

Abstract
Using a yeast two-hybrid method, we searched for amyloid precursor protein (APP)-interacting molecules by screening mouse and human brain libraries. In addition to known interacting proteins containing a phosphotyrosine-interaction-domain (PID)—Fe65, Fe65L, Fe65L2, X11, and mDab1, we identified, as a novel APP-interacting molecule, a PID-containing isoform of mouse JNK-interacting protein-1 (JIP-1b) and its human homolog IB1, the established scaffold proteins for JNK. The APP amino acids Tyr682, Asn684, and Tyr687 in the G681YENPTY687 region were all essential for APP/JIP-1b interaction, but neither Tyr653 nor Thr668 was necessary. APP-interacting ability was specific for this additional isoform containing PID and was shared by both human and mouse homologs. JIP-1b expressed by mammalian cells was efficiently precipitated by the cytoplasmic domain of APP in the extreme Gly681–Asn695 domain-dependent manner. Reciprocally, both full-length wild-type and familial Alzheimer's disease mutant APPs were precipitated by PID-containing JIP constructs. Antibodies raised against the N and C termini of JIP-1b coprecipitated JIP-1b and wild-type or mutant APP in non-neuronal and neuronal cells. Moreover, human JNK1β1 formed a complex with APP in a JIP-1b-dependent manner. Confocal microscopic examination demonstrated that APP and JIP-1b share similar subcellular localization in transfected cells. These data indicate that JIP-1b/IB1 scaffolds APP with JNK, providing a novel insight into the role of the JNK scaffold protein as an interface of APP with intracellular functional molecules.
Keywords: JIP-1b/IB1, amyloid precursor protein, phosphotyrosine-interaction-domain, scaffolding protein, c-Jun N-terminal kinase
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease pathologically characterized by senile plaques in the brain. The major constituent of the plaques is Aβ, cleaved off from the transmembrane precursor, termed amyloid precursor protein (APP). Genetic studies of familial AD (FAD) (Hardy, 1992) demonstrated that structural alterations in APP cause AD, based on the finding that certain FAD patients carry V642I/F/G mutations or an NL mutation (K595N/M596L) in APP695, a 695 residue version (Kang et al., 1987). However, how these mutations cause pathophysiology in AD and how wild-type APP (wt APP) contributes to normal functions have been little understood. Yet multiple studies (Qiu et al., 1995; Coulson et al., 1997; Gillian et al., 1997; Perez et al., 1997) have shown that wt APP performs physiological functions on the surface of neurons relevant to neurite outgrowth, neuronal adhesion, and axonogenesis, aside from its pathological role as an Aβ precursor. Rohn et al. (2000) and Sudo et al. (2000) independently found that anti-APP antibody treatment causes death in neuronal cells. Consistently, considerable amounts of APP are found on the surface of neurons (Jung et al., 1996; Brouillet et al., 1999; Sudo et al., 2000).
Parallel to these studies on wt APP, FAD-associated V642 mutants of APP have been shown to induce neuronal apoptosis (Wolozin et al., 1996;Yamatsuji et al., 1996a; Zhao et al., 1997; Luo et al., 1999). Toxicity by FAD mutants of APP and presenilin (PS)-2 is most likely a controllable process, mediated by pertussis toxin (PTX)-sensitive G-proteins (PG) (Wolozin et al., 1996; Yamatsuji et al., 1996a,b;Giambarella et al., 1997; Hashimoto et al., 2000). Furthermore, V642I-specific toxicity does not occur when APP lacks the His657–Lys676region, termed Domain 20 (Yamatsuji et al., 1996a,b; Hashimoto et al., 2000). Okamoto et al. (1995, 1996) showed in vitro that wt APP and V642I-APP exert antibody-dependent and constitutive PG-activating functions, respectively, both through Domain 20. Sudo et al. (2000) confirmed that cell-surface wt APP triggers neuronal death antibody dependently in a PTX-sensitive manner. These findings suggest that APP, through PG, performs signaling functions mediated by Domain 20.
On the other hand, the study of Murayama et al. (1996) suggested that wt APP could also trigger PTX-resistant signaling pathways. Consistently, Hashimoto et al. (2000) found that FAD-associated NL mutant of APP not only exerted PTX-sensitive toxicity through Domain 20 but also exerted PTX-resistant neurotoxicity through the Met677–Asn695region, termed Domain 19. These findings suggest that APP has a PG-independent signaling function mediated by Domain 19. In accord with this idea, Lu et al. (2000) found that the Ala665–Asn695peptide, the C-terminal fragment generated through APP cleavage by caspases, is cytotoxic. This study was thus conducted to investigate how the C terminus of APP can contribute to the regulation of intracellular signals. Here we report a novel function of APP to interact with JIP-1b/IB1, which provides the scaffold with c-Jun N-terminal kinase (JNK).
MATERIALS AND METHODS
Oligonucleotides. The nucleotide sequences of oligonucleotides used were as follows: SM60, 5′-aattttacccatatgatgtgccagattatgcctctcccgaattcggatccc-3′; SM61, 5′-tcgagggatccgaattcgggagaggcataatctggcacatcatatgggtaa-3′; SM71, 5′-gatccgaattcgcggccgcgtcgactctagactcgagaagctt-3′; SM72, 5′-aattaagcttctcgagtctagagtcgacgcggccgcgaattcg-3′; SM74, 5′-ggccgaattccgcaagaagaaaccctatgg-3′; SM78, 5′-agctccgccatgggatacccttatgatgtgccagattatgccggatccccggaattc-3′; SM79, 5′-ctaggaattccggggatccggcataatctggcacatcataagggtatcccatggcgg-3′; SM108, 5′-ggccgaattcaagatggatgcagagttcggacatgattcaggatttgaag-3′; SM109, 5′-ggccgaattcaagaagaaacagtacacatccatcc-3′; SM110, 5′-ggccctcgagttagttctgcatttgctcaaagaacttg-3′; SM121, 5′-ggccgaattcaggaagaggcagtacgg-3′; SM122, 5′-ggccctcgagttaaatctgcatctgctccagg-3′; SM123, 5′-ggccctcgagtcagttctgctgcatcttggag-3′; SM124, 5′-gaattcaagaagaaacaggccacatccatccatcatg-3′; SM125, 5′-cgccgccgtggaaccagaggagc-3′; SM126, 5′-gatgcagcagaacggaggtgagaatccaacttac-3′; SM127, 5′-gcagaacggatatgaggctccaacttacaagttc-3′; SM132, 5′-cggatatgagaatccaactgccaagttctttgagc-3′; SM138, 5′-ggccgaattcagcgactggattgaccag-3′; SM139, 5′-ggccctcgagctactccaagtagatatcttctg-3′; SM143, 5′-gcagcccttgccaaaaacagctgtgtccttgagatcag-3′; SM241, 5′-gcctcgagtcaagcatccagacacttctggt-3′; and SM242, 5′-gcggatccttcgctgtgcgctcc-3′.
Plasmid construction. The sequences of all constructs were verified by dideoxy-termination using ABI310 sequencer (PE Applied Biosystems, Foster City, CA). PCR inserts were amplified by deep vent DNA polymerase (New England Biolabs, Beverly, MA) or KOD DNA polymerase (Toyobo, Osaka, Japan). Mouse brain cDNA used for RT-PCR was prepared from total RNA of ICR adult mouse, isolated using an RNeasy kit (Qiagen GmbH, Hilden, Germany) and reverse transcribed using Superscript II and an Oligo-dT primer (Life Technologies Oriental, Tokyo, Japan). pEG202-NLS polylinker portion was exchanged with the correspondingEcoRI–XbaI fragment of pEG202; then hemagglutinin (HA) epitope, encoding YPYDVPDYA, was inserted betweenEcoRI and XhoI using linkers SM60/SM61 (pEG202-NLS-HA). The coding sequences of APP, APLP1, and APLP2 and their variants were PCR amplified and cloned into pEG202 and pEG202-NLS-HA using EcoRI/XhoI. APP fragments were amplified from mouse APP695 with the following primers: APP649–695, SM109/SM110; APP595–695, SM108/SM110; and APP649–680, SM109/SM123. The cytoplasmic domain of APLP1 (APLP1607–653) was amplified with PCR from APLP1 plasmid obtained in another two-hybrid screening using SM74. The cytoplasmic domain of APLP2751(APLP2705–751) was cloned by RT-PCR using primers SM121/SM122. The point mutants of cytoplasmic domain fragments of APP were constructed by PCR-directed mutagenesis of APP649–695 and APP649–680and cloned into pEG202 and pEG202-NLS-HA using the following primers: Y653A, SM124; T668E, SM125; Y682G, SM126; N684A, SM127; and Y687A, SM132, respectively. Among the prey fusion plasmids used in the interaction mating, JIP-1493–660 was PCR constructed from JIP-1b493–707 using SM143. JIP-1b557–707 was PCR constructed using primes SM138/SM139. Other prey fusions were directly cloned in the screening. All prey fusions were cloned into EcoRI/XhoI of pJG4–5.
JIP-1b was cloned by RT-PCR and cloned intoKpnI/XbaI of pcDNA3.1-HisA (Invitrogen, Carlsbad, CA) (T7-JIP-1b). JIP-1 constructs were created using the prey fusion of JIP-1493–660 and T7-JIP-1b. T7-JIP-1b1–272 was constructed by deletingXhoI fragment from T7-JIP-1b. In constructing T7-JIP1271–660, T7-JIP-1b365–707, and T7-JIP-1365–660, corresponding fragments were cloned into pcDNA3.1-HisC (Invitrogen). pcDNA3 (Invitrogen) was modified using linkers SM78/SM79 and SM71/SM72 to insert Kozak's consensus sequence followed by HA epitope MGYPYDVPDYAGSP. IB1360–711 was cloned into theEcoRI/XhoI site of this vector (HA-IB1360–711). pGEX-2T (Amersham Pharmacia Biotech, Uppsala, Sweden) polylinker was modified by the insertion of the linker SM71/SM72, using BamHI/EcoRI (pGEX-2Tm). Glutathione S-transferase (GST)-fusion proteins used for bacterial protein expression were constructed by cloning the same PCR inserts used for yeast bait vectors inEcoRI/XhoI of pGEX-2Tm. Full-length mouse wt APP and its FAD mutants were cloned into pEF-BOS (Mizushima and Nagata, 1990).
