

Abstract
Cyclin-dependent kinase 5 (Cdk5) plays a pivotal role in brain development and neuronal migration. Cdk5 is abundant in postmitotic, terminally differentiated neurons. The ability of Cdk5 to phosphorylate substrates is dependent on activation by its neuronal-specific activators p35 and p39. There exist striking differences in the phenotypic severity of Cdk5-deficient mice and p35-deficient mice. Cdk5-null mutants show a more severe disruption of lamination in the cerebral cortex, hippocampus, and cerebellum. In addition, Cdk5-null mice display perinatal lethality, whereas p35-null mice are viable. These discrepancies have been attributed to the function of other Cdk5 activators, such as p39. To understand the roles of p39 and p35, we created p39-null mice and p35/p39 compound-mutant mice. Interestingly, p39-null mice show no obvious detectable abnormalities, whereas p35–/–p39–/– double-null mutants are perinatal lethal. We show here that the p35–/–p39–/– mutants exhibit phenotypes identical to those of the Cdk5-null mutant mice. Other compound-mutant mice with intermediate phenotypes allow us to determine the distinct and redundant functions between p35 and p39. Our data strongly suggest that p35 and p39 are essential for Cdk5 activity during the development of the nervous system. Thus, p35 and p39 are likely to be the principal, if not the only, activators of Cdk5.
Keywords: p35, p39, Cdk5, development, lamination, neuronal migration, Nudel, neurofilaments
In the developing brain, proper neuronal migration depends on the activity of a serine/threonine kinase, Cyclin-dependent kinase 5 (Cdk5). Mice deficient in Cdk5 die perinatally and show widespread defects in migration in many brain compartments, including the cerebral cortex, hippocampus, and cerebellum. In the cerebral cortex, the laminar organization of neurons is inverted with a superficial ectopic subplate. The pyramidal neurons of the hippocampus are not organized in a discrete layer. The cerebellum lacks foliation and tripartite layering. In addition, motor neurons of the spinal cord and cranial nerve nuclei display ballooned perikarya characteristic of chromatolytic changes (Ohshima et al., 1996, 1999; Gilmore et al., 1998).
p35 was the first neuronal-specific regulatory subunit of Cdk5 identified via its physical interaction with Cdk5, activating Cdk5 after direct binding (Ishiguro et al., 1994; Lew et al., 1994;Tsai et al., 1994). Interestingly, mice lacking p35 show lamination defects in the cerebral cortex but experience only mild disruptions in the hippocampus and have fairly normal cerebella (Chae et al., 1997; Kwon and Tsai, 1998). In sharp contrast to the Cdk5-null mutants, p35–/– mice are viable and fertile. The striking differences in phenotypic severity between the two mutant mice have been attributed to the presence of other regulatory subunits for Cdk5. Indeed, p39, another Cdk5 regulator, was isolated by virtue of its homology to p35 and is a bona fide regulator of Cdk5 in neurons (Tang et al., 1995). p39 can modulate neurite outgrowth (Xiong et al., 1997; Paglini et al., 1998), and its association with the actin cytoskeleton suggests that the p39/Cdk5 complex may play a role in regulating actin cytoskeleton dynamics in cells (Humbert et al., 2000a). In addition, there is evidence that p39 is present at synapses and thus may play a role in synaptogenesis (Humbert et al., 2000b). Despite these studies, the specific role of the p39/Cdk5 kinase complex is not clear. To investigate the function of p39 in vivo during development of the nervous system, we created mice with a targeted deletion in the p39 locus. In addition, we generated p35/p39 compound mutants to test the hypothesis that p35 and p39 are the essential two activators for Cdk5 function and to elucidate further the roles of p35 and p39 during neurodevelopment.
In this study, we show that p39-deficient mice do not exhibit detectable abnormalities in neuronal positioning in the nervous system. However, p35–/–p39–/–double-null mutants display phenotypes that are identical to those of Cdk5–/– mice in all neuronal compartments analyzed, indicating that p35 and p39 are indeed essential activators of Cdk5. Comparative analysis of gene dosage effects of p35 and p39 in the single and compound mutants reveals that p35 and p39 play distinct roles in the regulation of Cdk5 activity and have differential compensatory capabilities in particular brain compartments. Moreover, we have novel data to suggest that the role of Cdk5 in neurodevelopment extends beyond the scope established previously.
MATERIALS AND METHODS
Generation of p39-null mice. The murine homolog of p39 was isolated and characterized (Nilden et al., 1998). Corresponding genomic clones were obtained by screening of a 129/Sv mouse genomic library (Stratagene, La Jolla, CA) with a PCR product containing the entire p39 open reading frame. To construct the targeting vector (see Fig. 1A), 3 kb of upstream sequence and 5.4 kb of downstream sequence were cloned in the pPNT vector (Tybulewicz et al., 1991). The 3 kb of 5′-flanking fragment consisting of a BglII–SmaI fragment isolated from one λ phage was subcloned into the XbaI site of pPNT. The 5.4 kbXhoI–NotI fragment from the same phage was cloned into the XhoI–NotI site of pPNT. Thus, the coding region for amino acids 1–346 of p39 and 100 bp of noncoding upstream sequence was replaced with the neocassette in the opposite transcriptional orientation. Negative selection against random integration was conferred on the construct by a herpes simplex virus thymidine kinase gene. The targeting construct was linearized with NotI (unique site) for electroporation.
Fig. 1.

Generation of the p39–/–mutant mice. A, Targeted deletion of the p39 open reading frame is shown. The p39 targeting vector carrying the 1.8 kbneo cassette in the reverse orientation replaced 1 kb of the 1.1 kb open reading frame of the p39 locus. Black boxes denote the HindIII–XbaI fragment external to the region of homology contained within the targeting construct that is used as a probe in Southern blot analysis, which detects 4.7 and 5.5 kbHindIII–XhoI fragments from the wild-type and mutant alleles, respectively. Light gray boxes denote the regions amplified by PCR genotyping.B, Genotyping of three 3-week-old littermates by Southern blot analysis of theHindIII–XhoI fragment of genomic DNA yields a wild-type band of 4.7 kb and a neo band of 5.5 kb, indicating homozygous wild-type and knock-out animals, respectively. The presence of both bands represents a heterozygous animal. C, Genotyping by PCR yields a wild-type band of 350 bp and aneo band of 650 bp. The presence of both bands denotes a heterozygous animal. D, Absence of p39 protein in p39–/– mice is shown. Western blot analysis of wild-type, p39+/–, and p39–/–whole-brain lysates used an affinity-purified rabbit polyclonal antibody for p39. p35 and Cdk5 proteins continue to be expressed in the absence of p39 protein. E, Absence of p39-associated kinase activity in p39–/–mice is shown. One milligram of brain lysates was immunoprecipitated with a polyclonal p39 antibody, a polyclonal p35 antibody, or a polyclonal Cdk5 antibody and assayed for histone H1 kinase activity. H,HindIII; HsV-tk, herpes simplex virus thymidine kinase gene; IP, immunoprecipitate;WB, Western blot; X,XbaI.
Tissue culture of J1 embryonic stem (ES) cells and conditions for electroporation of the targeting construct were performed as described previously (Chae et al., 1997). Selection with G418 at 400 μg/ml and fialuridine at 0.2 μm was initiated 24 hr after electroporation. After 8–10 d, resistant clones were picked, expanded, and screened for homologous recombination events by Southern blot analysis. Genomic DNA from the clones was digested withHindIII and XhoI and probed with aHindIII–XbaI 700 bp fragment that is external to the region of homology between the p39 locus and the targeting vector. The sizes of the HindIII–XhoI fragments from the wild-type allele and mutant allele were 4.7 and 5.5 kb, respectively. The presence of a single integration event was demonstrated by hybridization with neomycin phosphotransferase gene-specific probe. Two ES clones were chosen to produce chimeras by microinjection into C57BL/6 blastocysts and implantation into pregnant foster mothers. Germ-line transmission of the mutation was determined by mating male chimeric animals to C57BL/6 females and confirmed by Southern blot analysis of tail genomic DNA of agouti offspring. Homozygous animals for the null allele were generated by mating F1 heterozygous mice. All mice were genotyped by Southern blot using theHindIII–XbaI 700 bp probe.
Generation of p35/p39 compound-mutant mice. Compound-mutant mice were generated via a two- or three-stage cross. p35–/– and p39–/– mice were mated to produce p35+/–p39+/–F1 progeny. F1 progeny were self-crossed to obtain compound-mutant mice, 6.25% of which were expected to be double knockouts. An additional cross was implemented with F2 progeny of p35+/–p39–/–and p35–/–p39+/–genotypes to produce litters of which 25% of pups will be double knockouts.
Genotyping of p35/p39 compound-mutant mice. p35 genotyping was performed using PCR as described previously (Kwon and Tsai, 1998). The p39 genotype was determined by Southern blot (see above) and by PCR analysis of genomic DNA prepared from tails. Two sets of PCRs allowed for the determination of genotype. Reaction 1 amplifies the region of 350 bp present only in nonrecombinant genomic DNA at the p39 gene locus (primer “p39 up,” AGC TTC GGC GGC TCA GAG CAG CC; primer “p39 down,” AGC ACG GAC GGC CGT TTG AGC). Reaction 2 amplifies the region of 650 bp specific to the genomic deletion (primers p39 up and “neo,” GAT ATT GCT GAA GAG CTT GGC).
Genotyping of Cdk5–/– mice.Cdk5-null mutants were generated and genotyped by Southern blot analysis as described previously (Ohshima et al., 1996).
Harvesting and fixation of embryonic day 18.5 embryos.Embryos were harvested individually from anesthetized, live females. Embryonic day 18.5 (E18.5) time-pregnant females were given intraperitoneal injections of Avertin (10 ml/kg; Aldrich, Milwaukee, WI) for anesthesia as described previously (Papaioannou and Fox, 1993;Inglis and Semba, 1997). After each individual embryo was removed, it was subjected to tail-clamp stimulation to determine viability, and the tail was subsequently snipped for genotyping. The embryos were immersion fixed for 7–14 d in Bouin's Fixative (Polysciences, Warrington, PA) for histopathological analysis or for 3–5 d in 4% paraformaldehyde in 0.1 m sodium phosphate buffer for immunohistochemical analysis.
