tetanus (mV) | 10.54 ± 0.41 | 10.51 ± 0.41 (8) | 11.93 ± 0.75 | 9.77 ± 0.80 (8)1-160 | 10.28 ± 1.0 | 10.48 ± 1.0 (6) |
| EJP amplitude 1 min after tetanus (mV) | 1.53 ± 0.13 | 1.63 ± 0.11 (8) | 1.55 ± 0.18 | 0.79 ± 0.12 (8)1-160 | 1.03 ± 0.10 | 1.15 ± 0.14 (6) |
| EJP amplitude 5 min after tetanus (mV) | 0.68 ± 0.093 | 0.65 ± 0.063 (8) | 0.79 ± 0.17 | 0.38 ± 0.08 (8)* | 0.76 ± 0.09 | 0.74 ± 0.07 (6) |
| PTP decay time constant (min) | 4.84 ± 0.51 | 4.62 ± 0.41 (8) | 4.75 ± 0.31 | 3.22 ± 0.43 (8)1-160 | 4.12 ± 0.30 | 4.19 ± 0.33 (6) |
| Pretetanic [Ca2+]i (μm) | 0.15 ± 0.02 | 0.16 ± 0.03 (4) | 0.15 ± 0.02 | 0.15 ± 0.01 (5) | 0.15 ± 0.02 | 0.17 ± 0.03 (4) |
| Peak [Ca2+]i during tetanus (μm) | 1.54 ± 0.09 | 1.57 ± 0.13 (4) | 1.65 ± 0.16 | 1.34 ± 0.13 (5)1-160 | 1.53 ± 0.05 | 1.52 ± 0.06 (4) |
| [Ca2+]i1 min after tetanus (μm) | 0.78 ± 0.07 | 0.78 ± 0.08 (4) | 0.78 ± 0.02 | 0.55 ± 0.03 (5)1-160 | 0.77 ± 0.01 | 0.78 ± 0.01 (4) |
| [Ca2+]i 5 min after tetanus (μm) | 0.34 ± 0.04 | 0.34 ± 0.01 (4) | 0.40 ± 0.04 | 0.26 ± 0.043 (5)* | 0.36 ± 0.04 | 0.33 ± 0.03 (4) |
| Slow [Ca2+]i decay time constant (min) | 5.66 ± 0.62 | 5.63 ± 1.45 (4) | 5.71 ± 0.69 | 3.30 ± 0.71 (5)* | 5.07 ± 1.48 | 5.93 ± 1.03 (4) |
| Pretetanic [Na+]i(mm) | 7.93 | 7.67 | 7.91 ± 0.67 | 7.96 ± 0.75 (3) | ||
| Peak [Na+]i during tetanus (mm) | 80.6 | 80.5 | 80.6 ± 5.32 | 82.6 ± 3.27 (3) | ||
| [Na+]i 5 min after tetanus (mm) | 33.2 | 36.4 | 36.8 ± 4.34 | 57.7 ± 2.47 (3)* | ||
| [Na+]i decay time constant (min) | 7.66 | 7.19 | 8.52 ± 1.92 | 13.44 ± 2.08 (3)* | ||
Postsynaptic EJP amplitude and presynaptic [Ca2+]i or [Na+]iin boutons were measured before and after tetanic stimulation. Data: mean ± SE; numbers of measurements in parentheses; significant changes by Student's paired t test;
p < 0.05;
F1-160: p < 0.01.
Plasmalemmal Na+/Ca2+exchange does influence PTP
As one probe of plasma membrane Na+/Ca2+exchange we used the relatively specific blocker KB R7943. This agent is much more effective against the reverse mode of transport, in which Ca2+ enters in exchange for Na+ efflux (IC50 = 0.3–2 μm cytoplasmic concentration) (Iwamoto et al., 1996; Watano et al., 1996), than against forward transport, in which Ca2+ is extruded in exchange for Na+ influx (IC50 = 17–30 μm). However, at these higher concentrations KB R7943 (>30 μm) also affected currents through voltage-dependent ion channels, including Ca2+ channels (Watano et al., 1996). At 10 μm bath concentration, KB R7943 was without effect on PTP in our preparations. At concentrations of 50 μm and higher, baseline synaptic transmission and tetanic presynaptic [Ca2+]iaccumulation were strongly reduced, probably because of presynaptic Ca2+ channel block. At 20 μm, such nonspecific effects on baseline transmission were not observed, but there was a modest reduction in tetanic Ca2+ accumulation and tetanic EJP amplitude, and the post-tetanic decay of [Ca2+]i and EJP amplitude were accelerated (Fig. 2, Table1). To confirm inhibition by KB R7943 of Na+/Ca2+exchange, effects on [Na+]i were observed. KB R7943 (20 μm) had no clear effect on [Na+]iaccumulation during tetanic stimulation, but it did slow post-tetanic [Na+]i decay (Fig.2E,F, Table 1).
Fig. 2.

Effects of KB R7943 on [Ca2+]i, [Na+]i, and PTP. The first tetanus was given in normal saline and the second one in the presence of 20 μm KB R7943 (reverse mode plasmalemmal Na+/Ca2+ exchange inhibitor).A, B, Tetanic [Ca2+]i and EJP amplitude were reduced in 20 μm KB R7943. C, D, Running averages of five post-tetanic [Ca2+]i measurements and eight post-tetanic EJP amplitudes in control saline (●) and 20 μm KB R7943 (○). E, F, Tetanic [Na+]i and running average of three post-tetanic [Na+]i measurements in control crayfish saline (●) and 20 μm KB R7943 (○).
