

Abstract
Ataxia-telangiectasia (A-T) is a neurodegenerative syndrome resulting from dysfunction of ATM (ataxia telangiectasia mutated). The molecular details of ATM function in the nervous system are unclear, although the neurological lesions in A-T are probably developmental because they appear during childhood. The nervous systems of Atm-null mice show a pronounced defect in apoptosis that is induced by DNA damage, suggesting that ATM may function to eliminate DNA-damaged neurons. Here we show that Atm-dependent apoptosis occurs at discrete stages of neurogenesis. Analysis of γ-irradiated mouse embryos showed that Atm-dependent apoptosis occurred only in the postmitotic populations that were present in the neuroepithelial subventricular zone of the developing nervous system. Notably, Atm deficiency did not prevent radiation-induced apoptosis in multipotent precursor cells residing in the proliferating ventricular zone. Atm-dependent apoptosis required p53 and coincided with the specific phosphorylation of p53 and caspase-3 activation. Thus, these data show that Atm functions early in neurogenesis and underscore the selective requirement for Atm in eliminating damaged postmitotic neural cells. Furthermore, these data demonstrate that the differentiation status of neural cells is a critical determinant in the activation of certain apoptotic pathways.
Keywords: ataxia-telangiectasia, ATM, p53, subventricular zone, neurogenesis, apoptosis, ionizing radiation, DNA damage, DNA repair
Individuals with the autosomal recessive disorder ataxia-telangiectasia (A-T) manifest a diverse array of symptoms, including immune deficiency, predisposition to cancer, progressive neurodegeneration, and hypersensitivity to ionizing radiation (IR) (for review, see Sedgwick and Boder, 1991; Lavin and Shiloh, 1997; Crawford, 1998; Gatti et al., 2001). Cell lines derived from A-T individuals also show x-ray sensitivity, cell cycle checkpoint defects, premature senescence, and genomic instability (Lavin and Shiloh, 1997; Rotman and Shiloh, 1998). A-T results from dysfunction ofATM (ataxia telangiectasia mutated), a 370 kDa protein that has protein kinase activity with specificity for serine and threonine residues and C-terminal sequence similarity to the phosphatidyl-inositol-3-kinase (PI3K) family (Savitsky et al., 1995; Lim et al., 2000).
The most prevalent feature of A-T is progressive neurodegeneration, although the mechanism and the etiological agent that are responsible are unknown. Therefore, understanding ATM signaling in the nervous system is particularly relevant to the neuropathology of A-T. It is likely that ATM dysfunction impacts during development, because the neurological defects in A-T are apparent early in life (Sedgwick and Boder, 1991; Crawford, 1998). Furthermore, Atm is highly expressed in the developing mouse nervous system but expressed only at low levels in the adult CNS (Soares et al., 1998).
Insight into ATM function in the nervous system has come fromAtm-null mice. These mice recapitulate many of the features of the human disease, including cancer predisposition and severe intestinal toxicity after radiation (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996; Herzog et al., 1998; Borghesani et al., 2000). Additionally, cells derived from the Atm-null mouse have similar characteristics to human A-T cells, such as cell cycle checkpoint defects and replicative senescence (Rotman and Shiloh, 1998). Although overt ataxia is not present in these mice, neurological deficits have been reported. These include behavioral abnormalities, dopaminergic neuron loss in the substantia nigra, and altered brain electrophysiology (Barlow et al., 1996; Eilam et al., 1998; Chiesa et al., 2000). In contrast to the extreme radiosensitivity present in A-T individuals, Atm-null mice show a striking resistance to DNA damage-induced apoptosis in the nervous system (Herzog et al., 1998;Chong et al., 2000). These data suggest that ATM may function to eliminate neural cells that have incurred genomic damage (Herzog et al., 1998; Lee and McKinnon, 2000). Additional support for this assertion comes from the prevalence of neurodegeneration in many DNA repair-deficient syndromes (Rolig and McKinnon, 2000).
The current consensus is that ATM functions as a protein kinase, and inactivation of this kinase activity is responsible for A-T. Biochemical and genetic analyses have identified various substrates for ATM (Lim et al., 2000), and some of these will be important for ATM function in the nervous system. One well characterized substrate of ATM is p53 (Kastan et al., 1992; Kastan and Lim, 2000). For example, IR promotes Atm-dependent p53 stabilization leading to G1 arrest. ATM is required for the phosphorylation of serine-15 and serine-20 of p53 in a DNA damage-dependent manner that leads to stabilization and transcriptional activation (Banin et al., 1998; Canman et al., 1998; Khanna et al., 1998; Waterman et al., 1998; Ahn et al., 2000). Whereas serine-20 phosphorylation of p53 occurs via an ATM-dependent modification of Chk2, serine-15 phosphorylation may be a direct event. Thus, in vitro, ATM can affect p53 phosphorylation, and these events may contribute collectively to p53 function (Meek, 1999). Although these Atm-dependent modifications of p53 are uncharacterized in the nervous system, they are likely to be relevant because the defect in IR-induced apoptosis in Atm-null neurons is also present inp53-null mice (Enokido et al., 1996; Morrison et al., 1996;Herzog et al., 1998; Chong et al., 2000). Therefore, we assessed Atm function in the developing nervous system. Here we report that Atm functions at defined developmental stages and structures in the nervous system to regulate radiation-induced apoptosis.