Histopathological and immunohistochemical analysis. Heads and bodies of E18.5 mice were dehydrated and embedded in paraffin. Serial coronal, parasagittal, and transverse tissue sections were cut at 6–7 μm and stained with hematoxylin and eosin for histopathological analysis. Two to three specimens of each genotype were analyzed, and littermates were used for comparison whenever possible. Calbindin D-28-K (Sigma, St. Louis, MO; 1:3000) and calretinin (SWANT, Bellinzona, Switzerland; 1:1000) immunoperoxidase staining was performed on paraformaldehyde-fixed tissue according to established protocols using the avidin–biotin–peroxidase complex technique (ABC; Vector Laboratories, Burlingame, CA). SMI34 (Sternberger Monoclonals, Lutherville, MD; 1:10,000) immunoperoxidase staining was performed on Bouin's fixed tissue.
Western blot analysis. Whole adult and postnatal day 0 (P0) mouse brains were lysed in 500 μl of radioimmunoprecipitation (RIPA) buffer (150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mmTris, pH 8) or E1A lysis buffer (50 mmHEPES, pH 7.0, 0.1% NP-40, 250 mm NaCl, and 5 mm EDTA, pH 8) with protease and phosphatase inhibitors (1 mm Na3VO4, 50 mm NaF, 1 mm PMSF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 nm okadaic acid, 5 μm pyrophosphate, 1 mm DTT, and 50 μm β-glycerophosphate) in a homogenizing Dounce and then spun at 13,000 rpm for 15 min at 4°C. The supernatant was collected, and the amount of protein was determined by the Bradford procedure using the Bio-Rad protein assay solution (Bio-Rad, Hercules, CA). Fifteen micrograms of total proteins were loaded per lane on an 8% acrylamide/bis-acrylamide (30%/0.8%) gel and separated by SDS-PAGE. Proteins were then transferred to Immobilon-P transfer membrane (Millipore, Bedford, MA). Blots were then subjected to protein detection by incubating with an affinity-purified polyclonal p35 antibody (1:1000), an affinity-purified polyclonal p39 antibody (1:1000) (Humbert et al., 2000a), a monoclonal Cdk5 antibody DC-17 (1:10) (Tsai et al., 1993), and a polyclonal actin antibody (1:1000; Sigma). For analysis of Nudel and Dab1 proteins, P0 whole-brain lysates were separated on an 8% acrylamide/bis-acrylamide (30%/0.4%) gel. Nudel was detected using an affinity-purified polyclonal antibody as described by Niethammer et al. (2000). Tau phosphorylation on Ser 202 and Ser 205 was detected using AT8 monoclonal antibody (1:1000; Endogen, Woburn, MA), and phosphorylation on Thr 396 was detected by the PHF-1 monoclonal antibody (1:1000; gift of Peter Davies, Albert Einstein College of Medicine, Bronx, NY). Total Tau was detected using the monoclonal antibody Tau5 (0.1 μg/μl; Neomarkers, Fremont, CA). For analysis of neurofilament proteins, P0 spinal cord lysates were made in ice-cold Triton X-100 buffer (1% Triton X-100, 50 mmTris, pH 7.5, 25 mm KCl, 2 mm MgCl2, 5 mm EGTA, 5 mmdithiothreitol, and 0.25 mm phenylmethylsulfonyl fluoride) with phosphatase inhibitors (50 mm NaF, 10 mm β-glycerophosphate, and 30 mm pyrophosphate) and then centrifuged at 100,000 × g for 1 hr at 4°C (Tsuda et al., 2000). The Triton-soluble fraction was measured for protein concentration by the Bradford method. The Triton-insoluble pellet was dissolved in SDS sample buffer, and protein concentration was normalized by Coomassie blue staining. Both fractions were subjected to SDS-PAGE on a 7% gel. Monoclonal antibodies SMI34 (1:1000) and SMI32 (1:1000) (Sternberger Monoclonals) were used to detect phosphorylated and unphosphorylated neurofilament heavy chain protein isoforms, respectively.
Densitometric analysis. In the analysis of Nudel phosphorylation, quantitation of optical density (OD) was performed using NIH Image 1.62 of two exposures within the linear range from three distinct sets of whole-brain extracts prepared from three series of P0 pups of the relevant genotypes at P0. The equal measurement area was applied to each band of individual samples and adjusted to background levels of each lane. The OD ratios were calculated and averaged for each genotype, and SEMs were determined. The results (percentage of OD of the fastest-migrating major Nudel isoform) were plotted by CA-CricketGraphIII (Computer Associates International, Islandia, NY).
Immunoprecipitation histone H1 kinase assay. P0 whole-brain lysates (500 μg of total protein) in RIPA or ELB buffers were immunoprecipitated with polyclonal p35, p39, and Cdk5 (C8 antibody; Santa Cruz Biotechnology, Santa Cruz, CA) and glutathioneS-transferase (GST; Santa Cruz Biotechnology) antibodies, and kinase assays in the presence of histone H1 were performed as described previously (Tsai et al., 1993).
RESULTS
Generation and characterization of mice with a targeted deletion in the p39 locus
Characterization of the p39 genomic locus demonstrated that p39 has a continuous coding region without interruption by intron sequences (Nilden et al., 1998). To delete the genomic p39 locus by homologous recombination (Capecchi, 1989), we created a p39 targeting construct by replacing the majority of the p39 coding region with the neomycin resistance (neo) gene in the opposite transcriptional orientation (Fig. 1A). J1 ES cells were electroporated with the targeting vector DNA, and subsequently, 576 resistant clones were isolated and screened by Southern blot hybridization analysis. This revealed 21 clones positive for the predicted size of the targeted allele.
Two ES cell clones were chosen for microinjection into C57BL/6 blastocysts and implantation into pseudopregnant foster mothers to produce chimeras. Both clones generated high level of chimerism (129/Sv-derived agouti coat color pigmentation > 60%), and male chimeras successfully transmitted the p39-null mutation through the germ line. Heterozygous p39+/– mice were fertile, viable, and indistinguishable from their wild-type littermates. p39+/– mice were intercrossed to obtain homozygous p39-deficient mice (p39–/–), as demonstrated by Southern blotting (Fig. 1B). In addition, a PCR amplification product of 650 bp signaled the presence of the neo cassette, and a 350 bp product represented the presence of wild-type p39 (Fig.1C). The appearance of both bands indicated a heterozygous animal. Genotype analysis of F2 progeny from the F1 p39+/– intercrosses was consistent with a normal expected Mendelian distribution. Whole-brain lysates of each genotype were analyzed for the presence of p39 by Western blot analysis and histone H1 kinase assay. By the use of an affinity-purified rabbit polyclonal antibody to p39 in Western blotting (Humbert et al., 2000a), p39 is not detectable in p39–/– brain lysates (Fig. 1D). The abundance of p39 in the heterozygous mutant is half that of wild-type (Fig.1D). Similar results were obtained after immunoprecipitation of brain lysates derived from homozygous or heterozygous mutants followed by an in vitro kinase assay testing for phosphorylation of histone H1 (Fig. 1E). These results demonstrate that the p39 mutant allele is a null allele and that p39 protein is entirely absent in the p39–/– animals.
It is interesting to note that loss of p39 produced no significant difference in the levels of p35 and Cdk5 in whole-brain lysates (Fig.1D). Similarly, p35- and Cdk5-associated histone H1 kinase activity was the same in brain extracts derived from wild-type, p39+/–, and p39–/– animals (Fig.1E).
The p39–/– mice are viable and fertile and do not display any obvious outward phenotypes. Histopathological analysis of adult brains did not reveal any noticeable abnormalities in the cerebral cortex, cerebellum, hippocampal formation, eye, and spinal cord. Full necropsy and analysis of all other tissues and organs including muscle tissue revealed no obvious defects.
Generation of p35/p39 compound-mutant mice
The lack of any notable phenotype in the p39–/– mice, in concert with the fact that only p35 and p39 had been isolated as activators of Cdk5, hinted at both discrete functions and a mechanism of redundancy between the two activators. To address these issues, we generated p35/p39 double knock-out mice. The compound-mutant mice were generated via a two-stage cross; the first involved crossing the p35–/– mice (Chae et al., 1997) with the p39–/– mice. The progeny of the first stage cross (F1 double heterozygous p35+/–p39+/–mice) were then mated to yield F2 progeny of the genotypes of interest, namely, p35+/–p39–/–mice, p35–/–p39+/–mice, and p35–/–p39–/–double knockouts.
Analysis of viability of F2 progeny revealed that p35–/–p39–/–double knock-out mice exhibit perinatal lethality and do not survive after P0 (Table 1; n = 324). Only 2.8% of the total number of animals analyzed at P0 were p35–/–p39–/–, significantly less than the expected Mendelian frequency of 6.25% (p < 0.013). This decreased frequency could reflect compromised viability as well as maternal cannabilization of dead or less viable pups. Those that are found alive on P0 showed weakness of movement, were runted, and were often isolated from the rest of the litter. Increased numbers of p35–/–p39–/–mice were found at E18.5 (data not shown). At E18.5, double-null mutant pups are also smaller in size (Fig.2A). After cesarean delivery, pups were tested for response to tail-clamp stimulation. Although tail clamping evokes an immediate and obvious vocal and physical response from most pups, some pups responded very weakly, if at all, to subsequent stimulation. The lack of response to tail-clamp stimulation corresponded with the p35–/–p39–/–genotype (data not shown). The statistical distribution of other genotypes, including p35–/–p39+/–and p35+/–p39–/–genotypes, remains close to the expected values at both P0 and P20. The lethality of the double mutants parallels the perinatal lethality of Cdk5–/– mutant mice (Ohshima et al., 1996). Interestingly, the viability of p35–/–p39+/–mice appears to decrease with age, because mice of this particular genotype sporadically die beginning as early as P60.
Table 1.
Viability of p35/p39 compound-mutant mice at P0 and P150
| Genotype | Expected (%) | Observed at P0 (%) | Observed at P150 (%) | |
|---|---|---|---|---|
| p35 | p39 | |||
| + /+ | 6.25 | 7.4 | 8.0 | |
| +/+ | + /− | 12.5 | 13.0 | 13.6 |
| − /− | 6.25 | 7.4 | 8.0 | |
| + /+ | 12.5 | 13.6 | 15.2 | |
| +/− | + /− | 25.0 | 24.4 | 26.8 |
| − /− | 12.5 | 9.9 | 10.6 | |
| + /+ | 6.25 | 8.4 | 8.6 | |
| −/− | + /− | 12.5 | 13.0 | 8.6 |
| − /− | 6.25 | 2.8 | 0 | |
| Total mice analyzed: | 324 | 298 | ||
Mice of p35+/−p39+/− genotypes were crossed to generate p35−/−p39−/− mice. At P0, with the exception of the p35−/−p39−/−mice, most of the genotypes were found to distribute according expected Mendelian values. The p35−/−p39−/− mice, alive and dead, were found at a significantly lower frequency (2.8%) than expected (6.25%) (p < 0.013). The lower frequency may reflect both compromised viability and maternal cannabilization. Statistical significance was calculated using a χ2 test. Double knock-out mice were not found viable after P0. Beginning as early as 2 months, p35−/−p39+/− mice also show a decrease in viability manifested in sporadic early death.
