

Abstract
Glia undergo inflammatory activation in most CNS pathologies and are capable of killing cocultured neurons. We investigated the mechanisms of this inflammatory neurodegeneration using a mixed culture of neurons, microglia, and astrocytes, either when the astrocytes were activated directly with lipopolysaccharide (LPS) and interferon-γ (IFN-γ) or LPS/IFN-γ-activated microglia were added to mixed neuronal cultures. In either case, activated glia caused 75–100% necrotic cell death within 48 hr, which was completely prevented by inhibitors of inducible nitric oxide synthase (iNOS) (aminoguanidine or 1400W). Activated astrocytes or microglia produced nitric oxide (NO) (steady-state level ∼0.5 μm), which immediately inhibited the cellular respiration of cocultured neurons, as did authentic NO. NO donors also decreased ATP levels and stimulated lactate production by neurons, consistent with NO-induced respiratory inhibition. NO donors or a specific respiratory inhibitor caused rapid (<1 min) release of glutamate from neuronal and neuronal–astrocytic cultures and subsequent neuronal death that was blocked by an antagonist of NMDA receptor (MK-801). MK-801 also blocked neuronal death induced by activated glia. High oxygen also prevented NO-induced neuronal death, consistent with death being induced by NO inhibition of cytochrome c oxidation in competition with oxygen. Thus activated glia kill neurons via NO from iNOS, which inhibits neuronal respiration resulting in glutamate release and subsequent excitotoxicity. This may contribute to neuronal cell death in inflammatory, infectious, ischemic, and neurodegenerative diseases.
Keywords: nitric oxide, mitochondria, astrocytes, microglia, neurons, inflammation
Astrocytes and/or microglia become “activated” by inflammatory mediators in a wide range of CNS pathologies, including brain inflammation, trauma, ischemia, and stroke; brain infections such as AIDS dementia, meningitis, and malaria; neurodegenerative diseases such as Alzheimer's, Parkinson's, Huntington's, multiple sclerosis, amyotrophic lateral sclerosis; and normal aging (Eddleston and Mucke, 1993; Kreutzberg, 1996). Glial activation involves changes in cell phenotype and gene expression, including the de novo expression of major histocompatibility complex class I and II antigens, cell adhesion molecules, cytokines such as tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β), and the inducible isoform of nitric oxide synthase (iNOS).
Glial activation is thought to be protective via destruction of pathogens, removal of debris, and promotion of tissue repair; however, excess activation can be deleterious (Banati et al., 1993; Hewett et al., 1994; Bolanos et al., 1997). Activated glia can kill neurons in coculture (Chao, 1996; Hu et al., 1997; Kingham et al., 1999; Tanabe et al., 1999), and this may occur in vivo during brain trauma, inflammation, post-ischemia, and infection, and in neurodegenerative diseases (Loihl and Murphy, 1998; Bolanos and Almeida, 1999; Liberatore et al., 1999). The mechanisms by which activated glia kill neurons in culture have been suggested to include the release of nitric oxide (Chao, 1996; Bolanos et al., 1997; Hu et al., 1997; Loihl and Murphy, 1998), reactive oxygen species (Beckman et al., 1994; Chao et al., 1995b), glutamate (Barger and Basile, 2001), TNFα, and IL-1β (Chao et al., 1995a; Viviani et al., 1998). NO is released from activated astrocytes and microglia (Brown et al., 1995; Chao, 1996; Murphy, 2000), and neurons are remarkably sensitive to NO-induced cell death (Leist et al., 1997a; Wei et al., 2000). The mechanisms of NO neurotoxicity are still unclear, but have been proposed to include the following: (1) activation of poly (ADP ribose) polymerase followed by NAD+ and ATP depletion (Zhang et al., 1994), (2) induction of apoptosis by poorly defined mechanisms (Leist et al., 1997b; Tamatani et al., 1998; Uehara et al., 1999), and (3) glutamate release (Meffert et al., 1994; Trabace and Kendrick, 2000) and excitotoxicity (Hewett et al., 1994; Leist et al., 1997a) and inhibition of mitochondrial respiration (Heals et al., 1999).
NO can potently, acutely, and reversibly inhibit mitochondrial respiration at cytochrome c oxidase in competition with oxygen (Brown and Cooper, 1994; Brown, 1999). We have shown previously that NO from activated astrocytes reversibly inhibits the cellular respiration of those astrocytes (Brown et al., 1995) and that NO reversibly inhibits the respiration of isolated nerve terminals (synaptosomes) at nanomolar concentrations (Brown and Cooper, 1994), causing acute glutamate release from the nerve terminals caused by the inhibition of cytochrome c oxidase (McNaught and Brown, 1998). Thus a possible mechanism of glial-induced neuronal death is NO from activated glia causing inhibition of neuronal respiration, leading to release of glutamate, which subsequently causes excitotoxic death of the neurons. We set out to test this and related hypotheses here.
MATERIALS AND METHODS
Neuronal cell culture. Cerebellar granule cells (CGCs) were prepared from 7-d-postnatal Wistar rats as described previously by Cambray-Deakin (1995) with some modifications. In brief, the cerebella were dissociated in Versene solution (1:5000; Life Technologies, Paisley, UK) and plated at 0.25 × 106 cells/cm2in 24-well plates (in 500 μl of DMEM) coated with poly-l-ornithine hydrochloride (15 μg/ml; Sigma, Paisley, UK). Cultures were maintained in DMEM (Life Technologies) supplemented with heat-inactivated horse serum (5%, Life Technologies) and fetal calf serum (5%; Sigma), 2 mml-glutamine, 25 mm KCl, and 10 μg/ml gentamicin. Cytosine-d-arabinoside (10 μm; ara-C, Sigma) was added to some cultures 24 hr after plating to inhibit non-neuronal cell proliferation, which are called “neuronal cultures” here (0.5 ± 0.1% of astrocytes and 0.4 ± 0.2% of microglia). Cultures of CGCs untreated with ara-C (called “neuronal–astrocytic cultures”) contained 12.2 ± 2.8% of astrocytes and 3.1 ± 0.7% of microglia as assessed immunocytochemically using antibodies against glial fibrillary acidic protein (GFAP; marker of astrocytes) and OX-42 (marker of microglia). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2/95% air. Neurons were routinely used at 9 d in vitro (DIV) because formation of synapses and response to NMDA receptor stimulation in the culture of CGCs are high after 8 DIV (Leist et al., 1997a).
Astrocyte and microglial cultures. Primary, mixed glial cell cultures were prepared from the cerebral cortex of 7-d-old rats (Wistar) as described previously (Bal et al., 1994). The same rat brains were used for isolation of CGCs. Briefly, cells isolated from cerebral hemispheres were dissociated in HBSS containing 0.25% trypsin (Sigma) and 0.02 mg/ml deoxyribonuclease I (Sigma-Aldrich, Steinheim, Germany) and plated at a density of 0.1 × 106 cells/cm2in 75 cm2 culture flasks (Falcon) in DMEM with 10% fetal calf serum. At confluency (12–14 DIV), primary glial cultures were used to isolate microglial cells as described previously (Taupenot et al., 1996). Briefly, mixed glial cultures were shaken to dislodge microglia that were loosely attached to the astrocytes. Microglia were purified by preplating for 30 min into culture flasks (75 cm2) at a density of 0.1 × 106cells/cm2, and then the contaminating cells were removed by changing the medium. Microglia were maintained in astrocyte-conditioned medium (medium collected from astrocytic cultures after 2 d and spun down) mixed 1:1 v/v with fresh DMEM (containing 10% fetal calf serum). Microglial cultures were used 24 hr after plating. The purity of the microglial and astrocyte culture (after isolation of microglia) was determined immunocytochemically with OX-42 (microglia marker, an anti-CR3 complement receptor antibody; Serotec, Oxford, UK) and anti-GFAP antibody (an astrocytic marker; AutogenBioclear, Calne, UK). The cells were fixed in 4% paraformaldehyde (Sigma) and then incubated with OX-42 or anti-GFAP (all at 1:200 dilutions) and visualized using biotinylated anti-mouse IgG antibodies (1:200 dilution), avidin–biotin–horseradish peroxidase complex, and diaminobenzidine tetrahydrochloride (ABC staining System; Santa Cruz Biotechnology, Santa Cruz, CA). Of the cells in microglial cultures, 99.5 ± 0.3% were positive for OX-42, the marker for macrophage/microglial cell types (GFAP-positive cells were not present). In astrocyte culture, 97–98% cells were anti-GFAP positive and only 2–3% cells were OX-42 positive (microglia).
Activation of microglia and astrocytes in culture. Cultures of astrocytes (12–14 DIV, confluent, 75 cm2 culture flasks, after shaking off microglia) or microglia (24 hr after isolation from mixed glial cultures) were activated by exposure to lipopolysaccharide (LPS) fromSalmonella typhimurium (10 μg/ml, Sigma, St. Louis, MO) and interferon-γ (IFN-γ, 100 U/ml; Sigma) in the presence of arginase (0.2 U/ml) for 16–18 hr. After the activation time, the microglia or astrocytes were gently trypsinized (0.1%) for 2–3 min (at 37°C), and the cells were spun down and resuspended in DMEM or Krebs-HEPES buffer consisting of (in mm): 1.5 CaCl2, 5.6 glucose, 10 HEPES, 4.7 KCl, 1.2 KH2PO4, 1.1 MgSO4, 118 NaCl, pH 7.4.
Assessment of cell viability and morphology. The viability of CGCs was estimated by propidium iodide (20 μg/ml) (Sigma) and Hoechst 33342 (10 μg/ml) (Sigma) staining using a fluorescence microscope (Leica). Propidium iodide stained only the cells with disrupted plasma membrane integrity, so these cells were considered to be necrotic. The nuclear morphology of the cell (chromatin condensation and fragmentation) was studied using the cell-permeable DNA dye Hoechst 33342 to assess whether apoptotic cells were present. Cells with homogeneously stained nuclei were considered to be viable. Additionally, to distinguish between necrosis and apoptosis at the single-cell level, annexin-V (Roche Diagnostics GmbH, Mannheim, Germany) and propidium iodide staining were performed simultaneously. Cells positive for both annexin-V and propidium iodide were considered to be necrotic, and cells that were annexin-V positive only (propidium iodide negative) were considered to be apoptotic.
Necrotic and apoptotic neurons were counted in three microscopic fields in each well (three wells per treatment) and expressed as a percentage of the total number of neurons. Each exposure was repeated at least three times. To estimate the number of dead cells that disappeared in cocultures of CGCs with activated microglia, the total number of dead and live cells was quantified and compared with the total number of cells in sister (nontreated, control) cultures.