Corresponding portions of IB1, JIP-1b, and JIP-1 were cloned into pEBG (Sanchez et al., 1994), a mammalian expression vector of GST, in constructing GST-IB1360–711, GST-JIP-1b493–707, and GST-JIP-1b540–707. pEBG without insert was used as GST control. GST-JNK1β1 was constructed by cloning theBglII fragment of SRHis-JNK1β1 (Hirai et al., 1996) into the BamHI site of pEBG.
Corresponding portions of JIP-1b, mDab1, and X11, obtained in the two-hybrid screening, were cloned into pET28(a) (JIP-1b and mDab1) or pET28(c) (X11) (Novagen, Madison, WI). The region containing two PIDs of Fe65 was PCR amplified from a Fe65 clone obtained in the screening using SM241/SM242 and cloned into pET28(a). These pET constructs were used to produce His-tagged JIP-1b493–707, His-tagged mDab12–217, His-tagged X1137–680, and His-tagged Fe65370–662, respectively.
Antibodies. Anti-T7 antibody (Novagen), anti-HA antibody (12CA5: Roche Diagnostics, Basel, Switzerland), anti-APP antibody (22C11: Roche Diagnostics), and anti-GST antibody (MAB2510: Upstate Biotechnology, Lake Placid, NY) were used for immunoblotting. Rabbit antisera were raised for CGGAASPPAASPFLGLHIASPPNFR corresponding to residues 10–33 of JIP-1b (αJIPN) and RAFQQFYKQFVEYTCPTEDIYLE corresponding to residues 685–707 of JIP-1b (αJIPC).
Yeast two-hybrid method. A LexA yeast two-hybrid system was purchased from OriGene Technologies (Rockville, MD). Yeast two-hybrid screening was performed according to standard procedures (Ausubel et al., 1999), using the yeast transformation method of Gietz and Schiestl (1995). All bait constructs of APP and its mutants failed to activate LEU2 or LacZ reporters. EGY48 (MATα, trp1,his3, ura3, leu2:: 6 ops-LEU2) was used for the screening with pEG202-NLS-HA- APP595–695, and EGY188 (MATα, trp1, his3, ura3,leu2:: 2 ops-LEU2) was used for pEG202-NLS-HA-APP649–695. pSH18–34 (Ausubel et al., 1999) was used for the LacZ reporter. The positive control bait was pSH17–4 (GAL4 activation domain fused to LexA), and the negative control baits were pRFHM1 (homeodomain of bicoid fused to LexA) and pEG202-Max (Max fused to LexA), provided in the kit. The human adult AD patient library was constructed using the method of Gubler and Hoffman (1983), using cDNA obtained from a brain of a patient diagnosed as having AD by necropsy, according to institutional guidelines. The mouse adult brain library was purchased from Clontech (Palo Alto, CA). Yeast strains were cultured on YPAD plates, or on minimal plates with these combinations of the following conditions: containing glucose (SD plates), or containing galactose and raffinose (Gal plates), or containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), or lacking the combinations of histidine (-his), uracil (-ura), tryptophan (-trp), or leucine (-leu), as described in Ausubel et al. (1999). To extract yeast proteins, cells with the same optical density were collected and suspended with 2× sample buffer, frozen, and then boiled, according to the protocol of Ausubel et al. (1999). Yeast interaction mating was performed according to the methods ofFinley and Brent (1994) and Meroni et al. (1997). Briefly, the bait constructs were transformed into RFY206 (MATa, trp1Δ:: hisG, his3Δ200, ura3–52,leu2–3, lys2Δ201) simultaneously with the LacZ reporter and grown as patches on SD-his-ura plates. The various fragments of genes cloned into pJG4–5 prey vectors were transformed into EGY48 and grown as lawns on SD-trp plates. The mating was performed as follows. On the first day, the patches of RFY206 bait transformants and the lawns of EGY48 prey transformants were replica plated on the same YPAD plates. The next day, the mated yeast on the YPAD plates was replica plated on SD-his-ura-trp-leu and Gal-his-ura -trp-leu plates and incubated for an additional 48 hr to monitor cell growth reflecting galactose-induced LEU2 reporter activity, and on SD-his-ura-trp plates to select the mated yeast. On the third day, the patches grown on the replica plated SD-his-ura-trp plates were again replica plated on SD (X-gal)-his-ura-trp and Gal (X-gal)-his-ura-trp plates and incubated an additional 24 hr to monitor galactose-induced LacZ reporter activity.
Bacterial protein production and purification. GST fusion proteins were expressed using BL21-Gold (Stratagene, La Jolla, CA) and purified with glutathione Sepharose 4B (Amersham Pharmacia Biotech), according to the manufacturer's protocol. The purified proteins were dialyzed three times against 100 vol of buffer A (20 mm Tris/HCl, pH 8.0, 1 mmDTT, 1 mm EDTA), and frozen at −80°C until use. His-tagged proteins were expressed using BL21(DE3) (Novagen) and purified with Chelating Sepharose Fast Flow (Amersham Pharmacia Biotech) loaded with NiSO4, according to the manufacturer's protocol. The purified proteins were dialyzed against buffer A and frozen until use, as above.
Transfection and cell lysate. COS7 cells (1 × 106) were seeded in 10 cm dishes the day before the transfection in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin sulfate. The cells were transfected with 6 μg of DNA using DEAE-dextran. NT2 cells (Stratagene), human neuronal precursor cells, were seeded at 1 × 106 in 10 cm dishes the day before the transfection in 50% DMEM, 50% Ham's F-12 supplemented with 10% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 2 mml-glutamine. They were then transfected with 12 μg of DNA using LipofectAMINE (Life Technologies Oriental).
Forty-eight hours after the initiation of transfection, the cells were washed once with PBS, lysed for 30 min at 4°C in buffer B [20 mm HEPES/NaOH, pH 7.4, 1 mm DTT, 1 mm EDTA, 150 mm NaCl, 0.5% (w/v) Triton X-100], supplemented with 10% (v/v) glycerol, 1 mmphenylmethylsulfonylfluoride, 10 μg/ml of aprotinin, and 10 μg/ml of leupeptin with occasional gentle shaking. The lysed cells were collected and centrifuged at 20000 × g for 15 min, and cleared lysates were used for further analysis.
Bacterial GST pull-down. In pull-down experiments using His-tagged proteins, 6 μg of GST-fusion protein was incubated with 20 μl of glutathione beads in a total volume of 0.5 ml of buffer A for 1 hr at 4°C and washed once with buffer C [20 mmTris/HCl, pH 7.4, 1 mm DTT, 1 mm EDTA, 150 mm NaCl, 0.1% (w/v) Triton X-100]. His-tagged proteins of various concentrations were adjusted to 150 mm NaCl and 0.1% Triton X-100, mixed with beads immobilizing GST-fusion protein for 2 hr at 4°C with rotation, and washed three times with buffer C. The washed beads were mixed with 90 μl of 1× sampling buffer, boiled, and subjected to immunoblotting and Coomassie brilliant blue (CBB) staining. In the pull-down experiments using COS cell lysate, 30 μg of GST fusion proteins were immobilized on 30 μl of glutathione beads as above, and washed once with buffer B. Lysates (0.5 mg protein) were incubated with the beads immobilizing GST-fusion protein for 2 hr at 4°C with rotation, and washed three times with buffer B. The washed beads were mixed with 90 μl of 1× sample buffer, boiled, and subjected to further analysis as above.
Immunoprecipitation. Cell lysates (0.5 mg protein) were incubated with αJIPN or αJIPC or corresponding preimmune sera for 1 hr at 4°C, and mixed with 10 μl of Protein G-Sepharose 4 Fast Flow (Amersham Pharmacia Biotech) for an additional 1 hr with rotation at 4°C. Immunocomplexes were washed three times with buffer B, boiled in 1× sample buffer, and subjected to immunoblotting.
Immunoblotting. Protein samples were submitted to SDS-PAGE, transferred to nitrocellulose membranes (BA85; Schleicher & Schuell, Dassel, Germany) using a semidry system, and blocked overnight with TBS (50 mm Tris/HCl, pH 7.5, 200 mm NaCl) plus 5% skim milk. The membranes were incubated 1 hr at room temperature with the primary antibodies or antisera diluted in the same buffer, and then washed five times with TBS containing 0.05% Tween 20. The secondary antibodies, horseradish peroxidase-conjugated anti-rabbit goat polyclonal antibody (Bio-Rad, Hercules, CA) and horseradish peroxidase-conjugated anti-mouse rabbit polyclonal antibody (Bio-Rad), were diluted in TBS containing 5% skim milk. After washing, the membranes were visualized using an ECL kit (Amersham Pharmacia Biotech) and developed using x-ray films (Fujifilm, Tokyo, Japan).
Cell staining. COS cells (3 × 105) were seeded in 60 mm dishes on the previous day and transfected with 2 μg of plasmids using LipofectAMINE (Life Technologies Oriental), and reseeded on coverslips the next day. Forty-eight hours after the initiation of the transfection, cells were fixed with 0.1 mphosphate buffer, pH 7.4, containing 4% paraformaldehyde for 20 min at room temperature. After extensive washing with PBS, the fixed cells were incubated with αJIPN, αJIPC, preimmune sera, anti-APP antibody (22C11), or mouse IgG (Cortex Biochem, San Leandro, CA) in PBS containing 3% BSA and 0.1% Triton X-100 overnight at 4°C, and visualized by goat fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG or horse Texas Red-conjugated anti-mouse IgG (Vector Laboratories, Burlingame, CA) in PBS containing 3% BSA. DNA was stained by incubating coverslips in PBS containing 0.2 μm2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5′-bi-1H-benzimidazole (Hoechst 33342; Sigma-Aldrich Japan, Tokyo, Japan), and washed five times with PBS. Coverslips were mounted to slides with Vectashield (Vector Laboratories). Digital images were taken with an LSM310 laser scanning microscope (Carl Zeiss, Jena, Germany).