Fig. 2.

Absence of Cdk5-associated kinase activity in p35–/–p39–/– double-null brains. A, p35–/–p39–/– double-mutant mice isolated at E18.5 are consistently smaller than are wild-type and single-mutant littermates. B, Absence of both p35 and p39 protein in p35–/–p39–/–mice is shown. Western blot analysis of whole-brain lysates from wild-type, p35–/–, p39–/–, and p35–/–p39–/– P0 pups is shown. p39 is upregulated in the absence of p35, but p35 protein is unaffected by the loss of p39. C, p35–/–p39–/– mice show absence of p35- and p39-associated kinase activity, whereas single-mutant mice display residual kinase activity. Five hundred micrograms of brain lysates of wild-type, p35–/–, and p39–/– P0 pups were immunoprecipitated with polyclonal p35 and p39 antibodies and a polyclonal GST antibody as negative control and assayed for histone H1 kinase activity. p35–/– brain lysates exhibit histone phosphorylation when immunoprecipitated with p39 antibody; p39–/– brain lysates exhibit histone phosphorylation when immunoprecipitated with p35 antibody.D, Absence of Cdk5-associated kinase activity in p35–/–p39–/– mice is shown. Both p35–/– and p39–/– brain lysates continue to display Cdk5-associated kinase activity, whereas kinase activity is absent or equivalent to background (GST-immunoprecipitated) levels in p35–/–p39–/– brain lysates.WT, Wild type.
Cdk5-associated kinase activity is absent in p35–/–p39–/– mice
Analysis of Cdk5 kinase activity in p35–/–p39–/–mice was performed to determine how loss of the regulatory proteins impacts on kinase activity. P0 pups were collected, and after PCR and/or Southern blot genotyping, their brains were harvested for protein biochemistry. Western blot analyses show that in p35–/– and p39–/– single knockouts, only the single respective proteins are absent, whereas both p35 and p39 proteins are absent from p35–/–p39–/–brain lysates (Fig. 2B). p39 protein is consistently detected at higher levels in the p35–/–mice, whereas p35 protein levels remain unchanged when p39 protein is deficient. The ability of p35 and p39-associated Cdk5 to phosphorylate histone H1 was analyzed using brain lysates derived from different genotypes. As expected, histone H1 phosphorylation is apparent when p35 or p39 was immunoprecipitated from wild-type brain lysates but not when a polyclonal GST antibody was used as negative control (Fig.2C). In p35–/– brain lysate, p35-associated Cdk5 kinase activity is absent, whereas immunoprecipitated p39 continues to induce histone H1 phosphorylation. Conversely, p35-associated kinase activity remained intact whereas p39-associated kinase activity was absent in the p39–/– mice. Double knock-out brain lysates display complete loss of p35 and p39-associated kinase activity. In addition, immunoprecipitation of Cdk5 using the polyclonal C8 antibody confirms that although Cdk5 is present, Cdk5-associated kinase activity cannot be detected in double knock-out brain (Fig.2D). We conclude from these data that although Cdk5 activity continues to be present in the single-mutant mice, the combined elimination of p35 and p39 renders the Cdk5 kinase inactive in the brain.
Lamination and neuronal-positioning defects in the p35/p39 compound-mutant cerebral cortex
Although the lack of detectable Cdk5 kinase activity in the absence of p35 and p39 provides compelling evidence that p35 and p39 are essential for Cdk5 activity, we decided to analyze whether the p35–/–p39–/–phenotype would recapitulate the Cdk5–/–phenotype. Comparative histopathological analysis of neural tissue was performed using embryonic day 18.5 mice. Both wild-type and single-mutant specimens are presented for control and comparison when relevant. The established phenotypes of p35–/– and Cdk5–/– mice served as a guide for the characterization of the defects present in the compound mutants (Ohshima et al., 1996, 1999; Chae et al., 1997; Gilmore et al., 1998;Kwon and Tsai, 1998).
Organizational and structural defects in the double knock-out brain are immediately apparent on observation at low magnification of 6–7 μm sections processed with hematoxylin and eosin (H&E) (data not shown). Closer examination of the cerebral cortex at high magnification (20×) showed obvious lamination and neuronal-positioning defects in the double knockout. In the E18.5 wild-type cortex, the stratification of neurons has resulted in establishment of a distinct cell-sparse marginal zone, archetypal cortical plate and subplate regions, and the intermediate zone overlying the ventricular zone (Fig.3A). A comparable section of the p39–/– cerebral cortex is indistinguishable from the wild-type cortex (Fig. 3B), whereas the cortex of p35–/– mice shows mild disruptions in neuronal positioning (Fig. 3C). At E18.5 in p35–/– mice, an ectopic subplate can be detected underneath the most superficial cortical plate, with later-born neurons accumulated underneath (Kwon and Tsai, 1998). Neurons accumulated within the intermediate zone are found slightly clustered. Even though the p39–/– cortex appears normal, the double-null cortex is more disrupted when compared with that of p35–/– mice (Fig.3D). The clumping phenomenon is remarkably more severe and is manifest throughout most of the cortex. An increase in cell density is noticeable in the marginal zone, which is less demarcated and blends into the superficial aspects of the cortical plate. Interestingly, the cortex of the Cdk5–/– mouse is virtually identical to the double knock-out cortex, with similar obfuscation of the normally laminated cortical plate by the clumps of neurons (Fig.3E) and a less distinct marginal zone.
Fig. 3.
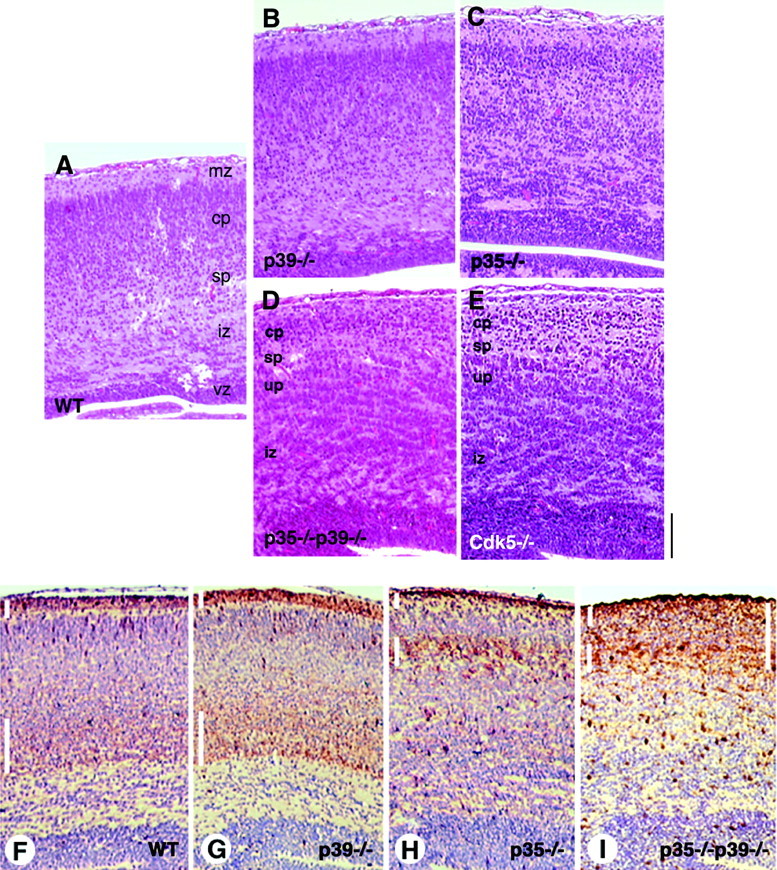
Neuronal positioning in the cerebral cortex is severely disrupted in p35–/–p39–/– brain and is comparable with that in the Cdk5–/– cerebral cortex. Coronal sections of embryonic day 18.5 mice were stained with hematoxylin and eosin. A, In wild-type cortex, discreet laminar organization is apparent, featuring a distinct marginal zone (mz), cortical plate (cp), subplate (sp), intermediate zone (iz), and ventricular zone (vz). B, C, p39-deficient mice (B) do not show any detectable abnormality in cortical lamination, whereas a mild disruption is visible in the p35–/– cortex (C). D, E, The p35–/–p39–/– double knock-out mice (D) exhibit severe disruptions in cortical neuronal positioning that is identical to that observed in Cdk5-deficient mice (E). An increase in cell density is apparent in the marginal zone, and neuronal rifts are more prominent in the cortex in both p35–/–p39–/– and Cdk5–/– mice. In Cdk5–/–mice, it was shown previously that the subplate is in an ectopic, superficial position, leaving an underplate (up) of cortical neurons underneath. The same pattern is observed in the p35–/–p39–/– cortex.F, Calretinin immunostaining, counterstained with hematoxylin, labels preplate derivatives, the marginal zone (short white vertical bar) and subplate (long white vertical bar), in the wild-type cortex. G, The preplate splits properly in the p39–/– mice to give rise to a marginal zone and appropriately located subplate.H, The p35–/– subplate is in a more superficial position as described previously. Despite the proximity of the subplate to the marginal zone in p35–/– mice, the two layers are clearly distinguishable. I, Disruptions in neuronal positioning in preplate-derived structures and the cortical plate in the p35–/–p39–/– mice are shown. The subplate of the p35–/–p39–/– cortex appears to run into an ill-defined marginal zone, because calretinin immunoreactivity is detected throughout the thin cortical plate amid the marginal zone and subplate (long white vertical bar,right). The subplate structure is more perceptible medially than laterally, suggesting that the defects are more pronounced in the lateral aspects of the brain. In F–I, dorsomedial is toward the left side of each panel. Scale bar, 150 μm.
