

Abstract
Large-conductance Ca2+-activated K+ channels (BK, also called Maxi-K orSlo channels) are widespread in the vertebrate nervous system, but their functional roles in synaptic transmission in the mammalian brain are largely unknown. By combining electrophysiology and immunogold cytochemistry, we demonstrate the existence of functional BK channels in presynaptic terminals in the hippocampus and compare their functional roles in somata and terminals of CA3 pyramidal cells. Double-labeling immunogold analysis with BK channel and glutamate receptor antibodies indicated that BK channels are targeted to the presynaptic membrane facing the synaptic cleft in terminals of Schaffer collaterals in stratum radiatum. Whole-cell, intracellular, and field-potential recordings from CA1 pyramidal cells showed that the presynaptic BK channels are activated by calcium influx and can contribute to repolarization of the presynaptic action potential (AP) and negative feedback control of Ca2+ influx and transmitter release. This was observed in the presence of 4-aminopyridine (4-AP, 40–100 μm), which broadened the presynaptic compound action potential. In contrast, the presynaptic BK channels did not contribute significantly to regulation of action potentials or transmitter release under basal experimental conditions, i.e., without 4-AP, even at high stimulation frequencies. This is unlike the situation in the parent cell bodies (CA3 pyramidal cells), where BK channels contribute strongly to action potential repolarization. These results indicate that the functional role of BK channels depends on their subcellular localization.
Keywords: calcium-activated potassium channels, BK channels, Slo, Maxi-K, presynaptic mechanisms, hippocampus, CA1, CA3, action potential repolarization, glutamatergic synapses, immunogold cytochemistry, BK-β4, KCNMB4
Large-conductance Ca2+-activated K+ channels (BK, also called Maxi-K orSlo channels) are found in neurons throughout the vertebrate nervous system (Hille, 1992; Knaus et al., 1996), but their functional roles in the brain are largely unknown (Storm, 1990; Sah, 1996; Vergara et al., 1998). In particular, it is not known whether functional BK channels exist in presynaptic terminals and whether they contribute to presynaptic action potential (AP) repolarization and control of transmitter release in the CNS. It seems plausible that they may do so, because BK channels are known to repolarize action potentials in certain peripheral nerve terminals (Robitaille et al., 1993; Blundon et al., 1995) and in some central neuronal somata (Storm, 1990; Sah, 1996;Shao et al., 1999), and to control secretion in gland cells (Petersen and Maruyama, 1984; Lingle et al., 1996).
Being both voltage and Ca2+ sensitive, BK channels seem well suited for negative feedback regulation of the Ca2+ influx and, hence, of transmitter release (Storm, 1987a; Robitaille et al., 1993). If present in presynaptic terminals, BK channels could activate during the action potential and accelerate its repolarization, as in neuronal somata (Adams et al., 1982; Storm, 1987a,b; Takahashi, 1990; Vergara et al., 1998; Shao et al., 1999). Thus, presynaptic BK channels would curtail the opening of voltage-gated Ca2+ channels during the spike, thereby reducing Ca2+influx and transmitter secretion. Such a mechanism may regulate transmission during variations in the intra-terminal calcium concentration ([Ca2+]i) or membrane potential, and could provide an “emergency brake” under conditions that cause excessive depolarization and Ca2+ accumulation in the terminals, e.g., brain ischemia or epilepsy.
Light microscopic (LM) data indicate that BK channels are widely expressed in the rat brain, with high levels in the cerebral cortex and hippocampus (Knaus et al., 1996; Wanner et al., 1999). By combining data from immunocytochemistry, in situ hybridization, and radioligand binding, it was inferred that many BK channels are probably targeted to axons and nerve terminals (Knaus et al., 1996; Wanner et al., 1999). However, with the limited resolution of LM it was not possible to determine whether BK channels are located in the presynaptic or postsynaptic membrane, nor was it possible to determine their distribution with respect to the synaptic cleft and release sites.
The goal of the present study was to test whether functional BK channels exist in presynaptic terminals and regulate transmitter release in brain synapses. We chose to study glutamatergic spine synapses in stratum radiatum of the CA1 area for the following reasons: (1) these synapses are rather typical cortical excitatory synapses; (2) the CA1 stratum radiatum contains a high density of BK channels [LM data: Knaus et al.(1996), Wanner et al. (1999)]; (3) BK channels repolarize spikes in CA1 pyramidal somata (Lancaster and Nicoll, 1987;Storm, 1987a,b, 1990; Shao et al., 1999); and (4) the stratified structure of the CA1 area facilitates recording of presynaptic and postsynaptic responses and identification of synapses by electron microscopy.
Our data indicate that functional BK channels exist in the presynaptic membrane facing the synaptic cleft and can regulate transmitter release. Furthermore, the functional role of BK channels differs between somata and terminals, indicating that their function depends on their subcellular localization.
Some of these results have been published previously in abstract form (Shao and Storm, 1997; Storm et al., 2001).
MATERIALS AND METHODS
Subjects. Male Wistar rats (Möllegaard, Ejby, Denmark) were deeply anesthetized with halothane before decapitation or fixation. Adult rats (6–10 weeks old) were used for morphological studies; 4- to 6-week-old rats were used for electrophysiology experiments. The care and use of animals were in accordance with institutional guidelines.
Antibodies for immunogold analysis. An antibody directed against residues 913–926 of the BK channel α-subunit was affinity purified as described previously (Knaus et al., 1996). The NMDA receptor antibodies (recognizing the C-terminal amino acids of NMDAR1 and NMDAR2A/B, respectively) were generously supplied by Dr. R. J. Wenthold (NIH, Bethesda, MD). All antibodies have been extensively characterized (Petralia et al., 1994a,b; Knaus et al., 1996)
Tissue preparation for electron microscopy. Five adult male Wistar rats (250–300 gm; Möllegaard) were deeply anesthetized by intraperitoneal injection of midazolam, fentanyl citrate, and fluanisone (3.8, 0.24, and 7.5 mg/kg body weight, respectively), before transcardial perfusion with 4% formaldehyde and 0.1 or 0.5% glutaraldehyde at pH 7.4 (Matsubara et al., 1996). Tissue blocks from the CA1 region of the hippocampus were freeze-substituted and embedded at low temperature in Lowicryl HM20 (Takumi et al., 1999).
Postembedding immunocytochemistry. Ultrathin sections (70–90 nm) were mounted on uncoated or Formvar-coated mesh grids and processed for immunogold cytochemistry (Matsubara et al., 1996). The sections were first immersed in a saturated solution of NaOH in absolute ethanol for 2–3 sec and then incubated in the following solutions (at room temperature): (1) 50 mmglycine in Tris buffer (5 mm) or phosphate buffer (20 mm), containing 50 mmNaCl and 0.1% Triton X-100 (T/PNT; 10 min), (2) 4% fish skin gelatin and 1% human serum albumin in T/PNT (blocking solution, 10 min), (3) affinity-purified anti-α(913–926) (Knaus et al., 1996) diluted 1:600 in the blocking solution overnight, (4) blocking solution (10 min), and (5) goat anti-rabbit Fab fragments coupled to 10 nm gold particles (GFAR10; British BioCell International, Cardiff, UK) diluted 1:30 in the blocking solution (2 hr). The sections were then rinsed in double-distilled water, dried, and subjected to formaldehyde vapor at 80°C for 1 hr (Ottersen et al., 1992). The sections were then subjected to a second immunogold incubation, using the same steps as above but with different primary antibodies (mixture of anti-NMDAR1 and anti-NMDAR2A/B, final concentrations 2.3 and 5.0 μg/ml, respectively) and gold reagents (15 nm goat anti-rabbit IgG diluted 1:20). The sections were counterstained and examined in a Philips CM 10 transmission electron microscope. Preabsorption of the antibodies with excess immunizing peptide removed all labeling.
Sampling and analysis of the EM data. Electron micrographs were taken from stratum radiatum of CA1b of the dorsal hippocampus (approximate anteroposterior level −3.0 mm) (Paxinos and Watson, 1998). Because symmetric synapses were virtually devoid of labeling, the sampling was restricted to asymmetric synapses with spines or dendritic stems. Only synapses with a distinct and well delimited postsynaptic density were included in the quantitative analysis. The distance between the gold particles and the outer margin of the presynaptic or postsynaptic membrane was measured in micrographs with a final magnification of ∼100,000× (verified by use of a calibration grid). Histograms and curves were produced by commercial software (SPSS by SPSS Inc.; bin width 8 nm).
Slice preparation for electrophysiology. Young male rats (4–6 weeks old) were deeply anesthetized with halothane before decapitation. Transverse hippocampal slices (400 μm thick) were prepared with a vibratome and maintained in an interface chamber filled with artificial CSF (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 1.25 KCl, 1.25 KH2PO4, 1.5 MgCl2, 1.0 CaCl2, 16 glucose, and saturated with 95% O2/5% CO2.
Recording and stimulation conditions. During the recordings, the slices were kept submerged in a chamber perfused with ACSF of the composition described above, except that the CaCl2 concentration was 2 mm. The ACSF was saturated with 95% O2/5% CO2 and heated to 34–36°C (whole-cell recordings: see Figs. 3, 4, 6, 9D) or 29–31°C (extracellular and sharp electrode intracellular recordings: see Figs. 5, 7, 9A,B, 10). Some experiments were also performed at room temperature (20–24°C). There was <1°C change during each recording. Bicuculline free base (10 μm) anddl-2-amino-5-phosphonopentanoic acid (dl-AP5; 100–200 μm) were routinely added to the medium to block inhibitory synaptic transmission and to prevent long-term potentiation (LTP) or long-term depression (LTD) mediated by NMDA-type glutamate receptors. Excitatory fibers were activated by electrical stimulation with a monopolar electrode (glass pipette filled with saline, or sharpened tungsten) placed in the middle of stratum radiatum of the CA1 area ∼100 μm from the site of recording (stimulation intensity: 100 μsec, 50–200 μA). In most experiments, trains of stimuli (two to five stimuli at 10–100 Hz) were delivered once every 30–60 sec.
Fig. 3.