MATERIALS AND METHODS
Animals. Mice were housed in an American Association of Laboratory Animal Care-accredited facility and were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures for animal use were approved by the institutional animal care and use committee at St. Jude Children's Research Hospital. The presence of a vaginal plug was designated as embryonic day 0.5 (E0.5) and the day of birth as postnatal day 0 (P0). All experimental groups contained at least three Atm or p53 knock-out mice along with wild-type littermates. Mice were irradiated with 14 Gy from a cesium irradiator (at a rate of 0.9 Gy/min) and killed at 1, 2, 4, 6, or 18 hr after irradiation. Littermate animals without irradiation were used as controls. Tissues were collected after transcardial perfusion with 4% paraformaldehyde in PBS. E12.5 and E15.5 embryos were submersed into paraformaldehyde solution immediately after dissection. Fixed tissues were cryoprotected in 25% buffered sucrose solution and cryosectioned at 12 μm with a HM500M cryostat (MICROM, Walldorf, Germany).
Histology and immunohistochemistry. Neutral red staining was performed with 1% Neutral Red (Aldrich Chemical, Milwaukee, WI) in 0.1m acetic acid, pH 4.8, for 1 min, followed by dehydration in ethanol; the slides were coverslipped with Permount (Fisher Scientific, Pittsburgh, PA). For immunohistochemistry the following antibodies were used: CM5 anti-p53 (at 1:1000 for postnatal brains and 1:500 for embryos; Novocastra Laboratories, Newcastle on Tyne, UK), anti-phospho-p53 (Ser15, at 1:250 for postnatal brains and 1:150 for embryos; New England Biolabs, Beverly, MA), anti-P27 (at 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), anti active-caspase-3 (CM1, at 1:1000; IDUN Pharmaceuticals, San Diego, CA), anti-Ki67 (at 1:1000; Novocastra Laboratories), and anti-β-tubulin III (Tuj1, at 1:1000; Babco, Richmond, CA). Antigen retrieval (Midgley et al., 1992) was used for all immunohistochemistry. Cryosections were incubated with antibodies overnight at room temperature after being quenched with endogenous peroxidase, using 0.6% hydrogen peroxide. Immunoreactivity was visualized with the VIP substrate kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's directions after the tissues were treated with biotinylated secondary antibody and avidin DH-biotinylated horseradish peroxidase-H complex (Vectastain Elite Kit, Vector Laboratories). Sections were counterstained with 0.1% methyl green (Vector Laboratories), dehydrated, and mounted in Permount.In situ end labeling (ISEL) staining was performed on cryosections with the Klenow-FragEL kit (Oncogene Research Products, San Diego, CA) according to the manufacturer's directions. For fluorescence detection the embryo sections were treated with blocking solution (5% donkey serum and 1% bovine serum) and then incubated with p53 antibody (CM5 at 1:500). Signals of p53 were visualized with TRITC-anti rabbit IgG raised in donkey (1:200). After intensive washing and blocking with 5% goat serum and 1% bovine serum, the sections were incubated with Ki67 antibody (1:500). FITC-anti rabbit IgG raised in goat (1:200) was used to detect Ki67 signals.
Nonradioactive in situ hybridization.pSPORT containing nucleotides 8345–8939 of mouse Atm was used to generate in situ probes (Soares et al., 1998). Digoxigenin (DIG)-labeled riboprobes were synthesized by in vitrotranscription according to the standard DIG-labeling reaction protocol from Roche Bioscience (Palo Alto, CA). T7 RNA polymerase was used to synthesize the sense probe, and SP6 RNA polymerase was used for the antisense probe. Cryosections were washed in PBS [containing (in mm) 140 NaCl, 2.7 KCl, 10 Na2HPO4, and 1.8 KH2PO4, pH 7.4] and deproteinized with 1 μg/ml proteinase K in Tris-EDTA buffer (100 mm Tris, 50 mm EDTA, pH 8.0) for 30 min. After brief post-fixation with 4% buffered paraformaldehyde and several washes, the sections were acetylated with acetic anhydride (0.25% in 0.1 m triethanolamine buffer, pH 8.0). Sections then were incubated with hybridization buffer (50% formamide, 50 μg/ml yeast tRNA, 50 μg/ml heparin, 1% SDS, and 5× SSC, pH 4.5) for 2 hr at 56°C. Finally, the sections were incubated with the hybridization buffer containing 0.6 ng/μl of antisense or sense probes at 72°C overnight in a humidified chamber. On the following day the sections were washed sequentially in 5× SSC, 0.2× SSC, and Tris buffer I (100 mm NaCl, 100 mm Tris, pH 7.5). For immunological detection the sections were incubated with 5% BSA and 5% normal sheep serum in Tris buffer I containing 0.1% Triton X-100 and then with a sheep anti-DIG alkaline phosphate antiserum (at 1:500; Roche) overnight at room temperature in a humidified chamber. To visualize the positive signals, we rinsed sections with Tris buffer I and subsequently with Tris buffer II, pH 9.5 (100 mm Tris, 100 mm NaCl, 50 mmMgCl2). The signal for Atm mRNA was developed with NBT/BCIP solution (Roche) in Tris buffer II, and the reaction was stopped by incubating the slides in 10 mm Tris and 1 mm EDTA, pH 8.1.
RESULTS
Atm-dependent apoptosis is a feature of differentiating cells in the retina
We used ISEL staining of P5 retina from Atm-nulland wild-type (WT) mice 18 hr after radiation treatment to identify the cells that were susceptible to apoptosis in this tissue. Although WT retina was susceptible to radiation-induced apoptosis, there was a pronounced resistance in Atm-null retina (Fig.1A). However, the resistance to apoptosis in the Atm-null retina was confined to the central region, because many neuroblasts in the periphery of the Atm-null P5 retina underwent apoptosis after radiation (Fig. 1A). A distinguishing characteristic of this peripheral cell population is that they are proliferating, undifferentiated cells, as distinct to the postmitotic central neuroblasts (Fig. 1B) (Young, 1985a,b). Thus, Atm is dispensable for radiation-induced apoptosis in cycling cells in the retina.
Fig. 1.

