

Abstract
Brain-derived neurotrophic factor (BDNF) modulates synaptic strength in hippocampal neurons, in addition to promoting survival and differentiation. To identify genes involved in trophic regulation of synaptic plasticity, we have used a multidisciplinary approach of differential display and family-specific slot blots in combination with whole-cell patch-clamp recordings of dissociated hippocampal neurons. Three hour exposure to BDNF elicited a 2.6-fold increase in synaptic charge and a concomitant induction of 11 genes as revealed by differential display, including the small GTP-binding vesicular trafficking protein Rab3A and the enzymeguanylate cyclase (GC). Slot blot analysis on a population of neurons confirmed an average of 3.1-fold induction of these clones. In contrast, individual pyramidal-like neurons that were first characterized electrophysiologically in the presence of BDNF and subjected to transcriptional analysis displayed more robust increases (4.8-fold), emphasizing the neuronal heterogeneity. Transcriptional changes of Rab3A and GC were accompanied by translational regulation, shown by Western blot analysis. Furthermore, a number of GC-associated and Rab3A effector molecules were induced by BDNF at either the gene or protein levels. The functional role of Rab3A in BDNF-induced synaptic plasticity was assessed using cells derived from Rab3A knock-out mice. These neurons failed to show an increase in synaptic charge in response to BDNF at 10 min; however a late response to BDNF was detected at 20 min. This late response was similar in time course to that induced by postsynaptic activation of glutamate receptors. Our results demonstrate a requirement for Rab3A and may reveal a temporal distinction between presynaptic and postsynaptic mechanisms of BDNF-induced synaptic plasticity associated with learning and memory.
Keywords: Rab3A, BDNF, synaptic plasticity, transcriptional analysis, hippocampus, culture
Evidence now indicates that neurotrophins exert a temporal continuum of synaptic actions, from acute regulation of transmission to longer-term effects on synapse development (Takei and Nawa, 1998; Lu and Chow, 1999; McAllister et al., 1999; Schuman, 1999). In the hippocampus, which is associated with learning and memory, brain-derived neurotrophic factor (BDNF) enhances long-term potentiation (LTP) (Korte et al., 1995; Figurov et al., 1996;Patterson et al., 1996) and subsequently increases synapse number (Takei and Nawa, 1998; Vicario-Abejon et al., 1998). We have found that BDNF rapidly increases synaptic responses in dissociated hippocampal neurons (Levine et al., 1995). However, underlying molecular mechanisms remain to be elucidated. There is evidence that both presynaptic and postsynaptic processes participate (Kim et al., 1994; Levine et al., 1995, 1998; Figurov et al., 1996; Gottschalk et al., 1998; Li et al., 1998; Crozier et al., 1999; Schinder et al., 2000). In the postsynaptic neuron, for example, BDNF increases synaptic strength, at least in part, via post-translational modification of NMDA receptor subunits (Levine et al., 1995, 1998; Suen et al., 1997; Crozier et al., 1999). Neurotrophin-induced synaptic plasticity results from such postsynaptic modifications, in conjunction with complex presynaptic mechanisms (Figurov et al., 1996; Gottschalk et al., 1998; Li et al., 1998;Schinder et al., 2000; Xu et al., 2000), the molecular basis of which is as yet unknown.
Binding of BDNF to the trkB receptor activates second messenger signaling cascades, resulting in both cytoplasmic and nuclear changes (Finkbeiner et al., 1997; Gottschalk et al., 1999). Acute post-translational modulation may constitute an early stage, followed by more stable changes involving altered gene expression and protein synthesis (Kang and Schuman, 1996; Finkbeiner et al., 1997; Bradley and Sporns, 1999). Indeed, prolonged treatment with neurotrophins promotes the development and maturation of synaptic sites (Wang et al., 1995;Takei and Nawa, 1998; Vicario-Abejon et al., 1998), probably involving regulation of vesicle proteins (Wang et al., 1995; Takei and Hatanaka, 1997; Pozzo-Miller et al., 1999). Moreover, mice with a targeted deletion of the BDNF gene exhibit impaired synaptic plasticity and depressed levels of several vesicle proteins. Exogenous BDNF restores both physiological responses and protein levels (Korte et al., 1995; Patterson et al., 1996; Pozzo-Miller et al., 1999). Thus, to date, studies of BDNF-induced gene expression have focused on candidate molecules; however the full spectrum of BDNF-regulated genes and their roles in synaptic plasticity have yet to be elucidated.
Here, we have combined whole-cell patch-clamp recordings in conjunction with differential display (DD) and single-cell transcriptional analysis to identify altered expression of known and novel genes in BDNF-induced plasticity. We are examining the hypothesis that differential gene regulation reflects the role(s) of the gene products in trophic regulation of synaptic plasticity. The potential functions of some of the identified genes have been defined using knock-out mice and pharmacological antagonists. We present the first demonstration of the requirement of a specific molecule, the vesicular trafficking protein Rab3A, in BDNF-induced synaptic plasticity. We have also further delineated temporal distinctions in early and late BDNF-induced synaptic plasticity. Using this multidisciplinary approach, we are beginning to define the previously uncharacterized molecular pathways in neurotrophin-induced plasticity.
MATERIALS AND METHODS
Cell culture preparation. Time-mated pregnant rats and mice were killed by CO 2 asphyxiation in accordance with institutional guidelines for the care and use of animals. Fetuses were removed by cesarean section and transferred to a sterile Petri dish with PBS. Fetal hippocampi were dissected from surrounding brain tissue, and meninges were completely removed. Low-density cultures of dissociated embryonic day 18 (E18) rat hippocampi (Sprague Dawley from Hilltop Laboratories, Scottdale, PA) were prepared as described previously (Levine et al., 1995). For some experiments, cultures were obtained from E17 wild-type orRab3A homozygous knock-out mice (The Jackson Laboratory, Bar Harbor, ME). Briefly, pooled tissue from each litter was mechanically dissociated in 7.5% nutrient medium and plated on poly-d-lysine-coated culture dishes at 200,000–350,000 cells/dish. Cultures were maintained in serum-free medium (Levine et al., 1995) for 10–13 d and contained virtually pure neurons.