RESULTS
Screening of molecules interactive with cytoplasmic domains of APP
Twenty million transformants of an adult mouse brain library were screened with the transmembrane plus cytoplasmic domain APP595–695 and the cytoplasmic domain APP649–695 (Fig.1A). All retransformation-positive clones carried a PID (Kavanaugh and Williams, 1994; Bork and Margolis, 1995; Kavanaugh et al., 1995) that was implicated in the binding to the NPXY motif. In addition to known APP-binding proteins—such as Fe65 (Fiore et al., 1995), Fe65L (Guenette et al., 1996), Fe65L2 (Duilio et al., 1998), X11 (Borg et al., 1996), and mDab1 (Trommsdorff et al., 1998)—a novel APP-interacting protein was found to be JIP-1b (Whitmarsh et al., 1998;Kim et al., 1999), a PID-containing form of JIP-1 (Dickens et al., 1997).
Fig. 1.

The bait used and clones recovered in the two-hybrid screening. A, An illustration of the regions of mouse APP695 used as bait. APP595–695starts from the two amino acids Lys595–Met596, mutated to Asn–Leu in the Swedish type of FAD. APP649–695encodes the entire cytoplasmic domain of APP, the amino acid sequence of which is conserved between human and mouse APP. The α-, β-, and γ-secretase cleavage sites are also indicated. B, The clones recovered from the screening and the domain organization of mouse JIP-1b and its human homolog IB1. The portions coded by the recovered plasmids are indicated as lines with their amino acid positions in parentheses. Theboxes indicate JNK binding domain (JBD), src-homology region 3 (SH3), and PID, with their amino acid positions in numbers.
Because the recovered plasmids from the two different series of screening were almost identical, and APP649–695showed stronger interaction with PID-containing clones than APP595–695 in a two-hybrid assay (data not shown), the human adult brain library of an AD patient was screened only with APP649–695. After screening 14 million transformants, we found three interacting molecules: Fe65, X11L (Tomita et al., 1999), and IB1 (Bonny et al., 1998). X11L is the homolog of mouse X11 (Tomita et al., 1999), and IB1 is the human homolog of mouse JIP-1b (Bonny et al., 1998). Because JIP-1b and IB1 were novel APP-interacting proteins, and cloned from both the adult mouse brain library and the human patient brain library, we further analyzed JIP-1b/IB1 in this study. The regions encoded by the recovered JIP-1b/IB1 plasmids are indicated in Figure 1B.
Requirement of PID in JIP-1b/IB1 and Y682, N684, and Y687 in G681YENPTY687 of APP
To determine the critical amino acids necessary for interaction of JIP-1b/IB1 with the APP cytoplasmic domain, various APP mutants (Fig. 2A) were constructed in both pEG202 and pEG202-NLS-HA bait vectors and transformed into RFY206 with LacZ reporter pSH18–34. When the transformants of pEG202-NLS-HA fusions were submitted to immunoblot analysis using anti-HA antibody, a single immunoreactive band corresponding to the expected size of the bait fusions was detected in each lane of transformants (Fig. 2B). Various JIP-1b/IB1 fusions in the prey vector pJG4–5 (Fig.2D) were transformed into an EGY48 yeast strain. The array of two patches of each RFY206 bait transformant harboring the bait constructs (Fig. 2C) was mated with the lawns of EGY48 prey transformants, and the mated cells were grown on minimal medium containing glucose or galactose and lacking histidine, uracil, tryptophan, and leucine, to assay galactose-induced LEU2 reporter activity, as shown in Figure 2D.
Fig. 2.
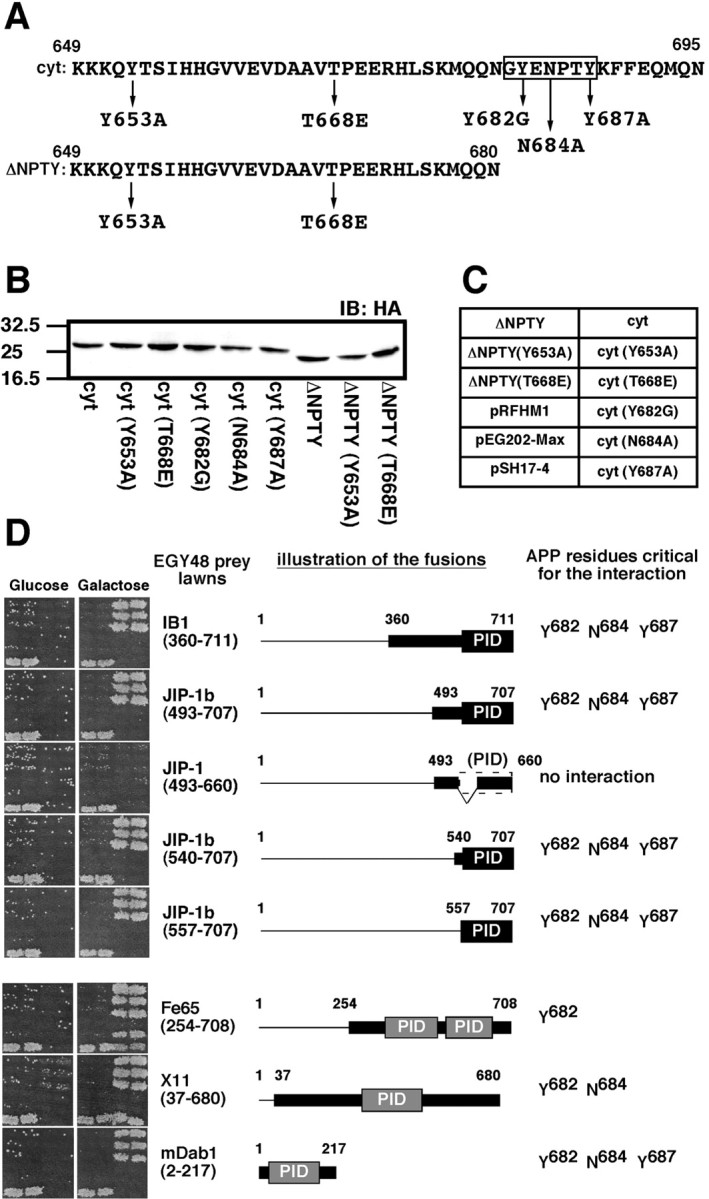
Requirement of both PID of JIP-1b/IB1 and G681YENPTY687 of APP for JIP-1b/IB1 interaction with APP. A, The indicated amino acid residues were changed by site-directed mutagenesis. APP649–695 and APP649–680 are designated as cyt and ΔNPTY, respectively. The G681YENPTY687 region isboxed. B, Proteins of RFY206 yeast cells transformed with fusions of various mutants of the APP cytoplasmic domain were probed with anti-HA antibody. The bait fusions used are identified under each lane. Their designations are as described in A. The numbers on theleft are the relative molecular weights of the size markers in kilodaltons. C, Two patches of each bait fusion transformant of the RFY206 strain were arranged as indicated. The names of APP bait variants are described in A. Two negative controls (pRFHM1 andpEG202-Max) and a positive control (pSH17–4) are also included.D, Interaction-mating of APP variants and JIP-1b/IB1 or other prey fusions. The left column of pictures shows the growth of mated yeast cells on glucose plates (Glucose), and the right column shows the growth of mated yeast cells on galactose and raffinose (Galactose) plates, as described in Materials and Methods. Galactose-dependent LEU2 activity was displayed by the difference in the growth of the mated cells between that in the glucose plates, which suppress the expression of the prey fusions, and that in the galactose plates, which induce it. The two negative controls (pRFHM1, pEG202-Max) showed no growth in any combination. The prey fusions are indicated at the right of thecolumns. The illustration of the fusions indicates the regions coded by the cDNA and the constructs as thin andthick lines, respectively. The first and last amino acids of cDNA and regions encoded by the constructs are indicated, except for mDab12–217, which starts at the second codon. PID regions were indicated as black boxes, except for JIP-1493–660, which has incomplete PID. APP residues critical for the interaction are noted at the right side, except for JIP-1493–660, which displayed no interaction with any APP variants. The GenBank accession numbers and amino acids corresponding to the PIDs were IB1, NM005456, 557–711; JIP-1b, AF054611, 557–707; JIP-1, AF003115, no PID; Fe65, AF206270, 364–505 and 535–660; X11, L34676, 297–460; mDab1, Y08381, 37–175.L34676 is a partial clone that lacks N-terminal 69 amino acids in its human or rat counterpart.
The fusions of IB1360–711, JIP-1b493–707, JIP-1b540–707, and JIP-1b557–707, all of which contain intact PID, interacted with APP649–695 (cyt) and its Y653A and T668E mutants [cyt (Y653A), cyt (T668E)]. In contrast, no interaction was observed with the other mutants of APP649–695 [cyt (Y682G), cyt (N684A), and cyt (Y687A)] or APP649–680 (ΔNPTY), nor with any of its mutants [ΔNPTY (Y653A), ΔNPTY(T668E)] (Fig.2D). This indicates that (1) the extreme C-terminal 15 amino acids of APP harboring G681YENPTY687are necessary for the interaction of JIP-1b/IB1 with APP; (2) Tyr682, Asn684, and Tyr687, contained in this region, are all critical for this interaction; and (3) the 151 amino acid PID region contained in the shortest construct, JIP-1b557–707, is sufficient for the interaction with APP. In further support of this observation, the fusion of JIP-1493–660, which is equivalent to the JIP-1b493–707 lacking 47 amino acids in the PID region, exhibited no interaction with the cytoplasmic domain of wt APP or APP mutants (Fig. 2D). This observation suggests that the PID region was sufficient and necessary for the interaction of JIP-1b with APP.