Atm is required for apoptosis that is induced by ionizing radiation in the differentiating zone of the retina.A, ISEL-positive signal occurs throughout the P5 wild-type (WT) retina but only in the undifferentiated field of the Atm-null retina 18 hr after radiation. B, Schematic diagram shows the stage of cell differentiation in the P5 retina; a is proliferative, whereas b is the differentiating region. In both A and B thearrow demarcates the boundary between proliferation and differentiation. C, At E15.5, cell death, shown by ISEL labeling, is widespread in the WT (e, f) but markedly reduced in theAtm−/− animals (g, h). The irradiatedp53−/− E15.5 retina (i, j) is indistinguishable to the unirradiated WT (c, d). Proliferative populations in the E15.5 retina are shown by Ki67 (a), and differentiating fields are shown by TuJ1 immunoreactivity (b).
To define Atm involvement in retinal cell populations further, we examined apoptosis after radiation at embryonic stages of retinal development. We used markers for proliferating cells, such as Ki67 (Gatter et al., 1986), and differentiating neurons, such as Tuj1 (a neuron-specific β-tubulin III), to distinguish these populations in the retina (Fig. 1Ca,Cb). Although WT retina showed extensive apoptosis after radiation (Fig. 1Ce,Cf), Atm deficiency resulted in a marked abrogation of IR-induced apoptosis in the neuroblasts of the developing E15.5 retina (Fig.1Cg,Ch). However, consistent with the P5 situation, some apoptosis was observed in the proliferative layer of theAtm−/− retina (Fig.1Cg,Ch). Essentially no apoptosis, as judged by ISEL staining, was seen without radiation treatment in the WT (Fig.1Cc,Cd) or Atm−/−or p53−/− (results not shown). There was a pronounced lack of apoptosis after IR in the Tuj1-positive layer of Atm-null neuroblasts compared with extensive death through this layer in WT retina. Apoptosis in theAtm−/− was confined to regions harboring cells that reside in the proliferative layer. Consistent with a requirement of p53 for radiation-induced apoptosis, there was no cell death in the E15.5 p53-null retina after IR (Fig. 1Ci,Cj; see below).
Serine-15 phosphorylation of p53 after ionizing radiation requires Atm
Atm signaling to p53 is a critical step in IR-induced apoptosis in the postnatal cerebellum (Herzog et al., 1998); accordingly, p53 stabilization and apoptosis after IR are reduced greatly in the P5 cerebellum of Atm-deficient animals (Fig.2). However, p53 stabilization still occurs, although at very reduced levels compared with WT (Fig.2e). Because ATM is known to modify serine-15 of p53 selectively after DNA damage (Banin et al., 1998; Canman et al., 1998), we assessed serine-15 phosphorylation of p53 after IR as a measure of Atm activation. We used phospho-specific antisera against phosphorylated serine-15 of p53 to detect phosphorylation of the equivalent residue (serine-18) of mouse p53. We compared stabilization and phosphorylation of p53, before and 1 hr after IR, in the P5 cerebellum in WT and Atm-null mice (Fig. 2). We found that serine-18 phosphorylation of p53 was absent 1 hr after IR inAtm-null animals, compared with controls (Fig.2d,f). Thus, phosphorylation of p53 serine-18 after radiation is Atm-dependent in the developing cerebellum and reflects Atm signaling to p53. These data also indicate a portion of p53 stabilization after IR is independent of serine-18 phosphorylation (compare Fig. 2e,f). Therefore, we also included anti-serine-15 phospho-specific immunohistochemistry in the following experiments to evaluate specific Atm signaling precisely in the developing nervous system.
Fig. 2.
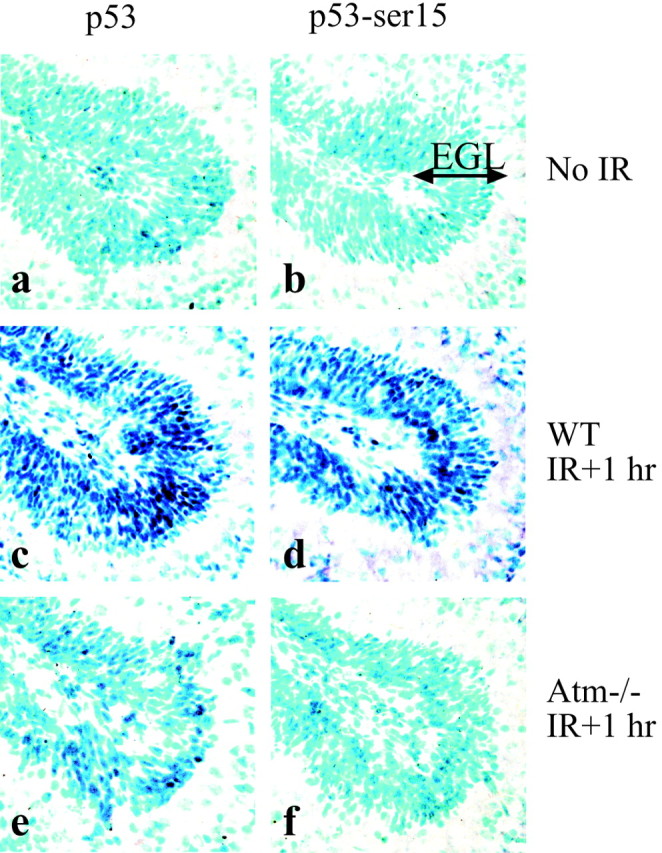
Serine-18 phosphorylation of p53 after radiation in the P5 cerebellum requires Atm. The external granule layer (EGL) of the wild-type P5 cerebellum shows increased amounts of p53 (c) and phosphorylated p53 (serine-18; d) 1 hr after radiation. In the Atm-deficient P5 cerebellum, p53 stabilization is reduced dramatically (e), and serine-18 phosphorylated p53 is not detected (f). No p53 or serine-18 phosphorylated p53 is detected in unirradiated tissue (a, b).