The acceleration of post-tetanic [Ca2+]i decay and slowing of [Na+]iremoval are consistent with a block of Na+/Ca2+exchange occurring in reverse mode. However, the effects on Ca2+ accumulation and tetanic EJP amplitude could be attributable to a reduction of Ca2+ influx through Ca2+ channels. This could result in reduced mitochondrial Ca2+ accumulation and therefore a reduction in both PTP and in post-tetanic [Ca2+]i (Tang and Zucker, 1997), but the effect on Na+removal is difficult to explain this way and suggests instead an action on Na+/Ca2+exchange. To distinguish effects on tetanic Ca2+ influx from effects on Na+/Ca2+exchange, KB R7943 was applied at the end of the tetanus. A solution containing 20 μm KB R7943 was washed in during the last minute of tetanic stimulation. The peak EJP amplitude (12.7 mV in the second tetanus vs 11.9 mV in the first tetanus) and [Ca2+]iaccumulation (1.09 μm in the second tetanus vs 1.07 μm in the first tetanus) were not influenced. Nevertheless, KB R7943 still accelerated Ca2+ removal (τslow = 3.41 min in the second tetanus vs 4.69 min in the first tetanus) and PTP decay (τ = 2.79 min vs the control value of 3.4 min) (Fig. 3). These effects are similar to those occurring when KB R7943 was present throughout the second tetanus (Fig. 2, Table 1).
Fig. 3.

KB R7943 application delayed until the end of tetanic stimulation. A, B, Tetanic [Ca2+]i and EJP amplitude were unchanged when applying 20 μm KB R7943 at the end of a 20 Hz tetanus. C, D, Running averages of three post-tetanic [Ca2+]imeasurements and two post-tetanic EJP amplitudes in control saline (●) and 20 μm KB R7943 (○). E,F, Tetanic [Na+]i and post-tetanic [Na+]i measurements in control crayfish saline (●) and when applying 20 μm KB R7943 at the end of the tetanus (○).
Taken together our results show that plasma membrane Na+/Ca2+exchange operates in reverse mode (admitting Ca2+ into nerve terminals) during and for some minutes after tetanic stimulation, attributable to the presynaptic accumulation of Na+, contributing to tetanic [Ca2+]iaccumulation and EJP potentiation and slowing post-tetanic Ca2+ removal and PTP decay. Blocking this process then reduces tetanic [Ca2+]iaccumulation and EJP potentiation and speeds Ca2+ removal and PTP decay.
Tetanic Na+ accumulation reverses plasmalemmal Na+/Ca2+ exchange and prolongs PTP
The operation of Na+/Ca2+exchange in reverse mode in PTP is further supported by our measurements of [Na+]i and [Ca2+]i changes during and after tetanic stimulation (Fig.4, Table 1). We have performed such measurements on small individual presynaptic boutons and with greater accuracy on larger preterminal nerve branches. Tetanic stimulation causes [Na+]i to rise from 7 to 80 mm and [Ca2+]i to rise from 0.13 to 1.15 μm. [Ca2+]i recovers to only slightly above resting levels within 2–5 min after the tetanus (Tang and Zucker, 1997), whereas [Na+]i relaxes more slowly to pretetanic levels (τNa = 8.52 ± 1.92 min, p < 0.05, vs τCa = 5.71 ± 0.69 min).
Fig. 4.

Relationship between the membrane potential (Vm) and calculated Na+/Ca2+ exchange equilibrium potential (ENa/Ca) before, during, and after 10 min, 20 Hz tetanic stimulation. Top panelshows average [Na+]i (○,n = 7) and [Ca2+]i(●, n = 8) in primary and secondary branches of the exciter axon (left) or in terminal boutons (right). Bottom panel shows the calculated ENa/Ca (○) and measuredVm (■, n = 4); it indicates that during the tetanus and PTP phase, the Na+/Ca2+ exchange operates in reverse mode. Tetanic and PTP periods are indicated by different shading patterns (tetanic period ▨; PTP ▧).
Na+/Ca2+exchange involves the movement of three Na+ ions in one direction in exchange for one Ca2+ ion in the opposite direction. Because there is a net transport of charge, the process is not electroneutral and depends on membrane potential as well as the Na+ and Ca2+gradients across the membrane (Blaustein and Lederer, 1999). The direction of Ca2+ flux can be outward (normal mode) or inward (reverse mode), according to whether the difference (Δν) between membrane potential (Vm) and the equilibrium potential for Na+/Ca2+exchange (ENa/Ca) is negative or positive:
where ENa/Ca depends on the Nernst potentials for Na+(ENa) and Ca2+(ECa):
From measurements of changes in [Na+]i, and [Ca2+]i during and after stimulation, and with [Na+]o = 205 mm and [Ca2+]o = 13.5 mm, ENa/Ca can be calculated. When compared with measurements ofVm (Fig. 4), it can be seen that the Δν is negative at rest (when Vm = −70 mV and ENa/Ca = −42 mV, Δν = −28 mV), but rapidly goes positive during tetanic stimulation (when Vm = −80 mV andENa/Ca = −177 mV, Δν = +93 mV). The hyperpolarization of nerve terminals during tetanic stimulation is caused by operation of the Na+/K+ pump (Wojtowicz and Atwood, 1985), which extrudes three Na+ ions for each two K+ ions admitted. Δν remains positive for some 15 min after stimulation, even rising slightly immediately post-tetanically because [Ca2+]i recovers more rapidly than [Na+]i. These data show that the plasma membrane Na+/Ca2+ exchanger should run in normal mode at rest, extruding Ca2+ from cytoplasm, but in reverse mode during and for some time after tetanic stimulation, admitting Ca2+ ions from outside, increasing the tetanic rise in [Ca2+]i, potentiating synaptic transmission, slowing the post-tetanic recovery of [Ca2+]i, and prolonging PTP.