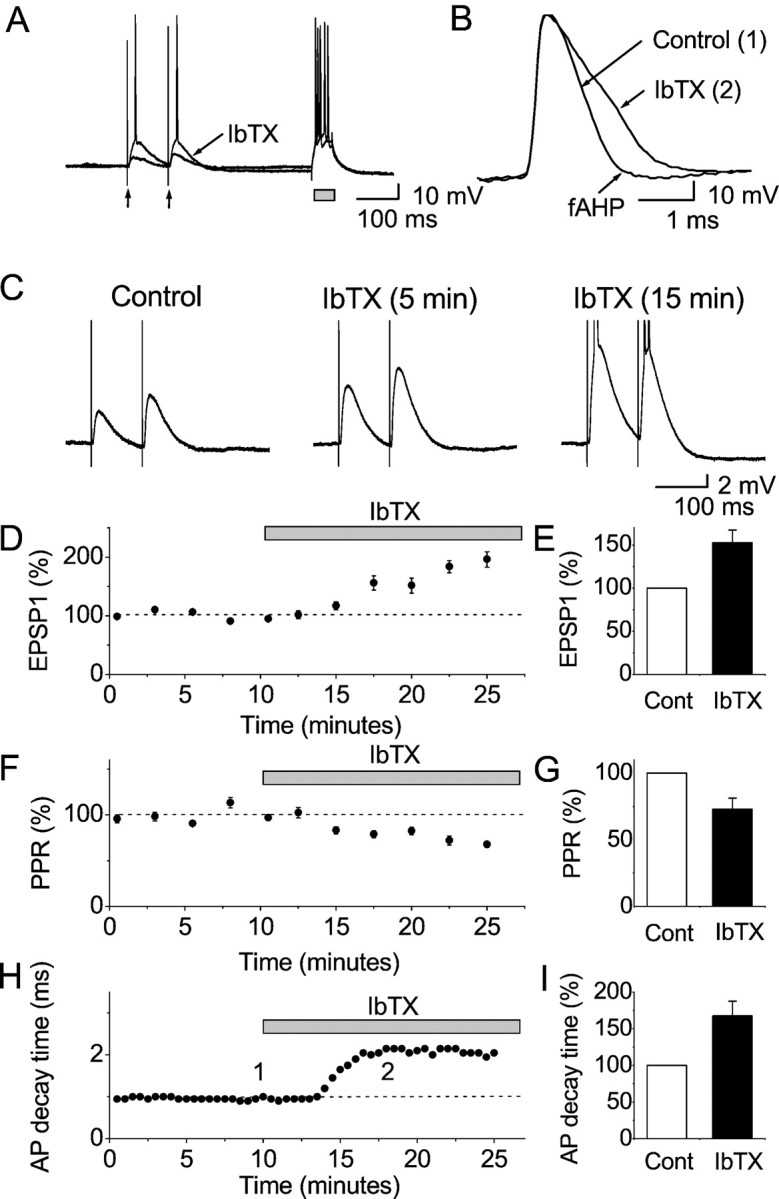
Effects of BK channel blockade by iberiotoxin (IbTX) on synaptic transmission and action potentials in CA1 pyramidal cells, after K+ channel blockade by 4-AP; whole-cell recording. A, EPSPs in response to activation of presynaptic fibers in stratum radiatum by a pair of electrical stimuli (arrows), recorded in the continuous presence of 100 μm 4-AP. The resting membrane potential was adjusted to −70 mV by steady current injection, and action potential trains were evoked by injecting a 50-msec-long depolarizing current pulse (gray box). The stimuli were repeated once every 30 sec. Representative single traces before and after bath application of 100 nm IbTX are shown superimposed. B, Representative records of the first action potential in trains evoked by depolarizing current pulses, as shown in A, before and after application of 100 nm IbTX, shown superimposed. Note the characteristic spike broadening by IbTX. C, Typical EPSPs from a cell before and after (5 and 15 min) application of 100 nmIbTX (average of 5 consecutive records). IbTX caused a progressive increase in the EPSP amplitudes, until action potentials were evoked.D, Summary time course of the EPSP amplitude (first EPSP in the pair, normalized to the amplitude at the beginning of the control period) in response to 100 nm IbTX for all whole-cell recordings in 4-AP (n = 5 cells). Stimulation was repeated once every 30 sec, and the average responses for 2.5 min periods are plotted. When EPSPs triggered spikes (in IbTX), the maximal subthreshold EPSP amplitude was used. E, Summary data (mean ± SEM) of the EPSP amplitude (first EPSP in the pair, normalized to the control period) in response to 100 nm IbTX, showing a significant difference (p = 0.011; n = 5 cells). F, Summary time course of the paired-pulse ratio (PPR) (second EPSP/first EPSP, normalized to the ratio during the control period) in response to 100 nm IbTX for all whole-cell recordings (n = 5 cells). Stimulation was repeated once every 30 sec, and the response for each 2.5 min period is plotted. G, Summary diagram of the effect of IbTX on the PPR, showing a significant difference (p = 0.025; n = 5 cells). H, Time course of the spike broadening caused by IbTX, expressed as the action potential 90–10% decay time, for the cell illustrated inA–C. Values are plotted for the first action potential in each spike train, evoked every 30 sec by a depolarizing current pulse (A, B).I, Summary data of the action potential decay time (first spike in the train, normalized to the control period) in response to 100 nm IbTX, showing a significant difference (p = 0.017; n = 5 cells). All diagrams (D–I) show mean ± SEM normalized to the control period.
Fig. 4.

Effects of IbTX on synaptic transmission after postsynaptic BK channel suppression by BAPTA, in the continuous presence of 100 μm 4-AP. A, Comparison between typical somatic action potentials (evoked by depolarizing current injection during whole-cell recording, as in Fig.3A) in two CA1 pyramidal cells, one recorded without BAPTA and the other with BAPTA in the patch pipette. Note the characteristic spike broadening and block of the fast AHP (fAHP) caused by BAPTA. B, Typical EPSPs from a cell before and after application of 100 nmIbTX (average of 5 consecutive EPSP records). IbTX increased the EPSP amplitudes. In these experiments, EPSP-evoked action potentials were prevented by adjusting the membrane potential to −80 mV with steady current and using moderate stimulus intensities. Thereby the full effect on the EPSPs could be measured in the absence of spikes.C, Summary time course of the EPSP amplitude (first EPSP in the pair) in response to 100 nm IbTX for all whole-cell recordings with BAPTA (n = 6 cells). Simulation was repeated once every 30 sec, and each response is plotted. The decline after 27 min may be caused by vesicle depletion. D, Summary data for the EPSP amplitude (first EPSP in the pair) in response to 100 nm IbTX, showing a highly significant difference (p = 0.0096;n = 6 cells). E, Summary time course of the paired-pulse ratio (PPR), normalized to the control value, in response to 100 nm IbTX for all whole-cell recordings with BAPTA (n = 6).F, Summary data for effect of 100 nm IbTX on the PPR for all cells recorded with BAPTA, showing a significant difference (p = 0.013; n= 6). All diagrams (D–F) show mean ± SEM, normalized to the value during the control period.
Fig. 6.

In the absence of 4-AP, IbTX broadened the action potentials but had no apparent effect on synaptic transmission and in CA1 pyramidal cells; whole-cell recording. A, Representative records of the first action potentials evoked by current pulses (Fig. 3A), before and after application of 100 nm IbTX. B, EPSPs in response to paired stimulation of presynaptic fibers in stratum radiatum. The resting membrane potential was adjusted to −70 mV by steady current injection, and action potentials were evoked by injecting a 100-msec-long depolarizing current pulse once every 30 sec (as in Fig. 3). Representative single traces before and after bath application of 100 nm IbTX are shown. C–D, Summary data showing the effects of 100 nm IbTX on the EPSP amplitude (first EPSP in the pair, normalized; n = 8 cells). Unlike in Figure 3, the EPSPs never triggered action potentials. There was no significant effect of IbTX on the EPSP amplitude (p = 0.36; n = 8). E, F, Summary data showing the effects of 100 nm IbTX on the paired-pulse ratio (PPR, normalized; n = 8 cells). There was no significant effect of IbTX on the PPR (p = 0.079; n = 8).G, Time course of the action potential broadening caused by 100 nm IbTX (90–10% decay time) for the cell illustrated in A and B. Values for first action potential in each spike train are plotted (Fig.3A). H, Summary diagram showing the effect of 100 nm IbTX on the action potential decay time (first spike in the train; n = 8 cells). The effect of IbTX on the action potential decay time was highly significant (p = 0.000031; n = 8). All diagrams (C–H) show mean ± SEM, normalized to the control period.
Fig. 9.

Effects of BK channel blockade by paxilline in hippocampal slices. A, Intracellular recording (sharp electrode) from a CA1 pyramidal cell, showing the effects of cumulative application of the BK channel blockers 100 μm IbTX, 5 μm paxilline, and 1 mm TEA on action potentials (A1) and on low-frequency (A2, 20 Hz) and high-frequency (A3, 100 Hz) trains of EPSPs. The records are shown superimposed in B andC. IbTX or paxilline had no apparent effect on the EPSPs at either frequency, although they broadened the action potential. In contrast, TEA both enhanced the EPSPs and caused further broadening of the action potential. B, Same action potentials as inA1, shown superimposed on a fast time scale.C, Same EPSPs as in A2 andA3, superimposed on a fast time scale. D, Whole-cell patch-clamp recording from a CA1 pyramidal cell. Application of 10 μm paxilline broadened the action potential (D1) but had no apparent effect on high-frequency (100 Hz) trains of EPSPs (D2). E, Summary data for five experiments in which paxilline was applied to CA1 pyramidal cells. The areas for the EPSP trains under control conditions and after application of paxilline were compared (normalized; control = 100%). No difference was found (p = 0.79;n = 5). All experiments appeared negative for each of the EPSPs in the train, regardless of the recording method (whole cell, 3 cells; intracellular, 2 cells), paxilline concentration (2, 5, or 10 μm), or stimulation frequency (20, 25, 50, or 100 Hz). As in previous figures, the action potentials were evoked by current injection (only the first spike in a train was measured), and the EPSPs were evoked by a stimulation electrode placed in stratum radiatum.
Fig. 5.