Atm is required for IR-induced apoptosis in cell populations of the subventricular zone
Analysis of Atm in the developing retina indicates a distinct role in mitotic compared with differentiating cells in radiation-induced apoptosis. To determine the extent of the relationship of Atm to cellular differentiation, we examined Atm function after IR during neural development. To do this, we irradiated embryos at either E12.5 or E15.5 and assessed Atm function via p53 stabilization and serine-18 modification of p53. In the developing nervous system at E12.5 there is extensive cellular proliferation, but only limited differentiation. At this stage the majority of the CNS shows immunoreactivity for the proliferation marker Ki67 (Fig.3a) but a restricted staining for neuronal differentiation markers such as Tuj1 (data not shown). As reported previously (Soares et al., 1998) and shown here in Figure3b, Atm expression as determined by in situ hybridization is particularly abundant in the ventricular layers of the neuroepithelium during this stage of development.
Fig. 3.
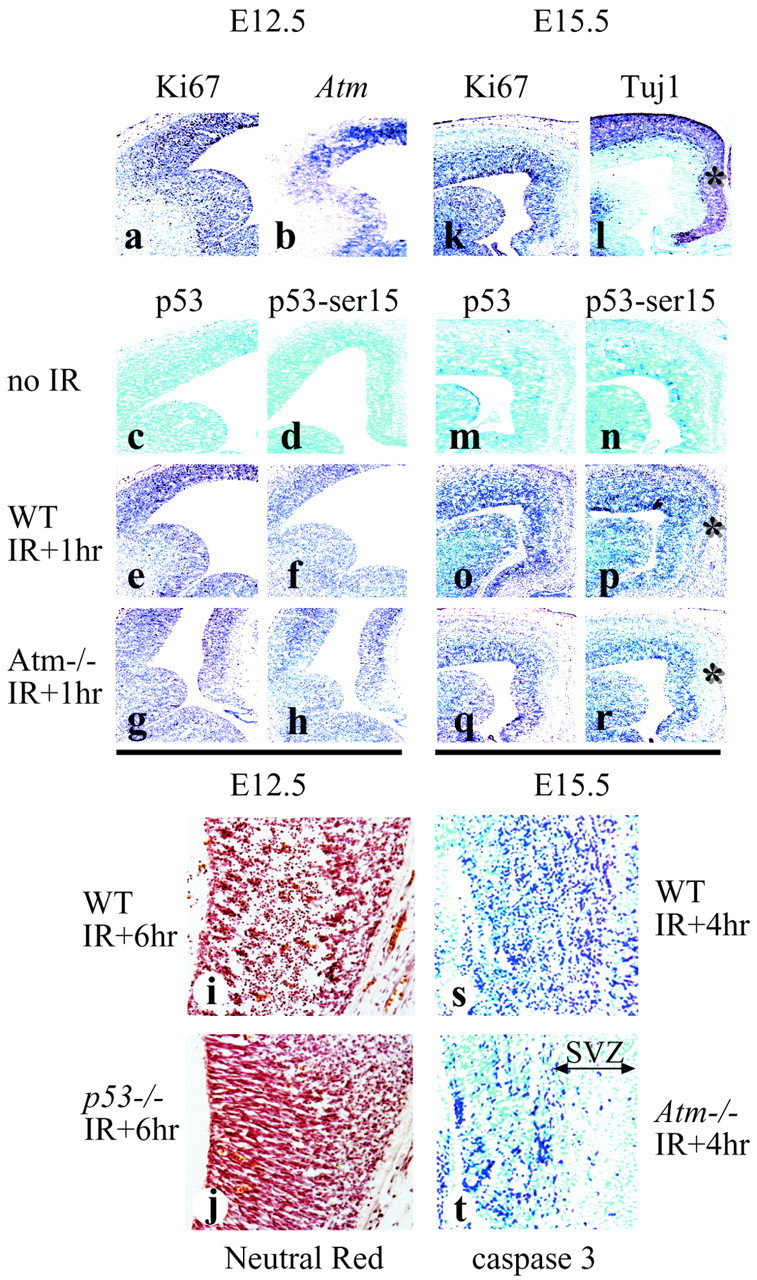
Atm function is distinct between the ventricular and subventricular zones. Comparative analysis of WT andAtm−/− embryos at E12.5 and E15.5 showed a marked reduction in radiation-induced death in theAtm−/− SVZ, as indicated by the asterisks. Regions of proliferation at E12.5 and E15.5 are indicated by Ki67 (a, k) and differentiation at E15.5 by Tuj1 (l) immunostaining.Atm expression at E12.5 is shown by in situ hybridization in b. In the ganglionic eminence and primordial plexiform layer at E12.5 (c–h), no differences in p53 immunostaining between WT andAtm−/− are observed: no irradiation (c, d), irradiated WT (e, f), or irradiatedAtm−/− (g, h). In the WT E15.5 neopallial cortex (m–r) p53 stabilization and serine-18 phosphorylation are widespread (o, p), whereas they are restricted to the ventricular zone inAtm−/− (q, r). Apoptosis is present in WT (i), but not in the ventricular zone of the E12.5p53−/− embryo (j). Regions of caspase-3 activation in E15.5 WT (s) andAtm−/−(t) coincided with p53 stabilization and phosphorylation.