Mitochondrial Na+/Ca2+exchange does not appear to participate in PTP
In addition to the plasmalemmal Na+/Ca2+exchanger, mitochondrial Na+/Ca2+exchange might also play a role in PTP. We probed this possibility with CGP 37157, a specific inhibitor of mitochondrial Ca2+ efflux (IC50 = 0.36 μm cytoplasmic concentration) (Cox et al., 1993). If mitochondrial Na+/Ca2+exchange regulates [Ca2+]i in PTP, then with sufficient tetanic Na+accumulation, mitochondrial Na+/Ca2+exchange might transport Ca2+ into the cytoplasm, increasing [Ca2+]iaccumulation and EJP potentiation and retarding the post-tetanic decay of [Ca2+]i and PTP. Block of this process would then reduce tetanic cytoplasmic [Ca2+]iaccumulation and synaptic transmission, and [Ca2+]i and PTP would decay more rapidly. Contrary to this scenario, we found that 25 μm CGP 37157 applied 20 min before a tetanus and left on until at least 20 min after the end of the tetanus was completely without effect on the pretetanic, tetanic, and post-tetanic [Ca2+]i changes and baseline transmission or PTP (Fig. 5, Table 1). Because the EJP during tetanic stimulation reflects facilitation and augmentation as well as potentiation (Zucker, 1989), these processes were all apparently unaffected.
Fig. 5.
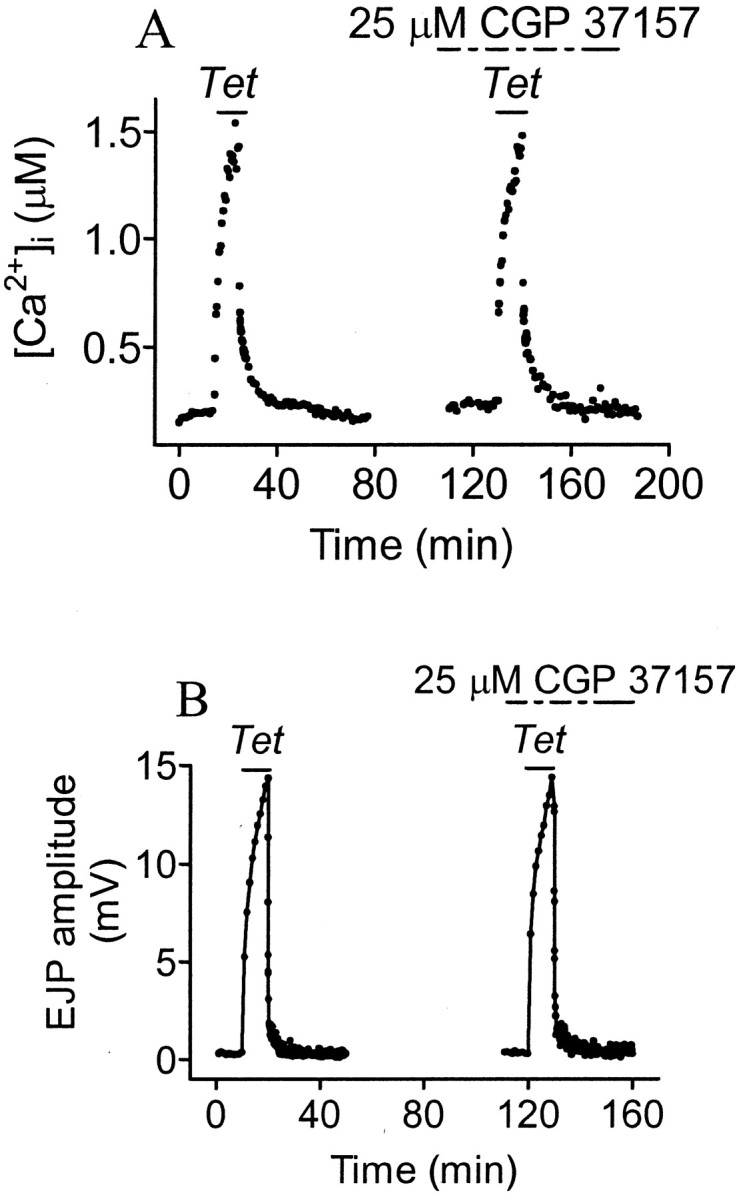
Effects of mitochondrial Na+/Ca2+ transport inhibitor CGP 37157 on [Ca2+]i and PTP. The first tetanus was given in normal saline and the second one in the presence of 25 μm CGP 37157. In 25 μm CGP 37157, [Ca2+]i (A) and EJP amplitude (B) were unchanged.
