

Abstract
The concept of a threshold of dopamine (DA) depletion for onset of Parkinson's disease symptoms, although widely accepted, has, to date, not been determined experimentally in nonhuman primates in which a more rigorous definition of the mechanisms responsible for the threshold effect might be obtained. The present study was thus designed to determine (1) the relationship between Parkinsonian symptom appearance and level of degeneration of the nigrostriatal pathway and (2) the concomitant presynaptic and postsynaptic striatal response to the denervation, in monkeys treated chronically with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine according to a regimen that produces a progressive Parkinsonian state. The kinetics of the nigrostriatal degeneration described allow the determination of the critical thresholds associated to symptom appearance, these were a loss of 43.2% of tyrosine hydroxylase-immunopositive neurons at the nigral level and losses of 80.3 and 81.6% DA transporter binding and DA content, respectively, at the striatal level. Our data argue against the concept that an increase in DA metabolism could act as an efficient adaptive mechanism early in the disease progress. Surprisingly, the D2-like DA receptor binding showed a biphasic regulation in relation to the level of striatal dopaminergic denervation, i.e., an initial decrease in the presymptomatic period was followed by an upregulation of postsynaptic receptors commencing when striatal dopaminergic homeostasis is broken. Further in vivofollow-up of the kinetics of striatal denervation in this, and similar, experimental models is now needed with a view to developing early diagnosis tools and symptomatic therapies that might enhance endogenous compensatory mechanisms.
Keywords: threshold for symptom appearance, early D2dopaminergic receptor upregulation, substantia nigra, striatum, dopaminergic homeostasis, compensatory mechanisms
Parkinson's disease (PD) is a progressive neurodegenerative disorder and is observed in 1% of the population over 55, the mean age at which the disease is first diagnosed (Hoehn and Yahr, 1967). PD was first characterized by James Parkinson (Parkinson, 1817), and consists of a syndrome including tremor, rigidity, postural abnormalities, and bradykinesia. The principal pathological characteristic of PD is the loss of pigmented neurons in the substantia nigra pars compacta (SNc) (Hassler, 1938). These pigmented neurons have since been identified as nigrostriatal dopamine (DA) neurons (Ehringer and Hornykiewicz, 1960).
Whereas the nature of the etiology of the process underlying clinical deterioration remains unknown, PD is characterized by its progressiveness (Hoehn and Yahr, 1967). It has been suggested that progression in PD is the consequence of linear age-related cell attrition superimposed on an SNc already damaged by transient exposure to a previous insult (Koller et al., 1991). An alternative view is that the onset and progression of idiopathic PD represents a novel ongoing degenerative process (McGeer et al., 1988) with an exponential decay (Fearnley and Lees, 1991). Until recently, the presymptomatic phase was thought to last at least 20 years (Hoehn and Yahr, 1967; Vingerhoets et al., 1994). However, others have suggested much shorter presymptomatic periods: Fearnley and Lees (1991) proposed 4.7 years, whereas Morrish et al. (1996) suggested 3.1 years. Although the length of the period preceding the first appearance of clinical signs remains open to debate, it is generally accepted that Parkinsonian signs appear when dopaminergic neuronal death exceeds a critical threshold, 70–80% of striatal nerve terminals and 50–60% of SNc perikarya (Bernheimer et al., 1973; Riederer and Wuketich, 1976). Although the concept of a threshold for onset of symptoms is widely accepted, it has never been determined experimentally in nonhuman primates, being essentially derived by extrapolating measurements of decreased striatal DA content in human postmortem tissue (Hornykiewicz and Kish, 1987) and mathematical projections of progression seen in human in vivo imaging studies (Morrish et al., 1996). The implications of this concept are great given that it defines a period in which DA depletion progresses without symptoms. This presymptomatic period provides an opportunity for presymptomatic therapeutic intervention and diagnosis.
The present study was performed in monkeys chronically treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) according to a regimen that produces a progressive Parkinsonian state (Bezard et al., 1997b, 2000, 2001b,c). It was designed to determine (1) the relationship between the appearance of Parkinsonian symptoms and the level of degeneration of the nigrostriatal pathway, and (2) the concomitant postsynaptic striatal response to the progressing denervation. The time course of changes in striatal DA content and metabolism, striatal DA transporter (DAT) binding, striatal DA receptor (DAR; D1-like and D2-like subtypes) binding, and number of both tyrosine hydroxylase-immunoreactive (TH-IR) and Nissl-stained neurons in the SNc was assessed.
MATERIALS AND METHODS
Animals. Experiments were conducted on 25 female cynomolgus monkeys (Macaca fascicularis; Shared Animal Health, Beijing, China; mean age, 3.1 ± 0.3 years; mean weight, 2.8 ± 0.2 kg). Animals were housed in individual primate cages under controlled conditions of humidity (50 ± 5%), temperature (24 ± 1°C), and light (12 hr light/dark cycles, lights on 8:00 A.M.), food and water were available ad libitum, and animal care was supervised by veterinarians skilled in the healthcare and maintenance of nonhuman primates. Experiments were performed in accordance with European Communities Council Directive of November 24, 1986 (86/609/EEC) for care of laboratory animals. All efforts were made to minimize animal suffering and to use the minimum number of animals necessary to perform statistically valid analysis. To maximize data obtained from these animals, brain tissues acquired in the present experiment will be used for further experiments on the mechanism of the progressive nature of PD.
Experimental protocol. Five untreated monkeys were killed at the beginning of the study and were termed day 0 (D0), non-Parkinsonian controls. The remaining 20 were treated daily (9:00 A.M.) with MPTP hydrochloride (0.2 mg/kg, i.v.; Sigma, St. Louis, MO) dissolved in saline according to a previously described protocol (Bezard et al., 1997b). This protocol describes a reproducible MPTP cumulative dosing regime that leads to the first appearance of Parkinsonian clinical signs after 15 ± 1 injections (i.e., a cumulative dose of 3.0 ± 0.2 mg/kg). Five presymptomatic monkeys were killed at day 6 (i.e., after 6 injections; D6 group), five presymptomatic monkeys at day 12 (i.e., after 12 injections; D12 group), five at day 15 after appearance of overt symptoms (i.e., after 15 injections; D15 group), and the remaining five fully Parkinsonian monkeys at day 25 (i.e., after 15 injections and 10 d of symptom progression and stabilization; D25 group). All animals were killed by sodium pentobarbital overdose (150 mg/kg, i.v.), and the brains were removed quickly after death. Each brain was bisected along the midline, and the two hemispheres were immediately frozen by immersion in isopentane (−45°C) and then stored at −80°C. Tissue was sectioned coronally at 20 μm in a cryostat at −17°C, thaw-mounted onto gelatin-subbed slides, dried on a slide warmer, and stored at −80°C.
Behavioral assessment. To follow the progression of the syndrome, animal behavior was assessed daily (2:00 P.M.) on a Parkinsonian monkey rating scale using videotape recordings of monkeys in their cages and clinical neurological evaluation as previously described (Bezard et al., 1997a; Imbert et al., 2000). For each group, however, the pertinent data are the assessments done the day of killing. During each session, two examiners evaluated the animals' level of motor performance, coaxing them to perform various tasks by offering appetizing fruits. A third examiner, watching a video recording, made an independent and blind assessment. The minimal disability score was 0, and the maximum score was 25 (Imbert et al., 2000). Differences in rating were discussed regularly to eliminate observer idiosyncrasy (Taylor et al., 1994). Bradykinesia was tested objectively at the beginning of each session by assessing the mean time required to pick up three pieces of fruit positioned 5 cm apart as previously described (Bezard et al., 1997a). A maximum time of 60 sec was allowed to perform the test.
Neurochemical analysis. The extent of striatal DA denervation was assessed by measuring levels of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) in both the caudate nucleus and the putamen using high-pressure liquid chromatography with electrochemical detection as previously described (Bezard et al., 2001c). After sections have been freeze-dried (−60°C; 40.10−3 atmospheric pressure) for 2 hr, the putamen and caudate nucleus regions were separately scraped off and sonicated in 200 μl of HClO4 0.1N containing 3,4-dihydroxybenzylamine as an internal standard. The homogenates were then centrifuged at 27,000 × gav for 20 min at 4°C. Pellets were retained for quantification of protein content by the Bradford assay. The high-pressure liquid chromatography system consisted of a pump (Beckman, Fullerton, CA) connected to a stainless steel separation column packed with Hypersil 50DS (Beckman). Electrochemical detection was performed using a BAS LC-4B detector (Waters Milford, MA) with a glassy carbon working electrode, an Ag–AgCl reference electrode, and an amperometric detector. Detector potential was set at +0.8 V versus the reference electrode. Concentrations of DA and metabolites were calculated using a computing integrator (Gold Nouveau version 1.6; Beckman). Mean and SEM values were calculated for both putamen and caudate nucleus for each group.