After radiation at E12.5, p53 stabilization and serine-18 phosphorylation were found throughout the ventricular zones (VZ) of the CNS in both WT and Atm-null animals (Fig. 3e–h). No detectable p53 stabilization or serine-18 phosphorylation was seen without radiation (Fig. 3c,d). Cell death measured by ISEL was also apparent throughout the CNS 4 hr after radiation and was confined to regions that showed serine-18 phosphorylation, p53 stabilization, and activated caspase-3 immunoreactivity (data not shown). Because the extent of p53 activation occurring in WT andAtm-null embryos was indistinguishable, it is unlikely that Atm is required for IR-induced apoptosis in these proliferative zones. Thus, in multipotential proliferating cells in the E12.5 VZ, Atm status is unimportant for radiation-induced apoptosis.
However, in contrast to E12.5, at E15.5 there was a clear Atm dependency for IR-induced death in the subventricular zone (SVZ) of the developing nervous system. Atm was required for p53 stabilization, serine-18 phosphorylation, and caspase-3 activation in the SVZ of the neopallial cortex (Fig. 3q,r,t), whereas radiation led to widespread apoptosis in WT embryos (Fig. 3o,p,s). However, like E12.5, cell populations in the VZ of the E15.5 neopallial cortex in either WT or Atm-null are equally susceptible to radiation. Although Atm is not required for radiation-induced apoptosis in the VZ, p53 is essential in both this region and the SVZ (Fig.3i,j). We also found an absence of radiation-induced apoptosis throughout the nervous system of p53-null embryos at developmental stages between E12.5 and E18.5 (results not shown).
In other regions of the CNS, such as the cingulate cortex, a similar requirement of Atm for IR-induced death in postmitotic cells (p27- and Tuj1-immunopositive regions) of the SVZ was observed (Fig.4). Throughout the WT cingulate cortex IR induced p53 stabilization, caspase-3 activation, and cell death (Fig.4d,h–j). However, in the Atm-deficient embryos, the activation of p53 and caspase-3 after radiation occurred only in the VZ (Fig. 4f,k–m), with no significant increase in immunoreactivity for p53 or active caspase-3 in the SVZ of theAtm-null embryos.
Fig. 4.
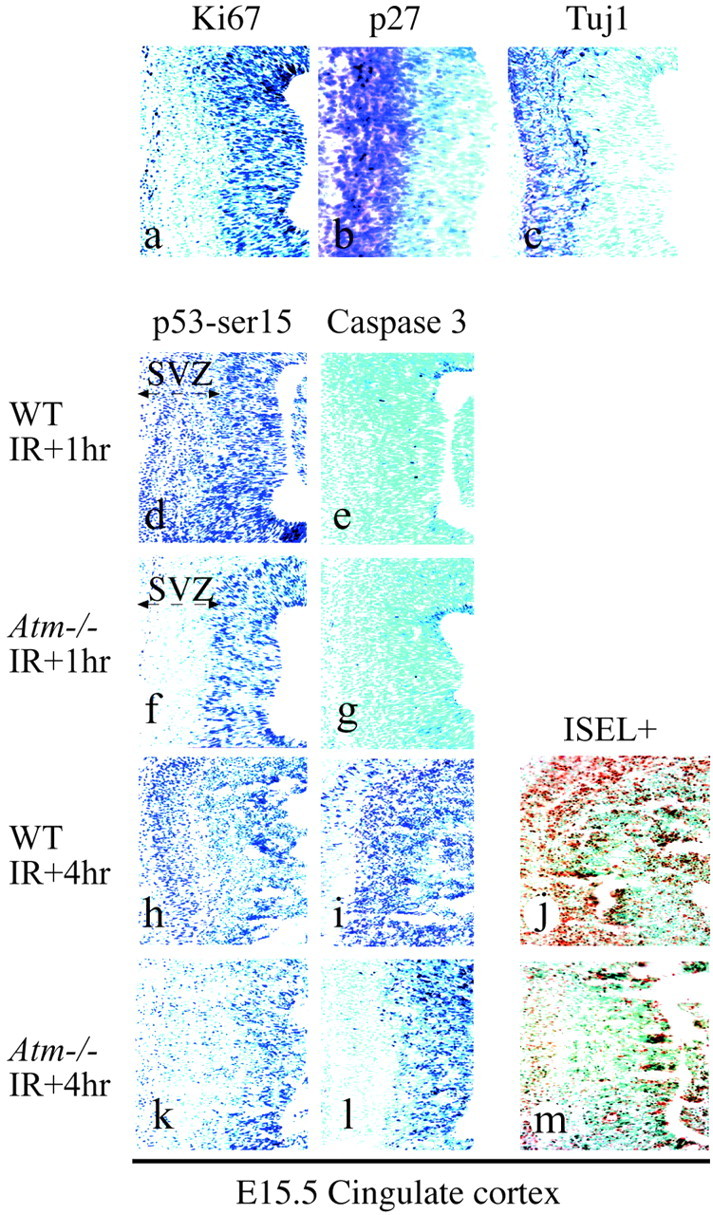
Postmitotic regions in theAtm−/− cingulate cortex are resistant to radiation-induced apoptosis. In theAtm−/− cingulate cortex at E15.5, the subventricular zone (SVZ) was markedly resistant to radiation-induced apoptosis. Proliferating cells were identified by Ki67 (a); to identify postmitotic regions that define the SVZ, we used p27 (b) and Tuj1 (c). The SVZ of WT embryos shows radiation-dependent serine-18 phosphorylation of p53 (d, h), whereas theAtm−/− SVZ does not (f, k). Activated caspase-3 immunoreactivity is shown at 1 hr (e, g) and 4 hr (i, l) after radiation. ISEL+ staining indicates that apoptosis occurs in WT (j), but not inAtm−/−(m), cingulate cortex SVZ.
Although Atm-dependent apoptosis coincided only with Tuj1- or p27-immunopositive regions in the SVZ, we used comparative colocalization of Ki67 and p53 after radiation in WT andAtm-null E15.5 embryos. Figure5 shows that the p53-positive signal in the Atm-null caudate nucleus was confined to regions that harbor Ki67-immunoreactive cells and was not present in the Ki67-negative cells (Fig. 5d–f), whereas the WT caudate nucleus showed a strong p53 signal in Ki67-negative regions (Fig. 5a–c).