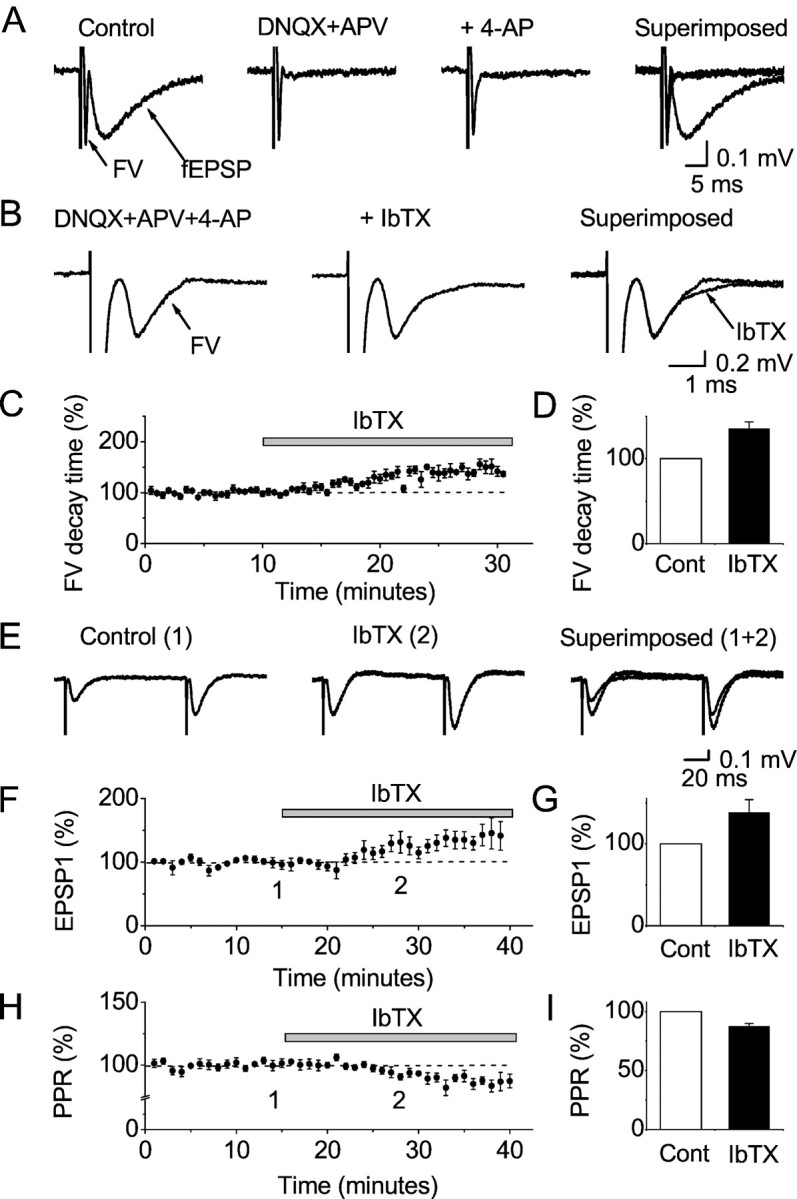
Extracellular field potential recordings of the effects of IbTX on the presynaptic compound action potential (fiber volley) and fEPSPs in CA1 stratum radiatum. A, Fiber volley and fEPSP in response to stimulation of presynaptic fibers in stratum radiatum. The glutamate receptor blockers DNQX (20 μm) and dl-AP5 (100 μm) blocked the fEPSP, thus isolating the fiber volley (FV). Application of 100 μm 4-AP broadened the fiber volley. B, Fiber volley recorded after DNQX, dl-AP5, and 4-AP (40 μm) application, as in A. Note the expanded time scale. Application of 100 nm IbTX broadened the volley further.C, Time course of the effect of IbTX on the fiber volley in the presence of DNQX, dl-AP5, and 4-AP (40 μm) (n = 5). The FV 90–10% decay time is plotted. D, Summary of the effect of IbTX on the fiber volley. As in C, the 90–10% decay time is plotted (mean ± SEM; n = 5 slices), showing a highly significant difference (p = 0.0041;n = 5). E, The effect of 100 nm IbTX of the fEPSP, recorded in 100 μm 4-AP and 100 μmdl-AP5, but without DNQX.F–I, Time courses (F,H) and summary diagram (G,I) of the effect of IbTX on the fEPSP (F, G) and PPR (H,I) in the presence of 100 μm 4-AP, but no DNQX. Mean ± SEM for five slices. The effects of IbTX on both the EPSP amplitude (G) and the PPR (I) were significant (p = 0.046 and p = 0.0078, respectively; n = 5).
Fig. 7.

Effects of IbTX on high-frequency trains of fEPSPs in CA1 stratum radiatum. Extracellular field potential recordings. A, Bath application of 100 nmIbTX failed to enhance a high-frequency (100 Hz) train of five fEPSPs (in the absence of 4-AP). Subsequent application of 20 μmDNQX suppressed the EPSPs, indicating that they were mediated mainly by non-NMDA-type glutamate receptors. B, Time course of the amplitude of the fifth EPSP in the train for five slices. Because the presynaptic Ca2+ accumulation is expected to be maximal during the last (5th) fEPSP in the train, this was chosen for plotting. However, IbTX had no apparent effect on any of the five fEPSPs in the train (1st–5th).C, The effect of 100 nm IbTX on the first to the fifth EPSP in the 100 Hz train. Summary data from five experiments (mean ± SEM) normalized to the control period for each of the five fEPSPs in the train; i.e., the amplitude of the first fEPSP in the train after IbTX application is expressed in percentage of the amplitude of the first fEPSP during the control period; the second fEPSP in IbTX is given in percentage of the second fEPSP, and so on. IbTX did not cause any significant increase of any of the five EPSPs in the train. Actually, the third and fourth EPSPs in the train were reduced in amplitude after IbTX application (p = 0.027 and 0.0022, respectively), probably because of rundown (p values for the 1st, 2nd, and 5th EPSP = 0.33, 0.060, and 0.073, respectively;n = 5 slices).
Fig. 10.

Different action potential repolarization mechanisms in soma and presynaptic axon terminals of CA3 pyramidal cells. A, Action potentials recorded intracellularly from the soma of a CA3 pyramidal cell, before and during bath application of IbTX (60 nm). The toxin slowed the repolarization and blocked the fast afterhyperpolarization (fAHP), indicating that BK channels dominate during most of the spike repolarization. B, Compound action potential recorded extracellularly from presynaptic axon terminals [i.e., fiber volley (FV) in response to stimulation of axons in stratum radiatum of CA1]. In contrast to the soma, the FV was not significantly affected by bath application of IbTX (60–100 nm). Summary data for all recordings are shown in C (n = 4;p = 0.0023) and D(n = 5; p = 0.17). Because the CA3 pyramidal cells give rise the majority of the CA1 stratum radiatum axons studied here, the contrasting results imply that two subcellular compartments within the CA3 pyramid cells use different spike repolarization mechanisms.
Extracellular field potential recording. Extracellular field potentials were recorded with a glass micropipette (filled with extracellular medium) that was placed in the middle of stratum radiatum of CA1. The electrical stimulation elicited compound action potentials from the presynaptic axons (fiber volley) followed by field EPSPs (fEPSPs). In experiments designed for measuring only the fiber volley, 20–100 μm DNQX (in addition todl-AP5 and bicuculline, see above) was added to the ACSF to eliminate the fEPSPs, which might otherwise contaminate the fiber volley and mediate epileptiform activity after K+ channel blockade by 4-AP. In a few experiments, the concentration of CaCl2 was raised to 4.0 mm (see Results). Only experiments with stable fEPSP and fiber volley responses before drug application were included in the analysis.
Whole-cell and intracellular recording. Whole-cell gigaseal recordings were obtained from CA1 pyramidal cells using the “blind” method. The patch pipettes were filled with a solution containing (in mm): 140 KMeSO4, 10 HEPES, 10 phosphocreatine Na salt, 2 ATP Na salt, 0.4 GTP Na salt, and 2 MgCl2, resulting in a pipette resistance of 4–7 MΩ. In some experiments, 10 mm BAPTA and 0–4.87 mm CaCl2 were added into the pipette solution. The cells were recorded with an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA) in the current-clamp bridge mode. The series resistance was 10–26 MΩ, and all potentials were corrected for the junction potential (−10 mV). Sharp microelectrodes were filled with 2 m potassium acetate (resistance 60–100 MΩ, pH 7.25). Intracellular recordings from somata of CA1 pyramidal cells were performed with an Axoclamp 2A amplifier (Axon Instruments) in current-clamp bridge mode (3 kHz low-pass filter). EPSPs were evoked by stimulating stratum radiatum of the CA1 area as described above. In addition, action potentials were elicited by a 50 msec depolarizing current injection (0.1–1 nA) once every 30 sec. Only cells with a stable resting membrane potential more negative than −60 mV and stable EPSP and action potential amplitudes were used for recording. To keep the conditions constant during the measurements, the cell membrane potential was manually clamped at a fixed potential near the resting potential for each cell (−65 to −70 mV).
Data acquisition, storage, and analysis. The data were acquired using pCLAMP 7.0 (Axon Instruments) at a sampling rate of 10 kHz and also digitized and stored on videotapes (Instrutec VR-10) and measured and plotted using pCLAMP 7.0 and Origin 5.0 (Microcal). Values are expressed as mean ± SEM. Two-tailed paired Student'st test was used for statistical analysis (α = 0.05). The p values are given in the figure legends.
Pharmacology of BK channels expressed in Chinese hamster ovary cells. Chinese hamster ovary (CHO) cells were transfected with cDNAs coding for hSloα and hSloβ4 subunits and green fluorescent protein (GFP) as described by Behrens et al. (2000). The cDNA ratios weresloα/hSloβ4/GFP = 2:5:3. This corresponds to a 7.5 molar hSloβ4 cDNA excess over hSloα, ensuring that hSloβ4-containing heteromultimers were the dominant channel type expressed. This was also confirmed by the kinetics of the recorded current, which was characteristic for α/β4 (Behrens et al., 2000). Current measurements at +80 mV were performed in the inside-out configuration of the patch-clamp technique, 12–24 hr after transfection, as described (Behrens et al., 2000). Ca2+concentration was adjusted to 11.0 μm. The concentrations were measured with ratiometric Ca2+ indicators Fura-2 and BTC (Molecular Probes, Leiden, The Netherlands). From a holding potential of 0 mV, a 100 msec hyperpolarizing prepulse to −140 mV was given before application of a 500 msec test pulse to +80 mV.
Chemicals and drugs. Iberiotoxin (IbTX) was produced by recombinant means in an Escherichia coli expression system and purified as described previously (Knaus et al., 1996). Some IbTX was also purchased from Peptide Institute (Tokyo, Japan). Paxilline was obtained from Sigma-Aldrich Norway AS (Oslo, Norway) and Alomone (Jerusalem, Israel). The remaining drugs were from Sigma-Aldrich Norway AS. Substances were bath applied by adding them to the superfusing medium. When applying IbTX, bovine serum albumin was also added to reduce peptide adhesion to the tubes and walls of the perfusion system. Paxilline was dissolved in methanol or DMSO to obtain 10 mm stock solutions that were diluted in the extracellular medium to a final concentration of methanol or DMSO <1:1000. Paxilline solutions were protected from light during preparation and experiments.
RESULTS
Immunogold analysis of BK channel distribution in hippocampal excitatory synapses
Pilot immunogold labeling experiments indicated that the BK channels were concentrated at synapses, mostly at asymmetric synapses with spines or dendritic stems (Fig. 1). Symmetric synapses at dendrites or pyramidal cell somata were rarely labeled. These initial observations raised two questions. (1) Are BK channels associated with glutamatergic synapses? (2) If so, are the BK channels expressed at presynaptic or postsynaptic membranes, or both? To resolve these issues we ran double-labeling experiments using small particles (10 nm) to identify BK channels and larger ones (15 nm) to identify NMDA receptors. Previous evidence indicates that NMDA receptors (but not AMPA receptors) are present in all asymmetric synapses on spines in the stratum radiatum and that the presynaptic elements in these synapses are enriched with glutamate (Takumi et al., 1999). Thus, the NMDA receptor should serve as a good marker of putative glutamatergic synapses.
Fig. 1.