Other PID-containing fusions obtained in the same screening were also examined for their ability to interact with cytoplasmic domain mutants of APP, as shown in Figure 2D. Fe65254–708, X1137–680, and mDab12–217 all displayed G681YENPTY687-dependent interaction with the cytoplasmic domain of APP, as evidenced by their positive interaction with cyt, cyt (Y653A), and cyt (T668E), and their negative interaction with ΔNPTY, ΔNPTY (Y653A), and ΔNPTY (T668E) (Fig. 2D). However, the critical APP residues necessary for the interactions were different in the three constructs. Fe65254–708 required Tyr682, but not Asn684 or Tyr687; X1137–680required Tyr682 and Asn684, but not Tyr687; mDab12–217required Tyr682 and Asn684, as well as Tyr687, the same residues necessary for JIP-1b/IB1 constructs containing PID (Fig. 2D). Therefore, in this experiment, the mode of APP interaction with JIP-1b/IB1 was distinct from that with Fe65 and X11, but indistinguishable from that with mDab1.
These observed interactions were confirmed by replica-plating the mated yeast cells to minimal plates, containing X-gal, and containing glucose or galactose, and lacking histidine, uracil, and tryptophan. The LacZ reporter activity visualized by the blue color development displayed the same patterns as in Figure 2D (data not shown). The same set of cytoplasmic domain mutants of APP fused to pEG202 bait vector displayed the same results as those of pEG202-NLS-HA fusion constructs (data not shown). These data indicate that JIP-1b/IB1 interacts with APP in a manner distinct from already known interactions of Fe65 or X11 with APP, and both PID of JIP-1b/IB1 and the G681YENPTY687region of APP, especially Tyr682, Asn684, and Tyr687, are essential for the interaction.
Purified JIP-1b protein binds to the cytoplasmic domain of APP with affinity comparable to that of mDab1, X11, and Fe65
To compare the binding affinity of JIP-1b PID with that of other PID-containing proteins, His-tagged JIP-1b493–707, which contains a part of SH3 domain and the entire PID (the same region illustrated in Fig.2D), was expressed in bacteria, purified, and subjected to pull-down experiments using bacterially produced recombinant APP cytoplasmic domain fused to GST (GST-cyt). His-tagged JIP-1b at various concentrations was mixed with glutathione beads immobilizing an equivalent amount of GST-cyt, and each precipitant (ppt) was probed for GST-cyt by CBB staining (Fig.3A, top panel), and for the bound His-tagged protein by immunoblotting using anti-T7 antibody (Fig. 3A, bottom panel). A similar amount of GST-cyt was detected in all precipitants (Fig. 3A). When the bound His-tagged JIP-1b was plotted against the concentration used for the binding experiment, the bound protein showed saturable binding to GST-cyt with half-maximal binding at ∼0.4 μm (Fig. 3A). His-tagged JIP-1b did not bind to GST at all concentrations tested, indicating that the binding of His-tagged JIP-1b to GST-cyt is specific (data not shown).
Fig. 3.

Purified JIP-1b binds to the cytoplasmic domain of APP with affinity comparable to those of other PID-containing proteins. A, His-tagged JIP-1b493–707, at the concentrations indicated under each lane, was precipitated with glutathione beads immobilizing the APP cytoplasmic domain fused to GST (GST-cyt). Precipitated GST-cyt was visualized with CBB staining (top panel), and bound His-tagged JIP-1b493–707 in the precipitants was detected with anti-T7 antibody (bottom panel). The numbers on the left are the relative molecular weights of the size markers in kilodaltons. The bound JIP-1b, calculated from the pixels of scanned image and a standard curve, was plotted against the concentration of JIP-1b used for the binding.B, Similar experiments were performed using His-tagged mDab12–217. The bands corresponding to mDab1 are faintly visible in CBB staining (top panel).C, Similar experiments were performed using His-tagged X1137–680. D, Similar experiments were performed using His-tagged Fe65370–662. The bands corresponding to Fe65 are visible in CBB staining (top panel). E, CBB staining of the purified His-tagged proteins used.
Similar experiments were performed using His-tagged mDab2–217 (the same region illustrated in Fig. 2D), His-tagged X1137–680 (the same region illustrated in Fig.2D), and His-tagged Fe65370–662 (the region containing its two PIDs). Again, a similar amount of GST-cyt was detected in all precipitants (Fig. 3B–D, top panels). When the bound PID-containing proteins were plotted against their concentration used for the binding, the binding curves showed saturable binding with the concentration of mDab1, X11, and Fe65 required for their half-maximal binding at ∼0.5, 0.5, and 0.15 μm, respectively (Fig.3B–D). None of these His-tagged proteins bound to GST at any of the concentrations tested, which confirmed the specificity of their binding (data not shown). The purity of the recombinant proteins used for these binding experiments as revealed by SDS-PAGE and CBB staining is shown in Figure 3E.
Therefore, bacterially expressed and purified JIP-1b bound to the cytoplasmic domain of APP in a specific and saturable manner, and it showed half-maximal binding at a submicromolar concentration, which is comparable with the half-maximal binding concentrations of other PID proteins—mDab1, X11, and Fe65. In addition, the data that purified recombinant JIP-1b bound to purified GST-cyt suggest that JIP-1b directly binds to the cytoplasmic domain of APP, as mDab1, X11, and Fe65. As assessed by the common T7-antigenicity, the maximal binding of JIP-1b to the cytoplasmic domain of APP was relatively lower than that of the other PID proteins, suggesting that the APP/JIP-1b complex might be a minority among all APP/PID-protein complexes under the conditions in which other PID proteins are also present. However, considering its comparably high affinity to those of other PID proteins, this feature of APP/JIP-1b interaction could be useful for signal transduction of high time resolution.
C terminus of APP precipitates JIP-1b/IB1 in a G681YENPTY687-dependent manner
To confirm the protein interaction of JIP-1b/IB1 with APP, the lysate of COS cells transfected with HA-IB1360–711 were mixed with various GST fusion proteins immobilized on glutathione beads, and the lysate and precipitants were analyzed by immunoblotting using anti-HA antibody (Fig. 4B). HA-IB1360–711 was detected in the precipitant of GST fused with the cytoplasmic domain of APP (GST-cyt), but not in those of GST alone, GST fused with the cytoplasmic domain of APP lacking C-terminal 15 amino acids (GST-ΔNPTY), or GST fused with the cytoplasmic domain of APLP1 (GST-APLP1cyt). Minor immunoreactive bands of lower relative molecular weights were attributed to partial degradation of the expressed protein. A faint band was detected occasionally in the precipitant of GST fused with the cytoplasmic domain of APLP2 (GST-APLP2cyt) (Fig. 4B). It is thus highly likely that IB1360–711 binds to the cytoplasmic domain of APP, which contains G681YENPTY687shown to be necessary by the interaction-mating experiment (Fig.2D). It was also noted that IB1360–711 more preferentially bound to the cytoplasmic domain of APP than the cytoplasmic domains of APLP1 and APLP2, under the conditions that were used.
Fig. 4.

JIP-1b/IB1, not JIP-1, is precipitated by the cytoplasmic domain of APP, but little by those of APLP1 and APLP2.A, An illustration of the JIP/IB1 constructs used in the bacterial GST-fused protein pull-down experiments. HA and T7 tags areboxed. Other designations are the same as the illustration of the fusions in Figure 2D.B, The lysates prepared from the COS cells transfected with HA-IB1360–711, T7-JIP-1b365–707, and T7-JIP-1365–660 were precipitated with glutathione beads immobilizing GST, GST-APP649–695 (GST-cyt), GST-APP649–680(GST-ΔNPTY), GST-APLP1607–653 (GST-APLP1cyt), and GST-APLP2705–751 (GST-APLP2cyt), and the lysate and the precipitants were visualized with anti-HA and anti-T7 antibodies as indicated. The precipitants were also stained with CBB, which shows the presence of a similar amount of GST-fusion protein. Thenumbers on the left are the relative molecular weights of the size markers in kilodaltons. Each datum presented in Figure 4 is the representative of three independent experiments. C, The lysates prepared from mock-transfected COS cells were precipitated using GST-cyt. The lysates (lysate) and precipitants (ppt) were visualized with anti-HA and anti-T7 antibody, indicating the absence of nonspecific bands in GST-cyt precipitants.
This IB1-APP interaction was reproduced by using its mouse counterpart, T7-JIP-1b365–707, the homologous region encoded by HA-IB1360–711. When the lysate of COS cells transfected with T7-JIP-1b365–707 was again precipitated with GST-fused proteins immobilized on glutathione beads, a weak but significant band corresponding to JIP-1b365–707 was detected in the GST-cyt precipitant, but not in any of the other precipitants (Fig.4B). The weakness of the JIP-1b immunoreactivity in the GST-cyt precipitant was attributed to proportionally weak expression of T7-JIP-1b365–707. Therefore, JIP-1b bound to the cytoplasmic domain of APP in a manner similar to that observed for its human homolog IB1.
When T7-JIP-1365–660, equivalent to JIP-1b365–707 lacking 47 amino acids in the PID region, was examined by the same GST-cyt pull-down experiment, no anti-T7-immunoreactive band was observed in the precipitants, although the construct was expressed sufficiently in the lysate (Fig.4B). These results indicate that the PID region is essential for the JIP interaction with APP, the same observation obtained by the interaction-mating experiment shown in Figure2D. The amounts of GST-fusion proteins contained in the precipitants were similar in all experiments above. The typical CBB staining is shown (Fig. 4B, CBB).