Plasmalemma Ca2+ ATPase does influence PTP
Transport inhibitory peptide (XIP) is another blocker of Na+/Ca2+exchange (Li et al., 1991), which binds to a cytoplasmic autoinhibitory calmodulin-binding domain of the exchanger with IC50 = 1.7 μm (Xu et al., 1997). This agent must be injected into presynaptic nerve terminals, so it is impractical to measure [Ca2+]i and synaptic transmission in the same preparations before and after drug administration. We therefore compared [Ca2+]i responses with tetanic stimulation in two groups of experiments (different from those used to record effects on EJPs), in one of which XIP had been injected to an estimated concentration of ∼5–10 μm. If Na+/Ca2+exchange operates in reverse mode in a tetanus, we would expect block of this process to reduce [Ca2+]iaccumulation and EJP potentiation and speed post-tetanic Ca2+ removal and PTP decay. In fact, we observed just the opposite effects (Fig. 6A, Table2).
Fig. 6.

XIP and C28R2 increase [Ca2+]i accumulation and potentiation and retard [Ca2+]i decay in PTP.A, B, EJP amplitudes during the first control tetanus and the second tetanus after presynaptic injection of inhibitors of plasma membrane Na+/Ca2+ exchange and Ca2+ ATPase: XIP (A) or C28R2 (B). C, D, Running averages of five (C) or four (D) post-tetanic EJP amplitudes in control saline (●) and in the presence of inhibitory peptide (○).E, F, [Ca2+]i, averages with SEs in controls (●; with no presynaptic peptide injection;n = 7), XIP injections (○ in E;n = 4), or C28R2 injections (○ inF; n = 4).
Table 2.
Effects of XIP and C28R2 on presynaptic [Ca2+]i and short-term synaptic enhancement
| Measurement | XIP | C28R2 | ||
|---|---|---|---|---|
| Control | 5–10 μm | Control | 1–5 μm | |
| Pretetanic EJP amplitude (mV) | 0.22 ± 0.03 | 0.23 ± 0.03 (5) | 0.23 ± 0.05 | 0.24 ± 0.04 (4) |
| Peak EJP amplitude during tetanus (mV) | 9.45 ± 1.02 | 14.43 ± 1.28 (5)2-160 | 9.96 ± 1.55 | 14.94 ± 1.62 (4)2-160 |
| EJP amplitude 1 min after tetanus (mV) | 1.13 ± 0.19 | 2.39 ± 0.37 (5)2-160 | 1.20 ± 0.31 | 2.76 ± 0.42 (4)2-160 |
| EJP amplitude 5 min after tetanus (mV) | 0.69 ± 0.12 | 1.47 ± 0.26 (5)* | 0.68 ± 0.17 | 1.56 ± 0.37 (4) |
| PTP decay time constant (min) | 3.78 ± 0.46 | 7.01 ± 0.61 (5)2-160 | 3.89 ± 0.30 | 8.31 ± 0.33 (4)* |
| Pretetanic [Ca2+]i (μm) | 0.16 ± 0.05 (7)2-a | 0.15 ± 0.04 (4) | 0.16 ± 0.05 (7)2-a | 0.16 ± 0.04 (4) |
| Peak [Ca2+]i during tetanus (μm) | 1.47 ± 0.15 (7)2-a | 2.28 ± 0.13 (4)‡ | 1.47 ± 0.15 (7)2-a | 2.38 ± 0.13 (4)‡ |
| [Ca2+]i 1 min after tetanus (μm) | 0.84 ± 0.10 (7)2-a | 1.29 ± 0.20 (4)† | 0.84 ± 0.10 (7)2-a | 1.59 ± 0.21 (4)† |
| [Ca2+]i 5 min after tetanus (μm) | 0.41 ± 0.04 (7)2-a | 0.69 ± 0.10 (4)† | 0.41 ± 0.04 (7)2-a | 0.89 ± 0.12 (4)‡ |
| Slow [Ca2+]i decay time constant (min) | 4.77 ± 0.96 (7)2-a | 8.78 ± 1.04 (4)† | 4.77 ± 0.96 (7)2-a | 9.07 ± 1.15 (4)‡ |
Data: mean ± SE; numbers of measurements in parentheses; significance tested by Student's paired t test;
p < 0.05;
F2-160: p < 0.01.
Presynaptic [Ca2+]i measurements were obtained from boutons in different preparations from controls. Multiple comparisons between groups were tested for significance by one-way ANOVA with Tukey's HSD test:
p< 0.05;
p < 0.01 versus control.
One explanation of these results is that XIP also blocks the plasma membrane Ca2+ ATPase at similar levels (2.5 μm) to those blocking Na+/Ca2+exchange (Enyedi and Penniston, 1993). Block of Ca2+ efflux by this route should increase [Ca2+]iaccumulation and EJP potentiation and retard post-tetanic Ca2+ removal and PTP decay, as we observed. To test this explanation, we used a different inhibitory peptide, C28R2, an agent that is somewhat more effective in blocking the plasmalemma Ca2+ ATPase (IC50 = 1 μm) (Enyedi and Penniston, 1993) than Na+/Ca2+exchange (IC50 = 6.2 μm) (Xu et al., 1997). Presynaptic injection of this inhibitor to ∼1–5 μm had effects identical to those of XIP (Fig.6B, Table 2), suggesting that both act mainly on the Ca2+ ATPase to increase [Ca2+]iaccumulation and EJP potentiation and retard post-tetanic Ca2+ removal and PTP decay.