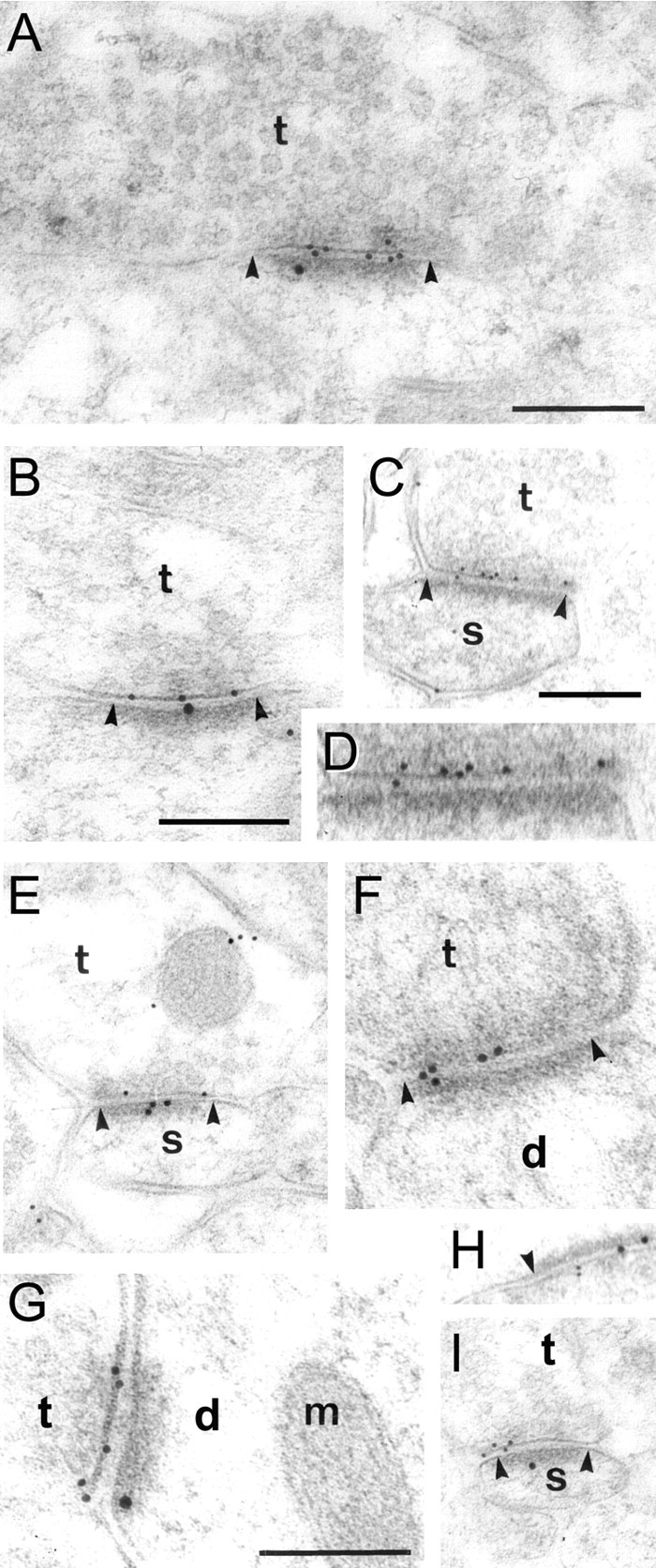
Electron micrographs showing the distribution of BK channels (first incubation, 10 nm particles) and NMDA receptors (second incubation, 15 nm particles) in double-labeled sections from the stratum radiatum of CA1. A,B, E, G, H,I, Both gold particle sizes are accumulated at asymmetric synapses (arrowheads). Small particles signaling BK channels are generally located presynaptic to the large particles signaling NMDA receptors. BK-immunoreactive terminals (t) are apposed to dendritic spines (s in C,E, I) or (less often) to dendritic stems (d in F, G). Particles are found along the entire extent of the presynaptic active zone (C, D, G). Some but not all of these synapses are immunopositive for the NMDA receptor (compare A, C). Scale bars: 200 nm (the same bars apply to C, E,H, I and to F andG, respectively). D is an enlarged part of C. In G, a mitochondrion is indicated by m.
The double-labeling experiments showed that particles signaling BK channels were often close to particles signaling NMDA receptors (Fig.1A,B,E,G–I), supporting the idea that BK channels are associated with glutamatergic synapses. Some BK-immunopositive, asymmetric spine synapses were NMDA receptor immunonegative (Fig.1C,D,F), but this is likely attributable to the inevitable reduction in sensitivity that follows the application of double-labeling protocols (Takumi et al., 1999).
The synaptic cleft separating the presynaptic and postsynaptic membranes was ∼12 nm wide in our material (average of 30 synapses; data not shown). Because a gold particle may end up as far as 30 nm from its epitope (Matsubara et al., 1996), reflecting the sizes of the interposed immunoglobulins or Fab fragments, it is impossible to attribute each individual particle to either the presynaptic or postsynaptic membrane. To resolve whether BK channels are expressed primarily presynaptically or postsynaptically, we therefore recorded the particle distribution along an axis perpendicular to the synaptic specialization. If the BK channels are exclusively postsynaptic, the particle distribution would be expected to show a peak that coincides with the peak of NMDA receptor immunoreactivity, previously shown to be restricted to the postsynaptic membrane (Takumi et al., 1999). In contrast, if the BK channels are mainly or exclusively presynaptic, one would predict that the gold particle distribution would peak further toward the presynaptic side.
Using the outer margin of the postsynaptic membrane as reference, the double-labeled preparations (Fig. 1) showed two distinct peaks (Fig.2C) with a peak-to-peak distance of 17 nm (at −5 and 12 nm; postsynaptic negative). The difference between the means was statistically significant atp < 0.0001 (Fig. 2, see legend).
Fig. 2.
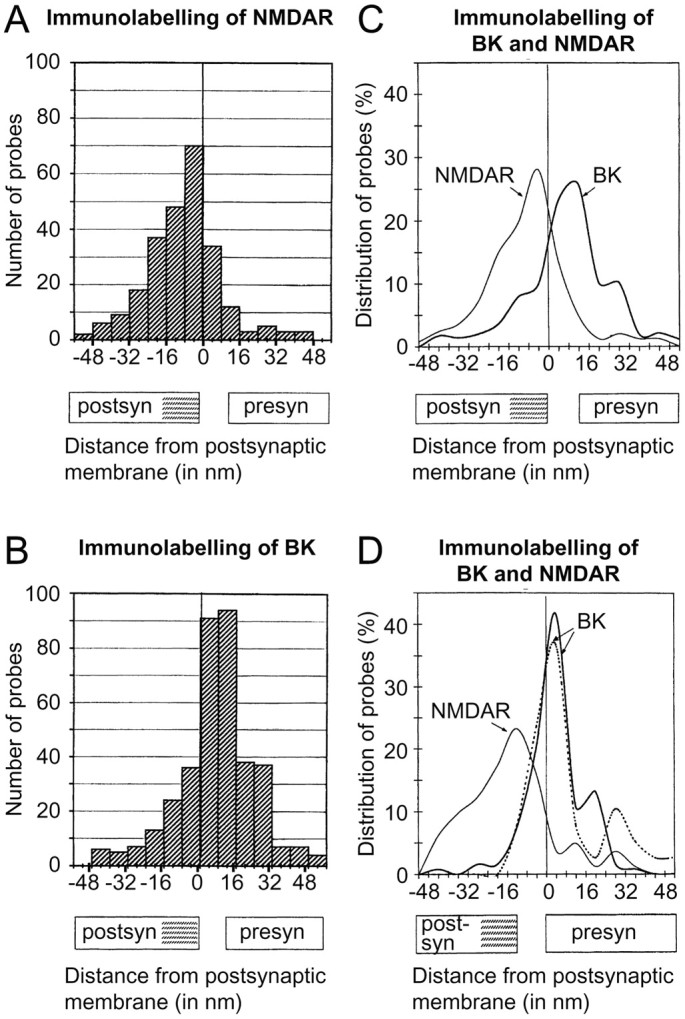
Quantitative analysis of gold particle distributions signaling BK channels (B–D) and NMDA receptors (A, C, D). The particle distribution was assessed along an axis perpendicular to the synaptic specialization (A–D) or extrasynaptic plasma membrane (D, dashed line). The approximate extent of the synaptic cleft and postsynaptic density (hatched) is indicated along theabscissa. Zero is defined as the outer margin of the postsynaptic (A–C) or presynaptic (D) membrane, and gold particles located postsynaptic to the reference line are assigned negative values. Compared with the gold particles signaling NMDA receptors (A), those signaling BK channels (B) are shifted in the presynaptic direction. This is evident in C, where the histograms inA and B have been transformed into curves. The number of gold particles is expressed in percentage to facilitate comparison between the two distributions. The peaks are located at −5 and +12 nm, i.e., over the postsynaptic and presynaptic membranes, respectively. The means were −8.52 ± 1.0 (n = 251) and 7.6 ± 0.9 (n = 366; p < 0.0001), and the kurtosis values were 1.29 and 1.19. D, Distribution of gold particles perpendicular to the presynaptic active zone (solid curves) and perpendicular to extrasynaptic parts of the presynaptic membrane (dashed curve). The analysis was performed exactly as in A–C except for the choice of reference line (outer margin of presynaptic rather than postsynaptic membrane). The BK immunogold distribution does not differ significantly between synaptic and extrasynaptic membrane domains (peaks 4.0 and 3.9 nm; 25 percentiles −1.4 and −1.1 nm). The linear density of gold particles in synaptic and extrasynaptic membranes was 7.0 and 0.26 particles per micrometer, respectively (measured along 41 and 256 μm; 144 synapses). No particles were observed along astrocyte plasma membranes (total length of sample 30 μm). Note that the variance in NMDA receptor immunogold distribution is larger in D than in C (506.6 vs 267.0), whereas the opposite is true for the BK signal (143.8 vs 295.8). This reflects the variability in the width of the synaptic cleft. Because the synaptic membranes are curved, the epitopes accessible for immunogold labeling will show a small shift in the postsynaptic direction relative to the reference line (hatched). Gold particles are indicated.
Because of variations in the width of the synaptic cleft and in the alignment of the synaptic membranes at the cut surface of the sections (where the epitopes are exposed for labeling), the exact location of the BK immunogold peak relative to the presynaptic membrane cannot be precisely determined by using the postsynaptic membrane as reference. Therefore, a second analysis was performed, this time with the outer margin of the presynaptic membrane as a line of reference. The distribution of BK immunogold particles showed a peak at +4 nm (Fig.2D), i.e., directly overlying the presynaptic membrane.
Although the peak was presynaptic to the reference line, the tail of the BK immunogold distribution extended farther than 20 nm in the postsynaptic direction, i.e., beyond the postsynaptic specialization (Fig. 2D). To resolve whether this tail could be explained by the length of the antibody “bridge” (see above), we made a corresponding analysis of the BK immunogold particles associated with the nerve terminal membrane lateral to the active zone. Thesolid and dashed curves in Figure2D (representing the active zone and extrasynaptic membrane domains, respectively) turned out to be almost identical with respect to peak and shape. These data are compatible with the idea that the BK channels are exclusively presynaptic.
