

Abstract
Tumor necrosis factor-α (TNF-α) expression has been documented extensively in animal models of traumatic spinal cord injury (SCI). However, the pathophysiological significance of TNF-α expression in the injured cord remains to be delineated. The TNF receptor (TNFR)–nuclear factor-κB (NF-κB) signal transduction pathway is important for maintaining cell viability. NF-κB exerts anti-apoptotic effects via an endogenous caspase inhibitory system mediated by cellular inhibitor of apoptosis protein 2 (c-IAP2). NF-κB transactivates c-IAP2 to inhibit caspase-3 activation. Progressive cell death, including morphological and biochemical features suggestive of apoptosis, has been noted after SCI. We explored the effects of TNFR1 or TNFR2 deletion on the apoptotic events downstream of NF-κB in relation to SCI pathology and functional recovery. Nuclear proteins from the injured cords of the TNFR1−/− mice had a reduced NF-κB binding activity compared with the wild-type controls. This decrease in NF-κB activation was accompanied by a reduction in c-IAP2 expression and an increase in the active form of caspase-3 protein. After SCI the TNFR1−/− mice had greater numbers of apoptotic cells, a larger lesion size, and worse functional recovery than wild-type mice. TNFR2-deficient mice had a similar, although not as pronounced, consequence as the TNFR1−/− mice. These findings support the argument that the TNFR–NF-κB pathway is beneficial for limiting apoptotic cell death after SCI and that a defective TNFR–NF-κB pathway results in a poorer neurological outcome. A worse functional outcome in TNFR−/− mice suggests that an endogenous apoptosis inhibitory mechanism mediated by TNFR activation, NF-κB, and c-IAP2 may be of pathophysiological importance.
Keywords: apoptosis, caspase-3, tumor necrosis factor, TNFR1, TNFR2, cytokine
Extensive demyelination and axonal destruction in traumatic spinal cord injury (SCI) are caused by a complex series of cellular and molecular events (Tator, 1995; Crowe et al., 1997; Rosenberg and Wrathall, 1997; Rosenbluth et al., 1997). In addition to neuronal and glial cell death, delayed waves of oligodendrocyte degeneration with morphological features suggestive of apoptosis have been described in the secondary white matter damage after SCI (Katoh et al., 1996; Li et al., 1996; Crowe et al., 1997; Liu et al., 1997; Springer et al., 1999). Tumor necrosis factor-α (TNF-α), a proinflammatory cytokine, is cytotoxic to oligodendrocytes (Louis et al., 1993; Mayer and Noble, 1994; D'Souza et al., 1995) and may contribute to white matter injury in CNS disorders (Probert et al., 1995). Increases in TNF-α expression after SCI have been documented extensively (Yakovlev and Faden, 1994; Wang et al., 1996;Bartholdi and Schwab, 1997; Xu et al., 1998). TNF-α has been implicated in the pathogenesis of CNS injury after traumatic or ischemic insults (Shohami et al., 1996; Barone et al., 1997).
TNF-α induces a wide spectrum of biological responses by interacting with two cell surface receptors, TNF receptor 1 (TNFR1) and 2 (TNFR2) (Smith et al., 1994). Recent studies have shown that TNFR activation is not necessarily deleterious. TNF-α and TNFR may prevent neuronal apoptosis induced by excitotoxin in vitro (Glazner and Mattson, 2000) and in vivo (Bruce et al., 1996), cerebral ischemia (Nawashiro et al., 1997), experimental allergic encephalomyelitis (Liu et al., 1998), and traumatic brain injury (TBI) (Scherbel et al., 1999). An important intermediate step in the TNFR-mediated signaling process is nuclear factor-κB (NF-κB) activation (Lenardo and Baltimore, 1989; Baeuerle and Henkel, 1994). NF-κB activation may prevent or enhance apoptosis under different experimental paradigms (Mattson et al., 2000). An anti-apoptotic role for the TNFR–NF-κB cascade has been reported (Wang et al., 1998;Tamatani et al., 1999; Chen et al., 2000). The cytoprotective-signaling events downstream of TNFR–NF-κB may involve cellular mammalian homologs corresponding to baculovirus inhibitors of apoptosis protein (IAPs). IAPs inhibit caspase activation (Xue and Horvitz, 1995; Urn et al., 1996; Roy et al., 1997; Deveraux et al., 1998; Wang et al., 1998). Among the IAPs, cellular inhibitor of apoptosis protein 2 (c-IAP2) is expressed after the activation of TNFR. c-IAP2 is transactivated by NF-κB (Chu et al., 1997) and forms a complex with TNFR-associated factor 2 (Rothe et al., 1995) to facilitate an anti-apoptotic action.
The TNFR–NF-κB cascade is activated in rat SCI models (Bethea et al., 1998; Xu et al., 1998). How this pathway regulates apoptosis in the injured spinal cord remains to be fully explored. Mice with a homozygous deletion of TNFR1 or TNFR2 have been generated and exhibit a defect in TNF-α-mediated signaling processes (Rothe et al., 1993;Erickson et al., 1994). In this study we examined the cellular events and morphological and functional outcome after SCI in mice with a TNFR1 or TNFR2 deficiency, in comparison to wild-type mice, to explore the pathophysiological significance of the TNFR–NF-κB cascade.
MATERIALS AND METHODS
Transgenic animals. TNFR1 (Rothe et al., 1993) and TNFR2 (Erickson et al., 1994) null-mutation mice on a C57BL/6 (B6, The Jackson Laboratory, Bar Harbor, ME) background were back-crossed with B6 mice for >12 generations and were maintained in the Washington University School of Medicine animal facility. B6 mice were used as the wild-type controls.
Spinal cord injury. Female mice, 8–12 weeks old, were anesthetized with an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg). During surgery the rectal temperature was monitored and maintained at 37.0 ± 0.5°C by a heating pad. The skin above the vertebral column was shaved and cleaned with antiseptic. A 15 mm midline skin incision was made, and the vertebral column was exposed. After the spinal thoracic region was exposed by separation of dorsal muscles to the side, the spinous processes of T8–T13 vertebrae were exposed. A laminectomy was performed at vertebral level T10 and one-half of T9, exposing the dorsal cord surface with the dura remaining intact. The vertebral column was stabilized with angled clamps on the T8 and T12 transverse processes. SCI was induced by a modified NYU Impactor (Gruner, 1992; Basso et al., 1996a,b; Liu et al., 1997). A 10 gm weight (1.5 mm tip in diameter) was allowed to drop from 3 mm onto the T10 segment. The impact rod was removed immediately, and the wound was irrigated. Muscles and incision were closed in layers, and the animals were placed in a temperature- and humidity-controlled chamber (ThermoCare, Incline Village, NV) for 3 hr. Sham-operated animals received the same surgical procedures but sustained no impact injury. Manual bladder expression was performed twice a day until reflex bladder emptying was established. All surgical procedures, interventions, and presurgical and postsurgical care followed the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Survival Surgery provided by the Animal Studies Committee of Washington University School of Medicine.
Behavioral tests. To evaluate the functional consequence of SCI, we performed the locomotor rating test with the Basso–Beattie–Bresnahan (BBB) scale, as described previously (Basso et al., 1996a). The BBB scale is readily applicable to mice without modifications (Jakeman et al., 2000) especially when the score is below 16 (G. M. Kim and C. Y. Hsu, unpublished observations). Before surgery the mice were placed individually in a molded plastic open field for 4 min to assure that all subjects consistently obtained a maximum score of 21 (n = 10–11 per group). For 28 d, beginning 1 d after injury, the mice were placed daily in an open field for 4 min, and well trained investigators scored the BBB scale in a blinded manner.