Increasing [Na+]i with ouabain acts on plasmalemmal Na+/Ca2+exchange
The results of Figures 2-4 and Table 1 suggest that tetanic Na+ accumulation activates the Na+/Ca2+exchanger in reverse mode, enhancing tetanic Ca2+ accumulation and prolonging its post-tetanic decay. Previous results also implicated Na+/Ca2+exchange as a target for Na+ action in PTP, but they did not distinguish mitochondrial from plasmalemmal Na+/Ca2+exchange (Mulkey and Zucker, 1992). It was shown, for example, that elevation of presynaptic [Na+]i by block of the Na+/K+ATPase with ouabain (Delaney and Tank, 1994) led to an increase in [Ca2+]i and EJP amplitude. We have now used the mitochondrial Na+/Ca2+exchange inhibitor CGP 37157 and the plasmalemmal Na+/Ca2+exchange inhibitors KB R7943 and XIP to identify the target of Na+ action on [Ca2+]i when ouabain is used to elevate [Na+]i (Fig.7).
Fig. 7.
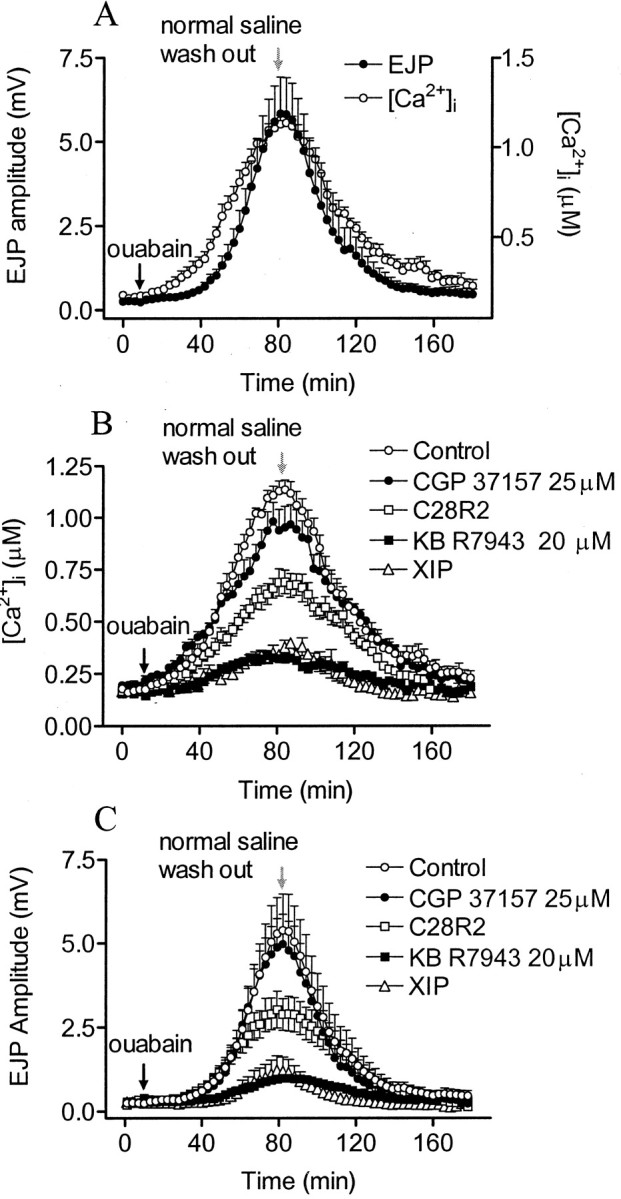
Effects of transport inhibitors on ouabain-induced [Ca2+]i accumulation and enhancement of synaptic transmission. A, Effects of 0.5 mm ouabain on presynaptic [Ca2+]i and EJP amplitude (n = 6). The preparations were stimulated at 2 Hz during the experiment, and [Ca2+]i was measured every 3 min. Arrows indicate ouabain application (black arrow) and subsequent washout (gray arrow). B, C, Response of [Ca2+]i and EJP amplitude to 25 μm CGP 37157 (●, n = 6), 20 μm KB R7943 (▪, n = 5), or after presynaptic injection of C28R2 (■, n = 5) or XIP (▵, n = 4), compared with controls (○, n = 6). Drugs blocked the [Ca2+]i elevation and EJP enhancement in proportion to their efficacy in blocking plasmalemmal Na+/Ca2+ exchange.