To allow direct comparison, the curves representing nerve terminal membrane domains were based on normalized values. However, it should be noted that the gold particle number per micrometer plasma membrane was far lower lateral to the active zone than in the active zone itself (0.26 vs 7.0 particles per micrometer). There was no evidence of a heterogeneous distribution of particles within the active zone, although an accurate analysis of this issue would require serial sections. The particle density over extrasynaptic spine membranes was close to zero and did not differ significantly from that over astrocytic plasma membranes (defined as the background value).
Taken together, these results indicate that BK channel α-subunits are targeted to the presynaptic active zone of hippocampal glutamatergic synapses.
Effects of BK channel blockade on synaptic transmission under conditions that promote BK channel activation
Having established the existence of BK channel α-subunits in the presynaptic membrane of CA1 synapses, we tested whether these subunits form functional presynaptic BK channels that can control transmitter release.
EPSPs were evoked by stimulation of presynaptic axons in the CA1 stratum radiatum in rat hippocampal slices. First, to test whether functional presynaptic BK channels are present, we added 4-AP (100 μm) to the extracellular medium. By blocking voltage-gated K+ channels (but sparing BK channels), 4-AP broadens the presynaptic action potential (Haas et al., 1983). This manipulation is expected to promote BK channel activation by prolonging the depolarization and increasing the Ca2+ influx produced by each action potential (Qian and Saggau, 1999). [Higher doses of 4-AP have been reported to also affect Ca2+ homeostasis in other ways, but there is no evidence for such an effect at 100 μm (Grimaldi et al., 2001)]. Blockers of NMDA and GABAA receptors were routinely added to the bath to suppress GABAergic inhibition and NMDA receptor-dependent synaptic plasticity.
Whole-cell voltage recordings were obtained from CA1 pyramidal cells. EPSPs were evoked by paired-pulse stimulation of presynaptic fibers in the stratum radiatum. Meanwhile, somatic APs were triggered by injecting depolarizing current pulses (Fig.3A). Bath application of 100 μm 4-AP enhanced the EPSP amplitude and broadened the AP (Storm, 1987b). Subsequent application of the specific BK channel blocker IbTX (60–100 nm) strongly increased the EPSP amplitude (Fig.3A,C–E). In parallel, IbTX blocked the fast afterhyperpolarization (fAHP) and slowed the repolarization of the somatic AP of the postsynaptic cell (Fig.3B,H,I). The toxin had no effect on the resting potential, input resistance, or AP amplitude (Fig. 3B). Finally, bath application of the AMPA-type glutamate receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μm) fully blocked the EPSPs that had been enhanced by IbTX (n = 5; data not shown) (see Figs. 5A,7A). These results indicate that BK channels can regulate glutamatergic synaptic transmission in CA1 pyramidal neurons.
To test whether the effect of IbTX was caused by presynaptic or postsynaptic BK channels, we plotted the paired-pulse facilitation (PPF) ratio (PPR), i.e., the ratio between the amplitudes of the second and first EPSPs in the pair. IbTX reliably reduced the PPR (Fig.3F,G), consistent with an IbTX-induced increase in the probability of transmitter release (Zucker, 1989), i.e., an effect mediated by presynaptic BK channels.
It should be noted, however, that the effect of presynaptic BK channels on PPF is likely to be complex. Because BK channel activation will be enhanced by the presynaptic [Ca2+]iaccumulation underlying PPF (Zucker, 1989), the BK current should tend to shorten the second AP and curtail the resulting Ca2+ influx and release to a larger extent than for the first AP, thus reducing PPF. On the other hand, the BK channels could indirectly enhance PPF in two ways: (1) by limiting the [Ca2+]iaccumulation that underlies PPF (because the BK current shortens the first AP and reduces Ca2+ influx) and (2) by limiting transmitter depletion in response to the first AP (because the BK-induced reduction in Ca2+ influx also reduces release), thus reducing the depression that counteracts PPF (Zucker, 1989). Therefore, the observed change in PPF is probably the net effect of competing mechanisms, i.e., probably an underestimate of the real impact of the presynaptic BK channels in transmission.
To test more directly whether the effect of IbTX was caused by to presynaptic or postsynaptic BK channels, we prevented the activation of postsynaptic BK channels by loading the target cell with the fast Ca2+ chelator BAPTA (10 mm in the whole-cell recording pipette) (Fig.4). In the BAPTA-loaded cells the AP was already broadened and the fAHP fully blocked within a few minutes of recording (Fig. 4A), confirming that BAPTA effectively prevented activation of the postsynaptic BK channels, as reported previously (Lancaster and Nicoll, 1987; Storm, 1987a). (The BAPTA-induced spike broadening was often more pronounced than with IbTX, suggesting that BAPTA may also affect channels other than BK.) Nevertheless, bath application of IbTX strongly enhanced the EPSP amplitude (Fig. 4B–D) and reduced the PPR in the BAPTA-loaded cells (Fig.4E,F), as it did in cells without BAPTA (Fig. 3). This result indicates that the effect of IbTX on synaptic transmission was caused by blockade of presynaptic BK channels. Similar results were obtained both with 10.0 mm BAPTA alone and with 10.0 mm BAPTA plus 4.87 mmCa2+, giving a calculated free [Ca2+]i of 100 nm in the intracellular medium (see Materials and Methods).
Effects of BK channel blockade on presynaptic compound action potentials
To further investigate the mechanism whereby presynaptic BK channels can regulate synaptic transmission, we recorded extracellular field potentials in the stratum radiatum of the CA1 area. This technique has the advantage that it allows both the presynaptic compound action potential (fiber volley) and the fEPSP to be recorded (Andersen et al., 1978; Laerum and Storm, 1994).
Figure 5A shows the presynaptic fiber volley (FV) and the fEPSP in response to stimulation of the axons in normal saline (Control). Application of the glutamate receptor blockers DNQX and dl-AP5 eliminated the fEPSP, leaving only the fiber volley. Subsequent application of 4-AP (40–100 μm) broadened the volley by slowing its decay. Figure 5B shows the fiber volley in the presence of DNQX,dl-AP5, and 4-AP at an expanded time scale. Application of IbTX (60 or 100 nm) slowed the late part of the repolarization of the fiber volley, thus increasing its 90–10% repolarization time. The average time course of the IbTX effect is plotted in C, and the summary data for the full effect of IbTX are shown in D (n = 8). The results indicate that after presynaptic spike-broadening by 4-AP, IbTX further slows the repolarization of the presynaptic AP, in particular the late part. This effect resembles the effect of BK channel blockers in CA1 pyramidal cell somata, where BK channels are responsible for the final spike repolarization (Lancaster and Nicoll, 1987; Storm, 1987a,b).
To test whether calcium influx was required to activate the presynaptic BK channels, we omitted Ca2+ from the extracellular medium and replaced it with Mn2+ ions (2 mm;n = 2), added 0.5 mmCd2+ to the Ca2+-containing medium (n= 2), or combined low [Ca2+] (0.5 mm) with high [Mg2+] (3.5 mm;n = 1). Under each of these conditions, 100 nm IbTX had no measurable effect on the fiber volley (n = 5; data not shown), indicating that, as in the soma (Storm, 1987a,b), activation of this channel type requires Ca2+ influx, probably through voltage-activated Ca2+ channels opened by the action potential (Storm, 1987a).
Extracellular fEPSP recording in stratum radiatum also has an advantage over somatic EPSP recordings, in that the fEPSPs are recorded closer to their site of origin (i.e. the dendritic synapses in stratum radiatum) and should therefore be less distorted by ionic conductances that attenuate and modify the EPSPs on their way from the dendrites to the soma (Hoffman et al., 1997; Johnston et al., 1999). Therefore, to test whether IbTX can enhance transmitter release, fEPSPs were recorded in the absence of DNQX (Fig. 5E–I), again using paired-pulse stimulation to monitor the facilitation. Figure5E shows a pair of fEPSPs before and after application of 100 nm IbTX. The toxin increased the amplitude of both fEPSPs in the pair, but the effect was largest on the first fEPSP (EPSP1), thus reducing the PPR. The time course of the effect is plotted in Fig. 5F (n = 5 experiments), and summary data are plotted in G (n = 5). Figure 5, H and I, shows that the PPR declined in response to IbTX (n = 5), in parallel with the increase in the EPSP amplitude (F) and the fiber volley decay time (C).
Results similar to those shown in Figure 5A–Dwere obtained also in experiments in which the EPSP was only partly blocked with lower doses of DNQX (2–10 μm), thus minimizing any possible overlap between the volley and EPSP and allowing both the volley and the EPSPs to be followed in the same recording (n = 3; data not shown).
These results support the hypothesis that IbTX slows the AP repolarization in the presynaptic terminals, thereby promoting Ca2+ influx and glutamate release. In conclusion, IbTX-sensitive BK channels can apparently contribute to presynaptic spike repolarization and regulation of transmitter release.
Effects of BK channel blockade on synaptic transmission under basal conditions
Having established that functional IbTX-sensitive BK channels exist in the glutamatergic terminals, we next asked whether these channels regulate transmitter release under basal experimental conditions without 4-AP. For this purpose, we again used whole-cell recording (Fig. 6). In the absence of 4-AP, IbTX (100 nm) had no significant effect on the EPSPs or the facilitation ratio (Fig.6B–F). In contrast, IbTX invariably caused a characteristic spike broadening in the postsynaptic cell (Fig.6A,G,H), confirming that an effective concentration of the toxin penetrated into the slice in each experiment. [The very slight, not significant, decline in the average EPSP amplitude that was observed during the late part of the recordings (Fig. 6C,D) probably reflects use-dependent transmitter depletion.] Similarly, during field potential recordings in the CA1 stratum radiatum, in the absence of 4-AP, there was no measurable effect of 100 nmIbTX on the fEPSPs or the facilitation ratio (n = 8; data not shown) or on the fiber volley (n = 5) (see Fig. 10B,D).
High-frequency stimulation
Our results with 4-AP and IbTX (Figs. 3-5) indicated that presynaptic BK channels can potentially regulate transmitter release, but that this does not occur during low-frequency stimulation under our normal experimental conditions (Fig. 6). One possible explanation for this observation is that BK channels may only activate during high-frequency burst discharges, which cause Ca2+ accumulation in the presynaptic terminals. This could be physiologically important because high-frequency bursts are a common firing pattern in hippocampal glutamatergic neurons in vivo (Fox and Ranck, 1981).
To test this hypothesis, we stimulated the presynaptic fibers with trains of five stimuli at 100 Hz given once every 60 sec (Fig.7). (Longer trains and higher frequencies were also attempted, but they induced progressive rundown of the EPSP amplitudes.) In field-potential recordings, each stimulus train elicited a series of fEPSPs with progressively increasing amplitude (Fig. 7A). There is good evidence that this facilitation is caused by cumulative Ca2+ accumulation in the terminals (Zucker, 1989; Wu and Saggau, 1994), a condition that should promote BK channel activation.