Dopamine transporter binding. The radiolabeling of [125I](E)-N-(3-iodoprop-2-enyl)-2β-carboxymethyl-3β-(4′-methylphenyl)-nortropane (PE2I) was performed from the stannyl precursor according to a previously described method to identify the dopaminergic nerve endings (Guilloteau et al., 1998). After purification, [125I]PE2I was obtained in a no-carrier-added form with a specific activity of 2000 Ci/mmol. [125I]PE2I was kept in ethanol at −20°C and is stable for 1 month in these storage conditions (Emond et al., 1997). Sections were incubated for 90 min at 25°C with 100 pm [125I]PE2I in pH 7.4 phosphate buffer (in mm: NaH2PO4 10.14, NaCl 137, KCl 2.7, and KH2PO4 1.76) as previously described (Chalon et al., 1999; Bezard et al., 2001a). Adjacent sections were incubated in the presence of 100 μm cocaine (Sigma) to define nonspecific binding. After incubation, sections were washed twice for 20 min in phosphate buffer at 4°C and then rinsed for 1 sec in distilled water at 4°C. After drying at room temperature, sections were exposed to β radiation-sensitive film (Hyperfilm βmax; Amersham Pharmacia Biotech, Buckingamshire, UK), together with calibrated [125I]microscales (Amersham) in x-ray cassettes, for 3 d to assess autoradiographically the radioactivity bound to regions of interest.
Dopamine receptor binding. Both the D1and D2 DARs were labeled using ligands specific for D1-like sites ([3H]SCH 23390; New England Nuclear, Paris, France; 75 Ci/mmol) or D2-like sites ([3H]YM-09151–2; New England Nuclear; 85 Ci/mmol). Binding experiments were performed as previously described (Delion et al., 1996): tissue sections were incubated for 1 hr at room temperature in a buffer solution (in mm: 50 Tris-HCl, 120 NaCl, 5 KCl, 2 CaCl2, and 1 MgCl2, pH 7.4) containing either 2 nm [3H]SCH 23390 or 0.3 nm[3H]YM-09151–2. Nonspecific binding was defined in the presence of 10 μm of (+)butaclamol for both subtypes of DAR. Incubations were terminated by rinsing in ice-cold 50 mm Tris-HCl, pH 7.4. Sections were then dipped for 1 sec in ice-cold distilled water. After drying at room temperature, sections were exposed to tritium-sensitive film ([3H]Hyperfilm; Amersham), together with calibrated [3H]microscales (Amersham) in x-ray cassettes, for 5–8 weeks to assess autoradiographically the radioactivity bound to regions of interest.
Analysis of autoradiographs. Densitometric analysis of autoradiographs (DAT and DAR bindings) was performed using an image analysis system (Image Pro Plus, version 3.0.01; Media Cybernetics, Atlanta, GA) as previously described (Henry et al., 1999; Bezard et al., 2001c). The optical density was assessed for the striatum at three rostrocaudal levels in accordance with the functional organization of the striatum (Morissette et al., 1999; Schneider et al., 1999) using a stereotaxic atlas (Szabo and Cowan, 1984): a rostral level including the caudate, putamen, and nucleus accumbens [anterior (A) 21.0]; a midlevel including the caudate, putamen, and globus pallidus pars externalis (A17.2); and a caudal level including the body of the caudate, the putamen, and both parts of the globus pallidus (i.e., pars externalis and pars internalis) (A14.6). Where appropriate, both caudate and putamen were divided into dorsolateral, dorsomedial, ventrolateral, and ventromedial quadrants for analysis. Four sections per animal, per striatal level were analyzed by an examiner blind with regard to the experimental condition. In addition, DAT binding was measured, in D0 and D25 groups, at a mesencephalic level where both SNc and VTA are present on adjacent sections to those used for TH immunohistochemistry. Slides were then stained with hemalun to allow further anatomical identification. Optical densities were averaged for each region in each animal and converted to amount of radioactivity bound by comparison to the standards. Mean radioactivity bound and SEM were then calculated for each group. Data are expressed in femtomoles per milligram of tissue equivalent.
Tyrosine hydroxylase immunohistochemistry. Mesencephalic sections were processed for TH immunohistochemistry and then counterstained with cresyl violet (Nissl staining) as previously described (Bezard et al., 1997b). Cell counts were performed using a computer-based image analyzer (Visioscan version 4.12; Biocom, Les Ulis, France). The boundaries of the SNc were chosen on three consecutive sections corresponding to a representative median plane of the SNc by examining the size and shape of the different TH-IR neuronal groups, cellular relationships to axonal projections, and nearby fiber bundles. The number of both TH-IR and Nissl-stained neurons per SNc representative plane was calculated three times by one examiner blind with regard to the experimental condition. Split cell counting error was corrected by using the formula of Abercrombie (1946). Mean cell number per plane and SEM were then calculated for each group of monkey.
Statistical analysis. For multiple comparisons of binding data, neurochemical data, cell counts, and time reaction data, one-way ANOVA was used to estimate overall significance followed by post hoc t tests corrected for multiple comparisons by the method of Bonferroni (Miller, 1981). For multiple comparisons of behavioral assessments, a Kruskal–Wallis nonparametric test was used to estimate overall significance followed by post hoc t tests corrected for multiple comparisons by the method of Dunn (Miller, 1981). All data were normally distributed, and significance levels of t test comparisons were adjusted for inequality of variances when appropriate. These analyses were completed using STATA program (Intercooled Stata 6.0; Stata Corporation, College Station, TX). Both regressions and best fitting correlations were done using Kaleidagraph program (version 3.5; Synergy Software, Reading, PA). A probability level of 5% (p < 0.05) was considered significant.
RESULTS
Changes in motor behavior
Repeated MPTP treatment had a significant effect on both the Parkinsonian rating score (Kruskal–Wallis = 23.5;p < 0.0001) and the bradykinesia test (F(4,20) = 184.7; p < 0.0001). As previously reported with this administration protocol (Bezard et al., 1997a,b, 1999), monkeys at D6 and D12 did not exhibit Parkinsonian motor symptoms (Parkinsonian score of 0 for all animals at both time points; p > 0.5 compared with D0). Furthermore, the mean duration of the bradykinesia test (2.4 ± 0.3 and 2.6 ± 0.5 sec, respectively) was equivalent to the performance of D0, i.e., non-Parkinsonian monkeys (3.0 ± 0.4 sec) (t = −0.6 and t = −0.4, respectively;p > 0.5). Both the D6 and D12 groups were thus considered as asymptomatic. Monkeys of both the D15 and D25 groups exhibited Parkinsonian motor abnormalities [median 11 (range 10–14) and median 17 (range 15–19) for D6 and D12, respectively; bothp < 0.05 vs D0]. The mean duration of the bradykinesia test was significantly increased in the D15 group (19.9 ± 9.1 sec) compared with D0 (3.0 ± 0.4 sec) (t = 16.8; p < 0.05). D25 monkeys could not perform the bradykinesia test, reflecting their inability to initiate a voluntary movement (60 sec; t = 57.0 vs D0,t = 57.6 vs D6, t = 57.4 vs D12, andt = 40.1 vs D15; all p < 0.05).
The transition between the presymptomatic and symptomatic periods thus occurred between days 12 and 15 of the intoxication protocol. A clinical score of 4 is necessary to decide that a monkey exhibits Parkinsonian motor abnormalities (Imbert et al., 2000). According to the regression applied to clinical scores between days 12 and 25, this score is reached at 13.2 d.