Electrophysiological recordings. Whole-cell recordings were performed after 10–13 d in culture. Currents were recorded with an Axoclamp 200 amplifier, digitized at 2.5 kHz with an INDEC IDA 15125 interface (INDEC, Capitola, CA), filtered at 5 kHz, and stored. Recording parameters and stimulus protocols were controlled by custom software written with Borland C2+ that uses device driver libraries supplied by INDEC. Data analysis programs were written with Microsoft Visual Basic. The external bath solution for current- and voltage-clamp recordings was (in mm): 1.67 CaCl 2, 1 MgCl 2, 5.36 KCl, 137 NaCl, 17 glucose, 10 HEPES, and 20 sucrose. The pipette solution contained (in mm): 105 Cs-methanesulfonate, 17.5 CsCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 Mg-ATP, 2 Na-ATP, 0.3 Na-GTP, and 20 phosphocreatinine, along with 50 U/ml creatinine phosphokinase. All recordings were made at room temperature. The typical range of pipette resistance was 3–5 MΩ. Cell capacitance was 10–20 pF, and access resistance was 7–20 MΩ. Each data point represents a cell from a separate dish, and at least three different platings were used for each condition.
Data analysis. Data were analyzed by integrating the synaptic currents for each sweep with conversion to synaptic charge. Experiments for population studies were kept for analysis if they showed less than ±10% change during a 10 min recording period. For Rab3A and guanylate cyclase (GC) experiments, the charge measurements for all sweeps in a 1 min period were averaged (binned). Baseline is considered the average synaptic charge during the 2 min period immediately before BDNF application. Percentage increases were determined by dividing the synaptic charge during BDNF exposure (20 ng/ml; Peprotech, Princeton, NJ) by the baseline. Wild-type mouse or vehicle-treated (DMSO) rat cell recordings were accepted if any 1 min bin during the BDNF treatment was twice the SEM above baseline, therefore indicating a response to BDNF. Rab3A mutant and LY83583-treated (Biomol, Plymouth Meeting, PA; 1 μm in DMSO) cells were rejected if either the 0–5 min binned time period or the 5–10 min binned time period during BDNF exposure was twice the SEM below baseline, indicating “run down.” One of 11 wild-type cells failed these criteria, and 3 out of 15 Rab3A cells failed. Student's t test was used for statistical comparisons (two-tailed with p < 0.05 indicating significance).
RNA isolation. Total cellular RNA was prepared from virtually pure neuronal cultures treated with either BDNF or NGF (50 ng/ml) or vehicle (water) for 3 hr by the guanidine isothiocyanate method (Thakker-Varia et al., 1998).
mRNA differential display. Display was performed essentially as described previously (Thakker-Varia et al., 1998) using the RNA image kit as per instructions (Gene Hunter, Boston, MA). Radiolabeled PCR products were analyzed by denaturing 6% PAGE. Gels were dried on Whatman paper and exposed to x-ray film.
Band recovery, cloning, and DNA sequencing. DNA from putative differentially expressed bands found in duplicate samples was reamplified by PCR using the appropriate primer combination for 40 cycles (Thakker-Varia et al., 1998). cDNA fragments were cloned intopCR II using the TA cloning kit (Invitrogen, Carlsbad, CA). Five separate clones from each fragment were sequenced on an Applied Biosystems 373A automatic sequencer. Comparisons of unknown nucleotide sequences with known sequences in the GenBank and European Molecular Biology Laboratory (EMBL) databases were performed using the BLAST program.
cDNA synthesis, amplified RNA amplification, and expression profiles from single isolated cells. During whole-cell patch-clamp recording, the electrode contained avian myeloblastoma virus (AMV) reverse transcriptase and oligo-dT24-T7 primer (Eberwine et al., 1995). Cellular contents were aspirated into the patch pipette with a small amount of suction after a 10 min recording. cDNA synthesis was completed in vitro at 37°C for 60 min. The cDNA was resuspended in 20 μl of H 2O, heated at 95°C for 3 min, and converted into double-stranded template as described previously (Eberwine et al., 1995). Twenty percent of the cDNA template was used to synthesize amplified RNA (aRNA) in 40 mm Tris, pH 7.4, 10 mmNaCl, 10 mm MgCl 2, 0.01 mm dithiothreitol, 500 μm ATP, GTP, CTP, and UTP, 20 U of RNAsin, and 100 U of T7 RNA polymerase at 37°C for 3–4 hr. aRNA was synthesized into double-stranded cDNA and used as a template for a second round of amplification as described previously (Eberwine et al., 1995). Ten nanograms of random hexamers with AMV reverse transcriptase were used to prime the first-strand cDNA synthesis. The second-strand cDNA was synthesized using 100 ng of the oligo-dT24-T7 amplification oligonucleotide as a primer with the Klenow fragment and T4 DNA polymerase. The ends of the double-stranded cDNA were made blunt with T4 DNA polymerase. The final aRNA synthesis was with the inclusion of 25 pmol of [α-32P]CTP (3000 Ci/mmol). The radiolabeled probe thus generated was then used to hybridize to cDNA slot blots or microarrays as described below.
Slot blots. A slot blot screening method was used to confirm the differential expression. Plasmid DNA from putative differential cDNA fragments cloned in pCR II vector (62.5 ng) was blotted onto nylon membranes using the Minifold II slot blot apparatus. Alternatively, commercially available gene-specific slot blots (Geno Tech., St. Louis, MO) were used. The membrane was differentially screened using 32P-labeled cDNA probes generated from total RNA of neurotrophin-treated or control neuronal cultures. The membranes were hybridized using PerfectHyb (Sigma, St. Louis, MO) at 68°C with denatured probe (1–5 × 106 cpm/ml) for 3 hr and washed to a high stringency. The washed membranes were exposed to Kodak Biomax-MR film with intensifying screens at −70°C for up to 5 d. Quantitation of band intensity was performed using Molecular Analyst (Molecular Dynamics, Sunnyvale, CA) software. Measured intensities were corrected using actin as the internal standard, and tRNA was used as a negative control.