It was reported previously that p35–/–and Cdk5–/– mice establish a marginal zone that can be detected by chondroitin sulfate proteoglycans and calretinin immunostaining, two markers for preplate derivatives that label Cajal-Retzius and subplate neurons and processes (Sheppard et al., 1991; Weisenhorn et al., 1994; Fonseca et al., 1995; Pearlman and Sheppard, 1996; Gilmore et al., 1998; Kwon and Tsai, 1998). Both Cdk5–/– and p35–/– mice at E18.5 and E16.5, respectively, display a superficially placed subplate that is eventually corrected in the p35–/– mouse during the early postnatal period (Gilmore et al., 1998; Kwon and Tsai, 1998). An age-matched comparative analysis between the relevant genotypes has not been performed to date.
In the E18.5 wild-type cortex, calretinin labeled the marginal zone and the subplate underneath the laminated cortical plate (Fig.3F). Calretinin staining in the p39–/– cortex marked a pattern identical to that of the wild-type cortex (Fig. 3G). In the p35–/– brain, the subplate was ectopically located in a more superficial position, as described previously (Kwon and Tsai, 1998), whereas the marginal zone appeared normal (Fig. 3H). Displaced cortical neurons are apparent below the subplate. A misplaced cortical plate is also apparent in p35–/–p39–/–mice beneath the superficial subplate structure, which is reminiscent of the defects in Cdk5–/– cortex. This dislocated cortical plate has been termed the underplate (Gilmore et al., 1998). The definition of the marginal zone and subplate was further distorted in p35–/–p39–/–mice, revealed via calretinin immunoreactivity throughout the superficial aspects of the cerebral wall. Although staining was more intense in the putative marginal zone and subplate structures, it was apparent throughout the thin cortical plate (Fig.3I). This blurring of the marginal zone and subplate was more severe in the lateral aspects of the brain, where axon tracts labeled by calretinin and neurofilament antibodies also coursed through the cortex in an aberrant manner, oblique to the pial surface (data not shown).
Hippocampal formation in p35/p39 compound-mutant mice
The hippocampal formation develops and matures over an extended period during the prenatal and postnatal stages, with complex proliferation and migration gradients (Bayer, 1980). The pyramidal neurons of the cornus Ammonis (CA) have already formed a distinct and compact layer at E18.5 (Fig.4A,A′), sandwiched between the earlier-born neurons of the stratum radiatum and stratum oriens, and will undergo further refinement postnatally (Bayer, 1980). The dentate gyrus is apparent at this stage (Fig.4A). In great contrast, the p35–/–p39–/–hippocampus suffers from a diffuse laminar organization of the CA pyramidal neurons with the appearance of cell-free rifts between clusters of cells (Fig. 4B). These clusters become clearly apparent under higher magnification (Fig.4B′). In addition, the dentate gyrus is indiscernible throughout the entire hippocampal formation. The Cdk5–/– hippocampus shows the same defects as the p35–/–p39–/–hippocampus (Fig. 4C,C′). As in the cerebral cortex, the p39–/– hippocampus is fairly normal (Fig. 4E), but the hippocampus of p35–/– mice at this stage shows signs of disorganization, although to a lesser degree of severity than that of the p35–/–p39–/–hippocampus (Fig. 4D). The p35–/– CA subfields, particularly CA3, are less compact than those in wild-type brain. The dentate gyrus appears underdeveloped as well.
Fig. 4.

p35 is the major Cdk5 activator in hippocampal development. Comparable coronal sections at the midhippocampal level of single and compound mutants demonstrate the contribution of p35 and p39 in hippocampal development. A, The wild-type hippocampus at E18.5 distinctly shows a tightly packed stratum pyramidale (SP) of the CA. The dentate gyrus (dg) can be distinguished at this stage.B, The p35–/–p39–/– hippocampus is severely disrupted. The pyramidal cell fields are diffuse, and neurons are often separated by the appearance of cell-free rifts. The dentate gyrus and hilus (h) are not clearly discernible.C, The phenotype of the p35–/–p39–/– hippocampus resembles that of the Cdk5–/– hippocampus in both morphology and cytoarchitecture. A′, Enlargement of theboxed area in A shows the organization of the stratum pyramidale of wild-type brains. B′, C′, Enlargement of boxed areas in B andC, respectively, shows the similar disorganization and clumping of p35–/–p39–/– and Cdk5–/– pyramidal neurons (arrowheads).D, The p35–/– hippocampus displays slight disorganization in the CA fields, with less densely packed neurons and some cell-free rifts. The dentate gyrus is also less distinct. E, The hippocampal formation in p39–/– mice is comparable with that of wild-type mice. ml, Molecular layer; SO, stratum oriens; SR, stratum radiatum. Scale bar inA′ applies to all panels: A–E, 600 μm;A′–C′, 200 μm.
Dosage-dependent differences for p35 and p39 in cortical and hippocampal development
To address the issue of genetic redundancy, we examined the phenotypes of mice expressing one allele of p35 or p39 in the absence of the other (hereafter referred to as 3:1 mice). The differences between the p35–/–p39+/–and the p35+/–p39–/–brains proved striking and revealed dosage-dependent differences between p35 and p39 function. In particular, the p35+/–p39–/–cortex is comparable with that of the wild type (Fig.5A), but the p35–/–p39+/–cortex features many characteristics of the double-null cortex and is markedly more disrupted than the p35–/–cortex (compare Figs. 5B, 3C). Despite the presence of the thin cortical plate, the obliquely oriented clumps are more prominent in the p35–/–p39+/–cerebral wall and resemble the patterns observed in the double-null mutants. However, there remain differences between the lamination defects of p35–/–p39+/–and p35–/–p39–/–brains (compare Figs. 5B, 3D). The marginal zone is even more obscured in the double knock-out cortex, the underlying cortical plate seemingly pushing into the area normally reserved for Cajal-Retzius neurons and early axonal processes.
Fig. 5.

p35 is the major activator of Cdk5 in the development of the cerebral cortex and hippocampus. Analysis of p35+/–p39–/– and p35–/–p39+/– brains reveals differential dosage-dependent effects of p35- and p39-null alleles.A, Expression of one allele of p35 in the absence of p39 is sufficient to direct gross normal neuronal positioning in the cerebral cortex. B, In contrast, the cortical phenotype in p35–/–p39+/– mice is remarkably more severe than that in either the p35–/– and p39–/– single-null mice and is reminiscent of the p35–/–p39–/– cortex. Note the increased cell density in the marginal zone and the rifts of clustered neurons oriented obliquely to the pial surface. C, Similar to the situation in the cerebral cortex, the hippocampus of p35+/–p39–/– mice resembles that of the wild-type, with a compacted stratum pyramidal and a discernible dg. D, The p35–/–p39+/– hippocampus exhibits defects similar to that of the p35–/–p39–/– and Cdk5–/– hippocampus. The cell-free rifts are less apparent than those observed in double-null mice. C′, D′, Higher magnifications of the stratum pyramidale inboxed regions of C and D, respectively, are shown. Again, one allele of p35 in the p39-null background is able to achieve a detectably normal hippocampal cytoarchitecture. ctx, Cortex;hc, hippocampus. Scale bar: A, B, 150 μm; C, D, 450 μm; C′, D′, 100 μm.
Similarly, the disruptions in the p35–/–hippocampus (Fig. 4D) became more prominent in the p35–/–p39+/–hippocampus, which closely resembles that of the p35–/–p39–/–mouse (Fig. 5D). There are slight differences, however. The cell-free rifts in the p35–/– p39+/–CA subfields appear less conspicuous than those found in double-null mutants (Fig. 5D′). The p35+/–p39–/–hippocampus is comparable with that of wild-type (Fig.5C,C′), demonstrating that one allele of p35 is sufficient for development of the hippocampus as well as of the cerebral cortex.
p35/p39 compound-mutant mice display cerebellar defects
Unlike the cerebral cortex, much of cerebellar development and maturation occurs during the postnatal period. By E18.5, however, a small structure featuring the rudiments of the stereotyped tripartite layering is already present, and the folia have begun to form as outlined by the four cardinal fissures (Fig.6A,G). An uniformly thick external granular layer (EGL) is clearly visible; the fuzzy matrix abutting the EGL marks the multicellular layer of Purkinje neurons (PCL). Strikingly, the cerebellum of a p35–/–p39–/–littermate shows complete lack of foliation and is often smaller in size (Fig. 6B,H). The EGL is visible, but the layer of Purkinje cells is absent. Purkinje cells are selectively labeled by calbindin immunostaining. Although calbindin staining reveals a conspicuous PCL in the wild-type cerebellum (Fig.6G), the labeled cells of the double knockout are found clustered within the cerebellum, near the posterior lobe (Fig.6H). Even though the EGL has formed a roof covering the cerebellum, it is thicker near the germinal trigone of the rhombic lip, where EGL neurons are born (Fig. 6B). The cerebellar phenotype of the p35–/–p39–/–mouse is identical to that described in the Cdk5–/– mouse (Ohshima et al., 1996,1999).
Fig. 6.
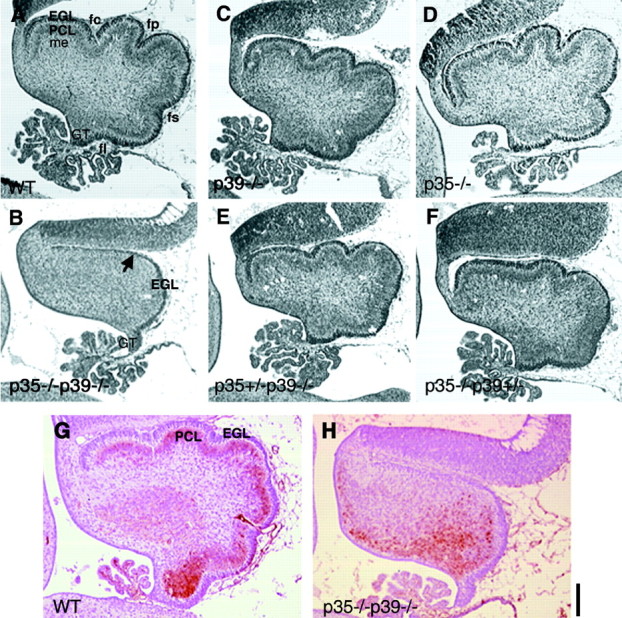
Development of the cerebellum in p35–/–p39–/– mice is severely disrupted. A, Midsagittal sections reveal that in theWT littermate at E18.5, the cerebellum displays rudimentary foliation specified by four cardinal fissures (fc, fp, fs, fl), and Purkinje cells have formed an elementary layer (PCL) beneath theEGL. B, Although the EGL is present in p35–/–p39–/– mice, the Purkinje cell layer is not perceptible in the unfoliated cerebellum. The EGL is thinner toward the anterodorsal aspects of the cerebellum (arrow) and thicker near the germinal trigone (GT) of the rhombic lip. C, D, The cerebellar morphologies of p39–/– and p35–/– mice appear comparable with that of theWT littermate. E, F, Additional genetic dissection of p35 and p39 function using p35+/−p39−/− (E) and p35−/−p39+/− mice (F) shows that although one p39 allele does not significantly rescue the cortical phenotypes observed in the double knockout (shown in Fig. 3), it is able to recover the Purkinje cell migration and foliation defects from that observed in the double knock-out cerebellum. G, Calbindin D28K staining labels the PCL beneath the EGL in a wild-type pup. H, In p35–/–p39–/– cerebellum, the labeled population of cells are clustered ectopically near the neuroepithelium facing ventricle IV and in the posterior lobe, near the germinal trigone. A–F show comparable parasagittal sections of brains from littermates. G andH show parasagittal sections of brains from different late-E18.5 littermates. fc, Preculminate fissure;fl, fissura posterolateralis; fp, fissura prima; fs, fissura secunda; me, medullary layer. Scale bar, 100 μm.