Kinetics of nigral degeneration
Repeated MPTP treatment had a significant effect on the number of both TH-IR cells (F(4,24) = 117.7;p < 0.0001) (Fig.1A) and the total number of surviving neurons, i.e., Nissl-stained cells (F(4,24) = 79.0; p < 0.0001) (Fig. 1B). From D6 onward, the number of TH-IR neurons decreased significantly in comparison with that of the D0 group (t = −169.0; p < 0.05) (Fig.1A). After D6, the number of TH-IR neurons was also significantly different to that of the preceding group (Fig. 1) (D12 vs D6, t = −191.4, p < 0.05; D25 vs D15,t = −368.4, p < 0.05). Whereas the number of TH-IR neurons was significantly reduced from D6 (D6 = −17.6%; D12 = −37.5%; D15 = −47.5%), the total number of surviving Nissl-stained cells in the SNc decreased significantly as compared with control D0, animals only from D15 onward (D6 = −2.1%; D12 = −6.7%; D15 = −28.4%, t = −307.4, p < 0.05). After D15, the number of Nissl-stained neurons became significantly different to that of the preceding group (Fig. 1) (D15 vs D12, t = −234.8,p < 0.05; D25 vs D15, t = −399.6,p < 0.05). The general gradient loss we observed begins by affecting the whole dorsal tier of the SNc and then its ventral tier (Fig. 1C). Indeed, the few remaining TH-IR neurons in the fully Parkinsonian animals are located within the ventral tier (Fig. 1C). The number of TH-IR neurons and of Nissl-stained cells in the fully Parkinsonian animals, i.e., in the group D25, were dramatically decreased by 85.8 and 65.3%, respectively.
Fig. 1.
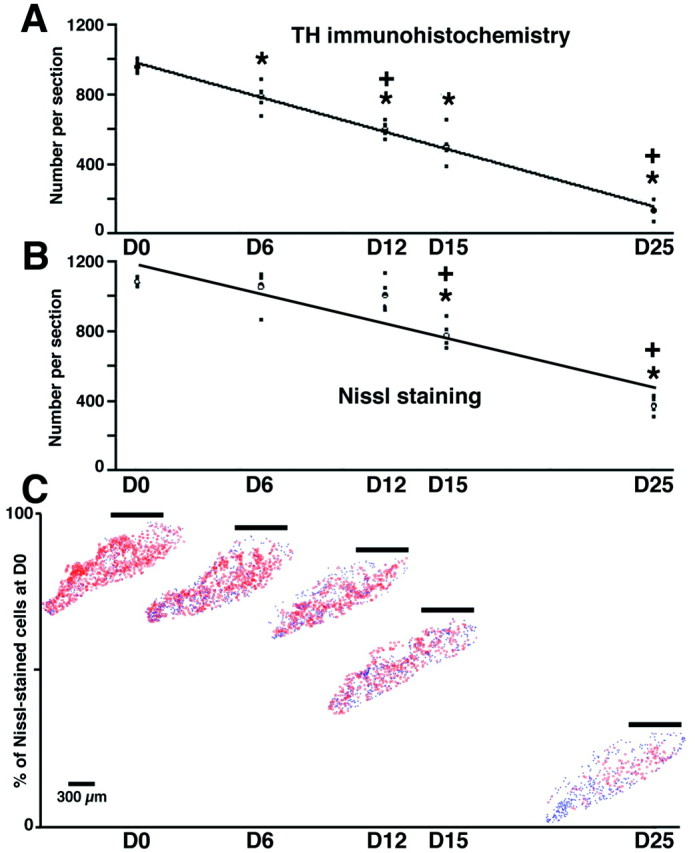
Time course of changes in the number of TH-IR neurons (A) and Nissl-stained cells (B) in the SNc. A,B,Filled squares represent the individual values (n = 5 in each group), theopen circle represents the mean value of each group, and the dark lines represent the kinetic fits. *Significant difference compared with D0, p < 0.05; +significant difference compared with the previous group,p < 0.05. C, Examples of cell-counting maps showing the typical patterns of degeneration in the SNc. TH-IR neurons are marked in red, whereas theblue symbols represent the Nissl-stained cells that were not TH-positive. The horizontal line above each map indicates the mean percentage of surviving cells (i.e., Nissl-stained). Note the selective disappearance of the dorsal tier of the SNc with time.
The severity of the mesencephalic lesion in D25 animals was also studied using DAT binding (F(1,9) = 411.9; p < 0.0001). DAT binding decreased significantly in the SNc from 42.4 ± 1.1 fmol/mg of tissue equivalent in D0 animals to 5.1 ± 1.4 fmol/mg of tissue equivalent in D25 monkeys (t = −37.4;p < 0.05). DAT binding thus demonstrated a lesion of 87.9%, equivalent to the severity revealed using TH immunohistochemistry.
Because the transition between the presymptomatic and symptomatic periods was calculated as occurring at 13.2 d, the level of degeneration at this time point is of critical interest. The kinetics of the decrease of TH-IR neurons followed a linear regression (y = 982.74–33.04x;r = 0.998) as the pattern of reduction in Nissl-stained cells did (y = 1199.9–29.167x;r = 0.927). Thus, according to these regressions, the threshold of nigral lesion required for clinical manifestation was calculated as being 75.2% of D0 of Nissl-stained cells (i.e., decreased by 24.8%), whereas the threshold number of TH-IR neurons would be 56.8% of D0 levels (i.e., decreased by 43.2%).
Despite both of these parameters followed linear regressions, the relationship between the number of TH-IR neurons (x) and Nissl-stained cells (y) is best represented by logarithmic equation (y = −1385.9 + 834.16log(x); r = 0.955; p< 0.05) rather than by a linear one. This dissociation suggests that, although SNc neurons lose their functional dopaminergic activity (earlier decrease in the number of TH-IR neurons), their cell bodies have not degenerated, at least in the early phase of the intoxication protocol.
Progression of striatal denervation
MPTP administration induced striatal dopaminergic denervation, as shown by decreases in DAT binding (Fig.2). This denervation was significant whether caudate or putamen or whichever rostrocaudal level or quadrant was considered (e.g., in the caudate nucleus at the rostral level: dorsolateral, F(4,23) = 69.8,p < 0.0001; dorsomedial,F(4,23) = 51.2, p < 0.0001; ventrolateral, F(4,23) = 24.4,p < 0.0001; ventromedial,F(4,23) = 30.8, p < 0.0001; and in the putamen at the rostral level: dorsolateral,F(4,23) = 83.1, p < 0.0001; dorsomedial, F(4,23) = 232.9,p < 0.0001; ventrolateral,F(4,23) = 121.3, p < 0.0001; ventromedial, F(4,23) = 93.5,p < 0.0001). From D6 onward, whichever level and quadrant was considered in the putamen, the DAT binding was significantly lower in comparison with the same region of group D0 (Fig. 2), as particularly shown in the dorsal putamen (rostral level: −35.2%, t = −47.9, p < 0.05 in the dorsolateral quadrant; −29.7%, t = −38.4,p < 0.05 in the dorsomedial quadrant; mid level: −36.4%, t = −46.6, p < 0.05 in the dorsolateral quadrant; −31.5%, t = −36.4,p < 0.05 in the dorsomedial quadrant; caudal level: −38.6%, t = −39.1, p < 0.05 in the dorsolateral quadrant; −26.1%, t = −49.8,p < 0.05 in the dorsomedial quadrant) (Fig. 2). A comparable decrease, compared with D0, was observed from D6 onward in the dorsolateral quadrant of the caudate nucleus (rostral level: −24.1%, t = −28.2, p < 0.05; mid level: −21.6%, t = −20.3, p < 0.05; caudal level: −33.1%, t = −39.1, p< 0.05). In contrast, for the other quadrants of the caudate nucleus, the decrease only became significant, compared with D0, from D12 onward (e.g., for the ventrolateral quadrant: at D6, −17.2%,t = −18.6, nonsignificant, whereas at D12, −49.9%,t = −54.6, p < 0.05, for the rostral level; at D6, −24.9%, t = −27.5, nonsignificant, whereas at D12, −54.4%, t = −59.6, p< 0.05 for the mid level).
Fig. 2.

A, Examples of DAT binding autoradiographs showing the progression of striatal denervation at the caudal level. Note the homogenous degeneration and the severe lesion in the D25 group. The horizontal line under each example indicates the mean percentage of DAT binding. Nonspecific binding is shown on the bottom left corner of the panel.B, DAT loss in the caudal putamen. Mean data (open circles) and individual data from all quadrants of all animals (dark circles;n = 20 in D0, D12, D15, and D25 groups and n = 16 in D6 group) are presented. Dark line represents the kinetic fits. Data are presented as femtomoles per milligram of tissue equivalent.