Histopathology. After receiving behavioral testing for 4 weeks, animals (n = 6–7 per group) were given a lethal overdose of pentobarbital and perfused intracardially with normal saline, followed by 4% paraformaldehyde in 0.1 mPBS, pH 7.4. For histological evaluation a 10 mm cord segment centered at the injury site was blocked from the vertebral column, placed in the same fixative overnight, and embedded in paraffin. Serial 10 μm cross sections were stained with hematoxylin and eosin. The presence of inflammatory cells, loss of neurons, and the existence of degenerating neurons, microcyst formation, and gliosis defined the rostrocaudal boundaries of the tissue damage. Total lesion length was determined by calculating the distance between the rostral and caudal boundary of the lesion area as determined by light microscopy (Kuhn and Wrathall, 1998).
Isolation of nuclear proteins. Nuclear protein extracts were prepared from sham-operated or injured spinal cord segments at 1, 4, and 24 hr after injury (n = 3 per time point) as described previously (An et al., 1993; Xu et al., 1998). An 8 mm cord segment (4 mm rostral and caudal from the epicenter) was dissected and frozen immediately after the animal was killed under anesthesia. Nuclear proteins were extracted according to a high-salt method (Dignam et al., 1983) with modifications (An et al., 1993; Yan et al., 1999; Xu et al., 2001a).
Electrophoretic mobility shift assay. NF-κB binding activity was assessed by electrophoretic mobility shift assay (EMSA) as described previously (X. Xu et al., 1998; J. Xu et al., 2001a), using the NF-κB consensus oligonucleotide 5′-AGTTGAGGGGACTTTCCCAGGC-3′ (Life Technologies, Rockville, MD). The NF-κB oligonucleotide was labeled with γ-32P-ATP according to Promega (Madison, WI) technical bulletin 106. The binding reaction was performed in a total volume of 20 μl containing the binding buffer [with (in mm) 10 Tris-HCl, 20 NaCl, 1 DTT, 1 EDTA, and 5% glycerol, pH 7.6], 0.0175 pmol of labeled probe (>10,000 cpm), 20 μg of nuclear protein, and 1 μg of poly(dI-dC). After incubation for 20 min at room temperature, the mixture was electrophoresed on a nondenaturing 6% polyacrylamide gel at 180 V for 2 hr under low-ionic-strength conditions. The gel was dried and subjected to autoradiography.
Western blot analysis. At 1, 4, and 24 hr after injury (n = 3 per time point), an 8 mm spinal cord segment (4 mm rostral and 4 mm caudal from the lesion epicenter) was dissected from control and injured mice and frozen immediately in liquid N2. The cord segment was homogenized in a buffer containing (in mm) 10 HEPES, 1.5 MgCl2, 10 KCl, and 0.5 DTT plus 1% NP-40, 1 μg/ml leupeptin, and 1 μg/ml aprotinin, pH 7.9, and then centrifuged at 14,000 × g. Protein (40 μg) from the supernatant of each sample was separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes by electrophoresis. The membranes were blocked in TBST buffer containing 20 mmTris-HCl, 5% nonfat milk, 150 mm NaCl, and 0.05% Tween 20, pH 7.5, for 1 hr at room temperature. Then the blots were incubated with either a primary polyclonal rabbit anti-c-IAP2 antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) or a rabbit anti-caspase-3 antibody (1:200; Santa Cruz Biotechnology), followed by a secondary alkaline phosphatase-conjugated anti-rabbit IgG antibody (1:5000; Promega). The Western blots were visualized with the Blot AP System (Promega).
Immunohistochemical staining. At 1 d after injury, mice (n = 3 per group) were overdosed by an intraperitoneal injection of 100 mg/kg sodium pentobarbital. Intracardial perfusion fixation was done with physiological saline, followed by 4% paraformaldehyde in 0.1 m PBS, pH 7.4. The spinal cord was dissected out carefully, and a 10 mm segment containing the injured epicenter was blocked, post-fixed for 2 hr in the same fixative solution, and transferred to a solution containing 30% sucrose in 0.1m PBS, pH 7.4, for diaminobenzidine (DAB; Sigma-Aldrich, St. Louis, MO) or immunofluorescence staining. Then the spinal cord segments were embedded in tissue-freezing medium (Sakura Finetek, Torrance, CA), cut horizontally or longitudinally at 10 μm on a cryostat into sections, and mounted on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA). Sections were permeabilized with 0.3% Triton X-100 and 4% normal goat serum in 0.01m PBS, pH 7.4, for 20 min and incubated overnight at 4°C with either a polyclonal rabbit anti-c-IAP2 antibody (1:250; Santa Cruz Biotechnology) or a rabbit polyclonal anti-caspase-3 antibody (1:100; PharMingen, San Diego, CA), followed by a biotin-labeled goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA). After washing, the sections were incubated further with ABC Elite complex (Vector Laboratories). The staining was visualized with DAB. Slides were washed, dehydrated, cleared in xylene, and mounted.
For the immunofluorescence labeling for myelin basic protein (MBP), mice were perfused at 7 d after injury (n = 3 per group). Transverse sections 3 mm rostral to the lesion epicenter were incubated with polyclonal rabbit anti-MBP antibody (1:100; Zymed Laboratories, South San Francisco, CA) at 4°C overnight, followed by fluorescein (FITC)-conjugated anti-rabbit IgG antibody (1:100; Vector Laboratories). Slides were washed, wet-mounted, and examined with theOlympus BX60 microscope (Olympus Optical, Tokyo, Japan). For negative controls the primary antibodies were replaced by normal rabbit serum in 0.01 m PBS, pH 7.4.
For the immunofluorescence double-labeling experiment, spinal cord sections were permeabilized with 0.3% Triton X-100 and 4% normal goat serum in 0.01 m PBS, pH 7.4, for 20 min. Polyclonal rabbit anti-c-IAP2 antibody (1:100; Santa Cruz Biotechnology) or rabbit anti-caspase-3 antibody (1:100; PharMingen) with either monoclonal mouse anti-NeuN antibody (1:500; Chemicon, Temecula, CA) or mouse anti-RIP antibody (1:100; Chemicon) was applied to the sections at 4°C overnight. Then the sections were incubated with an FITC-conjugated goat anti-rabbit IgG antibody (1:100; Vector Laboratories) and Texas Red-conjugated goat anti-mouse antibody (1:100; Vector Laboratories). Slides were washed, wet-mounted, and examined under fluorescent microscopy.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling staining. Spinal cord tissue (n = 4–6 per group) from mice with a 7-d-after-SCI survival time was examined via the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) method (Liu et al., 1997) with the Apoptag in situ kit (Oncor, Gaithersburg, MD). Negative control sections were treated similarly, but the TdT enzyme was omitted. TUNEL-positive nuclei were counted visually in five different regions at sampling sites 0, 1, and 2.5 mm rostral and caudal to the lesion center for each animal. Immunofluorescence double labeling with TUNEL staining and an oligodendrocyte cell surface marker, as detected with an anti-adenomatus polyposis coli antibody (1:400; Oncogene, San Diego, CA), was used to assess oligodendrocyte apoptosis after SCI.