Measurement of NO generation and oxygen consumption. Measurement of NO production and oxygen consumption of cells was performed using a Clark-type NO electrode (World Precision Instruments) inserted through the top of a thermostated, stirred, and sealed vessel with a Clark-type oxygen electrode in the base (Rank Brothers), permitting simultaneous measurement of NO and O2levels (Brown and Cooper, 1994). The NO electrode was calibrated with aliquots of NO-saturated water, assumed to contain 2 mm NO. After gentle scraping, CGCs (9 DIV), in sheets from 25 cm2 culture flasks (∼4.0 × 106 cells), were transferred into the above-described vessel maintained at 37°C in 0.5 ml of Krebs-HEPES buffer, pH 7.4, additionally supplemented with 2 mm glutamine and 25 mm KCl. After a few minutes (∼5 min), the activated astrocytes or microglia (∼0.5 × 106 of cells) were added to the vessel followed by the addition of arginine (200 μm; NOS substrate). At the end of the experiment, 10 μm hemoglobin (NO scavenger) was added to verify whether inhibition of neuronal respiration is reversible.
Measurement of intracellular ATP levels in neuronal and astrocyte cultures. ATP was determined luminometrically (Jade luminometer, Labtech International) using ATP Bioluminescence Assay Kit (Boehringer Mannheim) according to the provided protocol. Briefly, after the exposure of astrocyte culture (12–14 DIV, confluent, 24-well plate) or CGCs (in neuronal or neuronal–astrocytic cultures, 9 DIV, 0.25 × 106/cm2, cultured in 24-well plates) to NO donor, NOC-18 (500 μm), for various intervals of time (5, 10, or 20 min or 4 and 24 hr), the medium was withdrawn (stored for measurements of the lactate level) and the cells were exposed to lysed buffer mixed with dilution buffer for 5 min and than harvested by scraping. The aliquots of cellular extract were assayed for ATP content using the ATP dependency of the light-emitting luciferase-catalyzed oxidation of luciferin. ATP concentrations were expressed as percentage of control.
Determination of lactate accumulation. Aliquots of deproteinized culture medium were assayed for lactate using al-lactic acid kit (Boehringer Mannheim) according to the provided protocol. The amount of lactate was measured by monitoring the oxidation of l-lactic acid to pyruvate by NAD in the presence of l-lactate dehydrogenase. The equilibrium of the reaction was displaced in favor of pyruvate and NADH formation by glutamate-pyruvate transaminase in the presence of glutamate. NADH formation was monitored at 340 nm and was proportional to lactate concentration.
Measurement of glutamate content in neuronal conditioned medium. CGCs for measurements of glutamate release were cultured in 25 cm2 culture flasks at high density (0.6–1.0 × 106 of trypan blue-excluding cells per square centimeter) for 9 d in the presence or absence of 10 μm ara-C (neuronal and neuronal–astrocytic cultures, respectively). The volume of the medium in the culture flasks with CGCs was reduced to 2.5 ml just before exposure to NOC-18 (500 μm) or myxothiazol (2 μm) for various intervals of time (5, 10, or 20 min or 24 hr). After this time the deproteinized medium of cultured CGCs was assessed for levels of glutamate by a colorimetric method coupled to glutamate dehydrogenase and a formazan end product using a commercially available kit (Boehringer Mannheim). In brief, diaphorase, iodonitrotetrazolium chloride, and conditioned culture medium (after deproteinization) were combined (according to the provided protocol) and incubated for 2 min. Then 3.0 U of glutamate dehydrogenase solution was added, and the absorbability was measured at 492 nm after 15 min and then every 3 min until the reaction reached steady state. A standard curve was constructed by adding known concentrations of glutamate to culture medium.
Statistical analysis. Data are expressed as mean ± SD and were analyzed for significance using ANOVA.
RESULTS
Nitric oxide produced by activated microglia and astrocytes induces excitotoxic cell death of CGCs in culture
To determine whether activated microglia could kill neurons, we activated microglia with LPS/IFN-γ for 16–18 hr, washed them, then cocultured them with CGCs (85% neuronal/12% astrocytic cultures, 9 DIV, 0.25 × 106cells/cm2 in 500 μl of DMEM, 24-well plate) at three different densities (0.1, 0.2, and 0.3 × 106 cells of activated microglia per square centimeter) for 24 hr. After 24 hr the cell death of CGCs was assessed by propidium iodide and Hoechst 33342 staining and compared with a control culture (not exposed to activated microglia). Control cultures of CGCs (neuronal–astrocytic, 9 DIV) showed well differentiated neurons with an extensive neuritic network (Fig.1A) and very low necrotic cell death (Fig. 2). After addition of activated microglia (at any density), the first morphological change, evident after 3–4 hr, was retraction of all neurites (data not shown). After 24 hr of coculture of CGCs with activated microglia at the lowest density (0.1 × 106of cells per square centimeter), only 20 ± 5.0% of neurons were viable, and the rest were necrotic (propidium iodide-positive cells, 34.8 ± 5.4%), had condensed DNA (Hoechst-positive cells, 12.7 ± 2.4%), or had disappeared (32.5 ± 3.8%, as compared with the number of neurons in control cultures), presumably because of phagocytosis by activated microglia. Hoechst-positive cells displayed highly fluorescent nuclei with condensed chromatin, but mostly without fragmentation, a phenotype not present in control cultures. When CGCs where cocultured with activated microglia at the higher density (0.2 or 0.3 × 106cells/cm2), none of the neurons survived. At the density of 0.2 × 106cells/cm2(Fig. 1B), both necrotic and Hoechst-positive cells were observed, and some of the neurons disappeared (Fig. 2). At the highest density (0.3 × 106cells/cm2) of activated microglia, only necrotic cell death was observed (14.8 ± 3.8%), and the rest of the neurons disappeared (85.2 ± 10.8%), presumably because of phagocytosis by activated microglia.
Fig. 1.
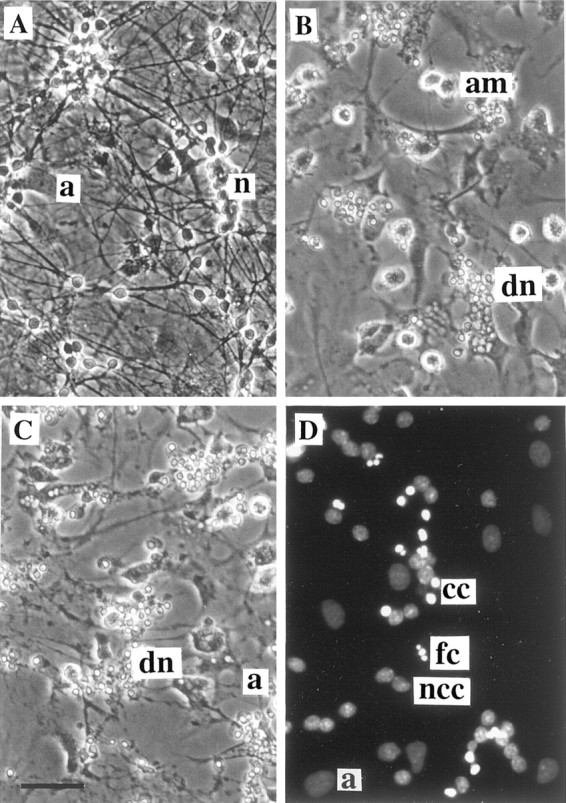
Cell death of CGCs (neuronal–astrocytic cultures, 9 DIV) induced by coculture with activated microglia or by direct exposure to proinflammatory cytokines or NOC-18 (NO donor).A, In the control culture of CGCs, note the phase-bright normal cell bodies and dense neuritic network. B, Coculture of CGCs (9 DIV, 0.25 × 106cells/cm2) with LPS/IFN-γ-activated microglia (0.2 × 106cells/cm2) for 24 hr induced cell death of all neurons. Note shrunken cell bodies and nuclei and loss of all neurites (phase-contrast photographs). C, Addition of LPS (4 ng/ml) and IFN-γ (100 U/ml) simultaneously for 48 hr to the CGCs, cultured in the presence of glial cells (9 DIV, nontreated with ara-C), caused severe neuronal cell death, but dead cells were not phagocytosed. D, Morphological analysis of nuclear chromatin in CGC culture (neuronal–astrocytic, 9 DIV) exposed to 500 μm NOC-18 for 4 hr using DNA-binding fluorochrome Hoechst 33342 (fluorescence microscope). Viable cells showed round nuclei with weak fluorescence, but some nuclei had strongly condensed chromatin (bright fluorescence), predominantly without fragmentation. However, a few nuclei with fragmented chromatin were also present. Particular cell or nuclear types are indicated by the following abbreviations (placed to the right of the cell): n, healthy neurons; a, astrocyte; am, activated microglia; dn, dead neurons; cc, condensed chromatin; fn, fragmented chromatin;ncc, non-condensed chromatin. Scale bar (shown inC): A–C, 40 μm;D, 20 μm.
Fig. 2.

Cell death of CGCs (neuronal–astrocytic cultures, 9 DIV) induced by addition of LPS/IFN-γ-activated microglia (0.2 × 106 cells/cm2) for 24 hr. The presence of activated microglia caused predominantly necrotic cell death (PI-positive cells); however, Hoechst 33342-stained (HS) neurons (with condensed chromatin, occasionally with fragmentation) were also present. Also, some CGCs disappeared completely (presumably because of phagocytosis by activated microglia). Values represent the means ± SD of three or more separate cultures. In each experiment, three wells per treatment were analyzed, and the cells in three fields per well were counted (∼80 ± 25 cells per field). **p < 0.01, ***p < 0.001 from control groups.
The neuronal death observed in the presence of activated microglia was mediated by nitric oxide because the presence of Hoechst-positive or propidium iodide-positive neurons was almost entirely blocked by the iNOS inhibitors, 200 μm aminoguanidine (Sigma) or 25 μm 1400W (Alexis Biochemicals, Lausanne, Switzerland) (Fig. 3A). The production of NO in the cocultures of CGCs with activated microglia was confirmed by the presence of nitrite in the medium (19.5 ± 0.6, 22.3 ± 1.2, and 27.8 ± 0.9 μm in the presence of 0.1, 0.2, and 0.3 × 106 activated microglia per square centimeter, respectively) as measured by the Griess reaction (Schmidt and Kelm, 1996). After addition of activated microglia (0.1 × 106cells/cm2) in the presence of iNOS inhibitors, 200 μmaminoguanidine or 25 μm 1400W, the level of nitrite in the medium was significantly decreased (5.6 ± 0.4 and 4.4 ± 0.3 μm,respectively; control level, without added microglia, 3.8 ± 0.4 μm). The death of CGCs was also prevented by the NMDA noncompetitive receptor antagonist, 10 μm MK-801 (dizocilpine maleate, Calbiochem), added 30 min before activated microglia (Fig. 3A), suggesting that microglial NO-induced neuronal death was mediated by glutamate.
Fig. 3.