As negative controls, the lysate of mock-transfected cells was precipitated with immobilized GST-cyt, and their lysates and the ppts were subjected to immunoblotting using anti-HA and anti-T7 antibodies. Anti-HA antibody detected faint bands in the lysate, which are different in size from those found in the lysate of cells transfected with HA-IB1360–711, but none in the precipitant. No immunoreactive bands were detected in either lysate or the precipitant with anti-T7 antibody (Fig. 4C). This confirms that the bands detected in the lysates or precipitants in Figure4B were derived from the transfected plasmids.
We were unable to perform interaction-mating assays using the bait fusions of the cytoplasmic domain of APLP1 and APLP2 because of their autoactivation of the LEU2 reporter gene (data not shown).
GST-fused JIP-1b/IB1 can precipitate full-length APP with or without FAD mutations
To further confirm the interaction of JIP-1b/IB1 with APP, reciprocal pull-down experiments were performed using GST-fused JIP-1b/IB1 constructs and full-length wt APP. When the lysates of COS cells transfected with wt APP and GST, GST-IB1360–711, GST-JIP-1b493–707, or GST-JIP-1b540–707, APP immunoreactivity was found in the glutathione-bead precipitants from lysates transfected with any GST-JIP-1b/IB1 construct but not in the precipitant from the lysate transfected with GST alone (Fig.5A). Similar amounts of APP were detected in lysates of these transfected cells (Fig.5A). Thus, GST-fused JIP-1b/IB1 constructs interacted with full-length wt APP.
Fig. 5.
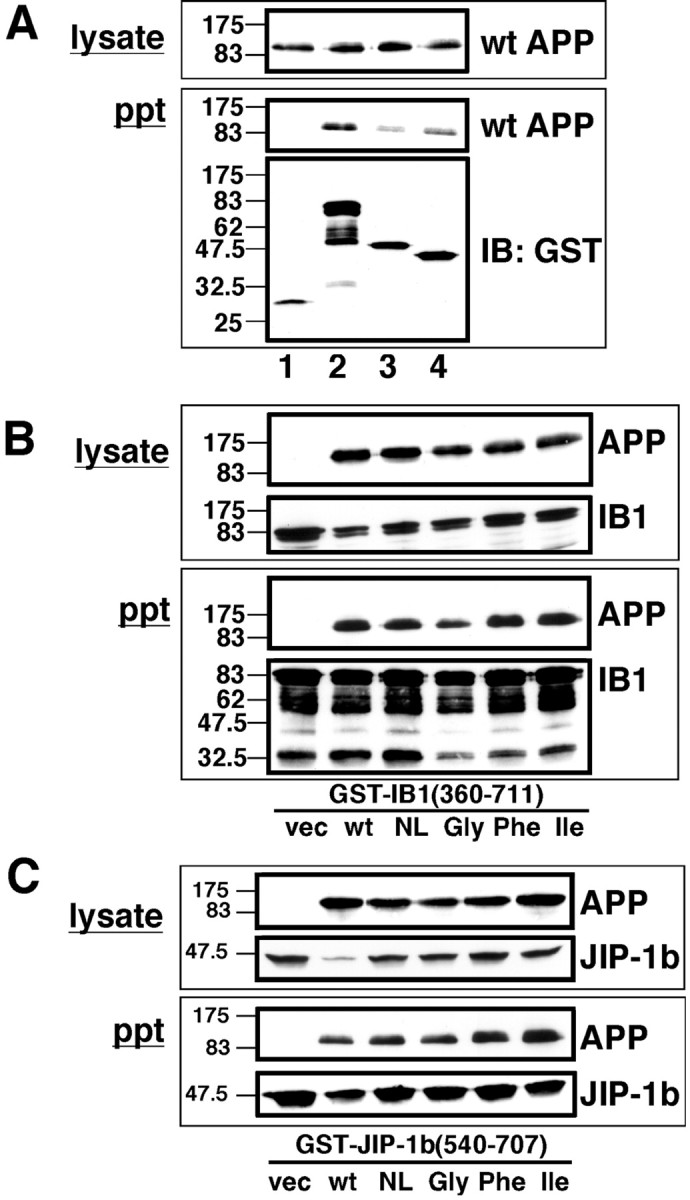
The C terminus of JIP-1b/IB1 precipitates wild-type and FAD mutant APPs. A, COS cells were cotransfected with wt APP with GST, GST-IB1360–711, GST-JIP-1b493–707, or GST-JIP-1b540–707(lanes 1–4, respectively) and precipitated with glutathione beads. The presence of APP and GST-fused proteins were probed with anti-APP antibody and anti-GST antibody, respectively. Thenumbers on the left are the relative molecular weights of the size markers in kilodaltons. Each datum presented in Figure 5 is the representative of three independent experiments. B, COS cells were cotransfected with GST-IB1360–711 with pEF-BOS (vec), wt APP (wt), NL-APP (NL), V642G-APP (Gly), V642F-APP (Phe), or V642I-APP (Ile) as indicated and precipitated with glutathione beads. Immunoreactivities of APP and GST-IB1360–711contained in the lysates (lysate) or precipitants (ppt) were detected by anti-APP antibody and anti-GST antibody, respectively. C, Similar experiments were performed with COS cells transfected with GST-JIP-1b540–707 instead of GST-IB1360–711. Other conditions and designations are the same as inB.
We next examined whether this interaction was affected by FAD mutations in APP. For this purpose, COS cells were transfected with GST-IB1360–711 and pEF-BOS, wt APP, or four FAD mutants of APP695 [K595N/M596L-APP (NL-APP), V642G-APP, V642F-APP, and V642I-APP] (Fig. 5B). The levels of expression of APP constructs were similar among wt APP and mutant APPs (Fig. 5B). When these lysates were precipitated with glutathione beads, APP immunoreactivity was detected in the precipitants from lysates transfected with either APP construct. This finding indicates that GST-IB1360–711 can interact, to similar degrees, with full-length APP with or without FAD-associated mutations. Similar results were obtained by a pull-down experiment using GST-JIP-1b540–707 instead of GST-IB1360–711 (Fig. 5C). JIP-1b/IB1 thus interacted similarly with wt APP and four FAD mutants (NL and V642I/F/G).
Anti-JIP-1b antisera precipitate full-length wt APP and APP mutants with JIP-1b expressed in COS cells and neuronal NT2 cells
When lysates of COS cells transfected with T7-JIP-1b1–272 or T7-JIP-1b1–707 were probed with rabbit antiserum raised against the N-terminal residues 10–33 of JIP-1b (αJIPN), the major immunoreactive bands at ∼40 and ∼120 kDa were those corresponding to the bands detected with anti-T7 antibody. In contrast, preimmune serum from the same rabbit failed to detect them (data not shown). Likewise, when lysates of cells transfected with T7-JIP-1271–660 or T7-JIP-1b1–707 were probed with rabbit antiserum raised against the C-terminal residues 685–707 of JIP-1b (αJIPC), the major immunoreactive bands at ∼70 and ∼120 kDa were those corresponding to the bands detected with anti-T7 antibody, but its preimmune serum could not detect them (data not shown). This indicates that both αJIPN and αJIPC antisera can specifically recognize transfected JIP-1b or its variants.
To further confirm the interaction of full-length JIP-1b with full-length APP, various combinations of plasmids were transfected into COS cells (Fig. 6A). When the lysates of transfected cells were analyzed by immunoblotting with anti-APP antibody, significant and similar amounts of wt APP and NL-APP were detected in the lysates of cells transfected with cognate constructs of APP, but little immunoreactivity of APP was detected in those transfected with the empty vector alone. Anti-T7 immunoreactivity of JIP-1b was detected in the lysates transfected with T7-JIP-1b cDNA, but little immunoreactivity was detected in those transfected with an empty vector control (Fig. 6A). These lysates were immunoprecipitated with the antisera, as indicated in Figure6B, using αJIPN or its preimmune serum. The precipitants were subjected to immunoblot analysis using anti-APP and anti-T7 antibodies to detect wt APP or NL-APP and T7-JIP-1b, respectively. Significant immunoreactivity of JIP-1b was detected in the αJIPN precipitants from all of the T7-JIP-1b-transfected lysates, but no immunoreactivity was detected in those from the empty vector-transfected cells, nor was it detected in the precipitants by preimmune serum (Fig. 6B). This indicates that αJIPN antiserum precipitated JIP-1b in the presence or absence of exogenous APP expression. When the precipitants were probed using anti-APP antibody, APP immunoreactivity was detected in the αJIPN precipitants of cells transfected with both T7-JIP-1b and either wt APP or NL-APP, but not in the precipitant by preimmune serum. No APP immunoreactivity was detected when empty vector controls were used in place of either JIP-1b or APP (Fig. 6B). This indicates that the immunoprecipitation of JIP-1b was specific and that exogenous JIP-1b expression was required for APP to be precipitated by αJIPN antiserum.
Fig. 6.