Ex vivo magnetic resonance imaging magnetic resonance imaging (MRI) analysis. At 1 d and 7 d after injury, mice were perfused with saline; 2 cm of spinal cord with vertebral column was removed immediately for magnetic resonance imaging (MRI). Diffusion-weighted images (DWI) maps were acquired. MRI studies were performed on an Oxford Instruments-200/400 (4.7 Tesla, 40 cm clear bore; Oxford, UK) magnet equipped with a 10 cm in inner diameter actively shielded gradient coil (60 G/cm, 200 μsec rise time; Magnex). The magnet and gradient coil and power supply were interfaced with a Varian UNITY-INOVA console controlled by a Sun Microsystems (Palo Alto, CA) Ultra-170 Sparc workstation. Excised mouse spinal columns were placed on a 2 × 3 cm plate for the imaging plan alignment. A birdcage coil of 2 cm in inner diameter was used as the transceiver. The DWIs were collected by using a conventional multislice spin echo sequence modified by adding a pair of Stejskal–Tanner diffusion-sensitizing gradients. The DWIs were acquired with TR 1.5 sec, TE 35 msec, slice thickness 0.2 mm, rectangular field of view 1.5 × 2.5 cm, and data matrix 128 ×128 (zero filled to 256 × 256). Diffusion-sensitizing gradients were applied along six directions of [Gx,Gy,Gz] = [1,1,0], [1,0,1], [0,1,1], [−1,1,0], [0,−1,1], and [1,0,−1]. Individual apparent diffusion coefficients of the diffusion tensor were determined by nonlinear squares curve fitting of the voxel amplitudes,A(b), of the six DWIs obtained at bvalue of 764 sec/mm2, and a T2-weighted image without diffusion-sensitizing gradient, A(b = 0), as described by Equation1:
| Equation 1 |
The diffusion-sensitizing factor, b, is defined as Equation 2:
| Equation 2 |
where γ is the gyromagnetic ratio, G is the amplitude of diffusion-sensitizing gradient, δ is the diffusion gradient pulse duration, and Δ is the time separation of the diffusion-sensitizing gradient pulse pairs.
Quantitation of spinal cord lesion area. At 7 d after injury, mice (n = 3 per group) were perfused intracardially. Frozen sections (16 μm thick) were permeabilized and blocked with 0.3% Triton X-100 and 3% normal donkey serum in 0.01m PBS, pH 7.4, for 15 min. To identify axons, we incubated the sections with a monoclonal mouse anti-SMI-31 antibody (1:1000; Sternberger Monoclonals, Lutherville, MD) at 4°C overnight, followed by a tetra-rhodamine (TRITC)-conjugated donkey anti-mouse antibody (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA). Sections were washed, mounted, and examined with Olympus BX60 microscopy (Olympus Optical). Control sections were incubated with normal mouse IgG as the primary antibody. The lesion area was identified by the boundary formed by SMI-31-positive axons. Three horizontal sections, one each at the central canal, 60 μm dorsally, and 60 μm ventrally, were selected for measurement with a Neurolucidasystem (MicroBrightField, Colchester, VT) attached to a Leica DRMB microscope; the values that were obtained were averaged.
Statistical analysis. Data are presented as mean ± SEM values. For BBB scores, histopathological analysis, TUNEL staining, and lesion area measurement, one-way ANOVA with post hoc Tukey test was used to determine statistical significance. A pvalue of <0.05 was considered statistically significant.
RESULTS
Functional assessment in TNFR-deficient mice
After traumatic SCI, all injured mice were paralyzed in both hind limbs. The functional recovery score was followed for 28 d with the BBB scoring system, a standard method for assessing the hind limb motor function after SCI in both rats (Basso et al., 1996a,b) and mice (Jakeman et al., 2000). Poor locomotor performance was identified in TNFR1−/− and TNFR2−/− mice (Fig.1A). At 4 weeks of injury, significantly lower BBB scores were noted in TNFR1−/− (8.6 ± 0.5;n = 10; p < 0.001) and TNFR2−/− (11.6 ± 0.9;n = 10; p < 0.01) mice compared with wild-type B6 mice (15.0 ± 0.8; n = 11). At 4 weeks after injury most of the TNFR1−/−mice were unable to bear weight on the hind limbs and had incomplete hind limb coordination.
Fig. 1.

Quantitative analysis of functional recovery and lesion length after traumatic SCI. A, TNFR1−/− and TNFR2−/− mice showed worse functional outcomes than the wild-type (WT) control B6 mice by BBB score at 4 weeks after injury (*p < 0.01 and **p < 0.001 vs wild-type). B, Lesion length measurements demonstrated that TNFR1−/− mice had more extensive tissue injury than B6 mice (*p < 0.02; n = 6–7 per group).
Histological analysis
At 4 weeks after SCI the lesion epicenter showed marked gliosis and white matter vacuolation as well as gross cord atrophy (data not shown). The lesion lengths in TNFR1−/−mice (3.71 ± 0.32 mm) were significantly longer than those in the wild-type controls (2.38 ± 0.27 mm; p < 0.01). The difference in lesion lengths between the TNFR2−/− mice (2.75 ± 0.26 mm) and the wild-type controls was not statistically significant (Fig.1B). Conventional spared tissue quantitation (Beattie et al., 1997; Liu et al., 1997) could not be used for mice in these experiments because of the lack of cavitation in the lesion center.
NF-κB binding activity
EMSA, performed with nuclear proteins extracted from the spinal cords of sham-operated mice at 1, 4, and 24 hr after injury, demonstrated a low basal level of NF-κB activity in wild-type as well as in TNFR1−/− and TNFR2−/− mice. SCI resulted in a significant time-dependent increase in NF-κB activity at 1 hr, with the activity reaching maximal levels at 4 hr and decreasing at 24 hr after SCI. TNFR1−/− and TNFR2−/− mice had a blunted NF-κB activation when compared with the wild-type controls, with the TNFR1−/− mice exhibiting the lowest levels of NF-κB binding activity (Fig.2A).
Fig. 2.

Effects of TNFR deletion on the NF-κB anti-apoptotic pathway after SCI. Samples from sham-operated and injured spinal cords were collected 1, 4, and 24 hr after SCI.A, EMSA showed a decrease in NF-κB binding activity in TNFR1−/− and TNFR2−/− mice at all of the time points that were tested. B, Western blot analysis showed a decrease in c-IAP2 expression after SCI in TNFR-deficient mice, especially TNFR1−/−.C, Western blotting shows that the expression of the active form of caspase-3 (17 kDa) was increased in TNFR-deficient mice, especially TNFR1−/−. The β-actin Western blot using the same spinal cord samples served as a control to demonstrate equal protein loading in each lane.
c-IAP2 and caspase-3 expression in TNFR-deficient mice
Because c-IAP2 is known to act downstream of the TNF-α–NF-κB cascade in preventing cell death (Chu et al., 1997), c-IAP2 protein expression in the spinal cord was examined by Western blot analysis. Basal levels of c-IAP2 in sham-operated wild-type controls were expressed at higher levels than in the TNFR1−/− and TNFR2−/− mice. After injury an increase in c-IAP2 expression was noted but was substantially less pronounced in the TNFR1 and TNFR2 mutants at 1 and 4 hr (Fig. 2B). However, at 24 hr the c-IAP2 protein levels remained low in the TNFR1−/− mice, whereas the TNFR2−/− mice expressed more c-IAP2 protein when compared with the wild-type mice. The expression of the active form of caspase-3 is a hallmark of apoptotic cell death (Kumar, 1997) and has been shown after SCI (Springer et al., 1999). The expression of caspase-3 was increased after injury and more pronounced in the TNFR1−/− and TNRF2−/− mice than in the wild-type controls (Fig. 2C). To ensure that the apparent increase in caspase-3 immunoreactivity represented an increase in the active form of caspase-3, we also did immunostaining with an antibody, CM1, specific for the caspase-3 active form (Han et al., 2000). Similar results were obtained with the CM1 antibody (data not shown). Consistent with the c-IAP2 Western blot result, c-IAP2 immunoreactivity was less intense in the injured cord in TNFR2−/− and especially in TNFR1−/− mice compared with the wild-type controls (Fig.3A–C). The immunoreactivity of caspase-3 was enhanced to a greater extent in TNFR1−/− mice compared with the wild-type B6 mice, whereas TNFR2−/− mice showed a similar intensity of staining as the wild-type controls (Fig.3D–F), confirming the results from the caspase-3 Western blot.