After D6, DAT binding continued to decrease progressively in both the putamen and caudate nucleus and was often significantly different from that of the preceding group (Fig. 2) as demonstrated, for example, in the dorsolateral quadrant of the putamen (rostral level: D12 vs D6,t = −35.5, p < 0.05; D15 vs D12,t = −22.8, nonsignificant; D25 vs D15,t = −28.4, p < 0.05; mid level: D12 vs D6, t = −29.5, p < 0.05; D15 vs D12, t = −18.2, nonsignificant; D25 vs D15,t = −30.6, p < 0.05; caudal level: D12 vs D6, t = −25.2, p < 0.05; D15 vs D12, t = −35.4, p < 0.05; D25 vs D15, t = −16.2, nonsignificant) (Fig. 2).
At the end of the protocol, i.e., in the group D25, global DAT binding in the striatum was dramatically decreased, by 96.6% (Fig. 2). In contrast with the linear decrease in the number of TH-IR neurons at the nigral level, the kinetics of striatal dopaminergic denervation followed an exponential regression. Figure 2B shows such a regression for the putamen at the caudal level, all quadrants of all animals being plotted [y = 192.7 exp(−0.155x); r = 0.942]. Taking 13.2 d as representing the time of transition between the presymptomatic and symptomatic periods, the threshold of striatal dopaminergic lesion required for clinical manifestation would be ∼19.7% of D0 levels (i.e., decreased by 80.3%). The correlation between the DAT binding (x) and TH-IR neurons (y) is thus best represented by a logarithmic equation [y = −20.31 + 421.69log(x); r = 0.918; p< 0.05]. The implications of this finding are that as the level of DAT binding fall, the number of TH-IR neurons does not fall as rapidly. Because decreases in DAT binding (x) and Nissl-stained cells (y) followed opposite patterns, with respect to the reduction in TH-IR neuronal number (examples shown in Figs.1B, 2B, respectively), it is not surprising that their correlation is best represented by a logarithmic equation [y = 260.4 + 408.99log(x);r = 0.910; p < 0.05].
Progression of striatal DA depletion
The impact of the progressive MPTP intoxication on striatal DA content was determined by measuring the levels of DA and its metabolites in the putamen and in the caudate nucleus. MPTP treatment significantly affected the DA, DOPAC, and HVA levels in both caudate nucleus (Fig. 3A) (DA:F(4,24) = 57.7, p < 0.0001; DOPAC: F(4,24) = 24.2,p < 0.0001; HVA:F(4,24) = 68.0, p < 0.0001) and putamen (Fig. 3B) (DA:F(4,24) = 62.9, p < 0.0001; DOPAC: F(4,24) = 27.5,p < 0.0001; HVA:F(4,24) = 65.3, p < 0.0001). In addition, there were no significant differences in DA levels between putamen and caudate nucleus (F(1,49) = 0.4; p > 0.5) (Fig. 3). When compared with D0 values (control), the level of DA in the putamen was significantly reduced by 41.7% in the D6 group (t = −59.6; p < 0.05) and by 58.2% in the D12 group (t = −82.8; p < 0.05). As noted above, both the D6 and D12 groups were asymptomatic. In the D15 group, striatal DA content was significantly decreased when compared with either D0 control animals (−85.8%; t = −122.1; p < 0.05) or asymptomatic D6 (t = −62.5; p < 0.05) and D12 groups (t = −39.2; p < 0.05). In the D25 group, DA depletion reached 97.9% of D0 levels (t = −139.4; p < 0.05) (Fig. 3B), and the DA level was significantly lower than in D6 (t = −79.9;p < 0.05) and D12 groups (t = −56.6;p < 0.05). Comparable significant decreases were observed for DOPAC and HVA levels in the putamen (data not shown) as well as for both DA and its two metabolites in the caudate nucleus (see Fig. 3A for DA decrease).
Fig. 3.

Progressive decrease in striatal DA content (in picograms per microgram of protein) measured in the caudate nucleus (A) and in the putamen (B).Filled circles represent the individual values (n = 5 in D0, D12, D15, and D25 groups andn = 4 in D6 group), the open circlerepresents the mean value of each group, and the dark lines represent the kinetic fits. *Significant difference compared with D0, p < 0.05; +significant difference compared with the previous group, p < 0.05. C, Levels of (DOPAC+HVA)/DA ratio in the putamen of the different groups.
The progression of striatal DA depletion followed an exponential regression in both the caudate nucleus [y = 199.85 exp(−0.192x); r = 0.964] (Fig.3A) and in the putamen [y = 206.18 exp(−0.157x); r = 0.953] (Fig.3B). Because the transition between the presymptomatic and symptomatic periods was calculated at 13.2 d, the threshold of dopaminergic depletion required for clinical manifestation would be ∼12.4% within the caudate nucleus (i.e., decreased by 87.6% compared with D0) and 18.2% within the putamen (i.e., decreased by 82.1% compared with D0). Levels of DA (x) and of DAT binding (y) within the striatum are linearly correlated in both the caudate nucleus (y = 13.70 + 0.76x; r = 0.915;p < 0.05) and the putamen (y = 7.15 + 0.78x; r = 0.944;p < 0.05). According to this linear relationship, the correlation between the striatal DA content and markers of nigral degeneration (i.e., TH-IR and Nissl counts) matched with those determined for the DAT, i.e., they show a logarithmic relationship [DA levels (x) and the number of TH-IR neurons (y): caudate nucleus, y = 250.72 + 279.06log(x), r = 0.875, p< 0.05; putamen, y = −54.3 + 432.67log(x),r = 0.931, p < 0.05] [DA levels (x) and Nissl-stained cells (y): caudate nucleus, y = 524.54 + 269.62log(x),r = 0.880, p < 0.05; putamen,y = 223.04 + 422.54log(x), r= 0.949, p < 0.05].
MPTP intoxication had a significant effect on the ratio of DA metabolites to DA, the (DOPAC + HVA)/DA ratio, in both caudate nucleus (F(4,24) = 3.2; p < 0.05) and putamen (Fig. 3C) (F(4,24) = 16.4; p < 0.0001). At all time points up to D15, the DA metabolites–DA ratio in the putamen was not significantly altered in comparison with D0. In contrast, in D25 animals, the (DOPAC + HVA)/DA ratio was significantly higher than at D0 (560,1% of D0 ratio, t = 4.5, p < 0.05), D6 (130.6% of D0 ratio, t = 4.2, p < 0.05), D12 (148.5% of D0 ratio, t = 4.0, p < 0.05), and D15 (232.8% of D0 ratio, t = 3.2,p < 0.05) (Fig. 3C). The relationship that that best describes the correlation between the DA metabolites–DA ratio and DA content, an exponential decay [y = 2.44 exp(−6.9 10−3x);r = 0.906; p < 0.05], underlines the need for a marked DA depletion before any increase in the DA turnover is reflected by the ratio.
D1-like dopamine receptor binding is not affected by dopaminergic denervation
MPTP-induced degeneration of the nigrostriatal pathway differentially affected striatal D1-like and D2-like DAR binding. No change in striatal D1-like DAR binding was observed in any rostrocaudal level either in the caudate nucleus (Fig.4) (e.g., at the rostral level: dorsolateral, F(4,23) = 1.3; dorsomedial, F(4,23) = 1.2; ventrolateral, F(4,23) = 0.3; ventromedial, F(4,23) = 2.3) or in the putamen (e.g., at the rostral level: dorsolateral,F(4,23) = 0.7; dorsomedial,F(4,23) = 2.1; ventrolateral,F(4,23) = 0.7; ventromedial,F(4,23) = 1.2).
Fig. 4.