Development of the cerebellum in the single knockouts and in the 3:1 littermates appears to be normal, with slight variations in size and degree of foliation (Fig. 6C–F). The 3:1 mice, in particular, seem to exhibit a decrease in cerebellar size. Interestingly, the fissura secunda in both genotypes is less prominent than that of the wild-type and single-mutant cerebella.
p35–/–p39–/– mice show disruptions in the olfactory bulb, thalamus, and brainstem
To collect additional evidence that p35 and p39 are essential activators of Cdk5, we analyzed other neuronal structures of p35–/–p39–/–and Cdk5–/– mice. The olfactory bulb, like the cortex, cerebellum, and hippocampus, also features characteristic layers of neurons. At E18.5, the most noticeable cell layer is the mitral cell layer, a formation of large neurons sandwiched between the molecular layer and the internal plexiform layer (Fig.7A). The lamination in the olfactory bulb is normal in the absence of p39 (Fig. 7B). There is a slight disorganization in the mitral cell layer in the p35–/– mouse; mitral cells are oftentimes disoriented and misaligned (Fig. 7C). Strikingly, the compact layer of mitral cells is completely missing in the p35–/–p39–/–as well as the Cdk5–/– olfactory bulbs. These large neurons are found scattered throughout the molecular layer (Fig. 7D; data not shown). These data suggest that Cdk5 also directs the laminar organization of olfactory bulb neurons.
Fig. 7.
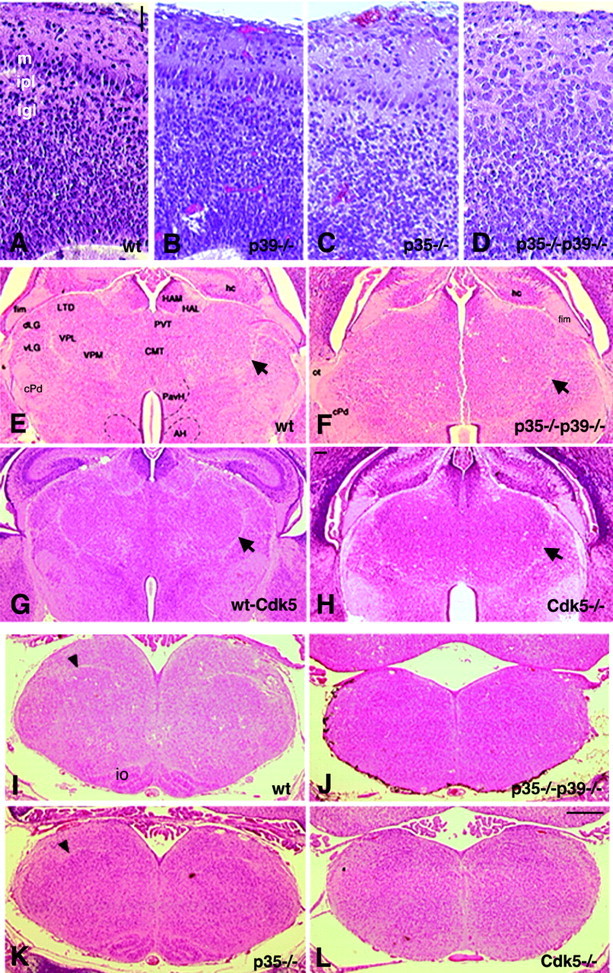
p35–/–p39–/– and Cdk5–/– mice exhibit severe defects in the olfactory bulb, thalamus, and brainstem. A–D, Parasagittal sections of the olfactory bulb stained with hematoxylin and eosin are shown. A, The wild-type olfactory bulb displays a visible lamination pattern, including a layer of large mitral cells (m), the internal plexiform layer (ipl), and the internal granular layer (igl). B, Olfactory bulb organization is intact in the absence of p39. C, The p35–/– olfactory bulb shows slight disruptions in the alignment and orientation of mitral cells but otherwise appears to be normal. D, In the absence of p35 and p39, mitral cells are disorganized and scattered throughout the ml, obscuring the internal plexiform layer as well. E–H, Coronal sections reveal diencephalic defects of p35–/–p39–/– and Cdk5–/– mice. E, F, The wild-type diencephalon (E) shows compartmentalization into thalamic and hypothalamic nuclei, whereas the double knock-out thalamus (F) is primarily uniform. Arrowsdenote the medial lemniscus. G, H, This similar defect is observed in the Cdk5–/– mice (H) in comparison with a wild-type littermate (G). I–L, Comparable coronal sections through the brainstem at the level of the caudal medulla show that the brainstem is hypoplastic and lacks distinct nuclei in the absence of p35 and p39. I, K, The inferior olive (io) and solitary tract (arrowheads) are visible in the wild-type (I) and p35–/–(K) brainstem. J, L, The overall morphology of the brainstem in p35–/–p39–/–(J) and Cdk5–/–(L) mice is abnormal. The organization observed normally in the brainstem is absent here, with the inferior olive clearly missing, as well as the solitary tract. Examination of adjacent serial sections in the rostral and caudal directions did not reveal any of these structures. AH, Anterior hypothalamic nucleus;CMT, centromedian thalamic nucleus; cPD, cerebral peduncle; dLG, dorsal lateral geniculate nucleus; fim, fimbria; HAL, lateral habenular nucleus; HAM, medial habenular nucleus;LTD, lateral dorsal thalamic nucleus;PavH, paraventricular nucleus of the hypothalamus;PVT, periventricular nucleus of the thalamus;vLG, ventral lateral geniculate nucleus;VPL, ventroposterolateral nucleus; VPM, ventroposteromedial nucleus. Scale bars: A–D, 20 μm;E–H, 150 μm; I–L, 350 μm.
Neuronal compartments comprised of multiple nuclei also appear to be severely disrupted in the absence of Cdk5 activity. In both p35–/–p39–/–and Cdk5–/– mice, the normal pattern of nuclear segregation in the thalamus is disturbed (Fig.7E–H), displaying an uniform distribution of cells and altered overall morphology. Although the medial lemniscus appears to be intact in the mutants, other fiber tracts are not clearly visible. This disruption of segregated nuclei and distinct tracts is manifest in brainstem structures such as the medulla (Fig.7I–L). This disorganization is evident in both coronal and parasagittal views of the brainstem. Most noticeably, the superior and inferior olivary nuclei are absent (Fig. 7J,L; data not shown). The solitary tract, a conspicuous bundle of axons seen in the coronal section of the caudal medulla (Fig. 7I,K), is lacking in p35–/–p39–/–and Cdk5–/– mice (Fig. 7J,L). Some nuclei are left intact, including the pontine nuclei, cochlear nuclei, and other cranial nerve nuclei (data not shown).
Motor neuron pathology in p35–/–p39–/– mice
Previously, large neurons of the cranial nerve nuclei and the motor neurons of the Cdk5–/– spinal cord were described as exhibiting ballooned perikarya and eccentric nuclei, suggesting chromatolytic changes in these large neurons (Ohshima et al., 1996). These phenomena are also evident in the p35–/–p39–/–spinal cord anterior horn at E18.5 (Fig.8D) and also in the cranial nerve nuclei (data not shown). Interestingly, p35–/– and p39–/– motor neurons may also exhibit some of these changes, although to a much lesser extent than that of the double knock-out and Cdk5–/– neurons (Fig. 8B,C).
Fig. 8.