Fig. 3.
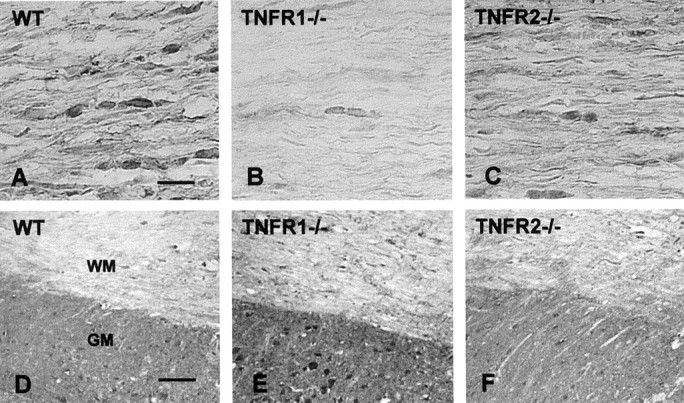
c-IAP2 and active caspase-3 immunoreactivity in B6, TNFR1−/−, and TNFR2−/−mice 24 hr after SCI. A–C, c-IAP2 expression was decreased in TNFR1−/− mice compared with B6 mice in the white matter tract 1 mm rostral to the lesion epicenter.D–F, Compared with B6 mice, caspase-3 expression was increased in TNFR1−/− mice in both the gray matter (GM) and white matter (WM). Longitudinal sections 1–2 mm rostral to the lesion epicenter are shown. Scale bars: A–C, 20 μm, D–F, 100 μm.
Cellular localization of c-IAP2 and caspase-3
To determine whether c-IAP2 and caspase-3 are present in neurons and oligodendrocytes, we performed an immunofluorescence double-labeling experiment on wild-type controls 1 d after SCI. c-IAP2 and the oligodendrocyte marker, RIP, showed cellular colocalization in the white matter tract in the injured cord (Fig.4A). Additionally, c-IAP2 colocalized with the neuronal nuclear filament maker, NeuN, in neurons in the gray matter (Fig. 4A) in the injured cord. In similar experiments caspase-3 immunoreactivity also was localized to neurons as well as to oligodendrocytes around the lesion epicenter area (Fig. 4B).
Fig. 4.
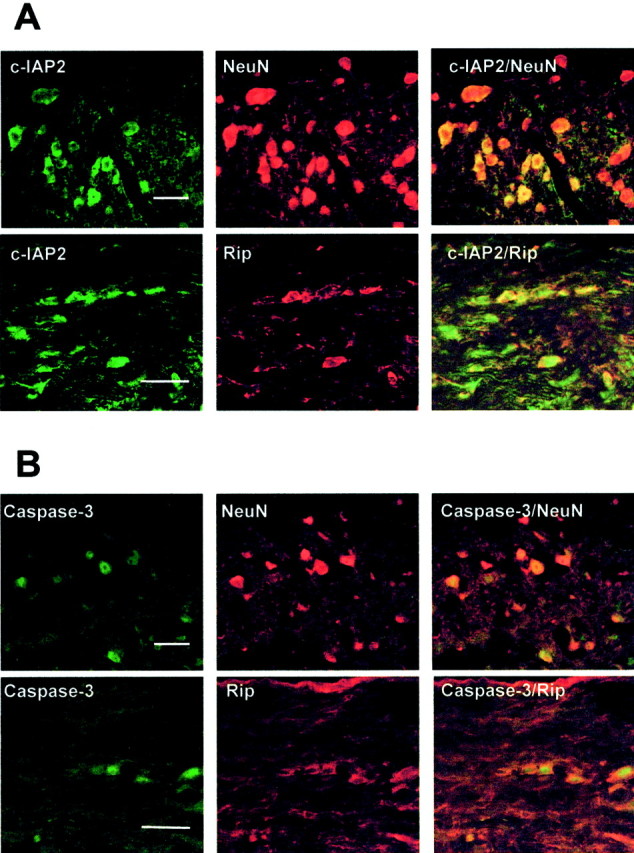
c-IAP2 and caspase-3 expression in neurons and oligodendrocytes 1 d after SCI in B6 mice. Longitudinal sections 1 mm rostral to the lesion epicenter are shown. c-IAP2 and caspase-3 were localized to neurons and oligodendrocytes as demonstrated by immunofluorescence double staining for an oligodendrocyte marker (Rip, red) or neuronal nuclear protein (NeuN, red) with either c-IAP2 (A, green) or caspase-3 (B, green). Scale bar, 20 μm.
TUNEL-positive cells in TNFR-deficient mice
Apoptosis of neurons, oligodendrocytes, astrocytes, and microglia has been demonstrated in the injured cord (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997). To test whether TNFR deletion affected the extent of cell death, we counted TUNEL-positive cells at five levels of the spinal cord (the lesion epicenter and 1 and 2.5 mm rostral and caudal) in TNFR1−/− and TNFR2−/− mice in comparison to the wild-type controls. It should be noted that TUNEL stain is not specific for apoptotic cells. Cells dying of necrosis also may be TUNEL-positive. There were significantly more TUNEL-positive cells in TNFR1−/− mice than in the wild-type mice at rostral 1 and 2.5 mm and caudal 2.5 mm levels (p < 0.05) (Fig.5A,B). Although the TNFR2−/− mice appeared to have a greater number of TUNEL-positive cells than TNRF1−/− or wild-type mice in the epicenter, the difference in the number of TUNEL-positive cells between the TNFR1 and TNFR2 mutants was not significant. Despite this exception in the epicenter, TNFR1−/− mice had significantly more TUNEL-positive cells than TNFR2−/− or wild-type mice. Across the five spinal cord regions that were examined (TNFR1−/−, 156.5 ± 23.2; TNFR2−/−, 116.6 ± 8.5; wild type, 73.9 ± 8.4), the TNFR2−/− mice exhibited an intermediate number that corresponded to the intermediate functional outcome after SCI, as shown in Figure 1. TUNEL-positive cells were located mostly in the white matter and expressed APC, an oligodendrocyte marker, indicating oligodendrocyte death (Fig.5C).
Fig. 5.