The Na+-dependent increases in [Ca2+]i and EJP amplitude were blocked almost completely by inhibitors of the plasma membrane exchanger but were affected very little by inhibition of mitochondrial Na+/Ca2+exchange. We also tested the effect of C28R2 on Na+-dependent [Ca2+]i elevation and EJP potentiation and found it to have a modest blocking effect (Fig. 7). C28R2 is most effective in blocking the plasmalemma Ca2+ ATPase, but it also inhibits plasmalemmal Na+/Ca2+exchange to some extent, as indicated above. In ouabain, with no tetanic stimulation, the only source for a rise in [Ca2+]i is by reverse mode Na+/Ca2+exchange, and its inhibition would prevent a rise in [Ca2+]i. Under these circumstances, with Ca2+accumulation reduced, inhibition of the Ca2+ ATPase should have little effect, and this accounts for our observation of reduced Ca2+ accumulation in ouabain in the presence of C28R2. This is the opposite of the effect of C28R2 on Ca2+ accumulation and EJP potentiation in a tetanus, where its major action is to reduce the Ca2+ ATPase-dependent extrusion of Ca2+ that has entered through voltage-dependent Ca2+ channels in the tetanus (Fig. 6).
DISCUSSION
Previous work (Tang and Zucker, 1997) showed that mitochondria accumulate Ca2+ during tetanic stimulation and release that Ca2+ back to the cytoplasm post-tetanically at crayfish neuromuscular junctions. That work also found no role for uptake or release of Ca2+ from endoplasmic reticulum in PTP. Assuming that CGP 37157 blocks mitochondrial Na+/Ca2+exchange in crayfish as effectively as in other preparations, the present study refines the role of mitochondrial Ca2+ transport by excluding Na+-dependent efflux of Ca2+ as a major route of Ca2+ flux in regulating the [Ca2+]iresponsible for PTP. Together the results suggest that Na+-independent modes of Ca2+ transport dominate in the tetanic uptake and subsequent post-tetanic release of Ca2+ by mitochondria. These modes include a Ca2+ uniporter and an apparently distinct rapid mode for uptake, and a Ca2+/H+exchanger and permeability transition pore for Ca2+ efflux (Gunter et al., 2000; Rizzuto et al., 2000). Our results do not distinguish among these processes.
Previous work (Mulkey and Zucker, 1992) also implicated [Na+]iaccumulation and activation of Na+/Ca2+exchange in elevating [Ca2+]iaccumulation in PTP and prolonging its removal. However, that work failed to distinguish mitochondrial from plasmalemmal Na+/Ca2+exchange. The present work identifies the plasma membrane Na+/Ca2+exchanger as the target of Na+action and shows that this exchanger operates in reverse mode to enhance tetanic [Ca2+]iaccumulation and EJP potentiation and to retard post-tetanic Ca2+ removal and the decay of PTP.
Mulkey and Zucker (1992) tested for operation of Na+/Ca2+exchange in reverse mode by tetanically stimulating in a Ca2+-free solution, then restoring external [Ca2+] and looking for a rise in [Ca2+]i. No such rise was observed, but it may be that by the time external Ca2+ reached normal levels, [Na+]i levels had already recovered sufficiently that Ca2+influx via Na+/Ca2+exchange was minimal.
Studies of Ca2+ regulation are limited by the imperfect selectivity of agents available to influence Ca2+ regulatory processes. Thus, in Figure2, KB R7943 could reduce both Ca2+ influx through voltage-dependent Ca2+ channels and the operation of the Na+/Ca2+exchanger in reverse mode. These actions were distinguished by administering this drug at the end of the tetanus, when most Ca2+ influx had already occurred normally (Fig. 3), leaving only effects attributable to block of Na+/Ca2+exchange. Similarly, XIP and C28R2 act on the plasmalemma ATPase, the dominant regulator of [Ca2+]i, to reduce its extrusion of Ca2+ ions during and after tetanic stimulation (Fig. 6). However, when ouabain is used to block Na+ extrusion without stimulation (Fig. 7), the rise in [Ca2+]i is more modest, and the Ca2+ ATPase is less strongly activated. Because the only source of a rise in [Ca2+]i under these circumstances is through reverse mode Na+/Ca2+exchange, it is not surprising that the main effect of XIP and C28R2 is in blocking this exchange, thus reducing [Ca2+]iaccumulation. All our findings are in accord with the reported relative effects of KB R7943, XIP, and C28R2 on Na+/Ca2+exchange, Ca2+ channels, and the Ca2+ ATPase, as indicated in detail in Results.
These results allow a qualitative, but not a quantitative, description of the regulation of [Ca2+]i in PTP at crayfish motor nerve terminals (Fig. 8). At rest (Fig. 8, 1), the plasma membrane Na+/Ca2+exchanger operates in the forward mode in concert with the Ca2+ ATPase to keep [Ca2+]i low, whereas the Na+/K+ ATPase maintains [Na+]ilevels. During high-frequency stimulation, presynaptic terminals load with Ca2+ and Na+ entering through voltage-dependent ion channels (Fig. 8, 2). As [Na+]i rises, plasmalemmal Na+/Ca2+exchange switches to reverse mode, and this becomes another source of Ca2+ entry. During this time the plasma membrane Ca2+ ATPase and mitochondrial uptake processes work to remove cytoplasmic Ca2+. Blocking mitochondrial fluxes with tetraphenyl phosphonium or carbonyl cyanidem-chlorophenyl-hydrazone (Tang and Zucker, 1997) or the plasmalemma Ca2+ ATPase with C28R2 or XIP leads to additional [Ca2+]iaccumulation and enhanced transmitter release, whereas blocking reverse mode Na+/Ca2+exchange with KB R7943 has the opposite effects.