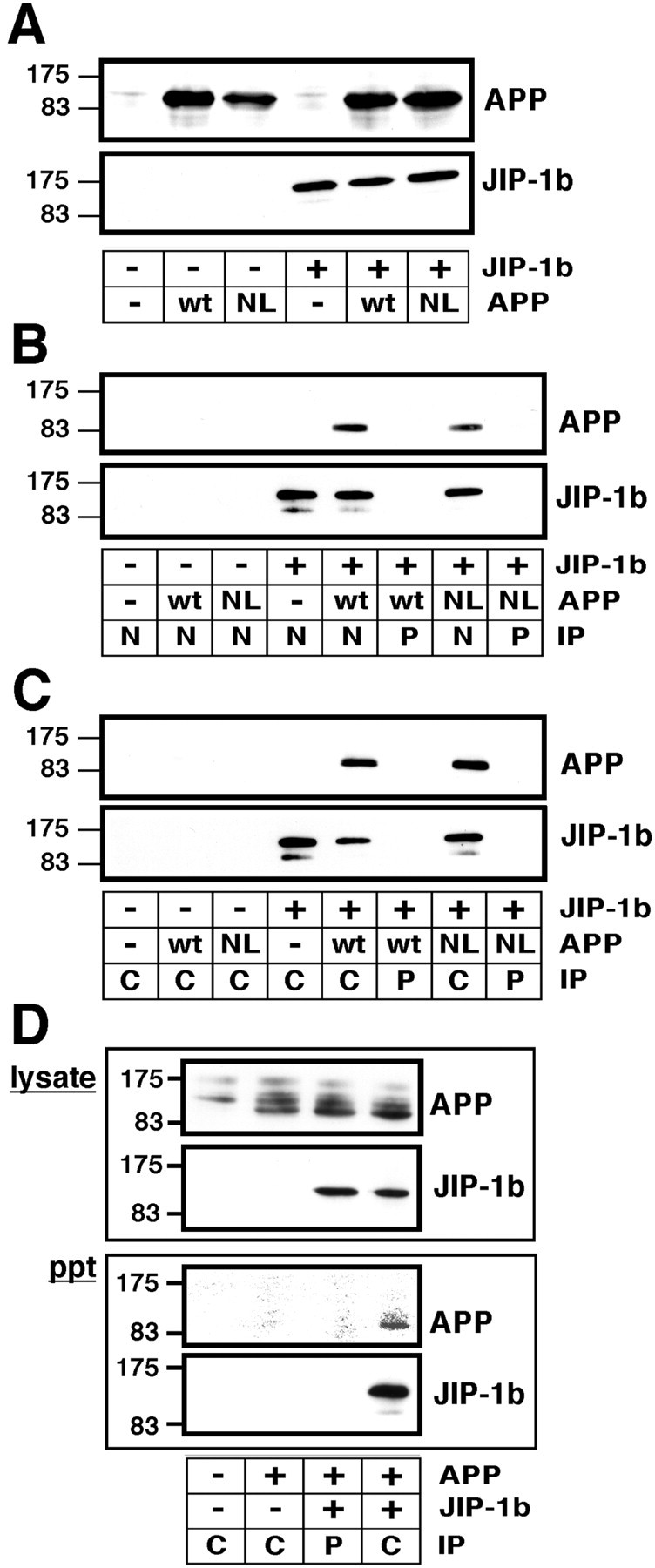
JIP-1b precipitates wild-type and FAD mutant APP in COS and NT2 cells. A, COS cells were transfected with the various combinations of wt APP, NL-APP, or their empty vector (wt, NL, or − in APP, respectively), and T7-JIP-1b or its empty vector (+ or − in JIP-1b). The presence of APP and JIP-1b in the lysates was detected with anti-APP and anti-T7 antibody, respectively. Thenumbers on the left are the relative molecular weights of the size markers in kilodaltons. Each datum presented in Figure 6 is the representative of three independent experiments. B, The same lysates prepared from the transfectants of indicated plasmids were immunoprecipitated with the antisera αJIPN (N) or its preimmune serum (P). APP and JIP-1b were detected as inA. Other designations are the same as inA. C, Similar experiments were performed with αJIPC (C) or its preimmune serum (P). Other conditions and designations are the same as in B. D, Similar experiments were performed using neuronal NT2 cells transfected with the indicated combinations of wt APP and T7-JIP-1b and αJIPC (C) or its preimmune serum (P). Their lysates (lysate) and precipitants (ppt) were probed for the presence of APP and JIP-1b. Other conditions and designations are the same as inC.
Essentially the same immunoprecipitation experiments were performed using αJIPC, yielding results similar to those obtained in the αJIPN experiments (Fig. 6C). Furthermore, similar results were obtained when similar immunoprecipitation was performed using V642I-APP instead of NL-APP (data not shown). Full-length JIP-1b thus interacted with full-length APP with or without FAD mutations.
To further confirm the interaction of JIP-1b and APP in neuronal cells, wt APP and T7-JIP-1b or their corresponding empty vectors were transfected to NT2 cells, human neuronal precursor cells (Pleasure and Lee, 1993), as indicated in Figure 6D. When the lysates were subjected to immunoblotting and probed for the presence of APP and JIP-1b using anti-APP and T7-antibodies, respectively, APP and JIP-1b were detected in the lysates of cells transfected with their cognate plasmids. Significant amounts of APP of higher molecular weights were also detected in all lysates, including that of empty vector-transfected NT2 cells. These lysates were immunoprecipitated using αJIPC and its preimmune serum, and the precipitants were again probed for the presence of APP and JIP-1b. As in the experiments using COS cells, both JIP-1b and APP could be detected in the precipitant from the lysate of NT2 cells transfected with both plasmids and precipitated with αJIPC, but not in the other combinations tested (Fig. 6D). The expression of JIP-1b in untransfected NT2 cells could not be detected using αJIPN or αJIPC (data not shown).
GST-fused JNK1β1 precipitates wild-type and FAD mutant APP in the presence of exogenously expressed JIP-1b
To investigate whether JIP-1b can scaffold JNK and APP, various combinations of plasmids were transfected to COS cells, and their lysates were probed with anti-APP, anti-T7, or anti-GST antibody to detect wt APP-, T7-JIP-1b-, and GST-fused proteins, respectively (Fig.7, lysate). The protein expression of wt APP and JIP-1b was confirmed in the lysates of cells transfected with corresponding plasmids, but not in empty vector controls. Likewise, GST and GST-JNK1β1 were detected in cells transfected with the encoding plasmids, as the bands of the corresponding sizes of their expressed proteins (Fig. 7,lysate).
Fig. 7.

GST-fused JNK1β1 can precipitate APPs in a JIP-1b-dependent manner. COS cells were transfected with the indicated combinations of wt APP or its empty vector (+ or − in wt APP), T7-JIP-1b or its empty vector (+ or − inJIP-1b), and GST orGST-JNK1β1, as indicated with + under eachlane, and their lysates were precipitated with glutathione beads. The presence of APP, JIP-1b, and GST-fused proteins in lysate (lysate) and precipitants (ppt) was detected with anti-APP, anti-T7, and anti-GST antibodies, respectively. Other designations are the same as in Figure 6. The data in this figure are representative of three independent experiments.
These lysates were precipitated with glutathione beads and subjected to immunoblot analyses (Fig. 7, ppt). GST or GST-JNK1β1 was detected in all precipitants from lysates transfected with the corresponding GST constructs. JIP-1b was detected in the precipitant from the lysate of cells transfected with both JIP-1b and GST-JNK1β1, but not in that from the lysate of cells transfected with GST alone nor the empty vector control of JIP-1b. This suggests that JIP-1b was specifically precipitated by JNK1b1. When the same precipitants were probed with anti-APP antibody, APP immunoreactivity was detected only when wt APP, JIP-1β, and GST-JNK1β1 were cotransfected. In contrast, no APP immunoreactivity was detected in either precipitant from lysates transfected in the other combinations. This provides evidence that wt APP forms a complex with JNK1β1 through the scaffolding by JIP-1b.
JIP-1b and APP share similar subcellular localization
To further confirm the interaction of JIP-1b with APP in vivo, subcellular localization of JIP-1b and APP was examined by immunocytochemical analysis using confocal microscopy. When COS cells transfected with wt APP cDNA were stained with anti-APP antibody and visualized with Texas Red-conjugated secondary antibody, cells showed cytoplasmic staining but not nuclear staining (Fig.8). No staining was observed when the same transfected cells were stained with control mouse IgG or when untransfected cells were stained with anti-APP antibody (data not shown).
Fig. 8.

APP and JIP-1b share similar subcellular localization. COS cells were transfected with APP, JIP-1b, or both. JIP-1b expression was detected with αJIPN and visualized with FITC-conjugated secondary antibody (green). APP expression was detected with 22C11 (anti-APP antibody) and visualized with Texas Red-conjugated secondary antibody (red). DNA was visualized with Hoechst 33342 (blue), as described in Materials and Methods. The yellow area in the merged section shows where JIP-1b and APP share similar subcellular localization. Scale bars, 10 μm. The same set of experiments was repeated at least three times and yielded similar results.
When T7-JIP-1b-transfected cells were stained with αJIPN antiserum and visualized with FITC-conjugated secondary antibody, JIP-1b staining was observed in the cytoplasm and absent in the nucleus. Staining using αJIPC yielded similar results (data not shown). No staining was observed when untransfected cells were stained with αJIPN or αJIPC or when corresponding preimmune sera were used to stain T7-JIP-1b-transfected cells (data not shown).
To investigate whether JIP-1b and APP share subcellular localization, cells were cotransfected with T7-JIP-1b and APP and doubly stained by αJIPN and anti-APP antibody (Fig. 8). The immunostaining of JIP-1b (stained green) and that of APP (stained red) significantly overlapped in the shared subcellular localization, as revealed by the yellow area in the merged picture. These observations are consistent with the notion that a certain fraction of JIP-1b interacts with APP in vivo.
DISCUSSION
We have herein identified JIP-1b/IB1 as a novel APP-interacting molecule that indicated an affinity for the cytoplasmic domain of APP comparable to those of other known PID-containing APP-interacting proteins. JIP-1 was initially characterized as a cytoplasmic inhibitor of JNK family kinases (Dickens et al., 1997) and subsequently found to interact with MKK7, MLK, DLK, and HPK-1 in addition to JNK (Whitmarsh et al., 1998). Coexpression of JIP-1 and JNK with MKK7 or MLK3 increased JNK activation (Whitmarsh et al., 1998). These findings have established that JIP-1 scaffolds the kinase components of the JNK signaling pathway (Davis, 2000). An additional isoform of JIP-1 has been reported in mouse (JIP-1b) (Whitmarsh et al., 1998; Kim et al., 1999), rat [islet-brain-1 (IB1)] (Bonny et al., 1998), and human (IB1) (Mooser et al., 1999). This isoform contains a 47-residue insertion that completes the PID region at the C terminus, which was originally identified in Shc interaction with NPXY in the cytoplasmic domain of the epidermal growth factor receptor (Kavanaugh and Williams, 1994; Kavanaugh et al., 1995). Neither the physiological nor the pathological role of the JIP-1 proteins has become totally clear, whereas expression of JIP-1 has been reported to transcriptionally activate the GLUT2 promoter (Bonny et al., 1998) and is implicated in the pathogenesis of a form of type 2 familial diabetes mellitus (Waeber et al., 2000) and in the cytoprotection of insulin-secreting cells (Bonny et al., 2000). The present study thus provides the first line of evidence that the JNK scaffold protein, abundant in the brain and in islet β-cells, could be relevant to Alzheimer's disease. Interestingly, it has been reported that in vivo, neurotoxicity by hippocampal administration of Ab1–42 occurs only in diabetic rats (Smyth et al., 1994).