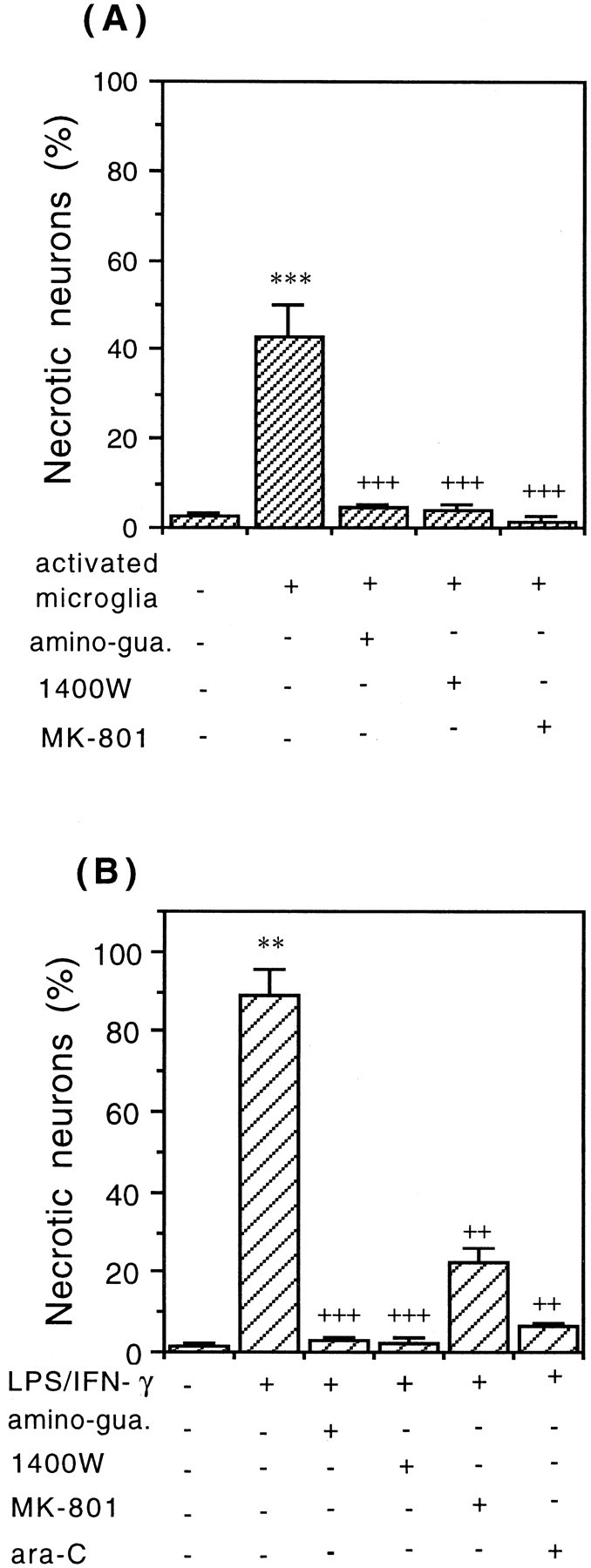
Necrotic cell death of CGCs (0.25 × 106cells/cm2) induced by activated microglia (A) or LPS/IFN-γ (B). Addition of 0.2 – 106cells/cm2 of LPS/IFN-γ-activated microglia for 24 hr (A) or LPS/IFN-γ for 48 hr (B) to the culture of CGCs (neuronal–astrocytic) caused necrosis (PI-positive cells). The necrotic neuronal cell death was completely prevented by iNOS inhibitors aminoguanidine (200 μm) or 1400W (25 μm), was wholly or partially blocked by an NMDA receptor antagonist (10 μm MK-801), and in the case of (B) was completely prevented by pretreatment with ara-C (to eliminate glial cells). Percentages of necrotic neurons (PI-positive cells) were calculated, and the values represent the means ± SD of at least three independent experiments. ***p < 0.001 from control groups and+++p < 0.001 from activated microglia (A) or ++p < 0.01, +++p < 0.001 from LPS/IFN-γ treatment (B).
To test whether activated astrocytes could kill neurons by similar mechanisms, neuronal–astrocytic cultures (9 DIV), untreated with ara-C (12.2 ± 2.8% of astrocytes and 3.1 ± 0.7% of microglia), were incubated with LPS (4 ng/ml) and IFN-γ (100 U/ml) for 48 hr (Fig. 1C). After that time only necrotic neuronal death was observed (propidium iodide-positive cells) and it was mediated by NO because the cell death was completely blocked by iNOS inhibitors (200 μm aminoguanidine or 25 μm 1400W) (Fig. 3B). The death of CGCs was also prevented by MK-801 (10 μm) (Fig.3B), suggesting again that glial NO-induced neurotoxicity was mediated by glutamate. When neuronal cultures (CGCs, 9 DIV, pretreated with ara-C to block the proliferation of glia; 0.5 ± 0.1% of astrocytes and 0.4 ± 0.2% of microglia as assessed immunocytochemically) were exposed to LPS/IFN-γ, no significant cell death was observed (Fig. 3B). This suggests that LPS/IFN-γ is not directly toxic to neurons, but rather toxicity is mediated by glia. The death of CGCs cultured in the presence of glia and LPS/IFN-γ was caused by NO released from activated glia cells because it could be prevented by iNOS inhibitors (Fig. 3B). In this case, activated astrocytes are the main source of NO because 9-d-old culture of CGCs contained 12.2 ± 2.8% of astrocytes and only 3.1 ± 0.7% of microglia. LPS or IFN-γ added to neuronal–astrocytic cultures separately did not cause any significant cell death.
NO-induced death of CGCs is prevented by MK-801
To test whether nitric oxide released from NO donors could reproduce cell death of neurons observed in the cocultures of CGCs with activated glia, CGCs (neuronal–astrocytic cultures) were exposed to either a “pure” NO donor NOC-18 (diethylenetriamine–nitric oxide adduct), also known as DETA-NONOate (RBI, Natick, MA; Sigma), or one capable of transnitrosylationS-nitroso-N-acetyl-dl-penicillamine (SNAP; Alexis) at different concentrations (50, 100, and 500 μm) for various intervals of time (4, 16, or 24 hr). The level of NO was measured using a NO-sensitive electrode. In the case of 0.5 mm SNAP, the release of NO was variable, but in most cases the steady-state level of NO measured by electrode was 0.5–0.75 μm in DMEM after 10–15 min. In the case of 0.5 mm NOC-18, the release of NO was less variable, and the steady-state level was between 0.4 and 0.6 μm in DMEM after 15–25 min.
Short incubations (4 hr) with either NO donor (NOC-18 or SNAP) at any concentration (50, 100, or 500 μm) caused little or no necrosis, as indicated by staining with propidium iodide. However, most of the cells at this time had strong chromatin condensation but mostly without nuclear fragmentation, as assessed by Hoechst 33342 staining (Fig. 1D). Additionally, only a small percentage of neurons (4–5%) bound annexin-V after the exposure to 500 μm NOC-18 or SNAP. These results suggest that NO-induced cell death had some apoptotic characteristic but without the classical morphological features (apoptotic-like cells). The presence of these apoptotic-like cells could not be prevented by a cell-permeable nonspecific caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (z-VAD-fmk; Alexis) at the concentration of 50 or 75 μm(100 μm was found to be toxic to the neurons), suggesting that these NO-induced changes may not be mediated by caspases. At this time (4 hr incubation), the presence of Hoechst-positive cells could be completely prevented by 10 μm MK-801 (noncompetitive inhibitor of NMDA receptor, added 30 min before NO donor), suggesting that NO-induced cell death of CGCs was mediated by glutamate (Fig.4A). After 16 hr of incubation with NO donors (SNAP or NOC-18), the number of Hoechst-positive cells was very low (between 3 and 5%; results not shown), but the presence of necrotic cells was significantly increased and almost completely prevented by MK-801 (10 μm) (Fig. 4B). After 24 hr of incubation with NO donors (NOC-18 or SNAP), only necrosis (presence of propidium iodide-positive cells) was observed. At that time, MK-801 (10 μm) could only partially prevent the presence of necrotic cells (Fig. 4C). Because the presence of Hoechst-positive and propidium iodide-positive cells was almost completely prevented by MK-801 (after 4 and 16 hr of incubation), this suggests that excitotoxicity was the main mechanism of granule cell death induced by nitric oxide.
Fig. 4.
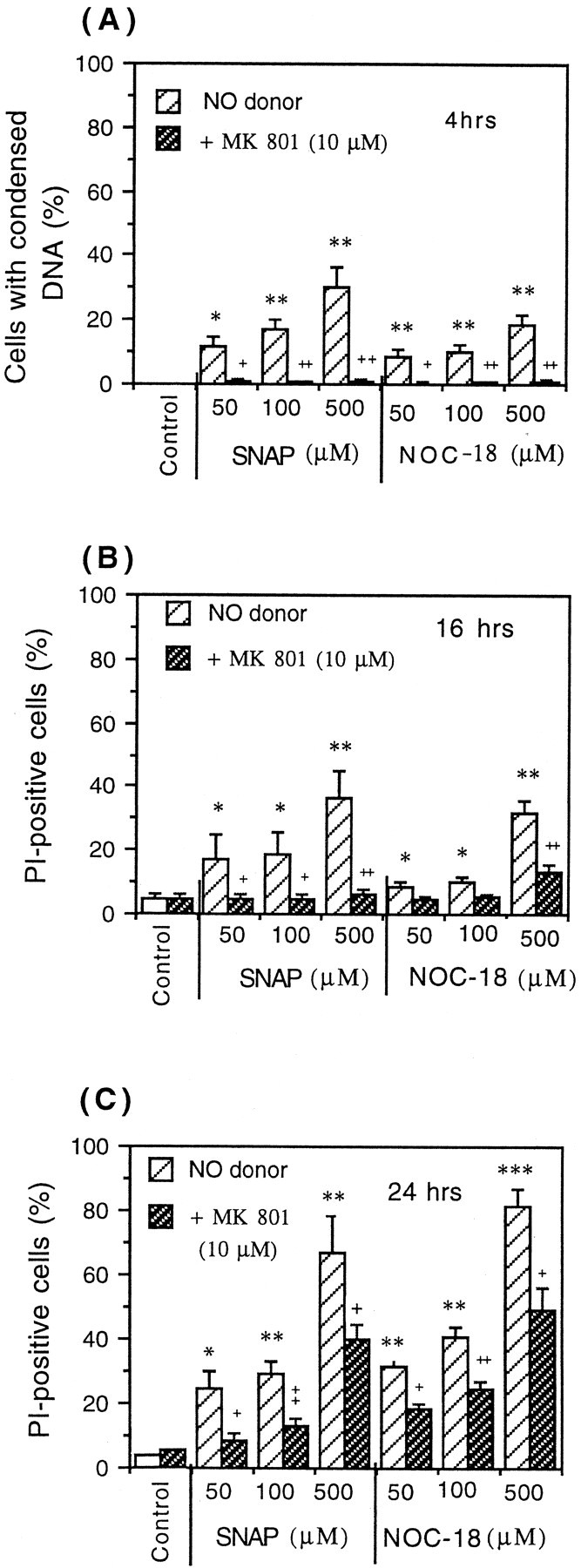
Induction of neuronal death in CGC cultures (neuronal–astrocytic, 9 DIV) by NO donors (NOC-18 or SNAP) after 4 hr (A), 16 hr (B), and 24 hr (C). After 4 hr of incubation with either NO donor (SNAP or NOC-18), Hoechst-positive neurons were observed (condensed chromatin, rarely with fragmentation) (Fig.1D), but no propidium iodide-staining cells were seen. After 16 or 24 hr of incubation, neurons died mainly by necrosis (PI-positive cells). Neuronal cell death induced by both NO donors (SNAP or NOC-18) was prevented by MK-801 (NMDA receptor antagonist) after 4 and 16 hr (but not 24 hr) of incubation. Values represent means ± SD (bars) of determinations made in three separate cultures. *p < 0.05, **p < 0.01, ***p < 0.001 from control groups and +p < 0.05,++p < 0.01,+++p < 0.001 from SNAP or NOC-18 treatment.