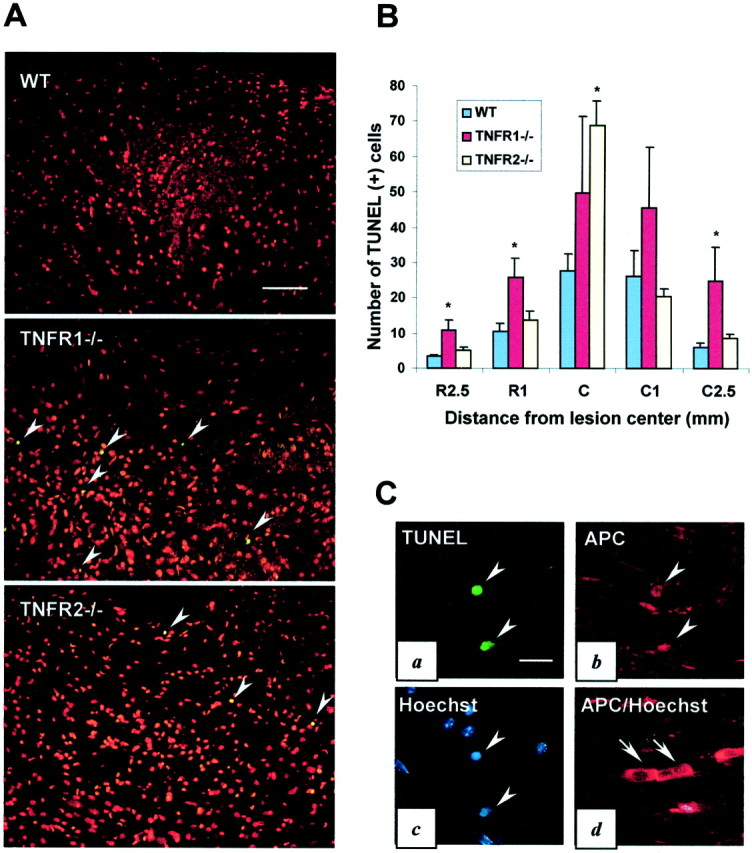
Enhanced apoptotic cell death in TNFR-deficient mice. A, Representative photomicrographs illustrating more TUNEL-positive cells (arrowheads,green) in TNFR1−/− and TNFR2−/− mice compared with B6 mice at 2.5 mm from the lesion center in the dorsal column area of the injured spinal cord 7 d after SCI. Note that nuclei from all of the cells are stained with propidium iodide (red). B, Quantitative analysis of TUNEL-positive cells 7 d after injury. TNFR1−/− mice showed a significantly larger number of TUNEL-positive cells compared with wild-type mice at 2.5 and 1 mm rostral and 2.5 mm caudal to the lesion epicenter (*p < 0.05; n = 4–6 per group). C, TUNEL (a), APC oligodendrocyte marker (b), and Hoechst 33342 (c) immunofluorescence staining of a section of the white matter from a wild-type mouse 1 mm rostral to the lesion epicenter shows that TUNEL-positive cells are oligodendrocytes. This is confirmed by Hoechst 33342 nuclear staining (c,arrowheads). Morphologically normal oligodendrocytes (APC, red) in the white matter 2.5 mm rostral from the lesion epicenter are not TUNEL-positive and have a normal nuclear chromatin Hoechst 33342 (blue) staining pattern (d). The arrows ina (TUNEL), b (APC), andc (Hoechst) point to the same cells (triple staining experiment). The arrows in d(APC/Hoechst) show live/normal oligodendrocytes double stained with APC and Hoechst. Scale bars: A, 100 μm;C, 20 μm.
Ex vivo MRI analysis after SCI
To examine the degree of spinal cord damage in the acute phase, we performed ex vivo MRI study 1 and 7 d after SCI. The sagittal MRI scan showed localized lesions in the spinal cord in the wild-type, TNFR1−/−, and TNFR2−/− mice (Fig.6A,B). The signal changes in spinal cord lesion area represent edema, hemorrhage, and tissue necrosis (Ohta et al., 1999; Bilgen et al., 2000). The lesion length determined by MR images is a reliable indicator of traumatic SCI severity (Hackney et al., 1994). A larger area of damage at the site of impact is apparent in both TNFR1−/− and TNFR2−/− mice. DWI measured at 7 d after SCI showed that the rostrocaudal spreading of the lesion was more extensive in both TNFR1−/− and TNFR2−/− mice.
Fig. 6.
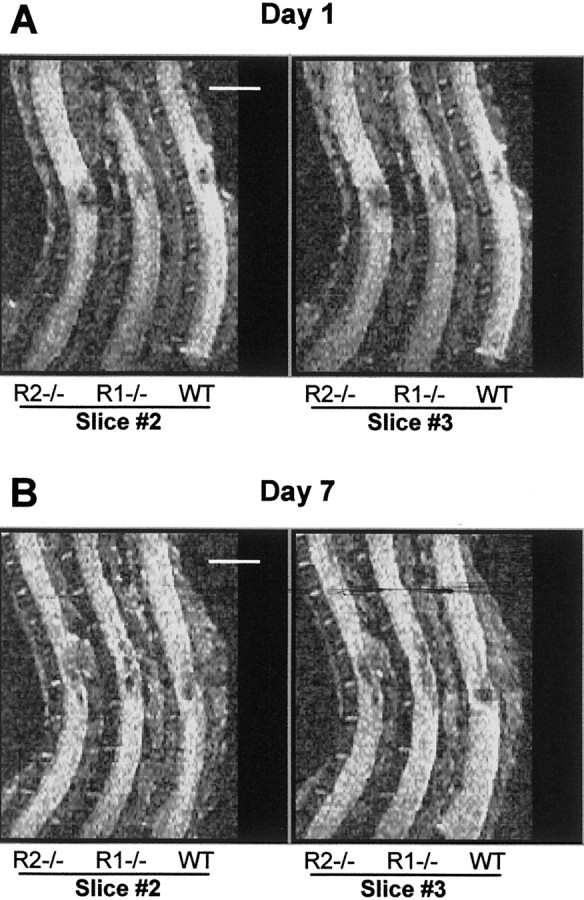
Representative MR images of damaged spinal cords after traumatic SCI. A, Spinal cord lesions in TNFR1−/−(R1−/−), TNFR2−/−(R2−/−), and wild-type (WT) mice as shown by DWI mapping 1 d after SCI. B, Lesions shown at 7 d. Two slice sections are shown, one from the center (Slice #3) and one that is 200 μm from the center (Slice #2).
Myelin and axonal damage in TNFR-deficient mice
The extent of myelin loss and axonal disruption was assessed by immunostaining with the use of an anti-MBP antibody (Fig.7) or axonal neurofilament marker, SMI-31 antibody (Fig. 8), respectively. Demyelination of white matter tract was observed in dorsal and lateral funiculi in TNFR1−/− mice (Fig.7C). Tissue destruction and microcyst formation were seen in the dorsal funiculus of TNFR1−/− mice (Fig. 7D) and TNFR2−/− mice to a lesser degree (Fig. 7F). The total area of tissue disruption at 7 d after injury as quantified on the basis of SMI-31 immunostaining showed more severe tissue damage in TNFR1−/− mice (3.21 ± 0.30 mm2) when compared with the wild-type controls (1.48 ± 0.23 mm2;p < 0.02) (Fig. 8B). The difference in the area of axonal damage between TNFR2−/− mice (2.43 ± 0.39 mm2) and the wild-type controls was not statistically significant.
Fig. 7.
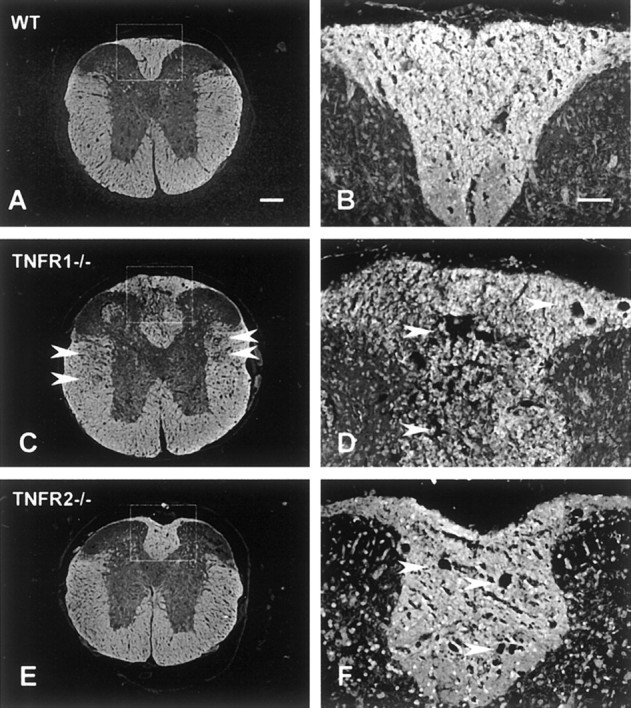
White matter demyelination demonstrated by MBP immunoreactivity counterstained with Hoechst 33342 at 7 d after SCI. Sections shown are 3 mm rostral to the lesion epicenter. At low magnification (A, C, E), white matter tract demyelination is observed in dorsal and lateral white matter in TNFR1−/− mice (arrowheads,C). At high magnification, tissue destruction and microcyst formation are more pronounced in the dorsal white matter in TNFR1−/− mice (arrowheads,D). Microcyst formation in the dorsal white matter is not as extensive in TNFR2−/− mice (arrowheads, F). Scale bars:A, C, E, 200 μm; B, D, F, 50 μm.