Fig. 5.

Radiation-induced stabilization of p53 occurs in proliferating cells. At 1 hr after radiation, p53 stabilization in the caudate nucleus from WT E15.5 is widespread (a–c), but in Atm−/− it is restricted to Ki67-expressing regions (d–f).c and f are merged images of a, b and d, e, respectively. Thearrows in c and f indicate the subventricular zone.
Thus, Atm is required for p53 activation in the SVZ, but not mitotic cells of the VZ. This is despite high Atm expression in these proliferating areas. Consequently, whereas Atm is important for IR-induced apoptosis in the SVZ, it does not appear to play a role in IR-induced apoptosis of the proliferative populations of the VZ.
Atm is required for IR-induced apoptosis in the developing peripheral nervous system
High levels of Atm expression have been found in dorsal root ganglia (DRG) throughout development and adulthood (Soares et al., 1998). However, no function has been reported for Atm in the PNS. To determine whether Atm function in the PNS is similar to that in the CNS, we examined E12.5 DRG after radiation. In theAtm-null DRG there was a pronounced reduction in cells undergoing IR-induced apoptosis compared with WT tissue (Fig.6b,d). Consistent with the report of heterogeneity of Atm expression in the DRG (Soares et al., 1998), the apoptotic response is not entirely uniform because there are some ISEL+ cells in the Atm-null DRG (Fig.6d). The resistance of Atm-null DRG to radiation-induced apoptosis occurred at all levels of the spinal cord (lumbar to sacral) in an indistinguishable manner to that shown in Figure 6 (results not shown). Consistent with other CNS regions, there was also widespread apoptosis in the spinal cord of the WT animal 18 hr after radiation, but not in the spinal cord of Atm-null mice (Fig. 6a,b). Other PNS regions, such as the trigeminal ganglion, also showed increased cell death in the wild-type, but not in the Atm-null, embryos after radiation (data not shown). Thus, consistent with Atm expression in the DRG, we demonstrate here that Atm is also important for radiation-induced apoptosis during development in this structure, broadening the Atm function to a role in the developing PNS.
Fig. 6.
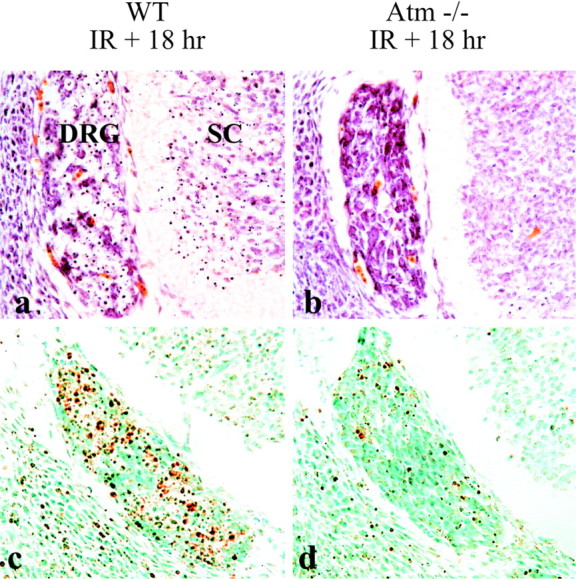
Atm is required for radiation-induced apoptosis in the peripheral nervous system. Widespread apoptosis in the WT (a, c), but not inAtm−/− (b, d), dorsal root ganglia (DRG) is shown both as neutral red staining (a, b) and ISEL staining (c, d). Neutral red-stained pyknotic nuclei are also apparent in the WT (a), but not inAtm−/−(b), spinal cord (SC).
DISCUSSION
In this report we showed that the developmental stage and differentiation status of the nervous system determine Atm-dependent apoptosis after radiation. Furthermore, Atm function appears to be important only in the SVZ, where it is required for apoptosis of neural cells after radiation. It is surprising that, whereas Atm is expressed in the VZ, it is not required for radiation-induced apoptosis in this region. Although it is possible that the death observed in theAtm−/− VZ was attributable to excessive radiation dose, coupled with the extreme sensitivity of proliferating cells to radiation, the striking resistance in theAtm−/− SVZ makes this explanation seem unlikely. Moreover, there is still a genetic basis for this apoptosis, becausep53−/− VZ cells do not undergo radiation-induced death. Therefore, an alternative effector must operate in the VZ that activates p53. Although the identity of this effector is speculative, it may be the ATM-related protein, ataxia telangiectasia Rad 3-related (ATR), which also can signal to p53 via serine-15 phosphorylation (Tibbetts et al., 1999; Shiloh, 2001). In contrast to the nervous system, during early development (∼E6.5) multipotential precursors in the mouse embryo show Atm-dependent apoptosis after IR (Heyer et al., 2000). This implicates a role for Atm during early stages of development and may have some bearing on the recent reports of early lethality in double knock-outs of Atm and DNA-PKcs (Gurley and Kemp, 2001), Atm and Ku (Sekiguchi et al., 2001), and Atm and PARP (Menisser-de Murcia et al., 2001).
The presence of apoptosis in the SVZ, but not in the VZ, after DNA damage has been reported in a number of different mouse models with inactivated DNA repair genes (Barnes et al., 1998; Gao et al., 1998;Deans et al., 2000; Gilmore et al., 2000; Sugo et al., 2000). However, it is likely that the actual DNA damage occurred in the proliferating cell populations of the VZ, possibly during S-phase when there is a greater opportunity for DNA strand breakage to occur (Haber, 1999). In these situations it follows that some sensor recognizes the incurred damage at an early stage after cell cycle exit. Furthermore, at least in some cases, this sensor is Atm (Lee et al., 2000; Sekiguchi et al., 2001). If endogenous DNA damage does occur in proliferating cells, then it is surprising that there is no apparent readout, such as p53 stabilization, until cell cycle exit. Perhaps this reflects relatively low levels of damage compared with radiation, in which the presence of p53 stabilization and apoptosis within the VZ points to a greater genomic insult than endogenous DNA damage.