The neuronal death of CGCs was also observed 24 hr after the addition of very low concentrations of NO-saturated water (1 or 2 μm NO) (Fig.5A). The presence of necrotic cells after the exposure of CGCs to NO-saturated water or NOC-18 was blocked by two different NO scavengers: 25 μm2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl 3-oxide (PTIO; Sigma) (Fig. 5A) and 50 μm hemoglobin (Sigma) (Fig. 5B). After the exposure of CGCs (24 hr) to SNAP, necrotic neuronal death (25 ± 3, 28.9 ± 4.2, and 66.9 ± 11.5% after 50, 100, or 500 μmSNAP, respectively) was also significantly blocked by both NO scavengers: 50 μm hemoglobin (14.3 ± 2.3, 15.6 ± 2.1, and 25.4 ± 4.3%) and 25 μm PTIO (18.4 ± 4.6, 19.0 ± 4.2, and 36.2 ± 6.3%).
Fig. 5.
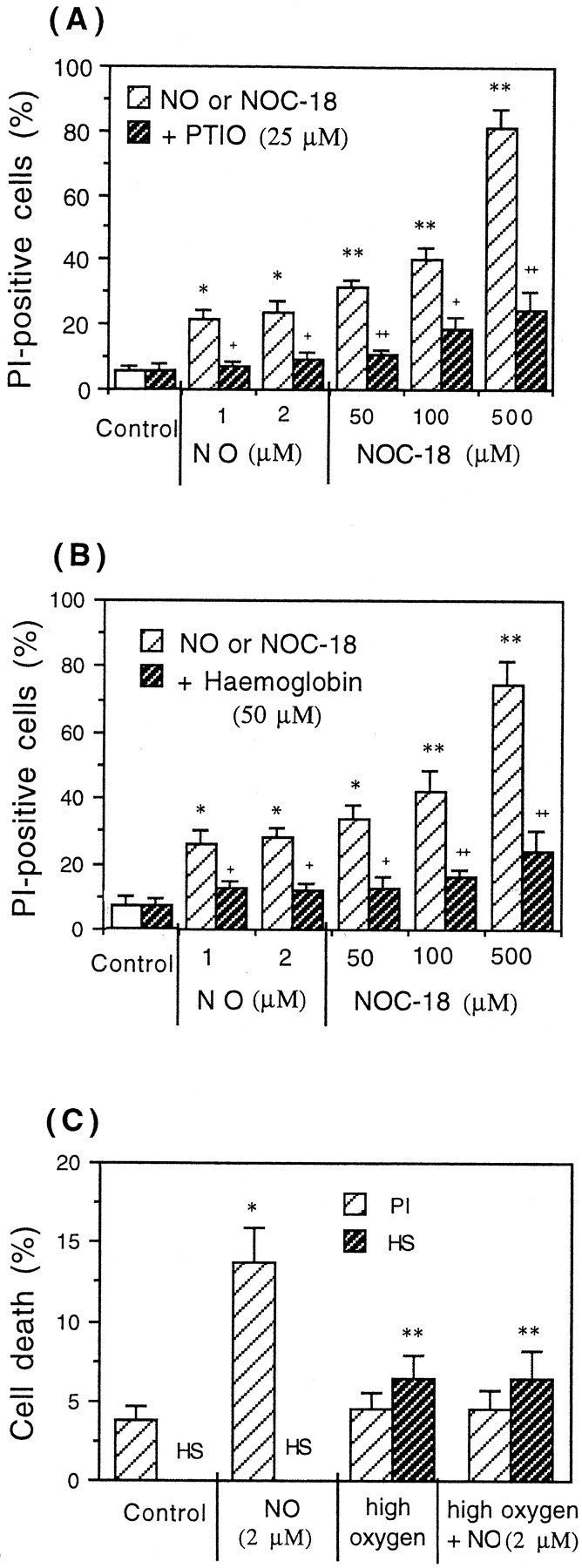
Necrotic cell death of CGCs (PI-positive cells) induced by NO-saturated water or NO donor (NOC-18) after 24 hr of exposure was prevented by NO scavengers PTIO (A) or hemoglobin (B) or by high oxygen (95% O2/5% CO2) (C). The data present the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 from control groups and+p < 0.05,++p < 0.01 from NO or NOC-18 treatment.
The applied concentrations of NOC-18 or SNAP caused cell death of neurons only, without affecting the viability of glial cells. The fact that a single addition of 1 or 2 μm of authentic NO caused significant cell death (18–22%) after 24 hr indicates that these neurons are very sensitive to NO-induced cell death and that because the NO breaks down rapidly (Fig.6A), the NO needs to be present for only a few minutes to initiate cell death. Interestingly, NO-induced necrosis of CGCs (after addition of NO-saturated water) was prevented when the medium was transiently oxygenated with 95% O2 and 5% CO2 before NO exposure (Fig. 5C). However, this high oxygen level on its own induced some apoptotic-like (Hoechst-positive) CGCs (Fig.5C). We have shown previously that oxygen antagonizes the NO-induced inhibition of respiration and glutamate release in synaptosomes, because nitric oxide binds competitively with oxygen to the same binding site on cytochrome oxidase (Brown and Cooper, 1994;McNaught and Brown,1998). Thus these results suggest that neuronal cell death could be mediated by NO-induced inhibition of mitochondrial respiration at cytochrome oxidase.
Fig. 6.
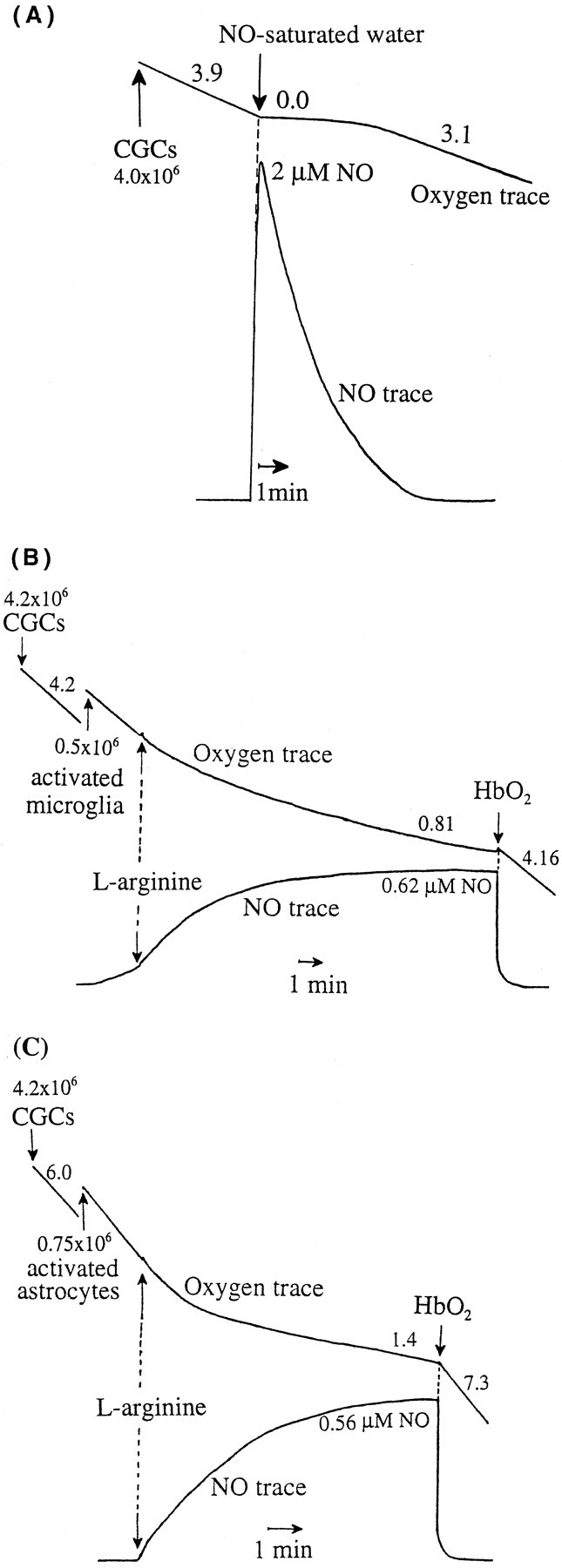
Rapid inhibition of mitochondrial respiration of CGCs by addition of 2 μm NO-saturated water (A) or NO produced by activated microglia (B) or activated astrocytes (C). A, Oxygen consumption (top trace) of CGCs (∼4.0 × 106 of cells) before addition of NO-saturated water was 3.9 natom of O/min/106 cells; after addition of NO-saturated water (2.0 μm NO), oxygen consumption was completely and immediately inhibited for the first few minutes, but the rate recovered as the NO level declined (3.1 natom of O/min/106 cells). B, Addition of activated microglia (∼0.5 × 106cells; NO level 0.62 μm) and arginine (200 μm; NOS substrate) caused rapid and strong inhibition of neuronal respiration (∼4.2 × 106cells; oxygen consumption, 4.2 natom of O/min/106 cells and 0.81 natom of O/min/106 cells before and after addition of activated microglia, respectively). The inhibition of neuronal respiration was reversible, because the addition of 10 μmhemoglobin (HbO2, NO scavenger) resulted in full activation of mitochondrial respiration (4.16 natom of O/min/106 cells). C, Addition of activated astrocytes (∼0.75 × 106 cells; NO level 0.56 μm) and arginine (200 μm) induced significant inhibition of oxygen consumption of CGCs (∼4.2 × 106 cells; oxygen consumption, 6.0 and 1.4 natom of O/min/106cells before and after addition of activated astrocytes, respectively), which was reversed by hemoglobin (HbO2) (7.3 natom of O/min/106 cells). Values placed next to the oxygen traces express oxygen consumption in natom of oxygen per minute per 106 cells.
Nitric oxide produced by activated microglia or astrocytes rapidly and reversibly inhibits mitochondrial respiration of surrounding neurons
We have shown previously that nanomolar concentrations of NO cause a rapid and reversible inhibition of mitochondrial respiration in brain synaptosomes (Brown and Cooper 1994) and astrocytes (Brown et al., 1995) caused by inhibition of cytochrome oxidase. We tested here whether NO could inhibit neuronal respiration in a similar way.