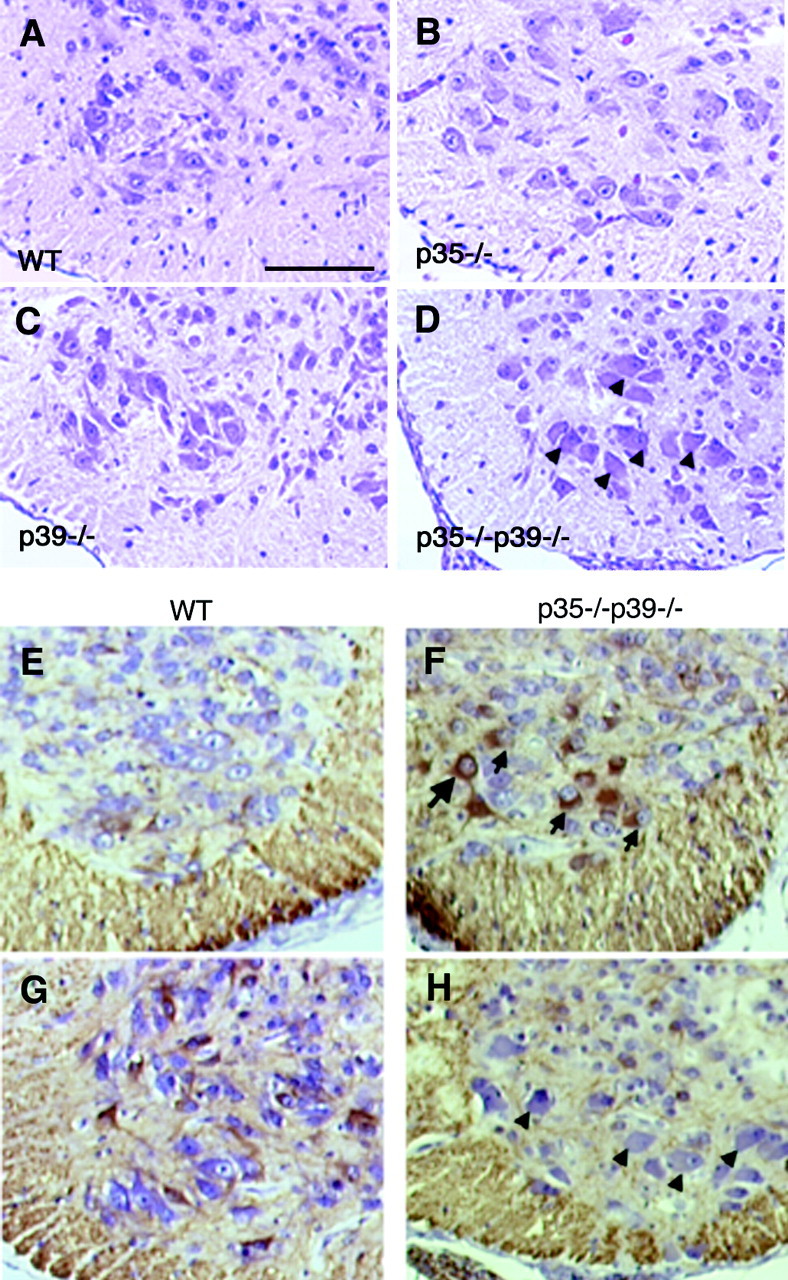
p35–/–p39–/– mice display motor neuron pathology. A, Transverse section through the lower thoracic spinal cord of a wild-type E18.5 pup shows large motor neurons in the anterior horn. B, C, Motor neurons of p35–/– (B) and p39–/– (C) mice appear primarily normal. D, In p35–/–p39–/– mice, the motor neurons exhibit ballooned perikarya and eccentric nuclei, which are hallmarks of chromatolysis. These chromatolytic changes are also seen in Cdk5–/– mice (reported previously).E–H, Immunostaining transverse sections of the wild-type (E, G) and p35–/–p39–/– (F, H) lower cervical (E, F) and lower thoracic spinal cord (G, H) with SMI34, a monoclonal antibody that detects a phosphorylated epitope of NF-H, shows that motor neurons of p35–/–p39–/– mice have aberrant accumulation in the soma of phosphorylated NF-H (F, arrows) that is normally found in axon fibers (E, G). SMI34-positive motor neurons, however, do not always display ballooned perikarya (F,large arrow). Conversely, ballooned neurons are not consistently labeled by SMI34 (H,arrowheads). This may reflect different stages in pathology and related changes in neurofilament phosphorylation. Scale bar, 150 μm.
Chromatolysis is often associated with aberrant accumulation of phosphorylated neurofilaments, particularly of phosphorylated neurofilament heavy chain (NF-H), in the cell soma. Under normal conditions, most phosphorylated NF-H is relegated to the axonal processes (Fig. 8E,G). Immunostaining comparative spinal cord transverse sections with SMI34, a monoclonal antibody that recognizes highly phosphorylated NF-H, shows that a population of double knock-out motor neurons in the ventral horn are positive for these hyperphosphorylated epitopes (Fig.8F,H). These data are consistent with that described previously for Cdk5–/– mice (Ohshima et al., 1996). Axon tracts of the white matter as well as dorsal and ventral roots continue to show phosphorylated NF-H even in the absence of Cdk5 activity (Fig. 8E–H; data not shown). Although a number of the chromatolytic cell bodies are marked by SMI34, it is interesting to note that not all ballooned cells consistently stain positive for SMI34. Conversely, not all cells positive for SMI34 display perikaryal swelling (Fig.8H). Quantification in two p35–/–p39–/–spinal cords reveals that ∼20% of ballooned neurons show phospho-NF-H accumulation (data not shown). In addition, it appears that phospho-NF-H accumulates more readily in motor neuronal soma in the cervical and upper thoracic spinal cord than in the lower thoracic and lumbar regions.
Phosphorylation of known Cdk5 substrates in p35–/–p39–/– mice
The phenotypes manifested in the p35–/–p39–/–mouse provide us with a glimpse of the developmental processes that require regulation of Cdk5 activity by p35 and p39. Such processes include the migration and lamination of cerebral cortex neurons, migration of Purkinje neurons of the cerebellum, and structural maintenance of motor neurons. Several proteins important in developmental and neurodegenerative events have been shown to be substrates of Cdk5. We used the p35–/–p39–/–mice to investigate how phosphorylation of three substrates, Nudel, NF-H, and microtubule-associated protein Tau, is altered in the absence of detectable Cdk5 activity in vivo.
There has been mounting evidence that regulation of transport is an important process during development of the nervous system (Yonekawa et al., 1998). Recently, we showed that Lis1, the protein encoded by the human type I Lissencephaly gene (Reiner et al., 1993), regulates cytoplasmic dynein and microtubule organization (Smith et al., 2000). Moreover, we showed recently that Cdk5 phosphorylation of Nudel, a novel Lis1-interacting protein homologous to Aspergillus nudulans NUDE, positively regulates dynein function (Niethammer et al., 2000). The phosphorylation profile of Nudel is clearly different in the p35–/–p39–/–mutants (Fig. 9A,B). In wild-type brain extracts, Nudel appears as multiple bands in Western blot analysis using an anti-Nudel affinity-purified polyclonal antibody. In the absence of p35 and p39, however, the loss of the slower-migrating species and compression into the higher mobility isoform are apparent and confirm that Nudel is phosphorylated by Cdk5in vivo (Fig. 9A). Densitometric analysis by NIH Image of three detectable Nudel isoforms of wild-type and p35–/–p39–/–mutants was performed by expressing the OD of each of the slower-migrating bands (B2 and B3; Fig.9B) as a percentage of the OD of the fastest-migrating major Nudel species (B1). The analysis revealed that in p35–/–p39–/–brains, the levels of the two slower-migrating Nudel phosphoisoforms are lower than those in the wild-type brains. The presence of Cdk5 in the double-null mutants confirms that absence of the regulatory subunits indeed affects Cdk5 substrate phosphorylation (Fig.9A, bottom).
Fig. 9.

Phosphorylation profiles of known Cdk5 substrates in p35/p39 compound mice suggest physiological and pathological roles for Cdk5. A, The phosphorylation profile of Nudel, a novel Lis1-interacting protein, differs in the p35–/–p39–/– brain lysates when compared with that of wild-type and single-mutant brain lysates. Nudel appears as three or more isoforms in brain extracts (arrowheads; B1–B3). The slower-migrating species of Nudel are less detectable in p35–/–p39–/– brains despite the presence of Cdk5. These data suggest that Nudel is a physiological substrate of the Cdk5 kinase complex during development.B, Quantification of optical density of Nudel isoforms by NIH Image 1.62 is shown. The optical densities of slower-migrating isoforms of Nudel (B2, B3 in A) are expressed as a ratio to the optical density of the fastest-migrating, major Nudel species (B1 in A). Results show that optical density ratios of B2 andB3 to B1 consistently differ betweenWT (n = 3) and p35–/–p39–/– [double knock-out (dKO); n = 3] brains.C, Phosphorylated and nonphosphorylated NF-H as detected by SMI34 and SMI32 antibodies, respectively, is not altered in p35–/–p39–/– spinal cord lysates in both the Triton-soluble and -insoluble fractions.D, Phosphorylation of microtubule-associated protein Tau is unchanged in the absence of Cdk5 activity. Monoclonal antibody AT8 detects phosphorylation on Ser 202 and Ser 205; monoclonal antibody PHF-1 detects phosphorylated Thr 396. Both sites were shown previously to be phosphorylated by Cdk5 in neurodegeneration. These data suggest that although Tau and NF-H were shown to be substrates of Cdk5, their phosphorylation by Cdk5 may be more relevant in pathological states than during development. P, Precipitated;S, soluble.
Phosphorylation is believed to modulate axonal transport of NFs, and depending on their phosphorylation states, NFs show different localization in neurons (for review, see Nixon and Sihag, 1991; Julien, 1999). The accumulation of phosphorylated NF-H in the motor neuron cell body as well as in neurons of the cranial nerve nuclei of p35–/–p39–/–and Cdk5–/– mice suggests that Cdk5 may play a role in regulating the distribution of NF-H (Ohshima et al., 1996). To begin to address this at a biochemical level, we harvested spinal cords from P0 pups of the relevant genotypes and lysed the tissue in a 1% Triton buffer in the presence of phosphatase inhibitors. Both the soluble and insoluble fractions of each genotype were subjected to SDS-PAGE electrophoresis and Western blot analysis with SMI34, which recognizes hyperphosphorylated NF-H, and SMI32, which recognizes unphosphorylated NF-H (Fig. 9C). We do not detect major differences in NF-H phosphorylation in both Triton-soluble and -insoluble fractions.
The microtubule-associated protein Tau is also known to be phosphorylated by Cdk5 (Kobayashi et al., 1993; Ishiguro et al., 1994;Patrick et al., 1999; Ahlijanian et al., 2000). We analyzed the phosphorylation profile of Tau at specific serine/threonine residues using phosphorylation-specific monoclonal antibodies in wild-type and p35–/–p39–/–whole-brain lysates. We did not observe significant changes in levels of phosphorylation at Ser 202 and Ser 205 using the AT8 antibody and at Thr 396 using the PHF-1 antibody (Fig. 9D).
DISCUSSION
The striking differences in the phenotypes of p35–/– and Cdk5–/– mice have been attributed to p39, a neuronal-specific p35 isoform and the only other Cdk5 activator isolated to date (Tang et al., 1995). The complementary spatiotemporal expression patterns of p35 and p39 make them seemingly perfect partners in developmental regulation of Cdk5 function (Cai et al., 1997; Delalle et al., 1997; Zheng et al., 1998; Wu et al., 2000). In addition, other activators of Cdk5 have not been isolated to date, provoking speculation on the essentiality of p35 and p39 function. We found that mice lacking p39 did not display noticeable defects, demonstrating a more complex relationship between p35 and p39. We provide evidence that deficiency of p35 and p39 is comparable with loss of Cdk5. p35–/–p39–/–animals display loss of Cdk5-associated kinase activity and exhibit a phenotype identical to that of the Cdk5–/– mice (Ohshima et al., 1996). These results indicate that p35 and p39 are essential activators of developmental Cdk5 function in that other activators cannot compensate for the joint loss of these two regulatory proteins. In addition, unlike Cdk5, the majority of p35 and p39 expression is neuronal. Therefore, the p35–/–p39–/–phenotypes demonstrate that phenotypes of Cdk5–/– mice are likely caused by loss of neuronal function. Our data correlate with a recent report stating that neuronal Cdk5 activity is critical for survival, because reconstituted Cdk5 transgene expression at the p35 promoter in Cdk5-null mutants rescues the defects (Tanaka et al., 2001).