The role of ATM in the nervous system is unresolved. However, one consistent feature of ATM is involvement in the response to selective DNA damage, such as double strand breaks. Indeed, as mentioned above, recent genetic data have highlighted the importance of the DNA damage response in nervous system development (Barnes et al., 1998; Gao et al., 1998; Deans et al., 2000; Gilmore et al., 2000; Sugo et al., 2000) and the essential role of Atm for apoptotic signal transduction associated with this response (Lee et al., 2000; Sekiguchi et al., 2001). Additionally, human diseases resulting from DNA repair or response abnormalities often are characterized by neurological lesions (Rolig and McKinnon, 2000). Given this clear requirement of DNA repair or DNA damage response for nervous system homeostasis, it seems likely that ATM is important in this capacity in the nervous system. The data reported here suggest a defined developmental period as critical for ATM function and suggest that ATM probably is involved in nervous system maintenance as early as initial neural differentiation. Less clear from these data is a role for ATM at later stages in the life of the nervous system. We have suggested previously that the progressive neurodegeneration seen in A-T is a result of cumulative damage during development, which impacts progressively as the nervous system ages (Herzog et al., 1998; Lee and McKinnon, 2000; Lee et al., 2000). However, it is also possible that ATM performs a non-DNA damage role in the nervous system and that this aspect of ATM function may contribute to nervous system maintenance later in life. ATM function in the nervous system has been linked to regulation of the oxidative load in the brain (Barlow et al., 1999), to a role in neurogenesis (Allen et al., 2001), to survival of dopaminergic neurons (Eilam et al., 1998), and to a possible involvement in vesicular transport (Lim et al., 1998) although, in each of these cases, the Atm-dependent mechanism is unclear.
The progression of neurodegeneration in individuals with A-T is apparent during early childhood and becomes increasingly severe with age, resulting in a requirement for a wheelchair before the early teens. If our hypothesis of accumulated genetic damage as the primary lesion in the nervous system of A-T individuals is correct, then it is likely that the progressive nature of A-T results from a dysfunction of damaged cells over time. However, given that all A-T individuals have an affected cerebellum, then there is some selectivity in the tissues that are affected, and the dysfunction may not be simply a stochastic event in the nervous system. There is still a need for a more precise description of the progressive pathology associated with A-T, because most data to date have been derived from autopsy samples, which provide limited details about disease progression. Advances in imaging techniques are likely to provide a detailed analysis of the progressive nature of A-T and insight into the neuropathology. These types of insight will generate a useful framework for resolving the progression and relative sensitivities of nervous system compartments affected by ATM inactivation.
Our data also highlight the selective nature of apoptosis as it relates to differentiation status. For example, p53 is required for the completion of apoptosis in proliferative and postmitotic cells after radiation, whereas Atm appears to be required for apoptosis only in postmitotic neural cells. However, between populations of different postmitotic neural cells that are Atm-dependent for apoptosis after radiation, different death effectors can be used. This is illustrated by comparing two regions that require Atm for IR-induced apoptosis, the developing retina and cerebellum, where Bax is essential for death in the cerebellum but is not required for death in retinal neuroblasts (Chong et al., 2000). Therefore, our data describing radiation-induced apoptosis in the nervous system have revealed a number of different levels of genetic control that are cell type-specific and cell stage-specific.
In summary, Atm function after DNA damage is linked intimately to the differentiation status of immature neural cells. Taken together, these data underscore an important role for Atm during early phases of development in response to select types of DNA damage. We hypothesize that this stage-specific function is a developmental checkpoint that monitors genomic integrity of neural cells as they exit the cell cycle and begin to differentiate. This damage, if allowed to consolidate during development, will lead to the progressive neurodegeneration that is seen in ataxia-telangiectasia.
Footnotes
These studies were supported by National Institutes of Health Grants NS-37956, NS-39867, and CA-21765 and by the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital. We thank Dr. Suzanne Baker for discussions and comments on this manuscript.
Correspondence should be addressed to Dr. Peter J. McKinnon, Department of Genetics, St. Jude Children's Research Hospital, 332 North Lauderdale, Memphis, TN 38105. E-mail: peter.mckinnon@stjude.org.