Fig. 8.
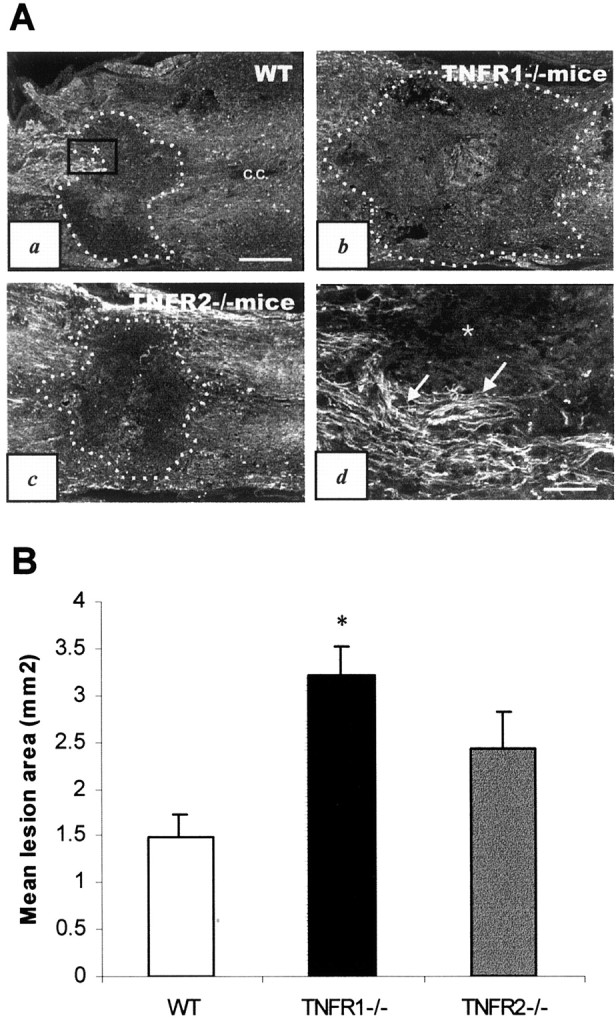
Axonal disruption in TNFR-deficient mice after SCI. A, Representative photomicrographs illustrating spinal cord lesion areas defined by neurofilament (SMI-31) immunostaining in B6 (a), TNFR1−/− (b), and TNFR2−/− (c) mice 7 d after SCI. All sections were cut longitudinally through the central canal (c.c.) for comparison. Dotted linesdelineating the lesion areas are based on SMI-31 immunoreactivity.d, High power view of the boxed area ina that demarcates the border between intact cord tissue (arrows) and the lesion area (asterisk). Scale bars: a–c, 400 μm; d, 100 μm.B, A histogram showing the mean lesion area among the three experimental groups (n = 3 per group). *Significant difference from the wild-type (WT) control (p < 0.02).
DISCUSSION
TNFR1−/− and TNFR2−/− mice, compared with the wild-type mice, demonstrated poor functional recovery and decreased NF-κB binding activity after SCI. In agreement with the decrease in NF-κB activation, c-IAP2 expression based on Western blot analysis also was decreased in TNFR1−/− mice compared with the wild-type mice after SCI. Consequent to a reduced c-IAP2 expression, active caspase-3 expression and the numbers of TUNEL-positive cells increased in the damaged spinal cords of TNFR1−/− mice.
TNF-α (Yakovlev and Faden, 1994; Wang et al., 1996; Bartholdi and Schwab, 1997; Xu et al., 1998) and TNFR (Li et al., 1998) are expressed in neurons and oligodendrocytes after traumatic SCI. A dual role can be played by TNF-α in CNS injury. TNF-α is known to be cytotoxic to oligodendrocytes in vitro (D'Souza et al., 1995). In addition, TNF-α has a detrimental role in TBI (Shohami et al., 1996) and experimental ischemic stroke (Barone et al., 1997; Yang et al., 1998), as shown by improved outcome measures that use anti-TNF-α strategies. Conversely, a protective role of TNF-α (Nawashiro et al., 1997) and a deleterious effect of TNFR deletion (Gary et al., 1998) have been demonstrated in cerebral ischemia, excitotoxic neuronal injury (Bruce et al., 1996), autoimmune-mediated demyelination (Liu et al., 1998), and TBI (Scherbel et al., 1999; Sullivan et al., 1999). In a recent study TNF-α−/− mice subjected to an extradural compression of the thoracic spinal cord showed no better recovery of hind limb function after SCI than the wild-type controls (Farooque et al., 2001). Recombinant murine interleukin-1β (IL-1β), IL-6, and TNF-α administered to the lesioned spinal cords of adult mice after SCI reduced the amount of tissue loss (Klusman and Schwab, 1997). Our data demonstrated more severe tissue damage and worse functional outcome in mice that were deficient in TNFR.
NF-κB was activated during the acute stage of SCI but was less pronounced in TNFR-deficient mice than in wild-type mice. The NF-κB heterodimer p50/RelA is known to upregulate various inflammatory mediator genes such as inducible nitric oxide synthase (iNOS), cytokines, cellular adhesive molecules, and proteases, which may be detrimental by perpetuating and intensifying an inflammatory reaction (Shimizu et al., 1990; Ledebur and Parks, 1995; Baldwin, 1996; Barnes, 1997; Barnes and Karin, 1997; O'Neill and Kaltschmidt, 1997; Schmedtje et al., 1997; Ghosh, 1999; Xu et al., 2001b). However, NF-κB also may exert a cytoprotective role (Beg and Baltimore, 1996; Grilli et al., 1996; Van Antwerp et al., 1996). Lack of the p50 NF-κB subunit increases the vulnerability of hippocampal neurons to excitotoxic injury (Yu et al., 1999). A cytoprotective action of NF-κB could be mediated via the induction of endogenous IAP genes. c-IAP2 expression is induced via NF-κB activation (Chu et al., 1997; Wang et al., 1998). c-IAP2, originally identified as a molecule recruited to the TNFR complex, contains two NF-κB-binding DNA consensus sequences within its promoter region (Hong et al., 2000).
IAP overexpression has been shown to suppress apoptosis induced by a variety of stimuli (Duckett et al., 1996; Liston et al., 1996; Deveraux et al., 1997). Overexpression of an X chromosome-linked IAP attenuated ischemia-induced caspase-3 activation and CA1 neuron degeneration in the rat hippocampus (Xu et al., 1999). Targeted expression of baculovirus p35 caspase inhibitor, an IAP analog, in oligodendrocytes protected mice against autoimmune-mediated demyelination (Hisahara et al., 2000). In the present study a decrease in NF-κB binding activity was accompanied by reduced expression of c-IAP2 in TNFR1−/− mice after SCI. This reduction in c-IAP2 expression in both TNFR1−/−and TNFR2−/− mice initially may cause more cell death after SCI. However, the delayed increase in c-IAP2 expression observed in TNFR2−/− mice may limit cell death in the injured cord, thus resulting in an intermediate level of functional outcome.