Oxygen consumption of CGCs alone (∼4.0 × 106 cells) was 3.9 natom of O/min/106 cells, but the addition of NO-saturated water (2 μm NO) caused immediate and complete inhibition of neuronal respiration (Fig.6A). However, it recovered once the NO levels in the medium were close to 0.0 μm (3.1 natom of O/min/106 cells) (Fig. 6A). Dramatic inhibition of neuronal respiration was also observed after addition of activated microglia (∼0.5 × 106 microglia added to 4.2 × 106 neurons), producing 0.55 ± 0.1 μm NO after addition of 200 μm arginine. Oxygen consumption was inhibited from 4.3 ± 0.4 in the absence of microglia to 0.75 ± 0.2 natom of O/min/106 in the presence of activated microglia, i.e., 82.0 ± 4.0% inhibition. This inhibition of respiration was reversible, because the addition of hemoglobin (NO scavenger) resulted in full activation of neuronal respiration (4.2 ± 0.2 nmol of O/min/106 cells) (means ± SD from nine independent experiments) (Fig. 6B, sample trace). Similar results were also observed when CGCs were incubated with activated astrocytes (Fig. 6C). Addition of activated astrocytes (∼0.75 × 106cells of astrocytes added to 4.2 × 106 neurons) producing 0.52 ± 0.16 μm NO caused strong and rapid inhibition of neuronal respiration (oxygen consumption before and after addition of activated astrocytes was 5.8 ± 0.26 and 1.2 ± 0.3 natom of O/min/106 cells, respectively, i.e., 79.0 ± 5.0% of inhibition), and the inhibition of respiration was completely reversed by hemoglobin (6.5 ± 0.8 natom of O/min/106 cells) (means ± SD from three independent experiments) (Fig. 6C, sample trace).
To test what levels of NO were produced by activated glial cells alone, activated microglia or astrocytes were incubated by themselves (∼1.0 × 106 cells), and the level of NO was measured using a NO electrode. They produced NO continuously to reach a steady-state level after 10–15 min of 1.1 ± 0.23 and 0.76 ± 0.35 μm (activated microglia and astrocytes, respectively) (data from three independent experiments).
Because NO produced by activated astrocytes or microglia inhibits neuronal respiration, this respiratory inhibition might contribute to neuronal cell death.
Respiratory inhibitors cause extensive neuronal cell death
To determine whether inhibition of mitochondrial respiration causes neuronal cell death, CGCs (neuronal–astrocytic cultures, 9 DIV) were exposed to specific respiratory inhibitors (myxothiazol, KCN, or azide) for various periods of time (4, 16, or 24 hr), and then the cell death was assessed by propidium iodide, Hoechst 33342, and annexin-V staining. After 4 hr of incubation with 2 μm myxothiazol (specific inhibitor of respiratory complex III) (Fig.7A), some necrotic cells were present (propidium iodide positive), but mainly Hoechst-positive neurons were observed (with bright and condensed chromatin, without fragmentation). The presence of Hoechst-positive cells could not be blocked by z-VAD-fmk. Only a very small percentage of cells was stained with annexin-V (between 1.0 and 2.0%). At this time of incubation, the presence of Hoechst-positive as well as propidium iodide-positive cells was almost completely prevented by MK-801 (NMDA receptor antagonist added 30 min before mitochondrial inhibitor) (Fig. 7A). After 16 hr of incubation, the number of necrotic cells increased, but MK-801 was less effective at blocking cell death (Fig. 7B). After 24 hr of incubation, almost all dead CGCs were necrotic (propidium iodide positive), and MK-801 was not effective at all in preventing cell death (Fig. 7C). Similar results were obtained after the exposure of CGCs to 2 mm azide or 2.5 mm KCN (inhibitors of cytochromec oxidase). In the case of azide, the percentage of Hoechst–positive cells was 64.5 ± 8.6, 32.9 ± 11.0, and 4.8 ± 1.5% in the absence of MK-801 and 0.5 ± 0.5, 29.0 ± 8.2, and 3.2 ± 1.8% in the presence of 10 μm MK-801 (after 4, 16, or 24 hr of incubation, respectively). Azide also caused necrotic cell death (propidium-positive cells): 8.9 ± 1.2, 52.1 ± 8.5, and 95.2 ± 2.3% in the absence of MK-801 and 2.8 ± 0.3, 54.4 ± 9.8, and 96.8 ± 3.4% of necrotic cells in the presence of MK-801 after 4, 16, or 24 hr of incubation, respectively. After exposure of CGCs to 2.5 mm KCN, Hoechst-positive cells were also present (54.0 ± 10.3, 26.4 ± 5.6, and 15.0 ± 3.8%, in the absence of MK-801), and these was partially blocked by 10 μm MK-801 (0.4 ± 0.3, 18.5 ± 4.9, and 14.8 ± 3.6% after 4, 16, and 24 hr of incubation, respectively). KCN also induced necrosis (12.1 ± 3.5, 73.6 ± 8.4, and 85.0 ± 9.8 in the absence of MK-801 and 2.3 ± 0.9, 45.2 ± 6.5, and 86.0 ± 8.4% in the presence of 10 μm MK-801 after 4, 16, and 24 hr of exposure, respectively).
Fig. 7.
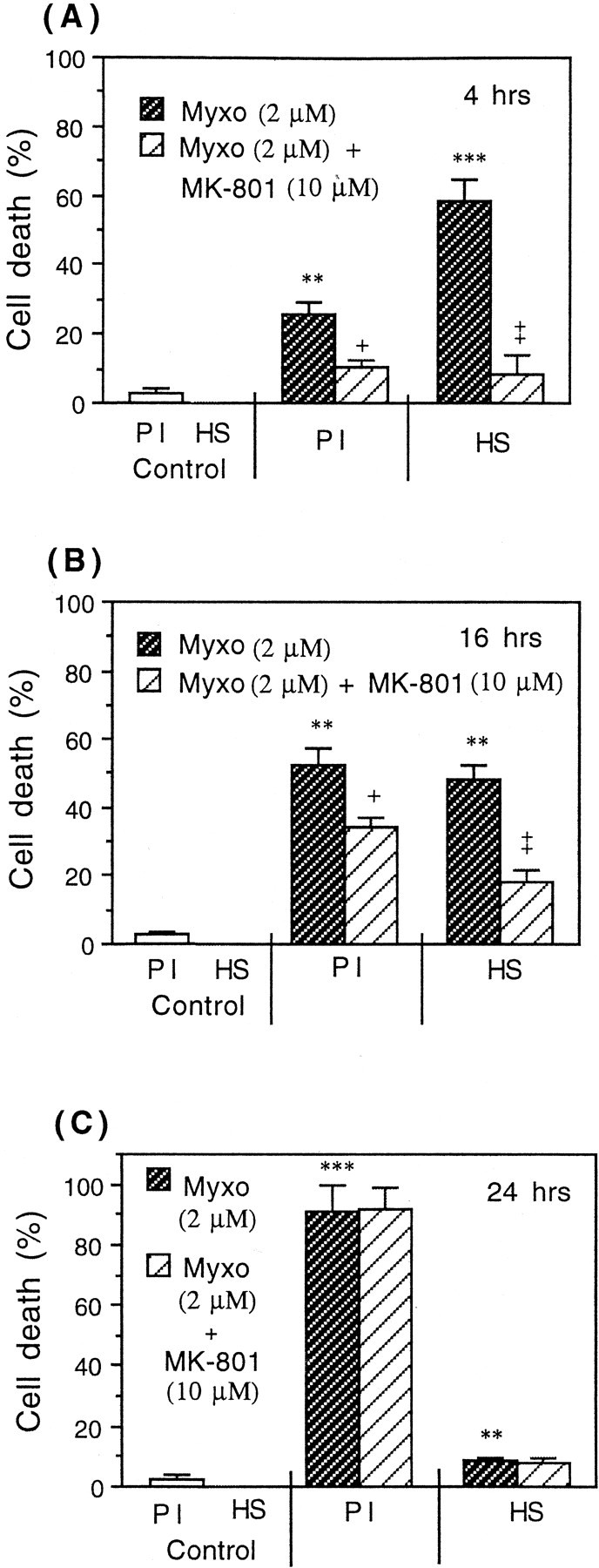
Induction of neuronal death in CGCs (neuronal–astrocytic cultures) by a specific mitochondrial inhibitor (2 μm myxothiazol) for 4 hr (A), 16 hr (B), and 24 hr (C) in the presence and absence of the noncompetitive NMDA receptor antagonist MK-801 (10 μm). A, After 4 hr of incubation with 2 μm myxothiazol (Myxo) mainly Hoechst-positive (HS) cells were observed (with condensed chromatin, without fragmentation). MK-801 prevented the neuronal death induced by myxothiazol after 4 hr (A) and partially after 16 hr of incubation (B) but not after 24 hr (C). Values represent means ± SD (bars) of determinations made in three separate cultures. **p < 0.01, ***p < 0.001 from control groups and +p < 0.05, ++p < 0.01 from myxothiazol treatment.
In general, the pattern of mitochondrial inhibitor- and NO donor-induced neuronal cell death was similar. Mitochondrial inhibitor- or NO donor-induced early condensation of DNA (4 hr incubation) was prevented by MK-801, but the later necrosis was only partially blocked by this NMDA receptor antagonist. In the case of respiratory inhibitors, MK-801 was generally less effective at blocking necrosis than in the case of NO donors. This might be because the respiratory inhibitor irreversibly inhibited neuronal respiration, whereas respiratory inhibition by NOC-18 was partially reversible with time.
Because NO inhibits respiration and specific respiratory inhibitors cause neuronal death of a similar type and time course (but with some differences), these results suggest that NO inhibition of mitochondrial respiration could be the main mechanism involved in triggering NO-induced neuronal death.
Nitric oxide rapidly depletes neuronal but not astrocytic ATP
Because NO-induced mitochondrial inhibition might result in energy depletion, ATP levels were studied in neuronal and neuronal–astrocytic cultures (CGCs, 9 DIV, treated or untreated with ara-C, respectively) and astrocytic cultures (12–14 DIV, confluent) exposed to NOC-18 (500 μm) for various intervals of time (5, 10, or 20 min or 4 and 24 hr). Severe and rapid ATP depletion (38.0 ± 7.0% in 20 min and 91.0 ± 1.0% after 24 hr) was observed in neuronal cultures (depleted of glial cells) (Fig.8A). In neuronal–astrocytic cultures, the decrease in ATP levels after exposure to NOC-18 (500 μm) was less dramatic but still significant (48.7 ± 1.8% after 24 hr) (Fig.8B). In contrast to neurons, astrocytes were much less sensitive to NO-induced ATP depletion. Indeed, short-time exposure to NOC-18 (500 μm) did not cause any ATP depletion (Fig. 8C), and 24 hr incubation caused only 10.0 ± 5.0% of ATP decrease when compared with the control culture (Fig. 8C). Even after 72 hr exposure to NOC-18 the ATP level had decreased by only 36.0 ± 4.0% (data not shown). However, astrocytes and neurons appeared to be equally sensitive to NO-induced inhibition of mitochondrial respiration (Brown et al., 1995) (Fig. 6A). These results are consistent with neurons being particularly dependent on mitochondrial energy production in contrast to astrocytes, which after NO-induced mitochondrial inhibition may be able to maintain ATP levels by glycolysis.
Fig. 8.