Analysis of subcellular localization using transfected cells indicates that JIP-1b and APP colocalized in the cytoplasm but both were not detected in the nuclei. Similar cytoplasmic localization of JIP-1b was reported by Dickens et al. (1997) and Whitmarsh et al. (1998), althoughBonny et al. (1998) reported that IB1 is a nuclear protein. Coffey et al. (2000) showed nuclear localization of JIP-1 proteins in cerebellar granule cells, and Meyer et al. (1999) showed that JIP-1 proteins localize in the cytoplasm in unpolarized NIE115 and PC12 cells but are concentrated at neurites when the cells are polarized. These differences in JIP-1 localization thus may reflect different functions of JIP-1 proteins assigned in different cell environments. Although the present study provides evidence that JIP-1b interacts with APP inside the transfected cells, it would be necessary to investigate whether endogenous APP and JIP-1b interact in nontransfected cells. Yet the notion that JIP-1b/IB1 colocalizes with APP is consistent with earlier studies (Becker et al., 1999; Kim et al., 1999; Yasuda et al., 1999;Marcinkiewicz and Seidah, 2000; Pellet et al., 2000) indicating that the subcellular and brain regional localizations of JIP proteins considerably overlap with those of APP. Because the putative α-secretase ADAM10 and the putative β-secretase BACE are expressed in the same neurons that express APP in the mouse brain (Marcinkiewicz and Seidah, 2000), APP cleavage by these putative secretases would lose the interaction of APP with JIP-1b/IB1, causing, in turn, a loss in the ability of JIP-1b/IB1 to specifically colocalize signaling molecules with APP. Although so far we have been unable to coimmunoprecipitate APP with IB1 from rat brain homogenates (data not shown), it remains unclear whether this failure is caused by inappropriate experimental conditions for specific immunoprecipitation of the APP/IB1 complex from solubilized brain homogenates or whether it implies that, with the APP/IB1 complex being a minor fraction, the majority of APP and IB1 in the brain does not complex with each other or form complexes with different partners. The latter notion is consistent, at least in part, with the observed relatively lower maximal binding of JIP-1b to the cytoplasmic domain of APP, as compared with those of the other PID-containing proteins tested.
By constructing deletion and point mutants, we have shown that the domains necessary for the APP/JIP-1b interaction are the cytoplasmic G681YENPTY687region in APP and the PID region in JIP-1b, completed by the insertion specific for this isoform. This accounts for the PID-nonbearing isoform JIP-1 not interacting with APP. As noted above, X11, Fe65, Fe65L, and mDab1 have been shown to interact with the C terminus of APP. The present study indicates that the APP/JIP-1b interaction requires Tyr682, Asn684, and Tyr687 contained in the G681YENPTY687region. This is different from the mode of APP interaction with Fe65 and X11 and similar to that with mDab1. The observed amino acids required for the interaction of Fe65 or X11 concur with a previous report (Borg et al., 1996). Interestingly, the JIP-1b isoform, which is interactive with APP, is the major transcript in the brain, and the noninteractive JIP-1 transcript is hardly detected (Coffey et al., 2000; Pellet et al., 2000), pointing to certain specific roles of the JIP-1b isoform in neuronal functions.
The mechanism underlying the observed JIP-1b/IB1 interaction with APP is thus consistent with the established NPXY motif interaction of PID in Shc (Kavanaugh and Williams, 1994; Kavanaugh et al., 1995; Songyang et al., 1995) and IRS-1(Pawson and Scott, 1997). Yet in the present GST pull-down experiments, the cytoplasmic domains of APP, APLP1, and APLP2, all of which contain the same NPXY structure GYENPTY, showed largely different binding intensities for JIP-1b/IB1, with APP being the strongest among them. These different binding characteristics might reflect the difference in the primary to ternary structures surrounding the NPXY motif, suggesting the presence of an additional structural requirement allowing NPXY to interact efficiently with PID. In support of this idea, the most recent literature, in which PID of JIP-1b is shown to interact with p190 rhoGEF (Meyer et al., 1999), indicates that the binding region of p190 does not contain the classical NPXY motif.
Because JIP-1b showed binding similar to full-length APP regardless of the presence of four different FAD mutations, JIP-1b is most likely involved in the basic function of APP. Although the binding of Fe65 or X11 to APP has been shown to affect Aβ secretion from APP (Borg et al., 1998; Sastre et al., 1998; Sabo et al., 1999; Tomita et al., 1999), so far we have not observed remarkable changes in Aβ42 secretion from NL-APP by cotransfection with JIP-1b (Z. Shao, S. Matsuda, and I. Nishimoto, unpublished observation). The JIP-1 proteins have been shown to serve as scaffold proteins for the organization of active JNK signaling complexes (Whitmarsh et al., 1998). In fact, we have shown in this study that APP associates with JNK via JIP-1b. It has also been established that APP interacts with the GTP-binding protein Go through the middle portion in the APP cytoplasmic domain adjacent to the NPXY-containing C terminus (Nishimoto et al., 1993; Okamoto et al., 1995; Brouillet et al., 1999). It is likely, therefore, that APP may serve as a membrane-anchoring protein that further scaffolds the JIP-scaffolding complex with other signaling molecules. Taking into account the recently cloned members of the JIP family, JIP2 and JIP3 (Yasuda et al., 1999; Kelker et al., 2000)—PID is contained in JIP2 but not in JIP3—it would deserve investigation whether APP might regulate the JNK signaling pathway through the binding of these various JIP proteins to the cytoplasmic domain of APP.
Footnotes
This work was supported in part by grants from the Ono Medical Research Foundation, the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and the Organization for Pharmaceutical Safety and Research (OPSR). We especially thank Yukiko Yoshimoto-Matsuda and Tomo Yoshida for excellent assistance and generous encouragement throughout the course of this study; Yumi and Yoshiomi Tamai for indispensable support; Masao Kaneki for cooperation; and Kazumi Nishihara and Dovie Wylie for expert technical assistance.
Correspondence should be addressed to Ikuo Nishimoto, Department of Pharmacology and Neurosciences, KEIO University School of Medicine, Shinjuku-ku, Tokyo 160-8582, Japan. E-mail:nisimoto@mc.med.keio.ac.jp.
REFERENCES
- 1.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. Wiley; New York: 1999. [Google Scholar]
- 2.Becker AJ, Gillardon F, Blumcke I, Langendorfer D, Beck H, Wiestler OD. Differential regulation of apoptosis-related genes in resistant and vulnerable subfields of rat epileptic hippocampus. Mol Brain Res. 1999;67:172–176. doi: 10.1016/s0169-328x(99)00060-1. [DOI] [PubMed] [Google Scholar]
- 3.Bonny C, Nicod P, Waever G. IB1, a JIP-1-related nuclear protein present in insulin-secreting cells. J Biol Chem. 1998;273:1843–1846. doi: 10.1074/jbc.273.4.1843. [DOI] [PubMed] [Google Scholar]
- 4.Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G. IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem. 2000;275:16466–16472. doi: 10.1074/jbc.M908297199. [DOI] [PubMed] [Google Scholar]
- 5.Borg J-P, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and Fe65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borg J-P, Yang Y, De Taddeo-Borg M, Margolis B, Turner RS. The X11α protein slows cellular amyloid precursor protein processing and reduces Aβ40 and Aβ42 secretion. J Biol Chem. 1998;273:14761–14766. doi: 10.1074/jbc.273.24.14761. [DOI] [PubMed] [Google Scholar]
- 7.Bork P, Margolis B. A phosphotyrosine interaction domain. Cell. 1995;80:693–694. doi: 10.1016/0092-8674(95)90347-x. [DOI] [PubMed] [Google Scholar]
- 8.Brouillet E, Trembleau A, Galanaud D, Volovitch M, Bouillot C, Valenza C, Prochiantz A, Allinquant B. The amyloid precursor protein interacts with Go heterotrimeric protein within a cell compartment specialized in signal transduction. J Neurosci. 1999;19:1717–1727. doi: 10.1523/JNEUROSCI.19-05-01717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulson EJ, Barrett GL, Storey E, Bartlett PF, Beyreuther K, Masters CL. Down-regulation of the amyloid protein precursor of Alzheimer's disease by antisense oligonucleotides reduces neuronal adhesion to specific substrata. Brain Res. 1997;770:72–80. doi: 10.1016/s0006-8993(97)00757-9. [DOI] [PubMed] [Google Scholar]
- 11.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 12.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 13.Duilio A, Faraonio R, Minopoli G, Zambrano N, Russo T. Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer's β-amyloid precursor protein. Biochem J. 1998;330:513–519. doi: 10.1042/bj3300513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finley R, Jr, Brent R. Interaction mating reveals binary and ternary connections between Drosophila cell cycle regulators. Proc Natl Acad Sci USA. 1994;91:12980–12984. doi: 10.1073/pnas.91.26.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 16.Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I. G protein βγ complex mediated apoptosis by familial Alzheimer's disease mutant of APP. EMBO J. 1997;16:4897–4907. doi: 10.1093/emboj/16.16.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gietz RD, Schiestl RH. Transforming yeast with DNA. Mol Cell Biol. 1995;5:255–269. [Google Scholar]