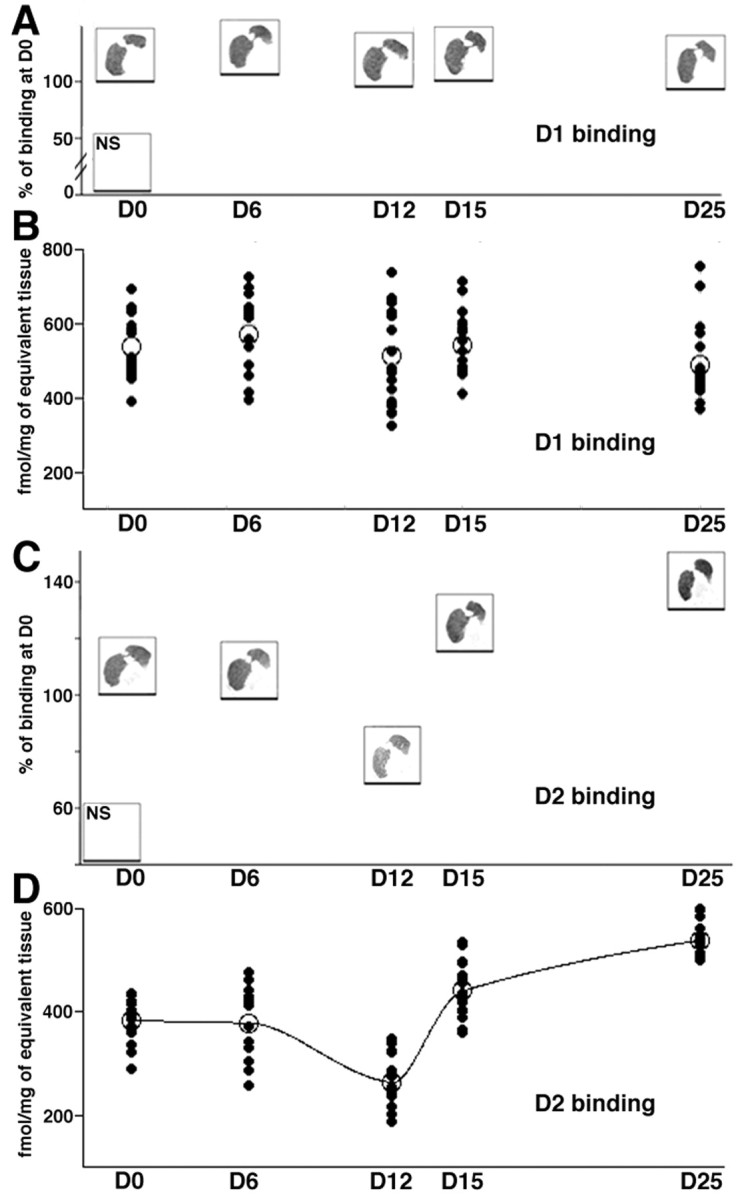
Examples of D1(A) and D2 (C) DAR binding autoradiographs at the caudal level of the striatum. Thehorizontal line under each example indicates the mean percentage of DAR binding. Nonspecific binding is shown on thebottom left corner of each panel. Below each of these examples, a scatter plot shows the mean binding data (open circles) of, respectively, D1(B) and D2 (D) ligands, measured in the putamen at the caudal level, all quadrants of all animals being plotted (dark circles,n = 20 in D0, D12, D15, and D25 groups andn = 16 in D6 group). Data are in femtomoles per milligram of tissue equivalent. D, Dark line interpolates the mean binding data of D2 ligand to underline the biphasic changes.
Biphasic regulation of D2-like dopamine receptor binding in relation with progression of the degeneration
MPTP-induced decreases in striatal DA afferents led to significant changes in D2-like DAR binding at all rostrocaudal levels and quadrants in both the caudate nucleus (e.g., at the rostral level: dorsolateral,F(4,23) = 53.8, p < 0.0001; dorsomedial, F(4,23) = 44.3,p < 0.0001; ventrolateral,F(4,23) = 9.9, p < 0.001; ventromedial, F(4,23) = 14.8,p < 0.0001) and in the putamen (e.g., at the rostral level: dorsolateral, F(4,23) = 32.7,p < 0.0001; dorsomedial,F(4,23) = 39.8, p < 0.0001; ventrolateral, F(4,23) = 25.8,p < 0.0001; ventromedial,F(4,23) = 31.7, p < 0.0001).
No significant change in D2-like DAR binding was observed in D6 animals compared with D0. In the D12 group (asymptomatic animals), the D2 binding was decreased in most of the quadrants, at all rostrocaudal levels, being particularly evident in the dorsal putamen (rostral level: 36.2% of decrease,t = −152.7, p < 0.05 in the dorsolateral quadrant; −32.6%, t = −127.6,p < 0.05 in the dorsomedial quadrant; mid level: −30.8%, t = −125.9, p < 0.05 in the dorsolateral quadrant; −25.7%, t = −98.6,p < 0.05 in the dorsomedial quadrant; caudal level: −29.4%, t = −111.2, p < 0.05 in the dorsolateral quadrant; −30.8%, t = −112.3,p < 0.05 in the dorsomedial quadrant) (Fig. 4).
At D15, when symptoms have appeared, D2-like binding is not different from D0 animals (e.g., in the rostral putamen: +21.0%, t = 88.5 in the dorsolateral quadrant, +16.6%, t = 64.8 in the dorsomedial quadrant, and in the caudal putamen: +18.1%, t = 66.5 in the dorsolateral quadrant; +14.1%, t = 54.5 in the dorsomedial quadrant) (Fig. 4). This apparent “normalization” of the D2 levels is, however, subsequent to the decrease observed in the asymptomatic D12 group. As a consequence, the binding of D2-like DAR ligand is significantly different at D15 from D12 (e.g., in the rostral putamen:t = 241.3, p < 0.05 in the dorsolateral quadrant; t = 192.5, p < 0.05 in the dorsomedial quadrant; mid level: t = 255.7,p < 0.05 in the dorsolateral quadrant;t = 226.5, p < 0.05 in the dorsomedial quadrant; caudal level: t = 174.6, p < 0.05 in the dorsolateral quadrant; t = 166.8,p < 0.05 in the dorsomedial quadrant) (Fig. 4). This obviously suggests a massive D2 upregulation between D12 and D15 (e.g., +67.2% at the caudal level), although no difference can be evidenced with D0 group.
The severe loss of DA terminals in D25 group was accompanied by a significant increase in the binding of D2-like DAR ligand in comparison with control D0 animals (e.g., in the rostral putamen: 47.7% of increase, t = 201.1,p < 0.05 in the dorsolateral quadrant; +40.6%,t = 158.9, p < 0.05 in the dorsomedial quadrant; mid level: +53.3%, t = 218.2,p < 0.05 in the dorsolateral quadrant; +56.4%,t = 216.5, p < 0.05 in the dorsomedial quadrant; caudal level: +48.9%, t = 176.1,p < 0.05 in the dorsolateral quadrant; +41.1%,t = 148.7, p < 0.05 in the dorsomedial quadrant) (Fig. 4). When compared with D12 group, the increase in D2-like DAR binding is huge (e.g., +104.4% at caudal level: t = 284.1, p < 0.05 in the dorsolateral quadrant; t = 261.0, p< 0.05 in the dorsomedial quadrant) (Fig. 4).
The timing of changes in D2 DAR binding did not follow a simple equation. The relationship between D2-like DAR binding (y) and both DAT binding and DA content (x) may be represented by second order polynomial equations (respectively, y = 551.75–5.42x + 3.4 10−2x2,r = 0.781, p < 0.05; y= 553.41–4.50x + 2.2 10−2x2,r = 0.797, p < 0.05). Such quadratic relationships imply synergistic action of two first order processes. D2-like DAR are located on both the presynaptic dopaminergic terminals and the postsynaptic striatal neurons. Thus, theses quadratic correlations suggest that the initial decrease in D2 DAR binding reflects the only disappearance of the dopaminergic terminals, whereas the subsequent increase represents mainly the compensatory answer of the postsynaptic side, the loss of remaining presynaptic D2 receptors being masked by its huge increase.
DISCUSSION
This study defined, in experimental Parkinsonism, the kinetics of nigrostriatal degeneration and determined the critical thresholds associated to symptom appearance (Table1). Symptom appearance was thought to require a 70–80% loss of striatal terminals, a 50–60% loss of dopaminergic neurons, and a 70–90% DA deficiency (Bernheimer et al., 1973; Riederer and Wuketich, 1976). Depletion of striatal markers we report fits with these predictions, whereas the nigral threshold is lower than expected (Table 1). Fearnley and Lees (1991), however, determined a threshold of 31% DA cell loss in human PD, whereas German et al. (1988) reported a decrease of 46% in mildly symptomatic MPTP-treated Macaca fascicularis. The general gradient loss we observed begins by affecting the whole dorsal tier of the SNc and then its ventral tier where there remained few TH-IR neurons in a fully Parkinsonian state (Fig. 1C), in accordance with previous reports (German et al., 1988, 1996).
Table 1.