REFERENCES
- 1.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- 2.Allen DM, van Praag H, Ray J, Weaver Z, Winrow CJ, Carter TA, Braquet R, Harrington E, Ried T, Brown KD, Gage FH, Barlow C. Ataxia telangiectasia mutated is essential during adult neurogenesis. Genes Dev. 2001;15:554–566. doi: 10.1101/gad.869001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 4.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 5.Barlow C, Dennery PA, Shigenaga MK, Smith MA, Morrow JD, Roberts LJ, 2nd, Wynshaw-Boris A, Levine RL. Loss of the ataxia telangiectasia gene product causes oxidative damage in target organs. Proc Natl Acad Sci USA. 1999;96:9915–9919. doi: 10.1073/pnas.96.17.9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 7.Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci USA. 2000;97:3336–3341. doi: 10.1073/pnas.050584897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 9.Chiesa N, Barlow C, Wynshaw-Boris A, Strata P, Tempia F. Atm-deficient mice Purkinje cells show age-dependent defects in calcium spike bursts and calcium currents. Neuroscience. 2000;96:575–583. doi: 10.1016/s0306-4522(99)00581-3. [DOI] [PubMed] [Google Scholar]
- 10.Chong MJ, Murray MR, Gosink EC, Russell HR, Srinivasan A, Kapsetaki M, Korsmeyer SJ, McKinnon PJ. Atm and Bax cooperate in ionizing radiation-induced apoptosis in the central nervous system. Proc Natl Acad Sci USA. 2000;97:889–894. doi: 10.1073/pnas.97.2.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawford TO. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–294. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 12.Deans B, Griffin CS, Maconochie M, Thacker J. Xrcc2 is required for genetic stability, embryonic neurogenesis, and viability in mice. EMBO J. 2000;19:6675–6685. doi: 10.1093/emboj/19.24.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eilam R, Peter Y, Elson A, Rotman G, Shiloh Y, Groner Y, Segal M. Selective loss of dopaminergic nigrostriatal neurons in brains of Atm-deficient mice. Proc Natl Acad Sci USA. 1998;95:12653–12656. doi: 10.1073/pnas.95.21.12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, Zhu C, Chaudhuri J, Davidson L, Ferrini R, Stamato T, Orkin SH, Greenberg ME, Alt FW. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 17.Gatter KC, Dunnill MS, Gerdes J, Stein H, Mason DY. New approach to assessing lung tumours in man. J Clin Pathol. 1986;39:590–593. doi: 10.1136/jcp.39.6.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gatti RA, Becker-Catania S, Chun HH, Sun X, Mitui M, Lai CH, Khanlou N, Babaei M, Cheng R, Clark C, Huo Y, Udar NC, Iyer RK. The pathogenesis of ataxia-telangiectasia. Learning from a Rosetta stone. Clin Rev Allergy Immunol. 2001;20:87–108. doi: 10.1385/CRIAI:20:1:87. [DOI] [PubMed] [Google Scholar]
- 19.Gilmore EC, Nowakowski RS, Caviness VS, Jr, Herrup K. Cell birth, cell death, cell diversity, and DNA breaks: how do they all fit together? Trends Neurosci. 2000;23:100–105. doi: 10.1016/s0166-2236(99)01503-9. [DOI] [PubMed] [Google Scholar]
- 20.Gurley KE, Kemp CJ. Synthetic lethality between mutation in Atm and DNA-PKcs during murine embryogenesis. Curr Biol. 2001;11:191–194. doi: 10.1016/s0960-9822(01)00048-3. [DOI] [PubMed] [Google Scholar]
- 21.Haber JE. DNA recombination: the replication connection. Trends Biochem Sci. 1999;24:271–275. doi: 10.1016/s0968-0004(99)01413-9. [DOI] [PubMed] [Google Scholar]
- 22.Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- 23.Heyer BS, MacAuley A, Behrendtsen O, Werb Z. Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. 2000;14:2072–2084. [PMC free article] [PubMed] [Google Scholar]
- 24.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 25.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 26.Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 27.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 28.Lee Y, McKinnon PJ. ATM-dependent apoptosis in the nervous system. Apoptosis. 2000;5:523–529. doi: 10.1023/a:1009637512917. [DOI] [PubMed] [Google Scholar]
- 29.Lee Y, Barnes DE, Lindahl T, McKinnon PJ. Defective neurogenesis resulting from DNA ligase IV deficiency requires Atm. Genes Dev. 2000;14:2576–2580. doi: 10.1101/gad.837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim DS, Kirsch DG, Canman CE, Ahn JH, Ziv Y, Newman LS, Darnell RB, Shiloh Y, Kastan MB. ATM binds to β-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci USA. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 32.Meek DW. Mechanisms of switching on p53: a role for covalent modification? Oncogene. 1999;18:7666–7675. doi: 10.1038/sj.onc.1202951. [DOI] [PubMed] [Google Scholar]
- 33.Menisser-de Murcia J, Mark M, Wendling O, Wynshaw-Boris A, de Murcia G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol Cell Biol. 2001;21:1828–1832. doi: 10.1128/MCB.21.5.1828-1832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Midgley CA, Fisher CJ, Bartek J, Vojtesek B, Lane D, Barnes DM. Analysis of p53 expression in human tumours: an antibody raised against human p53 expressed in Escherichia coli. J Cell Sci. 1992;101:183–189. doi: 10.1242/jcs.101.1.183. [DOI] [PubMed] [Google Scholar]
- 35.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- 37.Rotman G, Shiloh Y. ATM: from gene to function. Hum Mol Genet. 1998;7:1555–1563. doi: 10.1093/hmg/7.10.1555. [DOI] [PubMed] [Google Scholar]
- 38.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NGJ, Taylor AMR, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia-telangiectasia gene with a product similar to PI3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 39.Sedgwick RP, Boder E. Ataxia-telangiectasia. In: Vinken P, Bruyn G, Klawans H, editors. Handbook of clinical neurology. Elsevier; New York: 1991. pp. 347–423. [Google Scholar]
- 40.Sekiguchi J, Ferguson DO, Chen HT, Yang EM, Earle J, Frank K, Whitlow S, Gu Y, Xu Y, Nussenzweig A, Alt FW. Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc Natl Acad Sci USA. 2001;98:3243–3248. doi: 10.1073/pnas.051632098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 42.Soares HD, Morgan JI, McKinnon PJ. Atm expression patterns suggest a contribution from the peripheral nervous system to the phenotype of ataxia-telangiectasia. Neuroscience. 1998;86:1045–1054. doi: 10.1016/s0306-4522(98)00117-1. [DOI] [PubMed] [Google Scholar]
- 43.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- 47.Young RW. Cell proliferation during postnatal development of the retina in the mouse. Brain Res. 1985a;353:229–239. doi: 10.1016/0165-3806(85)90211-1. [DOI] [PubMed] [Google Scholar]
- 48.Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985b;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]