Contrary to the above hypothesis, however, bath application of IbTX (100 nm) caused no significant enhancement of any of the fEPSPs during the train (Fig. 7B,C) (n = 5). Likewise, similar high-frequency EPSP or EPSC trains evoked in CA1 pyramidal cells during whole-cell recording (n = 3) or intracellular recording with sharp electrodes (n = 2) were not measurably enhanced by IbTX (60–120 nm) (Fig.9A2,A3,B).
In a minority of the field potential (1 of 5) and intracellular recordings (1 of 5), however, IbTX application was followed by a small enhancement of the late EPSPs in the train, with a reasonably plausible time course. We do not know whether these results reflect genuine IbTX effects, brought out by certain unknown conditions, or whether they were merely accidental. As described above, the other experiments, including whole-cell recordings (n = 8), were all negative. Furthermore, in a series of pilot experiments, we were unable to find any drug-free condition under which effects of IbTX on EPSPs could reliably be observed, although several parameters were varied, including different stimulation frequencies (20–100 Hz), bath Ca2+ concentrations (2–4 mm), ages of animals (20 d to 3 months), and temperatures (20–34°C).
Thus it appears, paradoxically, that IbTX-sensitive presynaptic BK channels, although present near the release sites, do not contribute noticeably to control of transmitter release under basal experimental conditions.
Effects of BK channel blockade by paxilline
Recent data indicate that some BK channels, in particular those containing the neuronal accessory subunit β4 (KCNMB4), have a reduced sensitivity to the peptide toxins IbTX and charybdotoxin (ChTX) (Behrens et al., 2000; Meera et al., 2000). Because β4 is a neuronal subunit that is widely expressed in rat brain, including the hippocampus (Behrens et al., 2000; Brenner et al., 2000; Meera et al., 2000; Weiger et al., 2000), it is possible that β4-containing IbTX-insensitive BK channels exist in hippocampal presynaptic terminals. This might explain the lack of a synaptic IbTX effect under basal conditions. Furthermore, the access of bath-applied peptide toxins to presynaptic BK channels in the synaptic cleft may possibly be limited by a diffusion barrier. If so, BK channels located within or near the cleft might be less accessible to IbTX than somatic BK channels, which are readily blocked by this toxin (Fig. 3B) (Shao et al., 1999). To test for these possibilities, we used paxilline, a potent nonpeptidyl BK channel blocker. Because of its lipophilic nature, it can penetrate cell membranes that are impermeable to IbTX (Knaus et al., 1994) and would be expected to readily reach into the synaptic cleft.
Before testing paxilline in slice experiments, we needed to test whether this drug could block IbTX-resistant β4-containing BK channels. To this end, we coexpressed hSlo-α and hSlo-β4 subunits in CHO cells to produceSlo-α/β4 heteromultimeric BK channels, as described (Behrens et al., 2000). As illustrated in Figure8A–C, the resulting outward current was readily and reversibly blocked by 1 μm paxilline, with an average inhibition of 82.6% (n = 4). Similar results have recently been obtained independently by Martin Wallner and Meera Pratap (personal communication) on coexpressed hSlo-α and hslo-β4 in Xenopus oocytes. Figure8D–F shows that theSlo-α/β4 BK channels expressed in the CHO cells were resistant to both scorpion toxins, IbTX (100 nm) and ChTX (100 nm), but were fully blocked by tetraethylammonium (TEA, 5 mm). This is in accordance with the pharmacological properties that are typical ofSlo-α/β4 heteromultimeric BK channels (Behrens et al., 2000; Meera et al., 2000).
Fig. 8.

The effect of paxilline and scorpion toxins (IbTX and ChTX) on BK channels containing the β4-subunits, expressed in a mammalian cell line. CHO cells were transfected with cDNAs coding forhSloα and for hSloβ4 subunits as described in Materials and Methods. Current measurements at +80 mV were performed in inside-out patches, 12–24 hr after the transfection. The Ca2+ concentration was adjusted to 11.0 μm. From a holding potential of 0 mV, a hyperpolarizing 100 msec prepulse to −140 mV was applied before the 500 msec test pulse to +80 mV. A, Currents before (1) and during (2) bath application of 1 μm paxilline and after washout of the drug (3). B, Typical time course of the effect of 1 μm paxilline on the outward current amplitude (I) measured during the test pulse to +80 mV. The application of 1 μm paxilline is indicated by the bar. The numbers (1-3) indicate the times of the sample traces shown in A. C, Summary diagram of the current amplitudes (I) relative to control, from four experiments (mean ± SEM). Paxilline significantly reduced the current amplitude (p = 0.031;n = 4). D, Currents before [Control(1)] and during [IbTX(2)] bath application of 100 nm IbTX, followed by 5 mm tetraethylammonium [TEA(3)], washout [Wash(4)], application of 100 nmcharybdotoxin [ChTX(5)], 5 mm TEA again [TEA(6)], and washout [Wash(7)].E, Typical time course of the effects of IbTX, ChTX, and TEA on the outward current amplitude. The numbers (1-7) indicate the times of the sample traces shown in D. F, Summary diagram of the current amplitudes (I) relative to control, from four experiments (mean ± SEM). TEA reliably blocked the current (n = 9; p = 0.028), but IbTX (n = 4; p = 0.16) or ChTX (n = 9; p = 0.66) had no significant effect, which is typical for BK channels containing β4-subunits. For paxilline application, inside-out patches were used (to prevent dilution of the membrane-permeable paxilline by the toxin-free solution in the patch pipette), whereas outside-out patches were used for IbTX and ChTX, which bind on the outside.
Figure 9 illustrates the results of experiments with paxilline in hippocampal slices. Because pilot experiments using sharp electrode intracellular recordings from CA1 cells in slices from adult rats yielded some seemingly promising results with paxilline (L.-R. Shao and J. F. Storm, unpublished observations), we used this technique (Fig.9A–C) in addition to whole-cell recording from young animals (20–35 d) (Fig. 9D). However, paxilline (5–10 μm) failed to significantly enhance the EPSPs with both recording methods. This was true during both low-frequency (Fig. 9A2) (20 Hz; n = 5) and high-frequency (Fig. 9A3) (100 Hz; n = 5) stimulation. Similar results were also obtained with field potential recordings from both age groups (data not included in Fig. 9). In contrast, in the postsynaptic CA1 pyramidal cells, paxilline consistently induced a spike broadening characteristic of BK channel blockade, thus showing that the drug penetrated the slice in sufficient concentration (n = 7) (Fig.9A1,B,D). When applied after IbTX, however, paxilline caused no further broadening of the action potential (Fig. 9A–C) (n= 2). Thus, these experiments yielded no evidence for IbTX-resistant, paxilline-sensitive BK channels, either presynaptically or postsynaptically. Similar effects of paxilline were obtained both with sharp electrode (Fig. 9A–C) and whole-cell recording (Fig. 9D) and also in field potential recordings (n = 5; data not shown). Figure 9E compares the combined area of the five EPSPs in each train, before and after paxilline (5 or 10 μm) application (n = 5); no significant difference was found. Field recordings also showed that paxilline had no measurable effect on the fiber volley in the absence of 4-AP (n = 4; data not shown).
To test whether paxilline blocked presynaptic BK channels effectively and specifically, we added paxilline (5 μm) in the presence of 100 μm 4-AP during fEPSP recordings like those shown in Figure 5, E and F. Paxilline mimicked the effect of IbTX by enhancing the fEPSP (the first fEPSP amplitude increased by 25.8 ± 0.08%; p = 0.031;n = 5. Data not shown. The sixth experiment, in which the EPSP evoked action potentials after paxilline, was excluded from the analysis.). Furthermore, IbTX (100 nm) occluded the effect of paxilline. Thus, in the presence of 4-AP and 100 nm IbTX, paxilline had no further effect on the fEPSP (the mean first fEPSP amplitude was reduced by 4.3% after 5 μm paxilline was added; p = 0.45; n = 4; data not shown), thus indicating that the paxilline effect is specific. Finally, in one experiment, 100 nm IbTX added in the presence of 5 μm paxilline had no further effect on the fEPSPs (i.e., paxilline occluded the effect of IbTX), confirming that the effect of IbTX is also specific.
The observation that the effect of paxilline did not exceed the effect of IbTX (Fig. 9) may seem surprising in view of the evidence that the neuronal β4 subunit confers reduced IbTX sensitivity to BK channels and is densely expressed in rat brain (Behrens et al., 2000; Brenner et al., 2000; Meera et al., 2000; Weiger et al., 2000). However, even β4-containing BK channels can be blocked by long exposure to high IbTX concentrations (Meera et al., 2000). Therefore, our applications of 100 nm toxin over tens of minutes (Figs. 3-6) may have been sufficient to significantly block β4-containing channels.
Taken together, our results indicate that neither IbTX- nor paxilline-sensitive BK channels, including β4-containing BK channels, contribute substantially to presynaptic AP repolarization and control of glutamate release in CA1 stratum radiatum under normal experimental conditions.
In contrast to paxilline or IbTX, TEA (1 mm) consistently enhanced the EPSPs and broadened the action potential further, even when applied after IbTX and paxilline (Fig.9A–C). A similar effect was seen in field recordings of EPSPs (n = 3; data not shown). This observation shows that the failure of IbTX or paxilline to enhance EPSPs was not caused by either transmitter depletion or a basal release probability that was already close to 1 and could not be increased. It also indicates that TEA-sensitive K+ channels other than BK (most likely Kv channels) contribute to spike repolarization both in the soma and in presynaptic terminals. This would also explain previous observations of fiber volley broadening and fEPSP enhancement by 1 mm TEA in CA1 stratum radiatum (Laerum and Storm, 1994).
Comparison between BK channel function in soma and terminals of CA3 pyramidal cells
The CA3 cells give rise to the Shaffer collaterals which form the majority of the CA1 stratum radiatum glutamatergic axons and terminals studied here (Paxinos and Watson, 1998). We therefore tested the role of BK channels in the CA3 pyramidal somata (using intracellular recording to facilitate comparison with previous studies of BK channel involvement in spike repolarization) (Lancaster and Nicoll 1987; Storm, 1987a,b; Shao et al., 1999). As illustrated in Figure10, A and C, IbTX (60 nm) reliably broadened the CA3 somatic action potentials by slowing the final two-thirds of the spike repolarization in all cells tested (n = 4). This effect of BK channel blockade is very similar to that previously found in CA1 pyramidal cells (Fig. 3) (Lancaster and Nicoll, 1987; Storm, 1987a,b; Shao et al., 1999) but contrasts sharply with the lack of IbTX effect in the Shaffer collaterals (Fig. 10B,D) (n = 5) and the corresponding lack of IbTX effect on EPSPs under identical conditions (Figs. 6, 7). These results indicate that different action potential mechanisms dominate in the soma and terminals of the same cell type and that the functional role of BK channels depends on its subcellular localization within the CA3 pyramidal cells.