Thresholds for symptom appearance at 13.2 d and level of degeneration in fully Parkinsonian animals at D25 (% of D0 values ± SD)
| D13.2 symptom appearance | D25 full syndrome | |||||
|---|---|---|---|---|---|---|
| % of D0 | % of decrease | ± | % of D0 | % of decrease | ± | |
| Nissl-stained cells (SNc) | 75.2 | 24.8 | 6.3 | 34.7 | 65.3 | 4.3 |
| TH-IR neurons (SNc) | 56.8 | 43.2 | 6.2 | 14.2 | 85.8 | 4.6 |
| DAT binding (putamen) | 19.7 | 80.3 | 4.9 | 3.4 | 96.6 | 1.1 |
| DA content (putamen) | 18.2 | 81.8 | 5.6 | 2.1 | 97.9 | 0.9 |
Temporospatial lesion progression and nature of the initial pathological trigger
Acute administration of high doses of MPTP produces uniform striatal dopaminergic denervation both in monkey (Perez-Otano et al., 1994) and human (Snow et al., 2000) as it occurs in the present study. Some studies have demonstrated that either a single low-dose or chronic low-dose regimen of MPTP intoxication produces a greater depletion of dopaminergic markers in the putamen than in the caudate nucleus (Irwin et al., 1990; Moratalla et al., 1992), a pattern similar to that found in PD (Kish et al., 1988; Brooks et al., 1990). Damier et al. (1999b)proposed that the temporospatial lesion progression reflects differences in pathogenesis either of the MPTP-induced Parkinsonism or of PD. They identified compartmental subdivisions within the SNc (Damier et al., 1999a), each of them being differentially affected by progression of the disease (Hirsch et al., 1988; Damier et al., 1999b). Based on evidences suggesting a within-SNc origin for pathological process (Hirsch, 1999), they hypothesized that different localities would have different projection zones leading to a gradient in DA depletion with a higher loss in dorsal and caudal parts of the putamen than in the caudate nucleus. Because the active metabolite of MPTP is taken up by DAT (Gainetdinov et al., 1997), a within-striatum trigger would lead to a more uniform striatal denervation. Thus, uniformity of lesion could reflect the fundamental difference between the human disease and its closest animal model, i.e., the nature of the initial pathological trigger.
Increase of DA metabolism does not compensate in the early stages
Compensatory mechanisms are thought to mask the existence of PD before appearance of clinical symptoms (Zigmond et al., 1990). Their role is the maintenance of functional striatal DA concentration (so-called dopaminergic homeostasis) through optimization of both DA synthesis by surviving DA neurons and use by postsynaptic neurons (Zigmond et al., 1990). An increase in the value of the ratio of DA metabolites to DA would reflect actions that residual nigrostriatal neurons undertake to maintain dopaminergic homeostasis (Zigmond et al., 1990). The increase in the DA metabolites–DA ratio observed in D25 animals, associated with an exponential decay in the relationship between this ratio and DA content, is in agreement with previous reports (DiPaolo et al., 1986; Elsworth et al., 2000). This study confirms that an increase in DA metabolism requires an extensive lesion (Bernheimer et al., 1973; Elsworth et al., 2000). Moreover, because the metabolites–DA ratio is significantly different to D0 animals (controls) only in the D25 animals (Fig. 3), an increase in DA metabolism would not constitute an effective compensatory mechanism in early stages. Even mildly symptomatic animals (D15 group) do not show this purported compensatory mechanism.
DAT downregulation would be a purported compensatory mechanism
The increased DA efflux observed in partially denervated animals is attributed to a decrease in the rate at which DA is removed from extracellular fluid by remaining terminals, rather than to an increased DA release (Stachowiak et al., 1987; Snyder et al., 1990). The demonstration that DAT mRNA per DA neuron decreases in PD further supported this “downregulation of DA uptake” (Uhl et al., 1994). Although few TH-IR neurons remain in D25 animals, DAT binding is almost absent in the striatum. The relationship we show here between the decreases in TH-IR neurons, nigral Nissl-stained cells, and striatal DAT binding suggests that DA terminals degenerate before the soma (Sundström and Samuelsson, 1997). Both these results support the hypothesis that a decrease in the number of DAT per remaining dopaminergic nerve ending would enhance levels of DA. However, levels of DAT binding and striatal DA content are linearly correlated, suggesting a direct relationship (Figs. 2, 3). If DAT downregulation would occur, the relation between DAT binding and DA content should be best represented by a logarithmic equation highlighting the earlier decrease of the DAT compared with DA content. Our data would be more consistent with the lack of DAT downregulation. Direct electrochemical measurement of DA overflow are required before giving a definitive ruling on this question (May et al., 1988; Garris et al., 1997).
D2 upregulation would occur as early compensatory mechanism
The increase in the responsiveness of either D1 or D2 receptors on striatal neurons has been suggested as developing once striatal DA loss exceeds 75–80% (Thornburg and Moore, 1975; Lee et al., 1978). Previous studies on D1 binding in Parkinsonism lack consensus, differing considerably between authors and/or experimental approaches (Lee et al., 1978; Buonamici et al., 1986;Marshall et al., 1989). However, in the human Parkinsonian striatum, no modification in D1 density has been reported (Lee et al., 1978; Bokobza et al., 1984). This would suggest that functional D1 supersensitivity might be mediated through an increase in the activity of the downstream transduction pathways rather than simple elevations in receptor number (Walaas et al., 1984).
On the other hand, postmortem studies of experimental or human Parkinsonian brains have demonstrated that postsynaptic supersensitivity occurs through an increase of D2binding, as reported here at D25 (Creese et al., 1977; Lee et al., 1978; Falardeau et al., 1988; Todd et al., 1996). This increase in DAR binding is considered as representing an increase in postsynaptic DARs (Jaber et al., 1996) because the D2 autoreceptors are less numerous than the postsynaptics (Levey et al., 1993). As with the DA metabolism upregulation, postsynaptic supersensitivity is not observed in D15 group. A greater depletion of DA than has previously been supposed might be required to provoke this postsynaptic compensatory mechanism. Upregulation of postsynaptic D2 receptors would thus not be responsible for delaying the appearance of Parkinsonian symptoms although DA depletion increases because it is only significant in hardly symptomatic animals.
Before the present study, our understanding of PD pathophysiology has been predominantly gained from studies comparing the normal and fully-Parkinsonian states (Zigmond and Stricker, 1989; DeLong, 1990). With the present approach, changes in D2 binding in response to progression of DA depletion showed two phases. An initial decrease in D2 binding was followed by an increase that reaches a level far above the control situation (Fig.4D). The early decrease in presymptomatic animals was surprising but is not without precedent. At the beginning of MPTP era, monkeys were rendered hemi-Parkinsonian, and side-to-side differences were measured. However, the intracarotid administration of MPTP did induce partial lesions of nontreated side. For example, we reported a decrease in D2-like binding in the nontreated, partially-denervated side of hemi-Parkinsonian macaques (Graham et al., 1990). We hypothesized that this loss of binding on nontreated side represented a loss of presynaptic D2 binding sites situated on the dopaminergic terminals (Graham et al., 1990). We can speculate that the observed reduction in D2binding results from reducing presynaptic DAR. As the lesion progresses beyond D12, DA homeostasis may break down, and D2receptor upregulation is initiated. This would account for first in an apparent normalization of binding (D15) and then an obvious supersensitivity (D25). Evolution of D2 DAR binding changes is not linearly correlated with the progression of striatal denervation, as revealed by the relationship with both DAT binding and DA content. D2 upregulation would start immediately after D12 (in the presymptomatic stage when DA depletion is of 58–60%) and not after extensive lesion.
Breakdown of striatal dopaminergic homeostasis and symptom appearance
Such beginning of postsynaptic adaptive mechanism would reflect the breakdown of striatal dopaminergic homeostasis. Until D12, the decrease in DA terminals would be passively compensated, mainly through a shift from wiring to volume transmission (Zoli and Fuxe, 1996; Bezard and Gross, 1998). When this compensation breaks down, a compensatory increase in D2 density would occur throughout the progression from first symptoms to a full Parkinsonism. We have previously published evidence of dissociation between PD appearance and striatal dopaminergic homeostasis breakdown (Bezard et al., 2001b,c). We thus suggest that D2 upregulation would also begin before the Parkinsonian signs appearance. This adaptive mechanism would constitute an acute response to striatal denervation in the MPTP monkey model because D2 binding has recently been shown to return to normal levels in lesioned primates kept for months after their intoxication (Todd et al., 1996; Decamp et al., 1999).