Consistent with a reduced expression of c-IAP2, an increase in the expression of the active form of caspase-3, together with an increase in the number of TUNEL-positive cells, was observed in the injured cord in TNFR1−/− mice after SCI. The molecular machinery necessary for activation and execution of the caspase-3 apoptotic pathway is operational after traumatic SCI in rats associated with an intense expression of DNA fragmentation factor/caspase-activated DNase in oligodendrocytes (Springer et al., 1999). An increase in caspase-3 expression and the numbers of dead cells in TNFR1−/− mice followed a blunted NF-κB response and reduced c-IAP2 expression after SCI. These results are consistent with the contention that an endogenous inhibitory mechanism on cell death may be activated via the TNFR–NF-κB cascade. In the TNFR1−/−and TNFR2−/− mice, to a lesser extent, this anti-apoptotic mechanism may be deficient.
The extent of demyelination and axonal disruption in TNFR1−/− mice was increased after SCI. The loss of locomotor function from traumatic SCI is known to depend on the extent of demyelination and axonal destruction (Blight and Decrescito, 1986; Wrathall et al., 1996; Beattie et al., 1997). Lesion expansion through secondary demyelination of white matter tract has been attributed to delayed oligodendrocyte apoptosis distant from the lesion center (Crowe et al., 1997). Cycloheximide, known to inhibit apoptosis in the nervous system, produced substantial preservation of tissue, reduced central cavitation, and improved recovery of function after SCI (Liu et al., 1997). In addition, the inhibition of caspase-1 and -3 activity reduced post-traumatic lesion size and improved motor performance in a mouse SCI model (Li et al., 2000). Therefore, limiting apoptotic cell death in the spinal cord might reduce tissue damage and enhance functional recovery after SCI.
The individual contribution of TNFR1 or TNFR2 has not been elucidated clearly. Compared with the wild-type mice, TNFR2−/− and especially TNFR1−/− mice showed lower intensity of NF-κB activation as well as poorer functional recovery after SCI. Shared signaling pathways may account for the close functional relationship between TNFR1 and TNFR2. These two receptors have the same proximal adapter domain, TNFR-associated factor 2, which activates NF-κB-inducing kinase for NF-κB nuclear translocation (Hsu et al., 1996). The cooperative signaling by TNFR1 and TNFR2 may be explained by the “ligand-passing effect” of TNFR2. TNFR2 is known to regulate the rate of TNF-α association with TNFR1, possibly by increasing the local concentration of TNF-α at the cell surface via rapid ligand association and dissociation (Tartaglia et al., 1993; Riches et al., 1998). Results from the present study raise the possibility that TNFR1 and TNFR2 interact via either a cooperative cross-linking effect of individual TNFR or a shared anti-apoptosis signaling pathway.
In summary, results from the present study are the first to demonstrate an increase in c-IAP2 expression after SCI and support the contention that the TNFR–NF-κB–c-IAP2 cascade may mediate an important endogenous protective mechanism against apoptosis after SCI. Our findings suggest that TNFR1, and probably to a lesser extent TNFR2, plays a role in limiting cell death, tissue damage, and functional recovery after SCI. Results from our study infer that therapeutic strategies aimed at suppressing TNF-α production and/or NF-κB activation after SCI should be reevaluated. Moreover, a regimen directed at limiting apoptosis in the acute post-traumatic period also may be beneficial in improving functional outcome after SCI.
Footnotes
This study is supported by National Institutes of Health (NIH) Grants NS37230, NS36350, and NS40162, by the Paralyzed Veterans of America, by the International Spinal Research Trust, and by NIH spinal cord injury PPG Grant NS39577. We thank Dr. Y. Y. He for providing expert technical assistance in tissue preparation.
Correspondence should be addressed to Dr. Chung Y. Hsu, Department of Neurology, Washington University School of Medicine, Box 8111, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail:hsuc@neuro.wustl.edu.
REFERENCES
- 1.An G, Lin TN, Liu JS, Xue JJ, He YY, Hsu CY. Expression of c-fos and c-jun family genes after focal cerebral ischemia. Ann Neurol. 1993;33:457–464. doi: 10.1002/ana.410330508. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS. The NF-κB and I-κB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ. Nuclear factor-κB. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 6.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-α. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 7.Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse, an in situ hybridization study. Eur J Neurosci. 1997;9:1422–1438. doi: 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- 8.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996a;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 9.Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter animal spinal cord injury study. J Neurotrauma. 1996b;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- 10.Beattie MS, Bresnahan JC, Komon J, Tovar CA, Meter MV, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W. Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol. 1997;148:453–463. doi: 10.1006/exnr.1997.6695. [DOI] [PubMed] [Google Scholar]
- 11.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 12.Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-κB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilgen M, Abbe R, Liu SJ, Narayana PA. Spatial and temporal evolution of hemorrhage in the hyperacute phase of experimental spinal cord injury: in vivo magnetic resonance imaging. Magn Reson Med. 2000;43:594–600. doi: 10.1002/(sici)1522-2594(200004)43:4<594::aid-mrm15>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Blight AR, Decrescito V. Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience. 1986;19:321–341. doi: 10.1016/0306-4522(86)90025-4. [DOI] [PubMed] [Google Scholar]
- 15.Bruce AJ, Bolong W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 16.Chen C, Edelstein LC, Gelinas C. The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-xL. Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 19.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 20.Deveraux QL, Roy N, Stennicke HR, Arsdale TV, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Souza S, Alinauskas K, McCrea E, Goodyer C, Antel JP. Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J Neurosci. 1995;15:7293–7300. doi: 10.1523/JNEUROSCI.15-11-07293.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duckett CS, Nava VE, Gedrich RW, Clem RJ, Dongen JLV, Gilfillan MC, Shiels H, Hardwick JM, Thompson CB. A conserved family of cellular genes related to the baculovirus IAP gene and encoding apoptosis inhibitors. EMBO J. 1996;15:2685–2694. [PMC free article] [PubMed] [Google Scholar]
- 24.Erickson SL, de Sauvage FJ, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan KC, Schreiber RD, Goeddel DV, Moore MW. Decreased sensitivity to tumor necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 25.Farooque M, Isaksson J, Olsson Y. Improved recovery after spinal cord injury in neuronal nitric oxide synthase-deficient mice but not in TNF-α-deficient mice. J Neurotrauma. 2001;18:105–114. doi: 10.1089/089771501750055811. [DOI] [PubMed] [Google Scholar]
- 26.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S. Regulation of inducible gene expression by the transcription factor NF-κB. Immunol Res. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- 28.Glazner GW, Mattson MP. Differential effects of BDNF, ADNF9, and TNF-α on levels of NMDA receptor subunits, calcium homeostasis, and neuronal vulnerability to excitotoxicity. Exp Neurol. 2000;161:442–452. doi: 10.1006/exnr.1999.7242. [DOI] [PubMed] [Google Scholar]
- 29.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 30.Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–126. doi: 10.1089/neu.1992.9.123. [DOI] [PubMed] [Google Scholar]
- 31.Hackney DB, Finkelstein SD, Hand CM, Markowitz RS, Black P. Postmortem magnetic resonance imaging of experimental spinal cord injury: magnetic resonance findings versus in vivo functional deficit. Neurosurgery. 1994;35:1104–1111. doi: 10.1227/00006123-199412000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Han BH, D'Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM. BDNF blocks caspase-3 activation in neonatal hypoxia–ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 33.Hisahara S, Araki T, Sugiyama F, Yagami K, Suzuki M, Abe K, Yamamura K, Miyazaki J, Momoi T, Saruta T, Bernard CC, Okano H, Miura M. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J. 2000;19:341–348. doi: 10.1093/emboj/19.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong SY, Yoon WH, Park JH, Kang SG, Ahn JH, Lee TH. Involvement of two NF-κB binding elements in tumor necrosis factor α-, CD40-, and Epstein–Barr virus latent membrane protein 1-mediated induction of the cellular inhibitor of apoptosis protein 2 gene. J Biol Chem. 2000;275:18022–18028. doi: 10.1074/jbc.M001202200. [DOI] [PubMed] [Google Scholar]