DISCUSSION
To our knowledge, the present results provide the first direct evidence that Ca2+-activated K+ channels are located in the presynaptic active zone and can regulate synaptic transmission in the vertebrate brain. This represents a novel principle for negative feedback regulation of the intra-terminal Ca2+concentration ([Ca2+]i) and synaptic transmission that may be widespread in the CNS. Being enhanced by [Ca2+]i, like the transmitter release itself, the BK channels seem well suited for dynamic regulation of Ca2+ influx and transmitter secretion. Furthermore, we found that BK channels contribute strongly to action potential repolarization in the soma but not in the axon terminals of CA3 pyramidal cells under basal conditions, indicating that the functional role of BK channels depends on their subcellular localization.
Evidence that BK channels are located in the presynaptic membrane
Several lines of evidence support the conclusion that BK channels are located in the presynaptic membrane of CA1 glutamatergic synapses and can regulate transmitter release. (1) Our EM immunogold data indicate that the pore-forming BK channel subunit, theSlo α protein, is targeted to the presynaptic membrane. The results agree well with less direct light microscopy data from rat brain (Knaus et al., 1996; Wanner et al., 1999). (2) The selective BK channel blocker IbTX caused broadening of the presynaptic compound action potential and a parallel enhancement of synaptic transmission, indicating that BK channels mediate presynaptic spike repolarization, in the presence of 4-AP. (3) The IbTX-induced enhancement of synaptic transmission was accompanied by reduced paired-pulse facilitation, indicating an increased transmitter release probability. (4) Suppression of postsynaptic BK channel activation by intracellular BAPTA did not block the effect of IbTX on synaptic transmission.
In contrast to the presynaptic boutons, the postsynaptic dendritic membrane seemed to contain few, if any, BK channels. Our EM data gave no indication of BK channels on the postsynaptic side of the cleft. This result agrees with recordings from CA1 pyramidal dendrites that indicated little BK current in the apical dendrites (Poolos and Johnston, 1999). Because BK channels normally activate at potentials suprathreshold for spike generation (Storm, 1990; Shao et al., 1999), dendritic BK channels should hardly affect subthreshold EPSPs in any case.
The immunogold data
The double-labeling immunogold experiments served to determine whether there are BK channels at glutamate synapses, as well as their exact position relative to the synaptic cleft. Our data indicate that the BK channels are concentrated at asymmetric synapses that are NMDA receptor immunopositive and hence probably glutamatergic. Furthermore, the BK immunogold signal was shifted in the presynaptic direction relative to the NMDA receptor distribution, indicating that the BK channels are primarily or exclusively presynaptic.
The EM data indicate that the presynaptic BK channels are located in a strategic position next to the synaptic cleft, near the release sites (Figs. 1, 2). This resembles the arrangement in frog neuromuscular junction (Robitaille et al., 1993), whereas in chick ciliary ganglion BK channels were found on both the release face and non-release face of the presynaptic calyx (Sun et al., 1999). Being near the release sites, the BK channels may be colocalized with, and functionally coupled to, the voltage-gated Ca2+ channels that trigger transmitter release, which in these synapses are of the N and P/Q types(Wheeler et al., 1994; Malenka and Nicoll, 1999). Colocalization and coupling between BK and N-type Ca2+ channels have been reported in frog motor terminals (Robitaille et al., 1993) and hippocampal somata (Marrion and Tavalin, 1998).
Pharmacological properties of the BK channels
The possibility that some of the hippocampal BK channels might be IbTX insensitive because of β4 subunits (Behrens et al., 2000; Meera et al., 2000) was tested with the alternative BK channel blocker paxilline. Although paxilline suppressed IbTX-resistant β4-containing BK channels (Fig. 8), no evidence for native paxilline-sensitive IbTX-insensitive channels was obtained, either presynaptically or postsynaptically (Fig. 9). However, more detailed pharmacological experiments may be needed to fully test for β4 subunits. In addition to β4 subunits, Slack subunits have also been reported to associate with Slo-α and confer reduced IbTX sensitivity (Joiner et al., 1998). Because Slack is expressed in the hippocampus, and the paxilline sensitivity of Slack/Sloheteromultimers has not been reported, one may ask whether there may be presynaptic IbTX-insensitive Slack/Slo channels with an effect that went undetected. This seems unlikely, however, becauseSlack/Slo channels are reported to be blocked by 100 nm IbTX during depolarizations lasting up to ∼100 msec (Joiner et al., 1998). Therefore, our IbTX applications should have blocked any presynaptic Slack/Slo channels during action potentials.
BK channels and regulation of transmitter release
Although functional BK channels are evidently present in CA1 glutamatergic terminals, they appear not to regulate transmitter release under our basal experimental conditions. Furthermore, these channels seem not to substantially affect transmitter release even when presynaptic Ca2+ accumulation is enhanced by high-frequency stimulation (Figs. 7, 9).
Although presynaptic Ca2+-activated K+ channels have been described in some peripheral synapses (Gho and Ganetzky, 1992; Meir et al., 1999; Sun et al., 1999; Yazejian et al., 2000) and have often been assumed to control synaptic transmission, to our knowledge this has actually been demonstrated under normal conditions only in frog and lizard neuromuscular junction (Robitaille and Charlton, 1992; Robitaille et al., 1993; Blundon et al., 1995). Thus, the enigmatic silence of presynaptic BK channels may be a widespread phenomenon and could be caused by several factors. (1) These channels may be recruited only under extreme or rare conditions (see below), but nevertheless they have a sufficiently vital role to be favored by evolution. An analogy may be the cardiac long QT syndrome, caused by malfunction of human ether-a-go-go-related gene (HERG) and other cardiac channels. This has only minor consequences under normal conditions but predisposes for cardiac arrhythmia that can cause sudden death (Curran et al., 1995; Miller, 1996). (2) The channels may possibly perform functions other than those tested, or their main function may be at another developmental stage. (3) Some aspect of the artificial experimental conditions may suppress channel activity. For example, in the slice preparation, the BK channels may be deprived of modulatory influence from transmitters or hormones that may be required to boost their activity.
Under which conditions are presynaptic BK channels activated?
BK channels can be regulated by various factors, including discharge patterns, neuromodulators, and signaling cascades: transmitters, hormones, protein phosphorylation, etc. (Levitan et al., 1990; Gribkoff et al., 1997; Smith and Ashford, 1998; Valverde et al., 1999). Channel activation may also be modulated indirectly. For example, dephosphorylation of Kv1.4 voltage-gated K+ channels causes a 5–10 times faster inactivation (Roeper et al., 1997). Because Kv1.4 subunits are found in stratum radiatum, probably as parts of 4-AP-sensitive Kv1.1/Kv1.4 presynaptic K+ channels (Rhodes et al., 1997; Cooper et al., 1998), their enhanced inactivation could lead to a presynaptic spike broadening resembling the one that we obtained experimentally with 4-AP, which proved sufficient to recruit the BK channels. Further experiments are required to test these possibilities.
A particularly interesting possibility is that the presynaptic BK channels may serve as an emergency brake, protecting against hyperactivity, excitotoxicity, and cell death under pathological conditions. For example, these channels may be recruited by ischemia or other forms of metabolic stress. By causing depolarization (Hansen et al., 1982; Rosen and Morris, 1991; Martin et al., 1994; Krnjevic, 1997), such conditions may cause inactivation of voltage-gated K+ channels that normally contribute to presynaptic spike repolarization (e.g., Kv1.4, Kv1.1, Kvβ1) (Bielefeldt et al., 1992; Roeper and Pongs, 1996; Geiger and Jonas, 2000). The resulting depolarization-induced spike broadening (Storm, 1987b, their Figs. 8, 9; Cowan and Martin, 1992) may then allow the BK channels to activate and “take over” the repolarization, as they did after our 4-AP application. Because ischemia and hypoxia also can raise [Ca2+]i and stimulate BK channels (DiChiara and Reinhart, 1997; Krnjevic, 1997;Tekkok et al., 1999), their activation could be enhanced, thus limiting both vesicular glutamate release and reversal of glutamate transporters caused by depolarization (Rossi and Oshima, 2000). In accordance with this concept, BK channel openers recently were reported to provide significant cortical neuroprotection during acute brain ischemia (Gribkoff et al., 2001).
Different action potential mechanisms in soma and terminals of the same neuron
We found that hippocampal CA3 pyramidal somata closely resemble their relatives in area CA1, in that BK channels dominate during most of the spike repolarization (the final two-thirds) and the fast AHP in both cell types (Figs. 3, 10) (Storm, 1990; Shao et al., 1999). This suggests that the BK mechanism may be widespread among mammalian pyramidal somata. Furthermore, because the CA3 cells give rise to the Schaffer collaterals that form the majority of the CA1 stratum radiatum axons studied here, this implies that two subcellular compartments within the same cell (the CA3 pyramid) use different spike repolarization mechanisms under identical conditions: voltage- plus Ca2+-activated K+ channels in the soma, but only voltage-activated K+ channels in the terminals. Some differences between soma and terminal spike repolarization kinetics and mechanisms has been described previously, e.g., in neurosecretory cells (Bourque, 1990), and recently also in dentate granule cells (Geiger and Jonas, 2000). However, to our knowledge it has not been reported previously that Ca2+-activated K+ channels dominate the action potential repolarization in the soma but not in the terminals of the same neurons, an intriguing result in view of the predominantly presynaptic localization of BK channels.
Footnotes
This work was supported by the European Commission BIOMED-2 (Contract BMH4-97-2118), the MH Group Mechanism of Neuronal Communication–Norwegian Research Council, the Nansen, Langfeldt and Odd Fellow Foundations, and the Austrian Research Foundation (P12663-MED). The antibodies to NMDA receptors were generously supplied by Dr. R. J. Wenthold. We thank M. Wallner and M. Pratap, University of California Los Angeles, for generously sharing unpublished data regarding the paxilline sensitivity of β4-containing BK channels. Thanks are also due to Dr. T. Blackstad for help with EM analysis, and to S. Yousefi, K. M. Gujord, B. Riber, C. Knudsen, and G. Lothe for expert technical assistance.
Correspondence should be addressed to Johan F. Storm, Institute of Physiology, P.B. 1103, Blindern, N-0317 Oslo, Norway. E-mail:johan.storm@basalmed.uio.no.