SDS-PAGE and Western blot analysis. BDNF-treated and untreated hippocampal neurons were solubilized in lysis buffer (20 mm Tris, pH 8, 0.5% Triton X-100, and 0.5% SDS) containing proteinase inhibitors (Suen et al., 1997). Protein content was determined with the BCA protein assay kit (Pierce, Rockford, IL). Samples containing equal amounts of protein were denatured in Laemmli's sample buffer for 5 min and subjected to 4–12% or 8–16% gradient SDS-PAGE (Suen et al., 1997). The proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA) and blocked in milk powder plus normal serum. The PVDF membranes were incubated with antisera against Rab3A (United States Biochemicals, Cleveland, OH; 1:1000), GTPase-activating protein (Rab GAP; Biomol; 1:250), GDP–GTP exchange protein (Rab GEP; Y. Takai, Osaka, Japan; 1:250), GDP dissociation inhibitor (Rab GDI; A. Shisheva, Wayne, OH; 1:20,000), Rabphilin (Transduction Laboratories, San Diego, CA; 1:5000), Rab3-interacting molecule (RIM) (Transduction Laboratories; 1:1000), GC (Calbiochem, La Jolla, CA; 1:2000), and actin (Boehringer Mannheim, Indianapolis, IN; 1:1000) overnight at 4°C. Membranes were washed, followed by incubation with anti-rabbit or anti-mouse HRP-conjugated IgG (1:5000). The immunopositive bands were visualized by chemiluminescence using the ECL detection kit (NEN, Boston, MA). Quantification of intensity of the bands is performed using Molecular Analyst software and normalized to actin levels.
Immunocytochemistry. Cultures were fixed in 4% paraformaldehyde. A blocking step was performed in 30% normal goat serum in PBS containing 0.1% Triton X-100. Primary antibody was applied overnight at 4°C. Antibodies used were as follows: Rab3A (Biosource, Camarillo, CA; 1:500), Rab GAP (Biomol; 1:250), and Rab GDI (A. Shisheva; 1:20,000). No reliable antibody to GC was found for immunocytochemistry. Cultures were incubated in goat anti-rabbit biotinylated secondary antibody (Vector Laboratories, Burlingame, CA; 1:100), and staining was visualized using an ABC Vectastain detection kit (Vector Laboratories).
RESULTS
BDNF regulates gene expression at 3 hr
To elucidate molecular mechanisms underlying BDNF-induced synaptic plasticity, we used embryonic rat hippocampal neurons maintained in fully defined, serum-free medium (Levine et al., 1995). To allow sufficient time for gene induction, we chose a 3 hr BDNF exposure and then recorded synaptic responses under whole-cell voltage-clamp conditions of treated cells compared with control, untreated cells. Cells treated with BDNF (50 ng/ml) for 3 hr exhibited a 2.6-fold increase in synaptic charge (n = 8; t test,p < 0.05) (Fig.1A,B,D), although there was characteristic variability among individual neurons (Fig.1C). These data indicate that a 3 hr treatment is comparable with acute treatment (10 min) with BDNF in terms of synaptic response (Levine et al., 1995; Sherwood and Lo, 1999). The longer-term BDNF treatment, however, permits for transcription of immediate early genes as well as downstream genes that potentially play roles in synaptic plasticity.
Fig. 1.

Three hour BDNF treatment potentiates synaptic activity in dissociated hippocampal neurons. A, B, Example whole-cell voltage-clamp recordings (Vhold = −60 mV) from one control hippocampal neuron (A) and one neuron exposed to BDNF (20 ng/ml) for 3 hr (B).C, Effect of BDNF on synaptic charge in a population of neurons. Each triangle represents the average synaptic charge for one neuron during a stable 10 min recording period.D, Average of the individual cells shown inC. BDNF increased synaptic charge 2.6-fold compared with control cells (n = 8 each; *p< 0.05). Recordings were obtained from multiple platings.
Differentially expressed genes potentially involved in BDNF regulation of synaptic activity were identified by comparing patterns of mRNA expression in control cultures with those treated with BDNF or NGF for 3 hr. Figure 2 is a representative gel selected from over 20 different gels showing amplified subsets of mRNA by use of a combination of anchored and arbitrary primers. Amplified cDNAs in each experimental group are similar, indicating that the general gene expression profile remains unaltered. However, selective differences (←) among duplicates indicate induced expression of specific cDNAs after neurotrophin treatment (Fig. 2). Some genes appear to be induced by both NGF and BDNF, suggesting a general responsiveness to neurotrophins. We focused on genes regulated exclusively by BDNF, because NGF has not been implicated in synaptic plasticity (Levine et al., 1995, 1996).
Fig. 2.
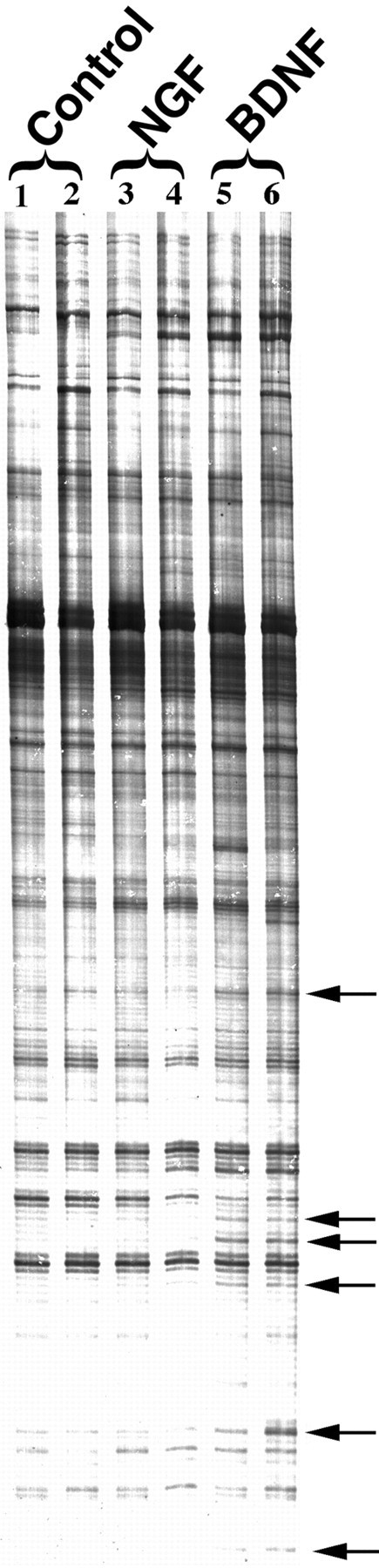
A number of genes are differentially expressed in BDNF-treated cultures. Total RNA from control (lanes 1, 2), NGF-treated (3 hr; 50 ng/ml; lanes 3, 4), and BDNF-treated (3 hr; 50 ng/ml; lanes 5, 6) sister cultures was subjected to differential mRNA analysis. A representative autoradiogram of amplified PCR products is shown for one set of primer pairs that identified distinct fragments (←) with differential expression in duplicate neurotrophin-treated groups.