- 18.Gillian AM, McFarlane I, Lucy FM, Overly C, McConlogue L, Breen KC. Individual isoforms of the amyloid β precursor protein demonstrate differential adhesive potentials to constituents of the extracellular matrix. J Neurosci Res. 1997;49:154–160. doi: 10.1002/(sici)1097-4547(19970715)49:2<154::aid-jnr4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 19.Gubler U, Hoffman BJ. A simple and very efficient method for generating cDNA libraries. Gene. 1983;25:263–269. doi: 10.1016/0378-1119(83)90230-5. [DOI] [PubMed] [Google Scholar]
- 20.Guenette SY, Chen J, Jondro PD, Tanzi RE. Association of a novel human FE65-like protein with the cytoplasmic domain of the β-amyloid precursor protein. Proc Natl Acad Sci USA. 1996;93:10832–10837. doi: 10.1073/pnas.93.20.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardy J. Framing β-amyloid. Nat Genet. 1992;1:233–234. doi: 10.1038/ng0792-233. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto Y, Niikura T, Ito Y, Nishimoto I. Multiple mechanisms underlie neurotoxicity by different types of Alzheimer's disease mutations of amyloid precursor protein. J Biol Chem. 2000;275:34541–34551. doi: 10.1074/jbc.M005332200. [DOI] [PubMed] [Google Scholar]
- 23.Hirai S, Izawa M, Osada S, Spyrou G, Ohno S. Activation of the JNK pathway by distantly related protein kinases, MEKK and MUK. Oncogene. 1996;12:641–650. [PubMed] [Google Scholar]
- 24.Jung SS, Nalbantoglu J, Cashman NR. Alzheimer's β-amyloid precursor protein is expressed on the surface of immediately ex vivo brain cells: a flow cytometric study. J Neurosci Res. 1996;46:336–348. doi: 10.1002/(SICI)1097-4547(19961101)46:3<336::AID-JNR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Kang J, Lemaire H-G, Unterback A, Salbaum JM, Masters CL, Grezeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 26.Kavanaugh WM, Williams LT. An alternative to SH2 domains for binding tyrosine-phosphorylated proteins. Science. 1994;266:1862–1865. doi: 10.1126/science.7527937. [DOI] [PubMed] [Google Scholar]
- 27.Kavanaugh WM, Turck CW, Williams LT. PTB domain binding to signaling proteins through a sequence motif containing phosphotyrosine. Science. 1995;268:1177–1179. doi: 10.1126/science.7539155. [DOI] [PubMed] [Google Scholar]
- 28.Kelkar N, Gupta S, Dickens M, Davis RJ. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol Cell Biol. 2000;20:1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim IJ, Lee KW, Park BY, Lee JK, Park J, Choi IY, Eom SJ, Chang TS, Kim MJ, Yeom YI, Chang SK, Lee YD, Choi EJ, Han PL. Molecular cloning of multiple splicing variants of JIP-1 preferentially expressed in brain. J Neurochem. 1999;72:1335–1343. doi: 10.1046/j.1471-4159.1999.721335.x. [DOI] [PubMed] [Google Scholar]
- 30.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 31.Luo JJ, Wallace W, Riccioni T, Ingram DK, Roth GS, Kusiak JW. Death of PC12 cells and hippocampal neurons induced by adenoviral-mediated FAD human amyloid precursor protein gene expression. J Neurosci Res. 1999;55:629–642. doi: 10.1002/(SICI)1097-4547(19990301)55:5<629::AID-JNR10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Marcinkiewicz M, Seidah NG. Coordinated expression of β-amyloid precursor protein and the putative β-secretase BACE and α-secretase ADAM10 in mouse and human brain. J Neurochem. 2000;75:2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- 33.Meroni G, Reymond A, Alcalay M, Borsani G, Tanigami A, Tonlorenzi R, Nigro CL, Messali S, Zollo M, Ledbetter DH, Brent R, Ballabio A, Carrozzo R. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997;16:2892–2906. doi: 10.1093/emboj/16.10.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer D, Liu A, Margolis B. Interaction of c-Jun amino-terminal kinase interacting protein-1 with p190 rhoGEF and its localization in differentiated neurons. J Biol Chem. 1999;274:35113–35118. doi: 10.1074/jbc.274.49.35113. [DOI] [PubMed] [Google Scholar]
- 35.Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mooser V, Maillard A, Bonny C, Steinmann M, Shaw P, Yarnall DP, Burns DK, Schorderet DF, Nicod P, Waeber G. Genomic organization, fine-mapping, and expression of the human Islet-Brain 1 (IB1)/c-jun-amino-terminal kinase interacting protein-1 (JIP-1) gene. Genomics. 1999;55:202–208. doi: 10.1006/geno.1998.5641. [DOI] [PubMed] [Google Scholar]
- 37.Murayama Y, Takeda S, Yonezawa K, Giambarella U, Nishimoto I, Ogata E. Cell surface receptor function of amyloid precursor protein that activates Ser/Thr kinases. Gerontology. 1996;42[Suppl 1]:2–11. doi: 10.1159/000213818. [DOI] [PubMed] [Google Scholar]
- 38.Nishimoto I, Okamoto T, Matsuura Y, Takahashi S, Okamoto T, Murayama Y, Ogata E. Alzheimer amyloid protein precursor complexes with brain GTP-binding protein Go. Nature. 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto T, Takeda S, Murayama Y, Ogata E, Nishimoto I. Ligand dependent G protein coupling function of amyloid transmembrane precursor. J Biol Chem. 1995;270:4205–4208. doi: 10.1074/jbc.270.9.4205. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto T, Takeda S, Giambarella U, Matsuura Y, Katada T, Nishimoto I. Intrinsic G-coupling function of amyloid precursor protein as a novel target of V642 mutations linked to familial Alzheimer disease. EMBO J. 1996;15:3769–3777. [PMC free article] [PubMed] [Google Scholar]
- 41.Pawson T, Scott JD. Signaling though scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 42.Pellet J-B, Haefliger J-A, Staple JK, Widmann C, Welker E, Hirling H, Bonny C, Nicod P, Catsicas S, Waeber G, Riederer BM. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur J Neurosci. 2000;12:621–632. doi: 10.1046/j.1460-9568.2000.00945.x. [DOI] [PubMed] [Google Scholar]
- 43.Perez RG, Zheng H, Van der Ploeg LHT, Koo EH. The β-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pleasure SJ, Lee VM. NTera 2 cells: a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J Neurosci Res. 1993;35:585–602. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 45.Qiu WQ, Ferreira A, Miller C, Koo E, Selkoe DJ. Cell-surface β-amyloid precursor protein stimulates neurite outgrowth of hippocampal neurons in an isoform-dependent manner. J Neurosci. 1995;15:2157–2167. doi: 10.1523/JNEUROSCI.15-03-02157.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohn TT, Ivins KJ, Bahr BA, Cotman CW, Cribbs DH. A monoclonal antibody to amyloid precursor protein induces neuronal apoptosis. J Neurochem. 2000;74:2331–2342. doi: 10.1046/j.1471-4159.2000.0742331.x. [DOI] [PubMed] [Google Scholar]
- 47.Sabo SL, Lanier LM, Ikin AF, Khorkova O, Sahasrabudhe S, Greengard P, Buxbaum JD. Regulation of β-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J Biol Chem. 1999;274:7952–7957. doi: 10.1074/jbc.274.12.7952. [DOI] [PubMed] [Google Scholar]
- 48.Sanchez I, Hughes RT, Mayer B, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 49.Sastre M, Turner RS, Levy E. X11 interaction with β-amyloid precursor protein modulates its cellular stabilization and reduces amyloid β-protein secretion. J Biol Chem. 1998;273:22351–22357. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- 50.Smyth MD, Kesslak JP, Cummings BJ, Cotman CW. Analysis of brain injury after intrahippocampal administration of β-amyloid in streptozotocin-treated rats. Neurobiol Aging. 1994;15:153–159. doi: 10.1016/0197-4580(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 51.Songyang Z, Margolis B, Chaudhuri M, Shoelson SE, Cantley LC. The phosphotyrosine interaction domain of Shc recognizes tyrosine phosphorylated NPXY motif. J Biol Chem. 1995;270:14863–14866. doi: 10.1074/jbc.270.25.14863. [DOI] [PubMed] [Google Scholar]
- 52.Sudo H, Jiang H, Yasukawa T, Hashimoto Y, Niikura T, Kawasumi M, Matsuda S, Takeuchi Y, Aiso S, Matsuoka M, Murayama Y, Nishimoto I. Antibody-regulated neurotoxic function of cell surface β-amyloid precursor protein. Mol Cell Neurosci. 2000;16:708–723. doi: 10.1006/mcne.2000.0910. [DOI] [PubMed] [Google Scholar]
- 53.Tomita S, Ozaki T, Taru H, Oguchi S, Takeda S, Yagi Y, Sakiyama S, Kirino Y, Suzuki T. Interaction of a neuron-specific protein containing PDZ domains with Alzheimer's amyloid precursor protein. J Biol Chem. 1999;274:2243–2254. doi: 10.1074/jbc.274.4.2243. [DOI] [PubMed] [Google Scholar]
- 54.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 55.Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Milklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet. 2000;24:291–295. doi: 10.1038/73523. [DOI] [PubMed] [Google Scholar]
- 56.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 57.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D'Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 58.Yamatsuji T, Okamoto T, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Nishimoto I. Neuronal DNA fragmentation by familial Alzheimer's V642 mutants of APP via heteromeric G proteins. Science. 1996a;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 59.Yamatsuji T, Okamoto T, Takeda S, Murayama Y, Tanaka N, Nishimoto I. Expression of V642 APP mutant causes cellular apoptosis as Alzheimer trait linked phenotype. EMBO J. 1996b;15:498–509. [PMC free article] [PubMed] [Google Scholar]
- 60.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP Group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao B, Chrest FJ, Horton WE, Jr, Sisodia SS, Kusiak JW. Expression of mutant amyloid precursor proteins induces apoptosis in PC12 cells. J Neurosci Res. 1997;47:253–263. [PubMed] [Google Scholar]