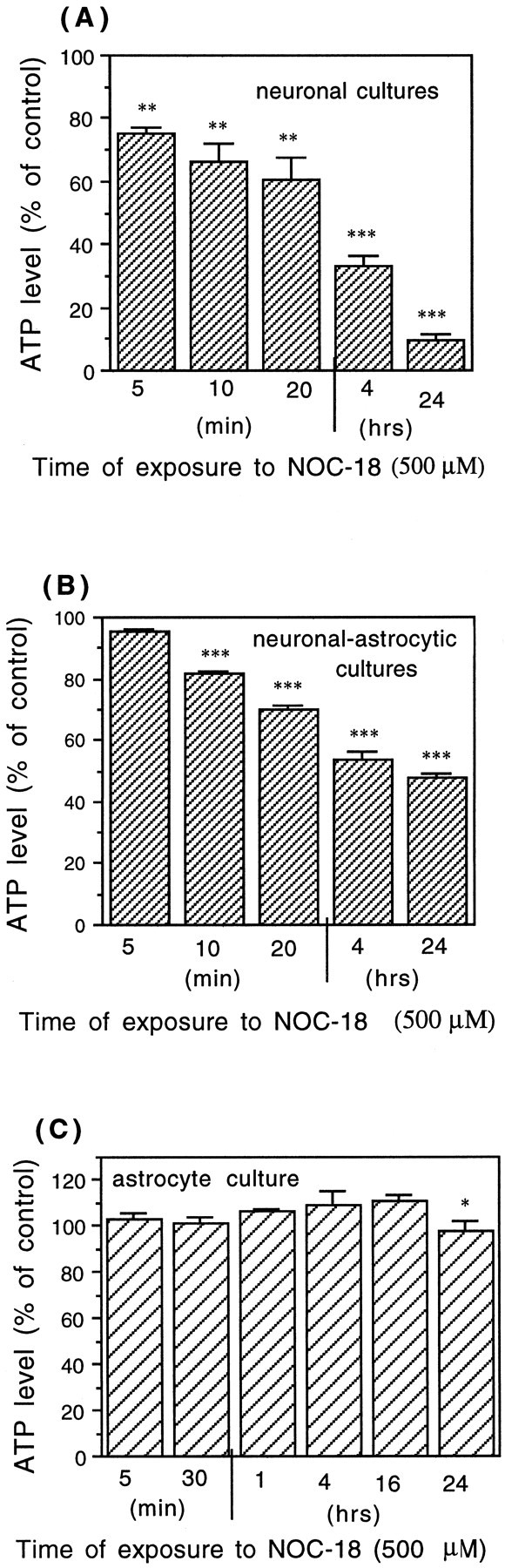
Depletion of ATP caused by 500 μmNOC-18 in neuronal cultures (CGCs treated with ara-C) (A), in neuronal–astrocytic cultures (CGCs cultured in the absence of ara-C) (B), and in pure astrocytic cultures (C). The data are expressed as percentage of the control (untreated) ATP levels, which were 1.51 ± 0.13 nmol of ATP/106cells in the neuronal cultures (A), 1.75 ± 0.1 nmol of ATP/106 cells in the neuronal–astrocytic cultures (B), and 7.1 ± 0.5 nmol of ATP/106 cells in the astrocytic cultures (C). Values represent means ± SD (bars) of determinations made in three separate cultures. *p < 0.05, **p < 0.01, ***p < 0.001 from control groups.
NO donors increase the level of l-lactic acid in neuronal and astrocytic cultures after prolonged exposure
To test whether NO-induced mitochondrial inhibition and subsequent ATP depletion in neuronal and astrocytic cultures would stimulate glycolysis, the level of l-lactic acid was measured in the medium of CGCs (9 DIV, 24-well plates) or astrocyte culture (12–14 DIV, confluent, 75 cm2 flasks) exposed to a NO donor (500 μm NOC-18) or a specific mitochondrial inhibitor (2 μm myxothiazol) for various intervals of time (5 min, 30 min, 4 hr, or 24 hr). Surprisingly, a short time incubation (5 or 30 min) with NOC-18 (but not with myxothiazol) caused a slight decrease in l-lactic acid levels, but longer exposure times (4 or 24 hr) significantly increased the level ofl-lactic acid in both types of cultures, neuronal (Fig.9A) and astrocytic (Fig.9B). However, in neuronal cultures (depleted of glia), the increased levels of l-lactic acid after the exposure to NOC-18 was only 28.3 ± 0.4% in contrast to astrocyte culture in which the level of lactate was increased 68.2 ± 0.8% when compared with the control culture.
Fig. 9.
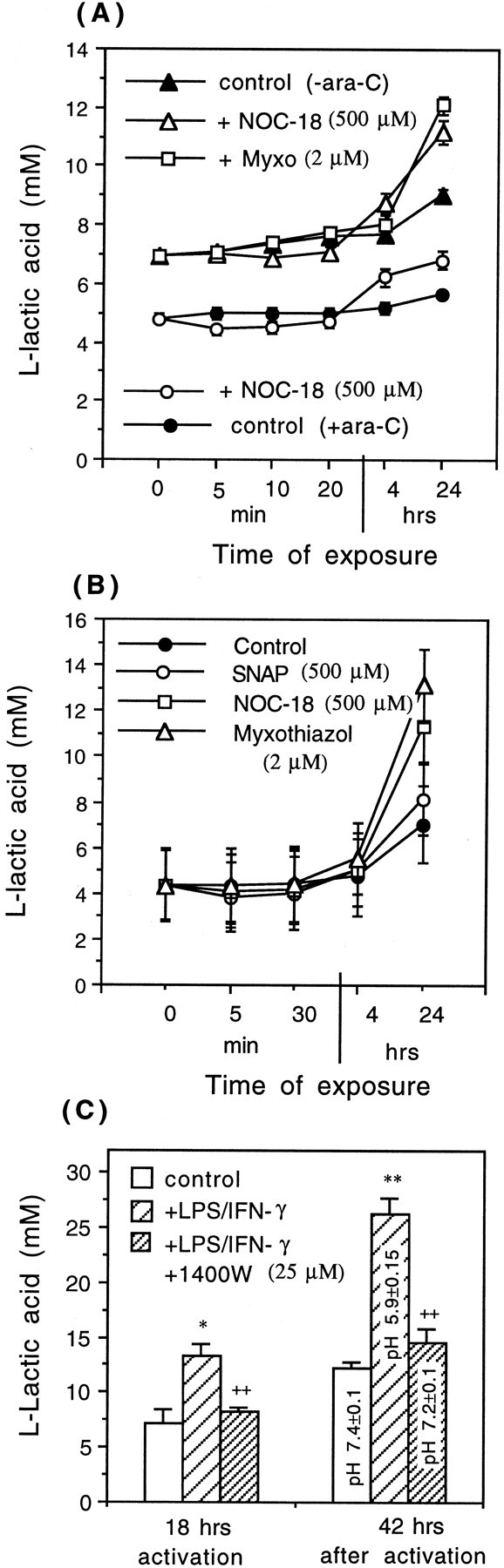
Levels of l-lactic acid in the medium of neuronal or neuronal–astrocytic (A) and astrocytic (B) cultures, exposed to NO donors (500 μm SNAP or NOC-18) or a specific mitochondrial inhibitor (2 μm myxothiazol). Neuronal cultures were cultured in the presence of ara-C (+ara-C), neuronal–astrocytic cultures in the absence of ara-C (−ara-C). C, Increased levels ofl-lactic acid and acidic pH (measured with pH electrode) in the medium of astrocytic cultures, activated for 18 hr with LPS/IFN-γ. Note the decreased levels of l-lactic acid (and increased pH) in the presence of an iNOS inhibitor (25 μm 1400W), suggesting that activation of glycolysis was mediated by NO. Values represent means ± SD (bars) of determinations made in three separate cultures. Statistical significance of the difference between l-lactic acid levels after 24 hr of exposure to NOC-18 was *p < 0.05 from control (+ara-C) and **p < 0.01 from control (−ara-C), and after exposure to myxothiazol it was **p < 0.01 from control (−ara-C) in A and **p < 0.01 after exposure to myxothiazol from control in B.
An activation of glycolysis was also observed in the culture of astrocytes after 18 hr of activation with LPS/IFN-γ because the level of l-lactic acid was significantly increased (control level, 7.18 ± 1.2 mm; after activation, 13.3 ± 1.15 mm) (Fig. 9C). This activation of glycolysis was probably mediated by NO-induced inhibition of respiration, because in the presence of an iNOS inhibitor (25 μm 1400W), levels of lactate were similar to the control levels (Fig. 9C). Forty-two hours after activation, the levels of lactate were even higher (26.2 ± 0.6 mm, pH 5.9 ± 0.15; control, 12.24 ± 0.5 mm, pH 7.4 ± 0.1), but in the presence of iNOS inhibitor (25 μm 1400W), the levels of lactate were only slightly higher (14.5 ± 1.2, pH 7.2) than in the control (Fig. 9C).
Nitric oxide causes glutamate release from neurons
Because neuronal cell death observed in the culture of CGCs exposed to activated glial cells (astrocytes or microglia) or to NO donors (SNAP or NOC-18) was prevented by MK-801 (NMDA receptor antagonist), suggesting that glutamate was involved in NO-induced cell death, we tested whether nitric oxide would cause glutamate release from cultured neurons. Indeed, NOC-18 (500 μm) caused immediate (<1 min) release of glutamate from neuronal–astrocytic (Fig. 10B) and neuronal cultures (Fig. 10A). Glutamate was undetectable in neuronal–astrocytic cultures before the addition of NO donor, but rose immediately after exposure to NOC-18 (500 μm) and was at the same level for the initial 30 min (8.2 ± 0.5 μm) and then decreased after longer incubation times (4 or 24 hr) (Fig.10B), possibly because of reuptake by astrocytes. These concentrations of glutamate in the medium are sufficient to activate NMDA receptors (Patneau and Mayer, 1990). In the case of neuronal cultures (CGCs cultures depleted of glial cells), NOC-18 also caused glutamate release, but the level increased with time (16.2 ± 2.4 μm after 24 hr) (Fig.10A), possibly because it could not be removed from the medium by astrocytes.
Fig. 10.