The identity of the differentially expressed genes induced exclusively by BDNF was derived from multiple gels and was determined by recovering cDNA products and reamplifying using corresponding primer sets. Partial sequence analysis and homology searches revealed that 4 of the 11 induced sequences corresponded to known genes with known functions, including ypt1/Rab3A, rat GC, ATP synthase, and an estrogen-induced gene (Table1). Six cDNAs corresponded to known expressed sequence tags (ESTs), and one of the cDNAs corresponded to a novel gene not cloned previously and of unknown function. The known functions of Rab3A and GC implicate them in synaptic transmission. Rab3A is a small GTP-binding protein involved in regulated exocytosis of neurotransmitters from synaptic vesicles (Bean and Scheller, 1997;Geppert and Sudhof, 1998). In addition, several lines of evidence suggest that the GC pathway is involved in synaptic plasticity (Zhuo et al., 1994; Arancio et al., 1995; Son et al., 1998). Moreover, in a separate study using DNA microarrays, Rab3A levels were increased by BDNF treatment (Thakker-Varia et al., 2000). Taken together, these findings validate the differential screening approach by isolation of at least two genes (Rab3A and GC) potentially relevant to BDNF-induced synaptic plasticity (Thakker-Varia et al., 1998).
Table 1.
Analysis of cDNA fragments identified by mRNA differential display
| Clone number | Fragment size | Regulation | Sequence homology | Accession number |
|---|---|---|---|---|
| C1e | 514 bp | Up | ypt1/Rab3A | X15747 |
| C6d | 325 bp | Up | EST | AA891880 |
| A1b | 584 bp | Up | EST | AA849956 |
| A1c | 584 bp | Up | EST | AI137233 |
| A3a | 350 bp | Up | Guanylate cyclase A | JO5677 |
| A3c | 356 bp | Up | Unknown | U25489 |
| A5d | 801 bp | Up | EST | AA997878 |
| A6c | 259 bp | Up | ATP synthase | AF115771.1 |
| G1b | 251 bp | Up | Estrogen-induced gene | S74324 |
| G2a | 578 bp | Up | EST | C81615 |
| G2c | 628 bp | Up | EST | AI117373 |
DNA from putative differentially expressed bands found in duplicate samples was reamplified by PCR using the appropriate primer combination. cDNA fragments were subcloned, and five separate clones from each fragment were sequenced. Comparisons of unknown nucleotide sequences to known sequences in the GenBank and EMBL data bases were performed using the BLAST program. The cDNAs ranged from 251 to 801 bp, and all of them were upregulated by BDNF.
Population and single-cell analysis
To confirm the effect of BDNF on putative clones, slot blot analysis was performed using RNA from a population of neurons. The putative cDNA clones identified by DD were immobilized on membranes and hybridized to probes generated from independent sets of treated or untreated whole cultures (Fig.3A). BDNF induced 9 of the 11 clones (average of 3.1-fold) including GC andRab3A.
Fig. 3.

BDNF induces more robust gene expression in electrophysiologically characterized individual pyramidal-like cells than in a population of hippocampal neurons. A, Slot blot on a whole population of hippocampal cells. cDNA from independent cultures treated with BDNF was synthesized and hybridized to the clones (62.5 ng) identified by differential display. Enhanced expression in the BDNF-treated samples was observed in 9 of the 11 clones. An average of 3.1-fold induction among the genes within the blot was observed.B, Example recordings from one control and one BDNF-treated cell and slot blot analysis on single pyramidal-like neurons characterized electrophysiologically. Whole-cell patch-clamp recordings from individual pyramidal-like cells in the absence (control) or presence of BDNF (3 hr; 20 ng/ml) were performed for 10 min, and cellular contents were aspirated into the patch pipette. Material from four control and four BDNF-treated cells was pooled. aRNA was synthesized and hybridized to the clones (62.5 ng). Enhanced expression in the BDNF-treated samples was observed in all 11 clones. tRNA served as a negative control. An average of 4.8-fold induction among the genes within the blot was observed when normalized to actin.
To examine gene expression in pyramidal-like individual cells that were characterized electrophysiologically, we performed transcriptional analysis at the single-cell level (Fig. 3B). The cell contents from four individual pyramidal-like, BDNF-treated (3 hr) or untreated neurons were harvested and pooled after each recording, and mRNA was amplified. Radioactively labeled amplified RNA obtained from the individual cells was used directly as a probe to hybridize to the slot blot (Fig. 3B). All 11 clones were induced by BDNF. BDNF elicited an average 4.8-fold induction among the 11 genes within the blot, normalized to actin. Therefore, the effect of BDNF was more robust in single cells that were identified as pyramidal-like neurons, compared with that of the whole cell population. Consequently, observations in a population of hippocampal neurons can be extrapolated to individual cells that exhibit responsiveness to BDNF.
GC has been implicated in synaptic transmission (Zhuo et al., 1994;Arancio et al., 1995; Son et al., 1998) and was among the 11 BDNF-induced sequences, rendering it appropriate to pursue genes in the GC pathway. Therefore to facilitate the discovery of additional BDNF-induced genes involved in the GC pathway, we used G-protein-specific slot blot. Radioactively labeled cDNAs prepared from control or BDNF-treated hippocampal cultures were hybridized to the blot; several genes were differentially expressed (Fig.4). Two genes related to the GC pathway increased dramatically; these are protein kinase G II (PKG II) and Gαolf, which transduces olfactory signals including GC and is expressed in the hippocampus (Zhuang et al., 2000). Two other genes involved in the GC pathway,PKGIA and PKGIB, were not expressed in either sample. In addition, two genes that are upregulated by BDNF are related to the adenylate cyclase pathway; these are the regulatory subunit of protein kinase A (PKA R2) and the anchor protein for PKA R2 (AKAP 84). MXA p78, an interferon-induced protein with GTPase activity, was also induced, whereas the β subunit of the G-protein complex (Gβ) was downregulated. Control housekeeping genes were not altered. These findings validate the initial screen using DD, in which GC was upregulated, and also implicate the adenylate cyclase pathway in BDNF-induced synaptic enhancement.
Fig. 4.

Slot blot analysis reveals BDNF-induced changes of G-protein family genes. RNA derived from control (A) or BDNF-treated (B) hippocampal cultures was used to prepare radioactively labeled cDNA. The cDNA was hybridized to a blot containing clones of G-protein-related genes at 2 or 20 ng of DNA. The expression of several genes increased in the BDNF-treated samples (↑). AKAP 84, Anchor protein for regulatory subunit of protein kinase A;Gαolf, protein involved in transduction of olfactory signals including the GC pathway; MXA p78, interferon-induced protein with GTPase activity; PKA R2, regulatory subunit of protein kinase A; and PKG II, protein kinase G II. The expression of one gene decreased in the BDNF-treated sample (↓): Gβ, the β subunit of the G-protein. Two housekeeping genes did not show differential expression:β-actin and GAPDH, glyceraldehyde phosphate dehydrogenase.