Fig. 8.

Multiple systems regulating presynaptic [Ca2+]i in PTP. Schematic represents the response of a presynaptic terminal, during four phases (1–4) of generating PTP (top panel). In the middle panel, [Na+]i, [Ca2+]i, and Δv (ENa/Ca −Vm) are depicted byblack, gray, and dashed lines, respectively. See Discussion for details. The [Na+]i and [Ca2+]i traces were normalized to the same peak amplitude (respective scales are shown in Fig. 4).NCX, Na+/Ca2+exchange; PMCA, plasma membrane Ca2+ATPase.
In the first 15 min after a strong tetanus (Fig. 8, 3), the slow removal of cytoplasmic Na+ keeps the plasma membrane Na+/Ca2+exchanger operating in reverse and admitting Ca2+. The plasma membrane Ca2+ ATPase works to reduce [Ca2+]i, whereas Ca2+ exits mitochondria by Na+-independent processes. If mitochondria are prevented from loading with Ca2+, the ATPase can remove cytoplasmic Ca2+ within seconds, even in the presence of Ca2+influx through the plasmalemmal Na+/Ca2+exchanger, and so no PTP is expressed (Tang and Zucker, 1997). Blocking reverse mode plasmalemmal Na+/Ca2+exchange speeds Ca2+ removal and shortens PTP, whereas blocking Ca2+ removal by the plasma membrane Ca2+ ATPase has the opposite effects. Finally, when PTP has fully decayed (Fig. 8,4) and [Ca2+]i is restored to resting levels, [Na+]i has recovered sufficiently that plasmalemmal Na+/Ca2+exchange reverts to normal mode and works with the plasma membrane Ca2+ ATPase to maintain [Ca2+]i at low levels. At some still undetermined point in period 3 or4, mitochondria are relieved of their excess Ca2+ and return to their resting state. The time course and magnitude of PTP depend, therefore, on an interplay between Na+-independent mitochondrial Ca2+ fluxes, the plasmalemmal Na+/Ca2+exchanger operating in both normal and reverse modes, the plasma membrane Ca2+ ATPase, and the plasmalemma Na+/K+pump.
Footnotes
The work was supported by National Institutes of Health Grant NS 15114. We thank Russell English for technical assistance.
Correspondence should be addressed to Robert S. Zucker, Department of Molecular and Cell Biology, University of California, Berkeley, CA, 94720-3200. E-mail:zucker@socrates.berkeley.edu.
REFERENCES
- 1.Alnaes E, Rahamimoff R. On the role of mitochondria in transmitter release from motor nerve terminals. J Physiol (Lond) 1975;248:285–306. doi: 10.1113/jphysiol.1975.sp010974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxter DA, Bittner GD. Intracellular recordings from crustacean motor axons during presynaptic inhibition. Brain Res. 1981;223:422–428. doi: 10.1016/0006-8993(81)91159-8. [DOI] [PubMed] [Google Scholar]
- 3.Baxter DA, Bittner GD. Synaptic plasticity at crayfish neuromuscular junctions: presynaptic inhibition. Synapse. 1991;7:244–251. doi: 10.1002/syn.890070309. [DOI] [PubMed] [Google Scholar]
- 4.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 5.Brodin L, Bakeeva L, Shupliakov O. Presynaptic mitochondria and the temporal pattern of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:365–372. doi: 10.1098/rstb.1999.0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox DA, Conforti L, Sperelakis N, Matlib MA. Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- 7.David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol (Lond) 1998;509:59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney KR, Tank DW. Calcium-dependent and calcium-independent enhancement of transmitter release at the crayfish neuromuscular junction studied with fura-2 imaging. Ann NY Acad Sci. 1991;635:452–454. doi: 10.1111/j.1749-6632.1991.tb36525.x. [DOI] [PubMed] [Google Scholar]
- 9.Delaney KR, Tank DW. A quantitative measurement of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J Neurosci. 1994;14:5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delaney KR, Zucker RS, Tank DW. Calcium in motor nerve terminals associated with post-tetanic potentiation. J Neurosci. 1989;9:3558–3567. doi: 10.1523/JNEUROSCI.09-10-03558.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enyedi A, Penniston JT. Autoinhibitory domains of various Ca2+ transporters cross-react. J Biol Chem. 1993;268:17120–17125. [PubMed] [Google Scholar]
- 12.Fossier P, Baux G, Tauc L. Presynaptic mechanisms regulating Ca2+ concentration triggering acetylcholine release at an identified neuro-neuronal synapse of Aplysia. Neuroscience. 1994;63:405–414. doi: 10.1016/0306-4522(94)90538-x. [DOI] [PubMed] [Google Scholar]
- 13.Friel DD. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium. 2000;28:307–316. doi: 10.1054/ceca.2000.0172. [DOI] [PubMed] [Google Scholar]
- 14.Garcia ML, Strehler EE. Plasma membrane calcium ATPases as critical regulators of calcium homeostasis during neuronal cell function. Front Biosci. 1999;4:D869–882. doi: 10.2741/garcia. [DOI] [PubMed] [Google Scholar]
- 15.Gleason E, Borges S, Wilson M. Control of transmitter release from retinal amacrine cells by Ca2+ influx and efflux. Neuron. 1994;13:1109–1117. doi: 10.1016/0896-6273(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 16.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 17.Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–296. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- 18.Habermann E. Palytoxin acts through Na+,K+-ATPase. Toxicon. 1989;27:1171–1187. doi: 10.1016/0041-0101(89)90026-3. [DOI] [PubMed] [Google Scholar]