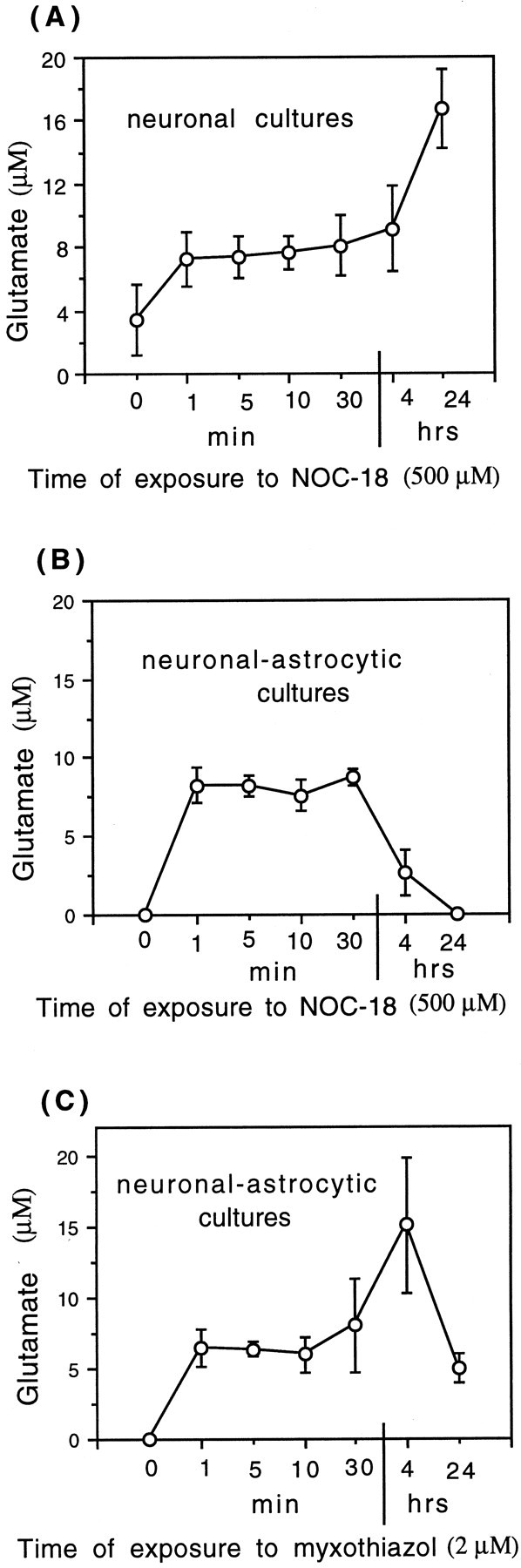
Release of glutamate from CGCs induced by NOC-18 (500 μm) or a specific mitochondrial inhibitor (2 μm myxothiazol). NOC-18 (500 μm) induced rapid release of glutamate from neuronal (A) or neuronal–astrocytic cultures (B) (treated and untreated with ara-C, respectively). C, Myxothiazol (2 μm; a specific mitochondrial inhibitor) also induced rapid glutamate release from neuronal–astrocytic cultures. Values represent means ± SD (bars) of determinations made in three separate cultures. Statistical significance of the difference between glutamate levels at time 0 and 10 min after treatment with NOC-18 was as follows:p < 0.05 in A,p < 0.001 in B, and after myxothiazol treatment p < 0.01 inC.
To test whether NO-induced inhibition of mitochondrial respiration could be the cause of glutamate release from CGCs, we studied the release of glutamate in neuronal–astrocyte cultures exposed to a specific mitochondrial inhibitor (2 μm myxothiazol) for various intervals of time (1, 5, 10, or 30 min and 4 or 24 hr). Similarly to the NO donor, myxothiazol also induced glutamate release from CGCs after a very short time of incubation (Fig. 10C). These results suggest that NO-induced inhibition of mitochondrial respiration could be the mechanism responsible for NO-induced release of glutamate from CGCs.
DISCUSSION
We have shown here (consistent with previous findings by Chao, 1996; Hu et al., 1997; Kingham et al., 1999; Tanabe et al., 1999) that inflamed glia have a remarkable capacity to kill cocultured neurons, even when present at relatively low concentrations. This has important implications for pathology because astrocytes, microglia, and macrophages become activated in virtually all CNS pathologies, including inflammatory, infective, post-ischemic, and neurodegenerative diseases, as well as aging (Eddleston and Mucke, 1993; Beal, 1995;Murphy, 2000). Activated glia may be directly causing neuronal death in these pathologies, although glial activation may also have protective roles, and neuronal death may contribute to glial activation. For example, in Alzheimer's disease, activated microglia and astrocytes expressing iNOS are found in the amyloid plaques surrounded by dead and dystrophic neurites (Wa et al., 1996; Wallace et al., 1997; Lee et al., 1999), β-amyloid can induce cultured glia to express iNOS and kill cocultured neurons via NO (Goodwin et al., 1995; Wisniewski et al., 1998), and anti-inflammatory drugs protect against Alzheimer's disease (McGeer and McGeer, 1995; McGeer et al., 1996; Lim et al., 2000).
In this study, activated microglia or astrocytes induced cell death of cocultured CGCs. The neuronal cell death was entirely mediated by glial NO from iNOS because it was completely prevented by two structurally unrelated iNOS inhibitors. This conclusion is supported by the finding that NO donors NOC-18 and SNAP (or NO-saturated water) producing steady-state levels of NO (0.5–0.75 μm) similar to those produced by activated glia (0.5–1 μm) (Fig.6B,C) caused a similar type of neuronal death, with a similar time course, that was similarly blocked by MK-801. Thus in our model of inflammatory neurodegeneration, neuronal death induced by activated glia was mediated by iNOS-derived NO alone, rather than other suggested mediators such as cytokines (Chao et al., 1995a; Viviani et al., 1998), proteases (Glockzin et al., 1999), or reactive oxygen species (Tanaka et al., 1994; Chao et al., 1995b).
One of the mechanisms by which NO causes neuronal death could be inhibition of mitochondrial respiration. In this study we found that NO from NO-saturated water or from activated astrocytes or microglia caused rapid, potent, and reversible inhibition of neuronal respiration. We have shown previously that such NO inhibition of respiration is caused by the reversible inhibition of cytochrome oxidase in competition with oxygen (Brown and Cooper, 1994; Brown et al., 1995; Brown, 1999). Others have shown similar inhibition in isolated mitochondria (Cleeter et al., 1994; Schweizer and Richter, 1994) and inhibition of energy metabolism in neurons (Brorson et al., 1999). We found here that a specific respiratory inhibitor (myxothiazol) replicated many of the effects of NO, including immediate respiratory inhibition, followed by rapid glutamate release, followed by neuronal cell death, blocked by MK-801 (at early incubation times). Moreover, preventing NO inhibition of respiration by transiently incubating the neurons with high oxygen during the exposure to NO-saturated water prevented subsequent NO-induced necrosis. However, we cannot rule out that this protection was mediated by more rapid breakdown of NO to NO2 or peroxynitrite at high oxygen levels, thus resulting in a shorter exposure time to NO. On the other hand, the fact that such short-term exposure to NO (1–5 min in Fig. 5C) results in significant neuronal cell death, which is blocked by high oxygen, indicates that NO2 or peroxynitrite is unlikely to mediate the neurotoxicity, and that whatever does mediate the actions of NO must happen within 1–2 min of NO exposure. Because NO caused inhibition of neuronal respiration within seconds, and NO or a specific respiratory inhibitor (myxothiazol) caused glutamate release within 1 min, and subsequent neuronal death is blocked by MK-801, NO-induced cell death is probably triggered by respiratory inhibition, followed by glutamate release and excitotoxicity. That NO inhibited respiration in the culture is supported by the finding that NO donors caused rapid ATP depletion in neuronal cultures (25% decrease within 5 min, 40% in 20 min, 90% in 24 hr) followed by activation of glycolysis (measured by lactate release), and that LPS/IFNγ-induced activation of astrocyte cultures caused increased lactate levels and acidification of the culture medium (Fig. 9C).
NO donors caused rapid (<1 min) glutamate release into the medium of mixed and pure neuronal cultures. A similar pattern of glutamate release was also observed after the exposure of mixed cultures to myxothiazol. Although the pattern differs after long-term treatment (4 and 24 hr), when the amount of NO released from NOC-18 may not be sufficient to fully inhibit respiration, the rapid release of glutamate caused by both NOC-18 and myxothiazol suggests that inhibition of mitochondrial respiration could be the mechanism responsible for glutamate release. We have shown previously that NO causes rapid glutamate release from synaptosomes caused by inhibition of cytochrome oxidase and probable reversal of the glutamate uptake carrier (McNaught and Brown, 1998). Others have shown NO-induced glutamate release in cultures and in vivo (Takita et al., 1997; Kingham et al., 1999; Trabace and Kendrick, 2000). The level of glutamate released into the culture medium by NO (∼8 μm glutamate) (Fig. 10A) has been shown to be sufficient to induce excitotoxic death of neurons (Patneau and Mayer, 1990). However, the NO-induced respiratory inhibition may also greatly increase the sensitivity of neurons to glutamate toxicity (because of depolarization and removal of the Mg2+ block of the NMDA receptor) (Vornov and Coyle, 1991). An antagonist of the NMDA receptor (MK-801) blocked neuronal death induced by activated astrocytes, microglia, NO donors, and myxothiazol (after short incubation time), suggesting that death was triggered at least in part by activation of NMDA receptors. MK-801 completely blocked DNA condensation at 4 hr (imaged by Hoescht staining), induced by NO donors and myxothiazol, and completely or partially blocked necrosis at 16 hr but was less effective at blocking necrosis at 24 hr. This may indicate either that 10 μm MK-801 was not completely effective at blocking NMDA receptors or that other mechanisms of NO-induced cell death become important after longer-term incubations with NO donors. Note that neuronal ATP was almost completely depleted at 24 hr (at least in the absence of MK-801) (Fig. 8A), which suggests that necrosis might have resulted from ATP depletion. However, MK-801 completely prevented neuronal death induced by exposure to activated microglia for 24 hr and very largely prevented that induced by activation of astrocytes in mixed culture for 48 hr, suggesting that cell death in these models is caused almost exclusively by excitotoxicity.
Death induced by activated astrocytes or microglia, NO, NO donors, or myxothiazol was confined exclusively to neurons; there was no significant cell death seen in astrocytes or microglia. This is consistent with our findings that NO donors caused no significant change in ATP levels in pure astrocyte cultures for up to 24 hr. The ATP level that we measured in mixed cultures probably had contributions from both neurons and glia. We have shown previously that astrocytes are sensitive to NO-induced inhibition of respiration (Brown et al., 1995), and we found here that glycolysis is activated in the astrocyte cultures by NO donors and LPS/IFN-γ, consistent with inhibition of respiration. However, astrocytes are known to have a higher glycolytic capacity than neurons (Pauwels et al., 1985; Peuchen et al., 1997) and are insensitive to excitotoxicity (Weiss et al., 1993).
We found previously in PC12 cells that NO donors induced necrosis caused by respiratory inhibition and ATP depletion if glucose was not present, but this was completely prevented by glucose, suggesting that cells with insufficient glycolytic capacity would be sensitive to NO-induce necrosis via respiratory inhibition. However, in the presence of glucose, NO induced caspase- and cytochrome c-mediated apoptosis (Bal-Price and Brown, 2000). The levels of NO donors used in the present study did not induce any cell death in the astrocytes. However, we found that higher levels of NO donors (1–2 mm NOC-18 for 48 or 72 hr) do induce apoptosis in astrocytes (DNA condensation and fragmentation; data not shown).
The neuronal cell death induced by activated glia, NO donors, and myxothiazol had some characteristics of apoptosis, but it was not classical apoptosis. Within 4 hr of NO donor treatment, most neurites were lost, the nuclei had shrunk, and the DNA was strongly condensed. However, the nuclear DNA was condensed but not fragmented, there was relatively little annexin-V staining, and a caspase inhibitor (zVAD) did not prevent DNA condensation or cell death. It is probably best to avoid trying to classify such death as either apoptotic or necrotic without further analysis.