- 35.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 36.Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT. Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma. 2000;17:299–319. doi: 10.1089/neu.2000.17.299. [DOI] [PubMed] [Google Scholar]
- 37.Katoh K, Ikata T, Katoh S, Hamada Y, Nakauchi K, Sano T, Niwa M. Induction and its spread of apoptosis in rat spinal cord after mechanical trauma. Neurosci Lett. 1996;216:9–12. doi: 10.1016/0304-3940(96)12999-2. [DOI] [PubMed] [Google Scholar]
- 38.Klusman I, Schwab ME. Effects of pro-inflammatory cytokines in experimental spinal cord injury. Brain Res. 1997;762:173–184. doi: 10.1016/s0006-8993(97)00381-8. [DOI] [PubMed] [Google Scholar]
- 39.Kuhn PL, Wrathall JR. A mouse model of graded contusive spinal cord injury. J Neurotrauma. 1998;15:125–140. doi: 10.1089/neu.1998.15.125. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S. The apoptotic cysteine protease CPP32. Int J Biochem Cell Biol. 1997;29:393–396. doi: 10.1016/s1357-2725(96)00146-x. [DOI] [PubMed] [Google Scholar]
- 41.Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-κB site and p65 homodimers. J Biol Chem. 1995;270:933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 42.Lenardo MJ, Baltimore D. NF-kB, a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 43.Li GL, Brodin G, Farooque M, Funa K, Holtz A, Wang WL, Olsson Y. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol. 1996;55:280–289. doi: 10.1097/00005072-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Li M, Ona VO, Chen M, Kaul M, Tenneti L, Zhang X, Stieg PE, Lipton SA, Friedlander RM. Functional role and therapeutic implications of neuronal caspase-1 and -3 in a mouse model of traumatic spinal cord injury. Neuroscience. 2000;99:333–342. doi: 10.1016/s0306-4522(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 45.Li Q, Yan P, Hsu CY, Xu J, He Y, Xu XM. Expression of tumor necrosis factor receptor (TNF-R) and nuclear factor-κB (NF-κB) in adult rats following traumatic spinal cord injury. Soc Neurosci Abstr. 1998;24:738. [Google Scholar]
- 46.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE, MacKenzie A, Korneluk RG. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- 48.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis JC, Magal E, Takayama S, Varon S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science. 1993;259:689–692. doi: 10.1126/science.8430320. [DOI] [PubMed] [Google Scholar]
- 50.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor-κB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 51.Mayer M, Noble M. N-acetyl-l-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro. Proc Natl Acad Sci USA. 1994;91:7496–7500. doi: 10.1073/pnas.91.16.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-α pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Ohta K, Fujimura Y, Nakamura M, Watanabe M, Yato Y. Experimental study on MRI evaluation of the course of cervical spinal cord injury. Spinal Cord. 1999;37:580–584. doi: 10.1038/sj.sc.3100881. [DOI] [PubMed] [Google Scholar]
- 54.O'Neill LA, Kaltschmidt C. NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 55.Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor-α. Proc Natl Acad Sci USA. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riches DW, Chan ED, Zahradka EA, Winston BW, Remigio LK, Lake FR. Cooperative signaling by tumor necrosis factor receptors CD120a (p55) and CD120b (p75) in the expression of nitric oxide and inducible nitric oxide synthase by mouse macrophages. J Biol Chem. 1998;273:22800–22806. doi: 10.1074/jbc.273.35.22800. [DOI] [PubMed] [Google Scholar]
- 57.Rosenberg LJ, Wrathall JR. Quantitative analysis of acute axonal pathology in experimental spinal cord contusion. J Neurotrauma. 1997;14:823–838. doi: 10.1089/neu.1997.14.823. [DOI] [PubMed] [Google Scholar]
- 58.Rosenbluth J, Schiff R, Liang WL, Menna G, Young W. Xenotransplantation of transgenic oligodendrocyte-lineage cells into spinal cord-injured adult rats. Exp Neurol. 1997;147:172–182. doi: 10.1006/exnr.1997.6583. [DOI] [PubMed] [Google Scholar]
- 59.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel RR, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 60.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2–TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 61.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci USA. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmedtje JF, Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-κB p65 transcription factor in human vascular endothelial cells. J Biol Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 64.Shimizu H, Mitomo K, Watanabe T, Okamoto S, Yamamoto K. Involvement of a NF-κB-like transcription factor in the activation of the interleukin-6 gene by inflammatory lymphokines. Mol Cell Biol. 1990;10:561–568. doi: 10.1128/mcb.10.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor α (TNFα) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 66.Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 67.Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 68.Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 69.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-κB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NF-κB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 71.Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75 kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55 kDa TNF receptor. J Biol Chem. 1993;268:18542–18548. [PubMed] [Google Scholar]
- 72.Tator CH. Update on the pathophysiology and pathology of acute spinal cord injury. Brain Pathol. 1995;5:407–413. doi: 10.1111/j.1750-3639.1995.tb00619.x. [DOI] [PubMed] [Google Scholar]
- 73.Uren AG, Pakusch M, Hawkins CJ, Puls KL, Vaux DL. Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc Natl Acad Sci USA. 1996;93:4974–4978. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 75.Wang CX, Nuttin B, Heremans H, Dom R, Gybels J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- 76.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB anti-apoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 77.Wrathall JR, Teng YD, Choiniere D. Amelioration of functional deficits from spinal cord trauma with systemically administered NBQX, an antagonist of non-N-methyl-d-aspartate receptors. Exp Neurol. 1996;137:119–126. doi: 10.1006/exnr.1996.0012. [DOI] [PubMed] [Google Scholar]
- 78.Xu D, Bureau Y, McIntyre DC, Nicholson DW, Liston P, Zhu Y, Fong WG, Crocker SJ, Korneluk RG, Robertson GS. Attenuation of ischemia-induced cellular and behavioral deficits by X chromosome-linked inhibitor of apoptosis protein overexpression in the rat hippocampus. J Neurosci. 1999;19:5026–5033. doi: 10.1523/JNEUROSCI.19-12-05026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP, Hsu CY. Amyloid-β peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001a;21:RC118. doi: 10.1523/JNEUROSCI.21-01-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu J, Kim GM, Ahmed SH, Xu J, Yan P, Xu XM, Hsu CY. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001b;21:92–97. doi: 10.1523/JNEUROSCI.21-01-00092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu X, Fan G, Chen S, Wu Y, Xu XM, Hsu CY. Methylprednisolone inhibition of TNF-α expression and NF-κB activation after spinal cord injury in rats. Mol Brain Res. 1998;59:135–142. doi: 10.1016/s0169-328x(98)00142-9. [DOI] [PubMed] [Google Scholar]
- 82.Xue D, Horvitz HR. Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature. 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- 83.Yakovlev AG, Faden AI. Sequential expression of c-fos protooncogene, TNF-α, and dynorphin gene in spinal cord following experimental traumatic injury. Mol Chem Neuropathol. 1994;23:179–190. doi: 10.1007/BF02815410. [DOI] [PubMed] [Google Scholar]
- 84.Yan P, Xu J, Li Q, Chen S, Kim GM, Hsu CY, Xu XM. Glucocorticoid receptor expression in the spinal cord after traumatic injury in adult rats. J Neurosci. 1999;19:9355–9363. doi: 10.1523/JNEUROSCI.19-21-09355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang GY, Gong C, Qin Z, Ye W, Mao Y, Bertz AL. Inhibition of TNFα attenuates infarct volume and ICAM-1 expression in ischemic mouse brain. NeuroReport. 1998;22:2131–2134. doi: 10.1097/00001756-199806220-00041. [DOI] [PubMed] [Google Scholar]
- 86.Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-κB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]