BDNF and protein expression
To determine whether altered gene transcription is accompanied by protein translation in this system, Western blot analysis was performed. Our initial observations of Rab3A and GC expression after a 3 hr BDNF treatment showed no obvious change in protein levels (data not shown). To allow additional time for protein translation, we used a 6 hr BDNF exposure. Under these conditions, Rab3A protein levels increased 2.0 ± 0.5-fold (n = 4), and GC increased 1.4 ± 0.1-fold (n = 3), normalized to actin and compared with control, vehicle-treated samples (Fig.5A,B).
Fig. 5.

BDNF induces translation of identified genes and their regulatory proteins in hippocampal neurons. A, Equal amounts (50 μg) of protein from control and BDNF-treated (6 hr; 50 ng/ml) cultures were loaded in each lane, electrophoresed, immunoblotted with antibodies, and visualized with ECL. GC (70 kDa) and Rab3A (25 kDa) are upregulated by BDNF treatment. Several regulatory proteins were also induced by BDNF: RIM (173 kDa), Rab GAP (120 kDa), Rabphilin (75 kDa), and Rab GDI (50 kDa). Rab GEP (210 kDa) was unaltered. Actin (42 kDa) levels remained unchanged. B, Quantitation of increase in protein levels in BDNF-treated samples relative to control cells after normalization to actin is shown (n = 4 except for GC, n = 3, and Rab GEP, n = 2). C–E, Identified proteins are localized to neurons. Hippocampal cultures were fixed and immunostained using avidin–biotin–horseradish peroxidase visualization. Rab3A (C) appears in a punctate pattern in neuronal processes. Rab GAP (D) and Rab GDI (E) staining is also localized to neurons. Note the cellular heterogeneity of staining intensity and localization to pyramidal-like neurons. Scale bar, 50 μm.
A number of molecules are known to regulate Rab 3A activity in synaptic vesicle trafficking (Novick and Brennwald, 1993). We analyzed their protein expression at 6 hr to evaluate potential involvement in BDNF-induced synaptic plasticity. Two proteins that regulate Rab3A activity, Rab GAP and Rab GDI, showed enhanced expression after BDNF application, whereas Rab GEP was not altered (Fig. 5A,B). In addition, Rabphilin and RIM, two proteins that interact with Rab3A to facilitate the anchoring of synaptic vesicles to the plasma membrane (Shirataki et al., 1993; Wang et al., 1997), were also upregulated by BDNF (Fig. 5A,B). Minimal transcriptional changes were observed for these regulatory proteins by reverse transcription-PCR (data not shown). Taken together, these data indicate that transcriptional changes of Rab3A by BDNF are also accompanied by translational regulation. Furthermore, as we have shown above for the GC pathway, molecules associated with the Rab3A pathway can be regulated by BDNF treatment and therefore may be involved in neurotrophin-induced synaptic plasticity.
To confirm the neuronal specificity of these protein products, immunocytochemical analysis in primary hippocampal neurons was performed. Rab3A and Rab GAP as well as Rab GDI were localized to subpopulations of neurons on the basis of morphological identification (Fig. 5C–E). Previous studies have demonstrated that all three of these proteins are concentrated at presynaptic nerve terminals (Novick and Brennwald, 1993; Oishi et al., 1998; Shisheva, 2001). A small percentage of nonpyramidal-like cells were immunonegative for Rab3A, Rab GAP, and Rab GDI, suggesting a heterogeneous population of hippocampal neurons with distinct functions, which may explain the variable synaptic as well as molecular responses to BDNF (Figs.1C, 3A,B).
Rab3A mutant mice
To evaluate the role of the identified genes Rab3A andGC in BDNF-induced synaptic plasticity, we used strategies to interfere with their functions. Previous studies have shown that mice with a targeted deletion of Rab3A have normal basal synaptic activity; however mossy fiber LTP in the hippocampal CA3 region cannot be induced (Geppert et al., 1994; Castillo et al., 1997). Consequently, Rab3A is apparently necessary for certain forms of synaptic plasticity. These reports, in conjunction with BDNF-induced transcriptional changes of Rab3A, led us to examine the effect of BDNF on hippocampal neurons isolated from wild-type andRab3A homozygous knock-out mice. Cells derived from wild-type mice exhibited a 1.9-fold increase in synaptic charge after 3–5 min of BDNF application (Fig.6A,B). This effect is comparable with our previous observations using rat hippocampal neurons (Levine et al., 1995). In contrast, cells derived from Rab3Amutant mice did not display a change in synaptic charge in response to BDNF in that time frame (Fig. 6A,B). Therefore, the synaptic vesicle trafficking protein Rab3A is required for BDNF-induced synaptic plasticity.
Fig. 6.