Distinct and compensatory functions of p35 and p39
Beyond the recapitulation of Cdk5–/– phenotypes, comparative analyses of p35/p39 single and compound mutants revealed findings that merit further attention because they add to our current understanding of p35 and p39 function. The increased severity and presentation of novel phenotypes in p35–/–p39–/–mutants suggest that there are distinct roles for p35 and p39. Discriminating p35 and p39 function required investigating the effects of reducing gene dosage of one protein in the context of complete loss of the other and examining the p35–/–p39–/–phenotypes that can be selectively rescued by one allele of p35 or p39. The analyses of these compound mutants, described below, brought to light the compensatory mechanisms accountable for the phenotypic discrepancies between p35–/–, p39–/–, and Cdk5–/– mice.
The exacerbation of neuronal-positioning defects in the p35–/–p39–/–cerebral cortex yields two possible explanations. First, it is plausible that adhesion is altered to a greater degree in the p35–/–p39–/–cortex, because it was shown that loss of p35 alone can alter neuronal adhesion via a β-catenin/N-cadherin mediated pathway (Kwon et al., 2000). Second, neuronal migration could be interrupted by further disruption of axon trajectories through the p35–/–p39–/–cortical plate in comparison with that described previously for p35–/– mice (Kwon et al., 1999). Indeed, staining axons with neurofilament and calretinin antibodies revealed that neuronal clumps are interposed between fibers coursing aberrantly through the p35–/–p39–/–cortex (data not shown). From these observations, p39 may either directly regulate these processes or compensate for p35 deficiency, but data from 3:1 mutants revealed p39 to have more of a compensatory role. One allele of p35 in a p39-deficient background can direct proper neuronal positioning and axonal fasciculation in the cerebral cortex. On the contrary, p35–/–p39+/–mice displayed cortical disruptions nearly identical to those found in p35–/–p39–/–and Cdk5–/– mice. A similar profile of alterations is manifest in the hippocampi of compound mutants. Thus, in neocortical and hippocampal development, the evidence points to p35 as the pertinent and major activator of Cdk5.
Neuronal migration in the cerebellum follows a markedly different pattern from that of the cerebral cortex and hippocampus but is also severely affected by loss of Cdk5 function. Proper migration of Purkinje and EGL neurons plays an important part in cerebellar morphogenesis (Altman and Bayer, 1997). The p35–/–p39–/–cerebellum is hypoplastic and unfoliated. Calbindin-positive Purkinje cells are clustered deep within the cerebellum with an extension toward the posterior lobe. The EGL is thinner across the anterodorsal surface. These two phenotypes point to a possible delay or arrest in cerebellar development. The Purkinje cell migration defect could result from disruption of guidance cues from EGL precursor neurons. In turn, EGL survival and proliferation are dependent on support from Purkinje cells in the form of the morphogen Sonic hedgehog (Baader et al., 1999;Dahmane and Ruiz-i-Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999). Thus, the EGL defects observed in p35–/–p39–/–mice may be a secondary disruption, because of a deficiency in trophic and proliferative support from the misplaced PCL. Interestingly, unlike in the cerebral cortex and hippocampus, one allele of p39 is able to direct the migration of Purkinje cells into a superficial layer and rescue the cerebellar phenotype. The differential rescue of one allele of p39 between the cerebral cortex and cerebellum suggests that p39 may be the relevant Cdk5 activator in normal cerebellar neuronal migration.
Additional evidence of compensatory mechanisms lies in the fact that in p35–/– whole-brain lysates, p39 protein is detected at a higher level. In contrast, p35 protein levels are insensitive to the loss of p39. Generally, p35 binds and activates Cdk5 more robustly than does p39 and is more abundant in the brain at both the protein and mRNA levels (R. Dhavan and L.-H. Tsai, unpublished data) (Zheng et al., 1998). These observations, taken together with the histological findings, argue that loss of p35 is more detrimental to Cdk5 function, particularly in the forebrain, and that the system compensates for that loss by upregulation of p39. The compensation is incomplete because p35–/– mutant cortices are abnormal. The contrary is true for loss of p39, which appears to be fully compensated for by p35. The removal of both is catastrophic, because it renders Cdk5 inactive, without available reparatory measures.
Novel roles for Cdk5 in development of the CNS
The increased marginal zone cell density of p35–/–p39–/–and Cdk5–/– cerebral cortices is a hitherto unreported phenotype hinting that Cdk5 function in corticogenesis is more intricate than supposed previously. Similar to Cdk5–/– mice, the splitting of the primordial plexiform layer into marginal zone and subplate does occur in p35–/–p39–/–mice; yet, these structures, identified histologically and via calretinin labeling, are strikingly less distinct when compared with wild-type and p35–/– cortices. Subplate formation entails a series of complex neuronal movements (Bayer and Altman, 1991). Our data suggest that beyond the primary partitioning, Cdk5 may regulate completion of compartmentalization in early cortical structures.
We also observed defects in areas of the nervous system that do not feature explicit laminar organization. The inferior olivary nuclei of the precerebellar system are absent in p35–/–p39–/–and Cdk5–/– mice. This defect may be caused by a disruption in the descending intramural migration of inferior olive neurons through the brainstem from the lower rhombic lip (Altman and Bayer, 1978; Altman and Bayer, 1987). Interestingly, the inferior olive sends the majority of its afferents to Purkinje cells and deep cerebellar nuclei (Altman and Bayer, 1997). The absence of such projections may be one of many factors that contribute to alterations observed in Purkinje cell positioning. Similarly, nuclear compartmentalization in many structures of the diencephalon and brainstem is grossly disorganized, if not altogether missing. Regulation of neuronal migration into discrete nuclei is not well understood but involves integration of a complex set of movements within other developing nuclei. Cdk5 is a crucial player in migration and the organization of many brain structures.
Linking form to function: physiological and pathological substrates of Cdk5
The motor neuron pathology and accumulation of phosphorylated NF-H in neuronal soma shed new light on the proposed kinase–substrate link between Cdk5 and NF-H (Starr et al., 1996; Sun et al., 1996; Pant et al., 1997). Changes in levels of NF-H phosphorylation cannot be detected in Western blots of spinal cord lysates from compound-mutant mice. Although it is possible that loss of Cdk5 activity leads to changes in NF-H phosphorylation that we cannot detect, the results are suggestive of changes in cellular NF-H distribution. Rather than a straightforward kinase–substrate relationship, Cdk5 may impact on NF-H behavior via an indirect mechanism during motor neuron development, such as transport. The ensuing accumulation of NF-H prompts a stress response and induces aberrant phosphorylation of NF-H by other kinases known to phosphorylate NF-H (Guidato et al., 1996; Bajaj and Miller, 1997; Veeranna et al., 1998).
Recent evidence uncovered an interaction between spinal cord neurofilaments and the microtubule motor proteins dynein and dynactin that mediates translocation along microtubules in vitro(Shah et al., 2000). A potential role for Cdk5 in regulating transport has surfaced recently with observations that Cdk5 phosphorylation of Nudel, a Lis1-interacting protein, may positively regulate dynein function (Faulkner et al., 2000; Feng et al., 2000; Niethammer et al., 2000; Sasaki et al., 2000; Smith et al., 2000). In p35–/–p39–/–brains, the phosphorylation profile of Nudel is altered, supporting Nudel as a physiological substrate of, but not exclusive to, Cdk5 during development.
We propose that Cdk5 substrates fall into two categories, those that are physiological substrates (such as Nudel) and those that are phosphorylated by Cdk5 under stress-induced or pathological conditions. The microtubule-associated protein Tau and NF-H may be such latter substrates, because their phosphorylation appears unchanged in perinatal p35–/–p39–/–whole brains and spinal cords, respectively. Tau is hyperphosphorylated when Cdk5 activity is deregulated as a result of stress-induced proteolytic cleavage of p35 to p25 (Patrick et al., 1999; Kusakawa et al., 2000; Lee et al., 2000; Nath et al., 2000). Similarly, NF-H phosphorylation by Cdk5 could be a neuropathological response, as implicated by evidence that NF-H is phosphorylated by Cdk5 in human and a mouse model for amyotrophic lateral sclerosis (Bajaj et al., 1999;Nguyen et al., 2001). p35–/–p39–/–mice along with viable p35/p39 compound-mutant mice will provide a powerful system in which to analyze and manipulate further both physiological and pathological roles of Cdk5.
Footnotes
This work was primarily supported by National Institutes of Health Grant GM53049. S.H. was supported by Association pour la Recherche sur le Cancer. We thank Li Zhang for expert technical assistance; Deanna Smith, Rani Dhavan, Ming-Sum Lee, and Janet Volker for critical reviews during the preparation of this manuscript and Martin Niethammer and Ramses Ayala for providing the Nudel antibody.
J.K. and S.H. contributed equally to this work.
Correspondence should be addressed to Dr. Li-Huei Tsai, Howard Hughes Medical Institute and Department of Pathology, 200 Longwood Avenue, Boston, MA 02215. E-mail: li-huei_tsai@hms.harvard.edu.