REFERENCES
- 1.Adams PR, Constanti A, Brown DA, Clark RB. Intracellular Ca2+ activates a fast voltage-sensitive K+ current in vertebrate sympathetic neurones. Nature. 1982;296:746–749. doi: 10.1038/296746a0. [DOI] [PubMed] [Google Scholar]
- 2.Andersen P, Silfvenius H, Sundberg SH, Sveen O, Wigstrom H. Functional characteristics of unmyelinated fibers in the hippocampal cortex. Brain Res. 1978;144:11–18. doi: 10.1016/0006-8993(78)90431-6. [DOI] [PubMed] [Google Scholar]
- 3.Behrens R, Nolting A, Reimann F, Schwarz M, Waldschutz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett. 2000;474:99–106. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- 4.Bielefeldt K, Rotter JL, Jackson MB. Three potassium channels in rat posterior pituitary nerve terminals. J Physiol (Lond) 1992;458:41–67. doi: 10.1113/jphysiol.1992.sp019405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blundon JA, Wright SN, Brodwick MS, Bittner GD. Presynaptic calcium-activated potassium channels and calcium channels at a crayfish neuromuscular junction. J Neurophysiol. 1995;73:178–189. doi: 10.1152/jn.1995.73.1.178. [DOI] [PubMed] [Google Scholar]
- 6.Bourque CW. Intraterminal recordings from the rat neurohypophysis in vitro. J Physiol (Lond) 1990;421:247–262. doi: 10.1113/jphysiol.1990.sp017943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- 8.Cooper EC, Milroy A, Jan YN, Jan LY, Lowenstein DH. Presynaptic localization of Kv1.4-containing A-type potassium channels near excitatory synapses in the hippocampus. J Neurosci. 1998;18:965–974. doi: 10.1523/JNEUROSCI.18-03-00965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowan AI, Martin RL. Ionic basis of membrane potential changes induced by anoxia in rat dorsal vagal motoneurones. J Physiol (Lond) 1992;455:89–109. doi: 10.1113/jphysiol.1992.sp019292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 11.DiChiara TJ, Reinhart PH. Redox modulation of hslo Ca2+-activated K+ channels. J Neurosci. 1997;17:4942–4955. doi: 10.1523/JNEUROSCI.17-13-04942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox SE, Ranck JBJ. Electrophysiological characteristics of hippocampal complex-spike cells and theta cells. Exp Brain Res. 1981;41:399–410. doi: 10.1007/BF00238898. [DOI] [PubMed] [Google Scholar]
- 13.Geiger JR, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- 14.Gho M, Ganetzky B. Analysis of repolarization of presynaptic motor terminals in Drosophila larvae using potassium-channel-blocking drugs and mutations. J Exp Biol. 1992;170:93–111. doi: 10.1242/jeb.170.1.93. [DOI] [PubMed] [Google Scholar]
- 15.Gribkoff VK, Starrett JEJ, Dworetzky SI. The pharmacology and molecular biology of large-conductance calcium-activated (BK) potassium channels. Adv Pharmacol. 1997;37:319–348. doi: 10.1016/s1054-3589(08)60954-0. [DOI] [PubMed] [Google Scholar]
- 16.Gribkoff VK, Starrett JE, Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- 17.Grimaldi M, Atzori M, Ray P, Alkon DL. Mobilization of calcium from intracellular stores, potentiation of neurotransmitter-induced calcium transients, and capacitative calcium entry by 4-aminopyridine. J Neurosci. 2001;21:3135–3143. doi: 10.1523/JNEUROSCI.21-09-03135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haas HL, Wieser HG, Yasargil MG. 4-Aminopyridine and fiber potentials in rat and human hippocampal slices. Experientia. 1983;39:114–115. doi: 10.1007/BF01960661. [DOI] [PubMed] [Google Scholar]
- 19.Hansen AJ, Hounsgaard J, Jahnsen H. Anoxia increases potassium conductance in hippocampal nerve cells. Acta Physiol Scand. 1982;115:301–310. doi: 10.1111/j.1748-1716.1982.tb07082.x. [DOI] [PubMed] [Google Scholar]
- 20.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 21.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 22.Johnston D, Hoffman DA, Colbert CM, Magee JC. Regulation of back-propagating action potentials in hippocampal neurons. Curr Opin Neurobiol. 1999;9:288–292. doi: 10.1016/s0959-4388(99)80042-7. [DOI] [PubMed] [Google Scholar]
- 23.Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci. 1998;1:462–469. doi: 10.1038/2176. [DOI] [PubMed] [Google Scholar]
- 24.Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, Sanchez M, Giangiacomo K, Reuben JP, Smith AB, Kaczorowski GJ, Garcia ML. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–5828. doi: 10.1021/bi00185a021. [DOI] [PubMed] [Google Scholar]
- 25.Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML, Sperk G. Distribution of high-conductance Ca2+-activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci. 1996;16:955–963. doi: 10.1523/JNEUROSCI.16-03-00955.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krnjevic K. Coupling of neuronal metabolism and electrical activity. In: Lassen NA, editor. Brain work: the coupling of function metabolism and blood flow in the brain. Munksgaard; Copenhagen: 1997. pp. 65–78. [Google Scholar]
- 27.Laerum H, Storm JF. Hippocampal long-term potentiation is not accompanied by presynaptic spike broadening, unlike synaptic potentiation by K+ channel blockers. Brain Res. 1994;637:349–355. doi: 10.1016/0006-8993(94)91260-2. [DOI] [PubMed] [Google Scholar]
- 28.Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol (Lond) 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levitan IB, Chung S, Reinhart PH. Modulation of a single ion channel by several different protein kinases. Adv Second Messenger Phosphoprotein Res. 1990;24:36–40. [PubMed] [Google Scholar]
- 30.Lingle CJ, Solaro CR, Prakriya M, Ding JP. Calcium-activated potassium channels in adrenal chromaffin cells. Ion Channels. 1996;4:261–301. doi: 10.1007/978-1-4899-1775-1_7. [DOI] [PubMed] [Google Scholar]
- 31.Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 32.Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 33.Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 34.Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP. Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci. 1996;16:4457–4467. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meera P, Wallner M, Toro L. A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc Natl Acad Sci USA. 2000;97:5562–5567. doi: 10.1073/pnas.100118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- 37.Miller C. The inconstancy of the human heart. Nature. 1996;379:767–768. doi: 10.1038/379767a0. [DOI] [PubMed] [Google Scholar]
- 38.Ottersen OP, Zhang N, Walberg F. Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience. 1992;46:519–534. doi: 10.1016/0306-4522(92)90141-n. [DOI] [PubMed] [Google Scholar]
- 39.Paxinos G, Watson C. The rat brain in stereotactic coordinates. Academic; San Diego: 1998. [Google Scholar]
- 40.Petersen OH, Maruyama Y. Calcium-activated potassium channels and their role in secretion. Nature. 1984;307:693–696. doi: 10.1038/307693a0. [DOI] [PubMed] [Google Scholar]
- 41.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994a;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994b;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poolos NP, Johnston D. Calcium-activated potassium conductances contribute to action potential repolarization at the soma but not the dendrites of hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:5205–5212. doi: 10.1523/JNEUROSCI.19-13-05205.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian J, Saggau P. Modulation of transmitter release by action potential duration at the hippocampal CA3-CA1 synapse. J Neurophysiol. 1999;81:288–298. doi: 10.1152/jn.1999.81.1.288. [DOI] [PubMed] [Google Scholar]
- 45.Rhodes KJ, Strassle BW, Monaghan MM, Bekele-Arcuri Z, Matos MF, Trimmer JS. Association and colocalization of the Kvβ1 and Kvβ2 β-subunits with Kv1 α-subunits in mammalian brain K+ channel complexes. J Neurosci. 1997;17:8246–8258. doi: 10.1523/JNEUROSCI.17-21-08246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 48.Roeper J, Pongs O. Presynaptic potassium channels. Curr Opin Neurobiol. 1996;6:338–341. doi: 10.1016/s0959-4388(96)80117-6. [DOI] [PubMed] [Google Scholar]
- 49.Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosen AS, Morris ME. Depolarizing effects of anoxia on pyramidal cells of rat neocortex. Neurosci Lett. 1991;124:169–173. doi: 10.1016/0304-3940(91)90086-9. [DOI] [PubMed] [Google Scholar]
- 51.Rossi DJ, Oshima TAD. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 52.Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 53.Shao LR, Storm JF. Iberiotoxin (IbTX) and charybdotoxin (ChTX) slow the somatic spike repolarization but fail to enhance the excitatory synaptic transmission in hippocampal pyramidal cells. Soc Neurosci Abstr. 1997;23:1482. [Google Scholar]
- 54.Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol (Lond) 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shao LR, Hu H, Storm JF. Contributions of four ionic currents (BK, SK, M and H) to the medium afterhyperpolarization (mAHP) and early spike frequency adaptation, and M- and H-current to the resting potential, in CA1 hippocampal neurons. Soc Neurosci Abstr. 1999;25:453. [Google Scholar]
- 56.Smith MA, Ashford ML. Mode switching characterizes the activity of large conductance potassium channels recorded from rat cortical fused nerve terminals. J Physiol (Lond) 1998;513:733–747. doi: 10.1111/j.1469-7793.1998.733ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Storm JF. Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res 435 1987a. 387 392[Erratum (1988) 443:410[. [DOI] [PubMed] [Google Scholar]
- 58.Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol (Lond) 1987b;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 60.Storm JF, Hu H, Shao L-R, Chavoshy S, Trieb M, Ottersen OP, Knaus H-G, Laake P (2001) Presynaptic BK channels in glutamatergic hippocampal terminals: their role in spike repolarization and regulation of transmitter release. Soc Neurosci Abstr, in press. [DOI] [PMC free article] [PubMed]
- 61.Sun XP, Schlichter LC, Stanley EF. Single-channel properties of BK-type calcium-activated potassium channels at a cholinergic presynaptic nerve terminal. J Physiol (Lond) 1999;518:639–651. doi: 10.1111/j.1469-7793.1999.0639p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi T. Membrane currents in visually identified motoneurones of neonatal rat spinal cord. J Physiol (Lond) 1990;423:27–46. doi: 10.1113/jphysiol.1990.sp018009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- 64.Tekkok S, Medina I, Krnjevic K. Intraneuronal [Ca2+] changes induced by 2-deoxy-d-glucose in rat hippocampal slices. J Neurophysiol. 1999;81:174–183. doi: 10.1152/jn.1999.81.1.174. [DOI] [PubMed] [Google Scholar]
- 65.Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, Mann GE, Vergara C, Latorre R. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science. 1999;285:1929–1931. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- 66.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 67.Wanner SG, Koch RO, Koschak A, Trieb M, Garcia ML, Kaczorowski GJ, Knaus HG. High-conductance calcium-activated potassium channels in rat brain: pharmacology, distribution, and subunit composition. Biochemistry. 1999;38:5392–5400. doi: 10.1021/bi983040c. [DOI] [PubMed] [Google Scholar]
- 68.Weiger TM, Holmqvist MH, Levitan IB, Clark FT, Sprague S, Huang WJ, Ge P, Wang C, Lawson D, Jurman ME, Glucksmann MA, Silos-Santiago I, DiStefano PS, Curtis R. A novel nervous system β subunit that downregulates human large-conductance calcium-dependent potassium channels. J Neurosci. 2000;20:3563–3570. doi: 10.1523/JNEUROSCI.20-10-03563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 70.Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yazejian B, Sun XP, Grinnell AD. Tracking presynaptic Ca2+ dynamics during neurotransmitter release with Ca2+-activated K+ channels. Nat Neurosci. 2000;3:566–571. doi: 10.1038/75737. [DOI] [PubMed] [Google Scholar]
- 72.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]