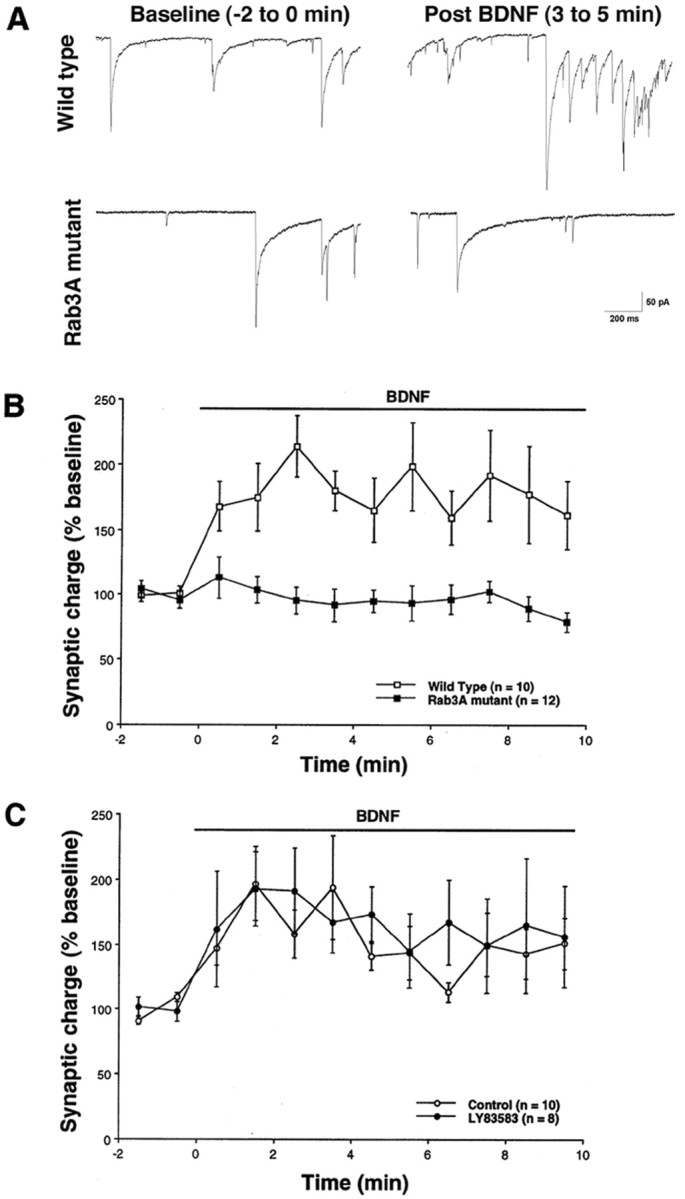
Neurons lacking Rab3A do not respond to BDNF within 10 min. A, Whole-cell patch-clamp recordings (Vhold = −60 mV) on wild-type or Rab3A knock-out mice during baseline recordings or 3–5 min after BDNF application are shown.B, Wild-type cells (■) demonstrated an approximate twofold increase in synaptic charge within 2 min of exposure to BDNF (20 ng/ml; exposure indicated by horizontal bar). Cells from Rab3A knock-out mice (▪), however, do not respond to BDNF (t test, p < 0.05). Representativetraces from wild-type and Rab3A mutant cells are shown.C, Rat neurons were tested for responsiveness to BDNF in the presence of LY83583 (1 μm), an antagonist to GC (●). No inhibition of BDNF-induced plasticity was observed compared with control, untreated cells (○) (t test,p > 0.05). Recordings were obtained from multiple platings. Error bars indicate SEM.
The role of GC in neurotrophin-induced synaptic enhancement was examined using pharmacological agents. An antagonist of GC, LY83583, has been shown to block induction of LTP in the CA1 region of the hippocampus (Zhuo et al., 1994; Arancio et al., 1995). Therefore, the GC pathway may be involved in synaptic plasticity. These findings, together with the induction of GC mRNA by BDNF, led us to study the effect of this GC inhibitor. The inhibitor did not antagonize the BDNF-induced increase in synaptic charge (Fig. 6C), suggesting that GC is not necessary for the effect of BDNF on transmission, although other perturbations may have to be pursued.
We have reported previously that the postsynaptic response to BDNF observed during iontophoretic application of glutamate becomes maximal only after 20 min of neurotrophin treatment (Levine et al., 1998;Crozier et al., 1999). To determine whether prolonged BDNF application has an effect on cells derived from Rab3A knock-out mice, recordings were performed for 30 min. A 1.8-fold increase in synaptic charge was observed after 20–22 min in the presence of BDNF (Fig. 7). Because the time course of the response to BDNF in Rab3A mutant cells is similar to that induced by iontophoresis of glutamate (Levine et al., 1998; Crozier et al., 1999), a postsynaptic locus may be implicated for the late response, although a presynaptic component may still be involved. These observations raise the possibility that the cells from the Rab3A mutants are predominantly deficient in the presynaptic component of the BDNF response, a hypothesis presently under investigation.
Fig. 7.

Cells lacking Rab3A display a late response to BDNF. Whole-cell voltage-clamp recordings (Vhold = −60 mV) were performed on neurons lacking Rab3A in the continuous presence of BDNF (20 ng/ml) for 30 min. There was an increase in synaptic charge detected only after 20 min of BDNF exposure (t test,p < 0.05). Recordings were obtained from multiple platings. Error bars indicate SEM.
DISCUSSION
We have used differential gene profiling to begin identifying molecules that may be involved in BDNF-induced synaptic plasticity. Our ultimate goals are to define mechanisms mediating acute versus long-term activation, presynaptic and postsynaptic mechanisms, and basal versus enhanced transmission and to characterize processes underlying differential cellular responses to trophins. We report here that Rab3A, which is upregulated by a 3 hr exposure to BDNF, is necessary for early enhancement of synaptic charge. Our observations support the hypothesis that altered expression of some genes by BDNF reflects the role of their products in synaptic plasticity.
BDNF exposure time and transcription
We initially examined a 3 hr exposure to permit adequate time to detect transcription of selected downstream genes while still maintaining an acute synaptic response (Sherwood and Lo, 1999). In fact, a 3 hr exposure increased synaptic charge ∼2.5-fold, comparable with that occurring after minutes (Levine et al., 1995; see alsoSherwood and Lo, 1999). Several studies have demonstrated that protein translation is required for enhanced transmission by neurotrophins (Kang and Schuman, 1996; Bradley and Sporns, 1999). Consequently, differential expression of genes in our studies may imply a requirement for de novo transcription for increased synaptic activity. However, because the effects of BDNF are extremely rapid (10 min) and our data indicate a requirement for Rab3A for this initial phase of plasticity, increased gene expression at 3 hr is more likely to be attributable to increased turnover of the molecules used during enhanced synaptic activity. Nevertheless, we cannot formally exclude the requirement for transcription until further studies are performed.
Although BDNF may have multiple functional effects depending on the duration of exposure (Sherwood and Lo, 1999), the trophin uniformly exerts presynaptic effects on vesicle proteins. Long-term (5 d) treatment of dissociated cortical neurons increases several vesicle proteins, including Rab3A, and increases terminal vesicle number (Takei et al., 1997). Moreover, synaptosomes from BDNF knock-out mice exhibit depressed levels of two vesicle proteins, synaptobrevin and synaptophysin, as well as reduced docked vesicles. The levels of the vesicle proteins are rescued by exogenous BDNF via a post-translational mechanism (Pozzo-Miller et al., 1999). Furthermore, synapsin is phosphorylated by BDNF via the mediation of MAP kinase, resulting in enhanced transmitter release (Jovanovic et al., 2000). In our study, using a limited random search approach, we have identified at least one vesicle protein, Rab 3A, that is upregulated by BDNF and is necessary for trophin-induced plasticity. Together, these studies suggest that BDNF regulates presynaptic vesicle protein function. Other vesicle proteins may also be required for the effect of BDNF, the subject of ongoing studies.