REFERENCES
- 1.Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altman J, Bayer SA. Prenatal development of the cerebellar system in the rat. II. Cytogenesis and histogenesis of the inferior olive, pontine gray, and the precerebellar reticular nuclei. J Comp Neurol. 1978;179:49–75. doi: 10.1002/cne.901790105. [DOI] [PubMed] [Google Scholar]
- 3.Altman J, Bayer SA. Development of the precerebellar nuclei in the rat. II. The intramural olivary migratory stream and the neurogenetic organization of the inferior olive. J Comp Neurol. 1987;257:490–512. doi: 10.1002/cne.902570403. [DOI] [PubMed] [Google Scholar]
- 4.Altman J, Bayer SA. Development of the cerebellar system in relation to its evolution, structure, and functions. CRC; Boca Raton, FL: 1997. The ascent and settling of Purkinje cells in relation to their time and site of origin. pp. 206–229. [Google Scholar]
- 5.Baader SL, Vogel MW, Sanlioglu S, Zhang X, Oberdick J. Selective disruption of “late onset” sagittal banding patterns by ectopic expression of engrailed-2 in cerebellar Purkinje cells. J Neurosci. 1999;19:5370–5379. doi: 10.1523/JNEUROSCI.19-13-05370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bajaj NP, Miller CC. Phosphorylation of neurofilament heavy-chain side-arm fragments by cyclin-dependent kinase-5 and glycogen synthase kinase-3alpha in transfected cells. J Neurochem. 1997;69:737–743. doi: 10.1046/j.1471-4159.1997.69020737.x. [DOI] [PubMed] [Google Scholar]
- 7.Bajaj NP, al-Sarraj ST, Leigh PN, Anderson V, Miller CC. Cyclin dependent kinase-5 (CDK-5) phosphorylates neurofilament heavy (NF-H) chain to generate epitopes for antibodies that label neurofilament accumulations in amyotrophic lateral sclerosis (ALS) and is present in affected motor neurones in ALS. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:833–850. doi: 10.1016/s0278-5846(99)00044-5. [DOI] [PubMed] [Google Scholar]
- 8.Bayer SA. Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol. 1980;190:87–114. doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- 9.Bayer SA, Altman J. Neocortical development. Raven; New York: 1991. The development of the primordial plexiform layer and its subsequent partitioning into layer I and the subplate (layer VII). pp. 65–72. [Google Scholar]
- 10.Cai XH, Tomizawa K, Tang D, Lu YF, Moriwaki A, Tokuda M, Nagahata S, Hatase O, Matsui H. Changes in the expression of novel Cdk5 activator messenger RNA (p39nck5ai mRNA) during rat brain development. Neurosci Res. 1997;28:355–360. doi: 10.1016/s0168-0102(97)00063-1. [DOI] [PubMed] [Google Scholar]
- 11.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 12.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 13.Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 14.Delalle I, Bhide PG, Caviness VS, Jr, Tsai LH. Temporal and spatial patterns of expression of p35, a regulatory subunit of cyclin-dependent kinase 5, in the nervous system of the mouse. J Neurocytol. 1997;26:283–296. doi: 10.1023/a:1018500617374. [DOI] [PubMed] [Google Scholar]
- 15.Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O'Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 16.Feng Y, Olson EC, Stukenberg PT, Flanagan LA, Kirschner MW, Walsh CA. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;28:665–679. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- 17.Fonseca M, del Rio JA, Martinez A, Gomez S, Soriano E. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol. 1995;361:177–192. doi: 10.1002/cne.903610114. [DOI] [PubMed] [Google Scholar]
- 18.Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guidato S, Tsai LH, Woodgett J, Miller CC. Differential cellular phosphorylation of neurofilament heavy side-arms by glycogen synthase kinase-3 and cyclin-dependent kinase-5. J Neurochem. 1996;66:1698–1706. doi: 10.1046/j.1471-4159.1996.66041698.x. [DOI] [PubMed] [Google Scholar]
- 20.Humbert S, Dhavan R, Tsai L. p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci. 2000a;113:975–983. doi: 10.1242/jcs.113.6.975. [DOI] [PubMed] [Google Scholar]
- 21.Humbert S, Lanier LM, Tsai LH. Synaptic localization of p39, a neuronal activator of cdk5. NeuroReport. 2000b;11:2213–2216. doi: 10.1097/00001756-200007140-00030. [DOI] [PubMed] [Google Scholar]
- 22.Inglis WL, Semba K. Discriminable excitotoxic effects of ibotenic acid, AMPA, NMDA and quinolinic acid in the rat laterodorsal tegmental nucleus. Brain Res. 1997;755:17–27. doi: 10.1016/s0006-8993(97)00101-7. [DOI] [PubMed] [Google Scholar]
- 23.Ishiguro K, Kobayashi S, Omori A, Takamatsu M, Yonekura S, Anzai K, Imahori K, Uchida T. Identification of the 23 kDa subunit of tau protein kinase II as a putative activator of cdk5 in bovine brain. FEBS Lett. 1994;342:203–208. doi: 10.1016/0014-5793(94)80501-6. [DOI] [PubMed] [Google Scholar]
- 24.Julien JP. Neurofilament functions in health and disease. Curr Opin Neurobiol. 1999;9:554–560. doi: 10.1016/S0959-4388(99)00004-5. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi S, Ishiguro K, Omori A, Takamatsu M, Arioka M, Imahori K, Uchida T. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993;335:171–175. doi: 10.1016/0014-5793(93)80723-8. [DOI] [PubMed] [Google Scholar]
- 26.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 27.Kwon YT, Tsai LH. A novel disruption of cortical development in p35(–/–) mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Kwon YT, Tsai LH, Crandall JE. Callosal axon guidance defects in p35(–/–) mice. J Comp Neurol. 1999;415:218–229. doi: 10.1002/(sici)1096-9861(19991213)415:2<218::aid-cne6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 29.Kwon YT, Gupta A, Zhou Y, Nikolic M, Tsai LH. Regulation of N-cadherin-mediated adhesion by the p35-Cdk5 kinase. Curr Biol. 2000;10:363–372. doi: 10.1016/s0960-9822(00)00411-5. [DOI] [PubMed] [Google Scholar]
- 30.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 31.Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 32.Nath R, Davis M, Probert AW, Kupina NC, Ren X, Schielke GP, Wang KK. Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun. 2000;274:16–21. doi: 10.1006/bbrc.2000.3070. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen MD, Lariviere RC, Julien J. Deregulation of cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- 34.Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee M, Morabito M, Tsai L. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 35.Nilden F, Backstrom A, Bark C. Molecular cloning and characterisation of a mouse gene encoding an isoform of the neuronal cyclin-dependent kinase 5 (CDK5) activator. Biochim Biophys Acta. 1998;1398:371–376. doi: 10.1016/s0167-4781(98)00073-6. [DOI] [PubMed] [Google Scholar]
- 36.Nixon RA, Sihag RK. Neurofilament phosphorylation: a new look at regulation and function. Trends Neurosci. 1991;14:501–506. doi: 10.1016/0166-2236(91)90062-y. [DOI] [PubMed] [Google Scholar]
- 37.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB. Migration defects of cdk5(–/–) neurons in the developing cerebellum are cell autonomous. J Neurosci. 1999;19:6017–6026. doi: 10.1523/JNEUROSCI.19-14-06017.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Caceres A. Evidence for the participation of the neuron-specific Cdk5 activator p35 during laminin-enhanced axonal growth. J Neurosci. 1998;18:9858–9869. doi: 10.1523/JNEUROSCI.18-23-09858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pant AC, Veeranna, Pant HC, Amin N. Phosphorylation of human high molecular weight neurofilament protein (hNF-H) by neuronal cyclin-dependent kinase 5 (cdk5). Brain Res. 1997;765:259–266. doi: 10.1016/s0006-8993(97)00561-1. [DOI] [PubMed] [Google Scholar]
- 41.Papaioannou VE, Fox JG. Efficacy of tribromoethanol anesthesia in mice. Lab Anim Sci. 1993;43:189–192. [PubMed] [Google Scholar]
- 42.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration [see comments]. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 43.Pearlman AL, Sheppard AM. Extracellular matrix in early cortical development. Prog Brain Res. 1996;108:117–134. [PubMed] [Google Scholar]
- 44.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 45.Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- 46.Shah JV, Flanagan LA, Janmey PA, Leterrier JF. Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell. 2000;11:3495–3508. doi: 10.1091/mbc.11.10.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheppard AM, Hamilton SK, Pearlman AL. Changes in the distribution of extracellular matrix components accompany early morphogenetic events of mammalian cortical development. J Neurosci. 1991;11:3928–3942. doi: 10.1523/JNEUROSCI.11-12-03928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 49.Starr R, Hall FL, Monteiro MJ. A cdc2-like kinase distinct from cdk5 is associated with neurofilaments. J Cell Sci. 1996;109:1565–1573. doi: 10.1242/jcs.109.6.1565. [DOI] [PubMed] [Google Scholar]
- 50.Sun D, Leung CL, Liem RKH. Phosphorylation of the high molecular weight neurofilament protein (NF-H) by Cdk5 and p35. J Biol Chem. 1996;271:14245–14251. doi: 10.1074/jbc.271.24.14245. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka T, Veeranna, Ohshima T, Rajan P, Amin ND, Cho A, Sreenath T, Pant HC, Brady RO, Kulkarni AB. Neuronal cyclin-dependent kinase 5 activity is critical for survival. J Neurosci. 2001;21:550–558. doi: 10.1523/JNEUROSCI.21-02-00550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 53.Tsai LH, Takahashi T, Caviness VS, Jr, Harlow E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development. 1993;119:1029–1040. doi: 10.1242/dev.119.4.1029. [DOI] [PubMed] [Google Scholar]
- 54.Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 55.Tsuda M, Tashiro T, Komiya Y. Selective solubilization of high-molecular-mass neurofilament subunit during nerve regeneration. J Neurochem. 2000;74:860–868. doi: 10.1046/j.1471-4159.2000.740860.x. [DOI] [PubMed] [Google Scholar]
- 56.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 57.Veeranna, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC. Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci. 1998;18:4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace VA. Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 59.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog [see comments]. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 60.Weisenhorn DM, Prieto EW, Celio MR. Localization of calretinin in cells of layer I (Cajal-Retzius cells) of the developing cortex of the rat. Brain Res Dev Brain Res. 1994;82:293–297. doi: 10.1016/0165-3806(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 61.Wu DC, Yu YP, Lee NT, Yu AC, Wang JH, Han YF. The expression of Cdk5, p35, p39, and Cdk5 kinase activity in developing, adult, and aged rat brains. Neurochem Res. 2000;25:923–929. doi: 10.1023/a:1007544106645. [DOI] [PubMed] [Google Scholar]
- 62.Xiong W, Pestell R, Rosner MR. Role of cyclins in neuronal differentiation of immortalized hippocampal cells. Mol Cell Biol. 1997;17:6585–6597. doi: 10.1128/mcb.17.11.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng M, Leung CL, Liem RK. Region-specific expression of cyclin-dependent kinase 5 (cdk5) and its activators, p35 and p39, in the developing and adult rat central nervous system. J Neurobiol. 1998;35:141–159. doi: 10.1002/(sici)1097-4695(199805)35:2<141::aid-neu2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]