Conclusions
The classic experimental approach, in which normal situation is compared with a fully-lesioned situation, can be complemented by the use of dynamic models that come closer to modeling the evolution of the disease (Bezard and Gross, 1998). The present study demonstrates (1) Parkinsonian symptom appearance with low level of SNc lesion, (2) an early D2 DAR upregulation before the end of the presymptomatic period, and (3) provides evidence that argues against the concept that an increase in DA metabolism could act as efficient adaptive mechanisms early in the disease progress. Further in vivo follow-up (single photon emission computed tomography and/or positron emission tomography) of the kinetics of striatal denervation in this, and similar, experimental models is now needed with a view to develop early diagnosis tools (possibly presymptomatic) and potential symptomatic therapies that might enhance endogenous compensatory mechanisms.
Footnotes
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Société de Secours des Amis des Sciences (E.B.), the Medical Research Council (UK), and the Parkinson's Disease Society (UK) (J.M.B., A.R.C.). The University of Manchester, the Centre National de la Recherche Scientifique (CNRS), and the Institut Fédératif de Recherche en Neuroscience (INSERM number 8; CNRS number 13) also funded this work.
Correspondence should be addressed to E. Bezard at his present address: Basal Gang, Laboratoire de Neurophysiologie, Centre National de la Recherche Scientifique Unité Mixte de Recherche 5543, Université Victor Segalen, 146 rue Léo Saignat, 33076 Bordeaux Cedex, France. E-mail: erwan.bezard@umr5543.u-bordeaux2.fr.
REFERENCES
- 1.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 2.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- 3.Bezard E, Gross CE. Compensatory mechanisms in experimental and human Parkinsonism: towards a dynamic approach. Prog Neurobiol. 1998;55:93–116. doi: 10.1016/s0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 4.Bezard E, Boraud T, Bioulac B, Gross C. Compensatory effects of glutamatergic inputs to the substantia nigra pars compacta in experimental Parkinsonism. Neuroscience. 1997a;81:399–404. doi: 10.1016/s0306-4522(97)00226-1. [DOI] [PubMed] [Google Scholar]
- 5.Bezard E, Imbert C, Deloire X, Bioulac B, Gross C. A chronic MPTP model reproducing the slow evolution of Parkinson's disease: evolution of motor symptoms in the monkey. Brain Res. 1997b;766:107–112. doi: 10.1016/s0006-8993(97)00531-3. [DOI] [PubMed] [Google Scholar]
- 6.Bezard E, Boraud T, Bioulac B, Gross C. Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur J Neurosci. 1999;11:2167–2170. doi: 10.1046/j.1460-9568.1999.00627.x. [DOI] [PubMed] [Google Scholar]
- 7.Bezard E, Boraud T, Bioulac B, Gross C. Evolution of the multiunit activity of basal ganglia in the course of a dynamic experimental Parkinsonism. In: Graybiel A, editor. The basal ganglia VI. Kluwer Academic; Norwell, MA: 2000. [Google Scholar]
- 8.Bezard E, Boraud T, Chalon S, Brotchie JM, Guilloteau D, Gross CE. Pallidal border cells: an anatomical and electrophysiological study in the MPTP-treated monkey. Neuroscience. 2001a;103:119–125. doi: 10.1016/s0306-4522(00)00546-7. [DOI] [PubMed] [Google Scholar]
- 9.Bezard E, Crossman AR, Gross CE, Brotchie JM (2001b) Structures outside the basal ganglia may compensate for dopamine loss in the pre-symptomatic stages of Parkinson's disease. FASEB J 10.1096:fj.00-0637fje. [DOI] [PubMed]
- 10.Bezard E, Ravenscroft P, Gross CE, Crossman AR, Brotchie JM. Upregulation of striatal preproenkephalin gene expression occurs before the appearance of Parkinsonian signs in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Neurobiol Dis. 2001c;8:343–350. doi: 10.1006/nbdi.2000.0375. [DOI] [PubMed] [Google Scholar]
- 11.Bokobza B, Ruberg M, Scatton B, Javoy-Agid F, Agid Y. 3H-spiperone binding, dopamine and HVA concentrations in Parkinson's disease and supranuclear palsy. Eur J Pharmacol. 1984;99:167–175. doi: 10.1016/0014-2999(84)90238-3. [DOI] [PubMed] [Google Scholar]
- 12.Brooks DJ, Ibanez V, Sawle GV, Quinn N, Lees AJ, Mathias CJ, Bannister R, Marsden CD, Frackowiak RS. Differing patterns of striatal 18F-dopa uptake in Parkinson's disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol. 1990;28:547–555. doi: 10.1002/ana.410280412. [DOI] [PubMed] [Google Scholar]
- 13.Buonamici M, Caccia C, Carpentieri M, Pegrassi L. D1 receptor supersensitivity in the rat striatum after unilateral 6-hydroxydopamine lesions. Eur J Pharmacol. 1986;126:347–348. doi: 10.1016/0014-2999(86)90072-5. [DOI] [PubMed] [Google Scholar]
- 14.Chalon S, Emond P, Bodard S, Vilar MP, Thiercelin C, Besnard JC, Guilloteau D. Time course of changes in striatal dopamine transporters and D2 receptors with specific iodinated markers in a rat model of Parkinson's disease. Synapse. 1999;31:1–6. doi: 10.1002/(SICI)1098-2396(199902)31:2<134::AID-SYN6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 15.Creese I, Burt DR, Snyder SH. Dopamine receptor binding: enhancement accompanying lesion-induced behavioural supersensitivity. Science. 1977;197:596–598. doi: 10.1126/science.877576. [DOI] [PubMed] [Google Scholar]
- 16.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain - I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D-28K immunohistochemistry. Brain. 1999a;122:1421–1436. doi: 10.1093/brain/122.8.1421. [DOI] [PubMed] [Google Scholar]
- 17.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain - II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999b;122:1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 18.Decamp E, Wade T, Schneider JS. Differential regulation of striatal dopamine D-1 and D-2 receptors in acute and chronic Parkinsonian monkeys. Brain Res. 1999;847:134–138. doi: 10.1016/s0006-8993(99)02015-6. [DOI] [PubMed] [Google Scholar]
- 19.Delion S, Chalon S, Guilloteau D, Besnard JC, Durand G. alpha-Linolenic acid dietary deficiency alters age-related changes of dopaminergic and serotoninergic neurotransmission in the rat frontal cortex. J Neurochem. 1996;66:1582–1591. doi: 10.1046/j.1471-4159.1996.66041582.x. [DOI] [PubMed] [Google Scholar]
- 20.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 21.DiPaolo R, Bédard P, Daigle M, Boucher R. Long-term effects of MPTP on central and peripheral catecholamine and indoleamine concentrations in monkeys. Brain Res. 1986;379:286–293. doi: 10.1016/0006-8993(86)90782-1. [DOI] [PubMed] [Google Scholar]
- 22.Ehringer H, Hornykiewicz O. Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des extrapyramidalen Systems. Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- 23.Elsworth JD, Taylor JR, Sladek JR, Collier TJ, Redmond DE, Roth RH. Striatal dopaminergic correlates of stable Parkinsonism and degree of recovery in Old-World primates one year after MPTP treatment. Neuroscience. 2000;95:399–408. doi: 10.1016/s0306-4522(99)00437-6. [DOI] [PubMed] [Google Scholar]
- 24.Emond P, Garreau L, Chalon S, Boazi M, Caillet M, Bricard J, Frangin Y, Mauclaire L, Besnard JC, Guilloteau D. Synthesis and ligand binding of nortropane derivatives: N-substituted 2β-carboxymethoxy-3β-(4′-iodophenyl) nortropane and N-(3-iodoprop-2E-enyl)-2β-carboxymethoxy-3β-(3′,4′-disubstituted phenyl) nortropane: new high-affinity and selective compounds for the dopamine transporter. J Med Chem. 1997;40:1366–1372. doi: 10.1021/jm960795d. [DOI] [PubMed] [Google Scholar]
- 25.Falardeau P, Bedard PJ, Di Paolo T. Relation between brain dopamine loss and D2 dopamine receptor density in MPTP monkeys. Neurosci Lett. 1988;86:225–229. doi: 10.1016/0304-3940(88)90575-7. [DOI] [PubMed] [Google Scholar]
- 26.Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 27.Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- 28.Garris PA, Walker QD, Wightman RM. Dopamine release and uptake rates both decrease in the partially denervated striatum in proportion to the loss of dopamine terminals. Brain Res. 1997;753:225–234. doi: 10.1016/s0006-8993(97)00003-6. [DOI] [PubMed] [Google Scholar]