Role of Rab3A in synaptic plasticity
Our current observations with mutant mouse neurons indicate that Rab3A is necessary for the early increase in synaptic charge induced by BDNF. The mechanisms via which Rab3A regulates vesicle trafficking are gradually emerging. The protein normally governs both efficient vesicle docking (Geppert et al., 1994) and fusion (Geppert et al., 1997; Lonart et al., 1998). The absence of Rab3A may inhibit BDNF-induced plasticity by preventing rapid replenishment of the vesicular pool at the presynaptic active zone necessary for enhanced transmission. In contrast to inhibition of the early synaptic response to BDNF in the mutants, a late response of Rab3A mutant cells to BDNF was observed and is temporally similar to that produced by pairing glutamate iontophoresis and BDNF exposure (Crozier et al., 1999). Therefore a postsynaptic locus may contribute to this late action of BDNF, although a presynaptic component may still be involved. This is the subject of ongoing study. We conclude that during a 30 min exposure to BDNF a two-component response may be manifested; an early component is dependent on Rab3A, and a later component is apparently Rab3A independent and may involve enhanced NMDA receptor function (Crozier et al., 1998). Further experiments examining the iontophoretic postsynaptic response of Rab3A mutant cells to BDNF will be useful to confirm this distinction.
Studies on Rab3A knock-out mice demonstrate that the molecule is essential for mossy fiber LTP in CA3 of the hippocampus (Castillo et al., 1997) but not for LTP in the CA1 region (Geppert et al., 1994;Geppert et al., 1997). Although controversial, it has been suggested that CA1 LTP, which is NMDA receptor dependent, consists of both presynaptic and postsynaptic components, whereas CA3 mossy fiber LTP, which is NMDA receptor independent, is primarily presynaptic (Bean and Scheller, 1997). Our data suggest that neurons with a Rab3A deletion are deficient only in the presynaptic component of the BDNF response, just as they are deficient only in CA3 LTP that is expressed presynaptically. Therefore, although BDNF-induced synaptic plasticity and CA3 LTP share some properties, our study is the first demonstration that both processes require the same synaptic vesicle protein.
Effects of BDNF on related pathways
The present results, in conjunction with mechanisms of mossy fiber LTP, provide additional insights into regulatory processes. Our finding that PKA R2 and AKAP 84 are upregulated by BDNF implicates the PKA pathway in the presynaptic modulation of synaptic plasticity. This is entirely consistent with the observations that CA3 LTP is cAMP dependent (Nicoll and Malenka, 1995; Goda and Stevens, 1996; Bao et al., 1998) and that synaptic potentiation by BDNF can be prevented by cAMP inhibitors (Boulanger and Poo, 1999). Finally, PKA phosphorylates two effectors of Rab3A, Rabphilin and RIM (Numata et al., 1994; Fykse et al., 1995; Lonart et al., 1998), further implicating the cAMP pathway. Taken together, these studies suggest that BDNF acts via the PKA pathway to phosphorylate Rab3A effectors, enhancing vesicular release.
Our studies suggest that Rab3A regulatory and GC-related molecules are also modulated by BDNF. Translation of the Rab3A accessory proteins was altered by the trophin, while transcription was unchanged. Altered translation is known to occur via local protein synthesis in the absence of transcription (Kang and Schuman, 1996; Steward, 1997). Our slot blot analysis indicated that several molecules in the GC pathway were also regulated by BDNF. Consequently, we have begun to define multiple signaling cascades that may mediate BDNF-induced synaptic plasticity.
Although GC and downstream genes were regulated by BDNF, a specific antagonist, LY83583, known to inhibit LTP (Zhuo et al., 1994;Arancio et al., 1995) did not alter plasticity in our studies. These results do not exclude a role for the GC pathway in BDNF-induced plasticity but do suggest that GC itself is not necessary for this process. Other downstream molecules in the GC pathway may be required; therefore we cannot definitively exclude the PKG pathway until other antagonists have been tested. Nevertheless, mice lacking PKGexhibit normal LTP (Kleppisch et al., 1999), supporting our current findings. We are presently investigating the role(s) of the other genes identified by our DD, including the ATP synthase,estrogen-induced gene, and the ESTs.
Population versus single-cell analysis
Our data suggest that observations in a population of hippocampal neurons can be extrapolated to individual cells that exhibit responsiveness to BDNF. Pyramidal-like electrophysiologically characterized neurons exhibited robust induction of the identified genes in contrast to the more modest increases observed on slot blots of whole populations. These observations highlight the heterogeneity of hippocampal populations (Figs. 1C, 5C–E) and emphasize the need to analyze genomic-electrophysiological relations at the single-cell level.
Mechanisms underlying differential responses of hippocampal neurons to BDNF may be attributable to a number of properties. For example, BDNF induced potentiation of evoked glutamate release when the postsynaptic cell was excitatory (glutamatergic) but not when it was inhibitory (GABAergic) (Schinder et al., 2000). In addition, highly active synapses may be potentiated more strongly by BDNF than are those with lower release probabilities (Gottschalk et al., 1998). However, this remains controversial because our studies (J. Alder, S. Thakker-Varia, and I. B. Black, unpublished observations) and those of Berninger et al. (1999) suggest that less active neurons respond more dramatically to BDNF than highly active ones; however, these differences may result from the different preparations used in these studies. Consequently, the basal state of the cell appears to affect the degree of synaptic potentiation by BDNF. We are currently combining single-cell transcriptional analysis with electrophysiology to determine whether this phenomenon correlates with levels of transcriptional change induced by BDNF. The present studies demonstrate the feasibility of combining whole-cell patch-clamp recording and transcriptional analysis at the single-cell level to identify genes necessary for synaptic plasticity.
Footnotes
This work was supported by grants from the National Alliance for Research on Schizophrenia and Depression, the Foundation of University of Medicine and Dentistry of New Jersey, the National Institute of Child Health and Human Development, and the New Jersey Commission for Science. We thank Y. Takai for the generous donation of the Rab GEP antibody and A. Shisheva for the Rab GDI antibody. We appreciate J. Eberwine's guidance with single-cell expression analysis. We thank S. Y. Lin for help with the Western blots and Kathryn Getzewich for her assistance.
S.T.-V. and J.A. contributed equally to this work.
Correspondence should be addressed to Dr. Ira B. Black, Department of Neuroscience and Cell Biology, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, 675 Hoes Lane, Center for Advanced Biotechnology and Medicine, Piscataway, NJ 08854-5635. E-mail: black@cabm.rutgers.edu.