In conclusion, we found that activated glia potently kill cocultured neurons via glial NO-inhibiting neuronal respiration, resulting in glutamate release causing excitotoxic death of neurons via NMDA receptors (Fig. 11). We have found recently that NO causes glutamate release from astrocytes (our unpublished data), and this might also contribute to excitotoxicity. NO inhibition of neuronal respiration might also activate NMDA receptors via plasma membrane depolarization. Calcium entry via NMDA receptors may induce neuronal death (possibly via mitochondrial damage) and might further increase NO production by activation of neuronal nitric oxide synthase.
Fig. 11.

Proposed scheme of inflammatory neurodegeneration mediated by glial NO. NO produced by activated microglia or astrocytes inhibits mitochondrial (mito) respiration of surrounding neurons, causing glutamate release (through glutamate transporters;GluT) from neurons (and possibly from astrocytes) and then stimulation of NMDA receptors (NMDAR). Activation of NMDA receptors by glutamate (possibly aided by respiratory inhibition-induced depolarization) triggers massive influx of Ca2+ into neurons, leading to apoptotic or necrotic cell death.
Footnotes
This work was supported by the Wellcome Trust.
Correspondence should be addressed to Anna Bal-Price, Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QW, UK. E-mail: akp26@mole.bio.cam.ac.uk.
REFERENCES
- 1.Bal A, Bachelot T, Savasta M, Manier M, Verna JM, Benabid AL, Feuerstein C. Evidence for dopamine D2 receptor mRNA expression by striatal astrocytes in culture: in situ hybridization and polymerase chain reaction studies. Mol Brain Res. 1994;23:204–212. doi: 10.1016/0169-328x(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 2.Bal-Price A, Brown GC. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J Neurochem. 2000;75:1455–1464. doi: 10.1046/j.1471-4159.2000.0751455.x. [DOI] [PubMed] [Google Scholar]
- 3.Banati RB, Gehramann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 4.Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JS, Chen J, Crow JP, Ye YZ. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeration. Prog Brain Res. 1994;103:371–380. doi: 10.1016/s0079-6123(08)61151-6. [DOI] [PubMed] [Google Scholar]
- 7.Bolanos JP, Almeida A. Role of nitric oxide in brain hypoxia-ischaemia. Biochim Biophys Acta. 1999;1411:415–436. doi: 10.1016/s0005-2728(99)00030-4. [DOI] [PubMed] [Google Scholar]
- 8.Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heals SJR. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68:2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- 9.Brorson JR, Schumacker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. J Neurosci. 1999;19:147–158. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 11.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 12.Brown GC, Bolanos JP, Heals SJ, Clark JB. Nitric-oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett. 1995;193:201–204. doi: 10.1016/0304-3940(95)11703-y. [DOI] [PubMed] [Google Scholar]
- 13.Cambray-Deakin MA. Cerebellar granule cells. In: Cohen J, Wilkin GP, editors. Neuronal cell culture. Oxford UP; New York: 1995. pp. 3–13. [Google Scholar]
- 14.Chao CC. Cytokine-stimulated astrocytes damage human neurons via a NO mechanism. Glia. 1996;16:276–284. doi: 10.1002/(SICI)1098-1136(199603)16:3<276::AID-GLIA10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 15.Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-d-aspartate receptors. Brain Behav Immunol. 1995a;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- 16.Chao CC, Hu S, Peterson PK. Modulation of human microglial cell superoxide production by cytokines. J Leukoc Biol. 1995b;58:65–70. doi: 10.1002/jlb.58.1.65. [DOI] [PubMed] [Google Scholar]
- 17.Cleeter MWJ, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AHV. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respirator chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 18.Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glockzin S, Knethen A, Scheffner M, Brune B. Activation of the cell death program by nitric oxide involves inhibition of the proteasome. J Biol Chem. 1999;274:19581–19586. doi: 10.1074/jbc.274.28.19581. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- 21.Heals SJR, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410:215–228. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- 22.Hewett SJ, Csernansky CA, Choi DW. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron. 1994;13:487–494. doi: 10.1016/0896-6273(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Ferreira A, Van Eldik LJ. S100 beta induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69:2294–2301. doi: 10.1046/j.1471-4159.1997.69062294.x. [DOI] [PubMed] [Google Scholar]
- 24.Kingham PJ, Cuzner ML, Pocock JM. Apoptotic pathways mobilized in microglia and neurons as a consequence of chromogranin A-induced microglial activation. J Neurochem. 1999;73:538–547. doi: 10.1046/j.1471-4159.1999.0730538.x. [DOI] [PubMed] [Google Scholar]
- 25.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 26.Lee SC, Zhao ML, Hirano A, Dickson DW. Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles, and senile plaques. J Neuropathol Exp Neurol. 1999;58:1163–1169. doi: 10.1097/00005072-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Leist M, Fava E, Montecucco C, Nicotera P. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci. 1997a;9:1488–1498. doi: 10.1111/j.1460-9568.1997.tb01503.x. [DOI] [PubMed] [Google Scholar]
- 28.Leist M, Volbracht C, Kuhnle S, Fava E, Ferrandomay E, Nicotera P. Caspase-mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide. Mol Med. 1997b;11:750–764. [PMC free article] [PubMed] [Google Scholar]
- 29.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 30.Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loihl AK, Murphy S. Expression of NOS-2 in glia associated with CNS pathology. Prog Brain Res. 1998;118:253–267. doi: 10.1016/S0079-6123(08)63213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeer EG, McGeer PL. Brain inflammation in Alzheimer disease and the therapeutic implications. Curr Pharm Des. 1995;10:821–386. [PubMed] [Google Scholar]
- 33.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 34.McNaught KSP, Brown GC. Nitric oxide causes glutamate release from brain synaptosomes following inhibition of mitochondrial function. J Neurochem. 1998;70:1541–1546. doi: 10.1046/j.1471-4159.1998.70041541.x. [DOI] [PubMed] [Google Scholar]
- 35.Meffert MK, Premack BA, Schulman H. Nitric oxide stimulates Ca+2-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 36.Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–14. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 37.Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pauwels PJ, Opperdoes FR, Trouet A. Effects of antimycin, glucose deprivation, and serum on cultures of neurons, astrocytes, and neuroblastoma cells. J Neurochem. 1985;44:143–148. doi: 10.1111/j.1471-4159.1985.tb07123.x. [DOI] [PubMed] [Google Scholar]
- 39.Peuchen S, Bolanos JP, Heals SJR, Almeida A, Duchen MR, Clark JB. Interrelations between astrocytes function, oxidative stress and antioxidant status within the central nervous system. Prog Neurobiol. 1997;52:261–281. doi: 10.1016/s0301-0082(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt HH, Kelm M. Determination of nitrite and nitrate by Griess reaction. In: Feelisch M, Stamler JS, editors. Methods in nitric oxide research. Wiley; New York: 1996. pp. 491–497. [Google Scholar]
- 41.Schweizer C, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem Biophys Res Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- 42.Takita M, Kaneko H, Suzuki SS, Akamatsu M. Lasting effect of NO on glutamate release in rat striatum revealed by continuous brain dialysis. NeuroReport. 1997;8:567–570. doi: 10.1097/00001756-199701200-00037. [DOI] [PubMed] [Google Scholar]
- 43.Tamatani M, Ogawa S, Niitsu Y, Tohyama M. Involvement of Bcl-2 family and caspase-3-like protease in NO-mediated neuronal apoptosis. J Neurochem. 1998;71:1588–1596. doi: 10.1046/j.1471-4159.1998.71041588.x. [DOI] [PubMed] [Google Scholar]
- 44.Tanabe K, Akanishi H, Maeda H, Nishioku T, Hashimoto K, Liou SY, Akamines A, Yamamoto K. A predominant apoptotic death pathway of neuronal PC12 cells induced by activated microglia is displaced by a non-apoptotic death pathway following blockage of caspase-3-dependent cascade. J Biol Chem. 1999;274:15725–15731. doi: 10.1074/jbc.274.22.15725. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka M, Sotomatsu A, Yoshida T, Hirai S, Nishida A. Detection of superoxide production by activated microglia using a sensitive and specific chemiluminescence assay and microglia-mediated PC12h cell death. J Neurochem. 1994;63:266–270. doi: 10.1046/j.1471-4159.1994.63010266.x. [DOI] [PubMed] [Google Scholar]
- 46.Taupenot L, Ciesielski-Treska J, Urlich G, Chasserot-Golaz S, Aunis D, Bader MF. Chromogranin A triggers a phenotypic transformation and the generation of nitric oxide in brain microglial cells. Neuroscience. 1996;72:377–389. doi: 10.1016/0306-4522(96)83172-1. [DOI] [PubMed] [Google Scholar]
- 47.Trabace L, Kendrick KM. Nitric oxide can differentially modulate striatal neurotransmitter concentrations via soluble guanylate cyclase and peroxynitrite formation. J Neurochem. 2000;75:1664–1674. doi: 10.1046/j.1471-4159.2000.0751664.x. [DOI] [PubMed] [Google Scholar]
- 48.Uehara T, Kikuchi Y, Nomura Y. Caspase activation accompanying cytochrome c release from mitochondria is possibly involved in nitric-oxide-induced neuronal apoptosis in SH-SY5Y cells. J Neurochem. 1999;72:196–205. doi: 10.1046/j.1471-4159.1999.0720196.x. [DOI] [PubMed] [Google Scholar]
- 49.Viviani B, Corsini E, Galli CL, Marinovich M. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271–276. doi: 10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- 50.Vornov JJ, Coyle JT. Enhancement of NMDA receptor-mediated neurotoxicity in the hippocampal slice by depolarization and ischemia. Brain Res. 1991;555:99–106. doi: 10.1016/0006-8993(91)90865-s. [DOI] [PubMed] [Google Scholar]
- 51.Wa J, Food MR, Gabathuler R, Rothenberger S, Yamada T, Yasuhara O, McGeer PL. Reactive microglia specifically associated with amyloid plaques in Alzheimer's disease brain tissue express melanotransferrin. Brain Res. 1996;712:122–126. doi: 10.1016/0006-8993(95)01407-1. [DOI] [PubMed] [Google Scholar]
- 52.Wallace MN, Geddes JG, Farquhar DA, Masson MR. Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp Neurol. 1997;144:266–272. doi: 10.1006/exnr.1996.6373. [DOI] [PubMed] [Google Scholar]
- 53.Wei T, Chen C, Hou J, Xin J, Mori A. Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim Biophys Acta. 2000;1498:72–79. doi: 10.1016/s0167-4889(00)00078-1. [DOI] [PubMed] [Google Scholar]
- 54.Weiss JH, Hartley DM, Koh J, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- 55.Wisniewski HM, Wegiel J, Wisniewski T. Pathogenesis of amyloid-β plaques: activated microglia the cause of fibrillar amyloid formation and neuropil degeneration. Neurosci News. 1998;1:30–34. [Google Scholar]
- 56.Zhang J, Dawson V, Dawson T, Snyder S. Nitric oxide activation of poly(ADP-ribose)synthase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]