- 19.Harootunian AT, Kao JP, Eckert BK, Tsien RY. Fluorescence ratio imaging of cytosolic free Na+ in individual fibroblasts and lymphocytes. J Biol Chem. 1989;264:19458–19467. [PubMed] [Google Scholar]
- 20.Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- 21.Juhaszova M, Church P, Blaustein MP, Stanley EF. Location of calcium transporters at presynaptic terminals. Eur J Neurosci. 2000;12:839–846. doi: 10.1046/j.1460-9568.2000.00974.x. [DOI] [PubMed] [Google Scholar]
- 22.Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi K, Tachibana M. Ca2+ regulation in the presynaptic terminals of goldfish retinal bipolar cells. J Physiol (Lond) 1995;483:79–94. doi: 10.1113/jphysiol.1995.sp020569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landò L, Zucker RS. Ca2+ cooperativity in neurosecretion measured using photolabile Ca2+ chelators. J Neurophysiol. 1994;72:825–830. doi: 10.1152/jn.1994.72.2.825. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Nicoll DA, Collins A, Hilgemann DW, Filoteo AG, Penniston JT, Weiss JN, Tomich JM, Philipson KD. Identification of a peptide inhibitor of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1991;266:1014–1020. [PubMed] [Google Scholar]
- 26.Luther PW, Yip RK, Bloch RJ, Ambesi A, Lindenmayer GE, Blaustein MP. Presynaptic localization of sodium/calcium exchangers in neuromuscular preparations. J Neurosci. 1992;12:4898–4904. doi: 10.1523/JNEUROSCI.12-12-04898.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgans CW, El Far O, Berntson A, Wassle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci. 1998;18:2467–2474. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulkey RM, Zucker RS. Post-tetanic potentiation at the crayfish neuromuscular junction is dependent on both intracellular calcium and sodium ion accumulation. J Neurosci. 1992;12:4327–4336. doi: 10.1523/JNEUROSCI.12-11-04327.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng Y. Ryanodine-sensitive component of calcium transients evoked by nerve firing at presynaptic nerve terminals. J Neurosci. 1996;16:6703–6712. doi: 10.1523/JNEUROSCI.16-21-06703.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng YY. Effects of mitochondrion on calcium transients at intact presynaptic terminals depend on frequency of nerve firing. J Neurophysiol. 1998;80:186–195. doi: 10.1152/jn.1998.80.1.186. [DOI] [PubMed] [Google Scholar]
- 31.Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- 32.Regehr WG. Interplay between sodium and calcium dynamics in granule cell presynaptic terminals. Biophys J. 1997;73:2476–2488. doi: 10.1016/S0006-3495(97)78276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol (Lond) 2000;529:37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scotti AL, Chatton JY, Reuter H. Roles of Na+-Ca2+ exchange and of mitochondria in the regulation of presynaptic Ca2+ and spontaneous glutamate release. Philos Trans R Soc Lond B Biol Sci. 1999;354:357–364. doi: 10.1098/rstb.1999.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simpson PB, Challiss RA, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends Neurosci. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 36.Smith AB, Cunnane TC. Ryanodine-sensitive calcium stores involved in neurotransmitter release from sympathetic nerve terminals of the guinea-pig. J Physiol (Lond) 1996;497:657–664. doi: 10.1113/jphysiol.1996.sp021797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 38.Tucker T, Fettiplace R. Confocal imaging of calcium microdomains and calcium extrusion in turtle hair cells. Neuron. 1995;15:1323–1335. doi: 10.1016/0896-6273(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 39.Watano T, Kimura J, Morita T, Nakanishi H. A novel antagonist, No. 7943, of the Na+/Ca2+ exchange current in guinea-pig cardiac ventricular cells. Br J Pharmacol. 1996;119:555–563. doi: 10.1111/j.1476-5381.1996.tb15708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wojtowicz JM, Atwood HL. Correlation of presynaptic and postsynaptic events during establishment of long-term facilitation at crayfish neuromuscular junction. J Neurophysiol. 1985;54:220–230. doi: 10.1152/jn.1985.54.2.220. [DOI] [PubMed] [Google Scholar]
- 41.Wojtowicz JM, Atwood HL. Long-term facilitation alters transmitter releasing properties at the crayfish neuromuscular junction. J Neurophysiol. 1986;55:484–498. doi: 10.1152/jn.1986.55.3.484. [DOI] [PubMed] [Google Scholar]
- 42.Xu W, Denison H, Hale CC, Gatto C, Milanick MA. Identification of critical positive charges in XIP, the Na/Ca exchange inhibitory peptide. Arch Biochem Biophys. 1997;341:273–279. doi: 10.1006/abbi.1997.9954. [DOI] [PubMed] [Google Scholar]
- 43.Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron. 2000;25:229–237. doi: 10.1016/s0896-6273(00)80885-5. [DOI] [PubMed] [Google Scholar]
- 44.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]