REFERENCES
- 1.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 2.Bao JX, Kandel ER, Hawkins RD. Involvement of presynaptic and postsynaptic mechanisms in a cellular analog of classical conditioning at Aplysia sensory-motor neuron synapses in isolated cell culture. J Neurosci. 1998;18:458–466. doi: 10.1523/JNEUROSCI.18-01-00458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bean AJ, Scheller RH. Better late than never: a role for rabs late in exocytosis. Neuron. 1997;19:751–754. doi: 10.1016/s0896-6273(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 4.Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem. 1999;6:232–242. [PMC free article] [PubMed] [Google Scholar]
- 5.Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- 6.Bradley J, Sporns O. BDNF-dependent enhancement of exocytosis in cultured cortical neurons requires translation but not transcription. Brain Res. 1999;815:140–149. doi: 10.1016/s0006-8993(98)01112-3. [DOI] [PubMed] [Google Scholar]
- 7.Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- 8.Crozier RA, Levine ES, Black IB, Plummer MR. The complex temporal profile of BDNF-enhanced synaptic transmission in hippocampal neurons. Soc Neurosci Abstr. 1998;24:800. [Google Scholar]
- 9.Crozier RA, Black IB, Plummer MR. Blockade of NR2B-containing NMDA receptors prevents BDNF enhancement of glutamatergic transmission in hippocampal neurons. Learn Mem. 1999;6:257–266. [PMC free article] [PubMed] [Google Scholar]
- 10.Eberwine J, Cao Y, Nair S, Miyashiro K, Mackler S, Finnell R, Surmeier J, Dichter M. Mechanisms of neuronal plasticity as analyzed at the single cell level. Prog Brain Res. 1995;105:117–126. doi: 10.1016/s0079-6123(08)63289-6. [DOI] [PubMed] [Google Scholar]
- 11.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 12.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 13.Fykse EM, Li C, Sudhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro. J Neurosci. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geppert M, Sudhof TC. RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 15.Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 16.Geppert M, Goda Y, Stevens CF, Sudhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- 17.Goda Y, Stevens CF. Synaptic plasticity: the basis of particular types of learning. Curr Biol. 1996;6:375–378. doi: 10.1016/s0960-9822(02)00499-2. [DOI] [PubMed] [Google Scholar]
- 18.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottschalk WA, Jiang H, Tartaglia N, Feng L, Figurov A, Lu B. Signaling mechanisms mediating BDNF modulation of synaptic plasticity in the hippocampus. Learn Mem. 1999;6:243–256. [PMC free article] [PubMed] [Google Scholar]
- 20.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- 21.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 22.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levine ES, Dreyfus CF, Black IB, Plummer MR. Selective role for trkB neurotrophin receptors in rapid modulation of hippocampal synaptic transmission. Brain Res Mol Brain Res. 1996;38:300–303. doi: 10.1016/0169-328x(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 27.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonart G, Janz R, Johnson KM, Sudhof TC. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 1998;21:1141–1150. doi: 10.1016/s0896-6273(00)80631-5. [DOI] [PubMed] [Google Scholar]
- 30.Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58:76–87. [PubMed] [Google Scholar]
- 31.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 33.Novick P, Brennwald P. Friends and family: the role of the Rab GTPases in vesicular traffic. Cell. 1993;75:597–601. doi: 10.1016/0092-8674(93)90478-9. [DOI] [PubMed] [Google Scholar]
- 34.Numata S, Shirataki H, Hagi S, Yamamoto T, Takai Y. Phosphorylation of Rabphilin-3A, a putative target protein for Rab3A, by cyclic AMP-dependent protein kinase. Biochem Biophys Res Commun. 1994;203:1927–1934. doi: 10.1006/bbrc.1994.2413. [DOI] [PubMed] [Google Scholar]
- 35.Oishi H, Sasaki T, Nagano F, Ikeda W, Ohya T, Wada M, Ide N, Nakanishi H, Takai Y. Localization of the Rab3 small G protein regulators in nerve terminals and their involvement in Ca2+-dependent exocytosis. J Biol Chem. 1998;273:34580–34585. doi: 10.1074/jbc.273.51.34580. [DOI] [PubMed] [Google Scholar]
- 36.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 37.Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- 39.Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–109. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- 40.Sherwood NT, Lo DC. Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J Neurosci. 1999;19:7025–7036. doi: 10.1523/JNEUROSCI.19-16-07025.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shirataki H, Kaibuchi K, Sakoda T, Kishida S, Yamaguchi T, Wada K, Miyazaki M, Takai Y. Rabphilin-3A, a putative target protein for smg p25A/rab3A p25 small GTP-binding protein related to synaptotagmin. Mol Cell Biol. 1993;13:2061–2068. doi: 10.1128/mcb.13.4.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shisheva A. Antibody and oligo nucleotide probes to distinguish intracellular expression and localization patterns of Rab GDP-dissociation inhibitor isoforms. Methods Enzymol. 2001;329:39–50. doi: 10.1016/s0076-6879(01)29064-8. [DOI] [PubMed] [Google Scholar]
- 43.Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- 44.Steward O. mRNA localization in neurons: a multipurpose mechanism? Neuron. 1997;18:9–12. doi: 10.1016/s0896-6273(01)80041-6. [DOI] [PubMed] [Google Scholar]
- 45.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takei N, Hatanaka H. Neurotrophin and synaptic plasticity (in Japanese). Tanpakushitsu Kakusan Koso. 1997;42:481–488. [PubMed] [Google Scholar]
- 47.Takei N, Nawa H. Roles of neurotrophins on synaptic development and functions in the central nervous system. Hum Cell. 1998;11:157–165. [PubMed] [Google Scholar]
- 48.Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- 49.Thakker-Varia S, Elkabes S, Schick C, Silverman GA, Peng L, Sherwood AC, Black IB. Gene expression in activated brain microglia: identification of a proteinase inhibitor that increases microglial cell number. Brain Res Mol Brain Res. 1998;56:99–107. doi: 10.1016/s0169-328x(98)00034-5. [DOI] [PubMed] [Google Scholar]
- 50.Thakker-Varia S, Alder J, Crozier RA, Plummer MR, Black IB. Gene expression profiling in BDNF-induced synaptic plasticity using microarrays. Soc Neurosci Abstr. 2000;26:123. [Google Scholar]
- 51.Vicario-Abejon C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 54.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuang X, Belluscio L, Hen R. Golfalpha mediates dopamine D1 receptor signaling. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-16-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]