- 29.German DC, Dubach M, Askari S, Speciale SG, Bowden DM. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonian syndrome in Macaca fascicularis: which midbrain dopaminergic neurons are lost? Neuroscience. 1988;24:161–174. doi: 10.1016/0306-4522(88)90320-x. [DOI] [PubMed] [Google Scholar]
- 30.German DC, Nelson EL, Liang C-L, Speciale SG, Sinton CM, Sonsalla PK. The neurotoxin MPTP causes degeneration of specific nucleus A8, A9, and A10 dopaminergic neurons in the mouse. Neurodegeneration. 1996;5:299–312. doi: 10.1006/neur.1996.0041. [DOI] [PubMed] [Google Scholar]
- 31.Graham WC, Clarke CE, Boyce S, Sambrook MA, Crossman AR, Woodruff GN. Autoradiographic studies in animal models of hemi-Parkinsonism reveal dopamine D2 but not D1 receptor supersensitivity. II. Unilateral intra- carotid infusion of MPTP in the monkey (Macaca fascicularis). Brain Res. 1990;514:103–110. doi: 10.1016/0006-8993(90)90440-m. [DOI] [PubMed] [Google Scholar]
- 32.Guilloteau D, Emond P, Baulieu JL, Garreau L, Frangin Y, Pourcelot L, Mauclaire L, Besnard J-C, Chalon S. Exploration of the dopamine transporter: in vitro and in vivo characterization of a high-affinity and high-specificity iodinated tropane derivative (E)-N-(3-iodoprop-2-enyl)-2b-carbomethoxy-3b-(4′-methylphenyl) nortropane (PE2I). Nucl Med Biol. 1998;25:331–337. doi: 10.1016/s0969-8051(97)00224-2. [DOI] [PubMed] [Google Scholar]
- 33.Hassler R. Zur Pathologie der Paralysis Agitans und des postenzephalitischen Parkinsonismus. J Psychol Neurol. 1938;48:387–476. [Google Scholar]
- 34.Henry B, Crossman AR, Brotchie JM. Effect of repeated l-DOPA, bromocriptine, or lisuride administration on preproenkephalin-A and preproenkephalin-B mRNA levels in the striatum of the 6-hydroxydopamine-lesioned rat. Exp Neurol. 1999;155:204–220. doi: 10.1006/exnr.1998.6996. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch EC. Mechanism and consequences of nerve cell death in Parkinson's disease. J Neural Transm [Suppl] 1999;56:127–137. doi: 10.1007/978-3-7091-6360-3_7. [DOI] [PubMed] [Google Scholar]
- 36.Hirsch EC, Graybiel AM, Agid Y. Melanized dopaminergic neurons are differentially affected in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 37.Hoehn HM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–442. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- 38.Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. In: Yahr M, Bergmann KJ, editors. Parkinson's disease. Raven; New York: 1987. pp. 19–34. [PubMed] [Google Scholar]
- 39.Imbert C, Bezard E, Guitraud S, Boraud T, Gross CE. Comparison between eight clinical rating scales used for the assessment of MPTP-induced Parkinsonism in the macaque monkey. J Neurosci Methods. 2000;96:71–76. doi: 10.1016/s0165-0270(99)00184-3. [DOI] [PubMed] [Google Scholar]
- 40.Irwin I, DeLanney LE, Forno LS, Finnegan KT, DiMonte DA, Langston JW. The evolution of nigrostriatal neurochemical changes in the MPTP-treated squirrel monkey. Brain Res. 1990;531:242–252. doi: 10.1016/0006-8993(90)90780-f. [DOI] [PubMed] [Google Scholar]
- 41.Jaber M, Robinson SW, Missale C, Caron MG. Dopamine receptors and brain function. Neuropharmacology. 1996;35:1503–1519. doi: 10.1016/s0028-3908(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 42.Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- 43.Koller WC, Langston JW, Hubble JP, Irwin I, Zack M, Golbe L, Forno L, Ellenberg J, Kurland L, Ruttenberger AJ. Does a long preclinical period occur in Parkinson's disease? Neurology. 1991;41:8–13. [PubMed] [Google Scholar]
- 44.Lee T, Seeman P, Rajput A, Farley IJ, Hornykiewicz O. Receptor basis for dopaminergic supersensitivity in Parkinson's disease. Nature. 1978;273:150–151. doi: 10.1038/273059a0. [DOI] [PubMed] [Google Scholar]
- 45.Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci USA. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall JF, Navarrete R, Joyce JN. Decreased striatal D1 binding density following mesotelencephalic 6-hydroxydopamine injections: an autoradiographic analysis. Brain Res. 1989;493:247–257. doi: 10.1016/0006-8993(89)91160-8. [DOI] [PubMed] [Google Scholar]
- 47.May LJ, Kuhr WG, Wightman RM. Differentiation of dopamine overflow and uptake processes in the extracellular fluid of the rat caudate nucleus with fast-scan in vivo voltammetry. J Neurochem. 1988;51:1060–1069. doi: 10.1111/j.1471-4159.1988.tb03069.x. [DOI] [PubMed] [Google Scholar]
- 48.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in Parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 49.Miller RG. Simultaneous statistical inference. Springer; New York: 1981. [Google Scholar]
- 50.Moratalla R, Quinn B, DeLanney LE, Irwin I, Langston JW, Graybiel AM. Differential vulnerability of primate caudate-putamen and striosome-matrix dopamine systems to the neurotoxic effects of 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA. 1992;89:3859–3863. doi: 10.1073/pnas.89.9.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morissette M, Grondin R, Goulet M, Bedard PJ, Di Paolo T. Differential regulation of striatal preproenkephalin and preprotachykinin mRNA levels in MPTP-lesioned monkeys chronically treated with dopamine D-1 or D-2 receptor agonists. J Neurochem. 1999;72:682–692. doi: 10.1046/j.1471-4159.1999.0720682.x. [DOI] [PubMed] [Google Scholar]
- 52.Morrish PK, Sawle GV, Brooks DJ. An [18F]dopa-PET and clinical study of the rate of progression in Parkinson's disease. Brain. 1996;119:585–591. doi: 10.1093/brain/119.2.585. [DOI] [PubMed] [Google Scholar]
- 53.Perez-Otano I, Oset C, Luquin MR, Herrero MT, Obeso JA, Del Rio J. MPTP-induced Parkinsonism in primates: pattern of striatal dopamine loss following acute and chronic administration. Neurosci Lett. 1994;175:121–125. doi: 10.1016/0304-3940(94)91094-4. [DOI] [PubMed] [Google Scholar]
- 54.Riederer P, Wuketich S. Time course of nigrostriatal degeneration in Parkinson's disease. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- 55.Schneider JS, Decamp E, Wade T. Striatal preproenkephalin gene expression is upregulated in acute but not chronic Parkinsonian monkeys: Implications for the contribution of the indirect striatopallidal circuit to Parkinsonian symptomatology. J Neurosci. 1999;19:6643–6649. doi: 10.1523/JNEUROSCI.19-15-06643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snow BJ, Vingerhoets FJG, Langston JW, Tetrud JW, Sossi V, Calne DB. Pattern of dopaminergic loss in the striatum of humans with MPTP induced Parkinsonism. J Neurol Neurosurg Psychiatry. 2000;68:313–316. doi: 10.1136/jnnp.68.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Snyder GL, Keller RW, Zigmond MJ. Dopamine efflux from striatal slices after intracerebral 6-hydroxydopamine: evidence for compensatory hyperactivity of residual terminals. J Pharmacol Exp Ther. 1990;253:867–876. [PubMed] [Google Scholar]
- 58.Stachowiak MK, Keller RW, Stricker EM, Zigmond MJ. Increased dopamine efflux from striatal slices during development and after nigrostriatal bundle damage. J Neurosci. 1987;7:1648–1654. doi: 10.1523/JNEUROSCI.07-06-01648.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundström E, Samuelsson E-B. Comparison of key steps in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in rodents. Pharmacol Toxicol. 1997;81:226–231. doi: 10.1111/j.1600-0773.1997.tb00051.x. [DOI] [PubMed] [Google Scholar]
- 60.Szabo J, Cowan WM. A stereotaxic atlas of the brain of the cynomolgus monkey (Macaca fascicularis). J Comp Neurol. 1984;222:265–300. doi: 10.1002/cne.902220208. [DOI] [PubMed] [Google Scholar]
- 61.Taylor JR, Elsworth JD, Roth RH, Sladek JR, Redmond DE. Behavioral effects of MPTP administration in the Vervet monkey, a primate model of Parkinson's disease. In: Woodruff ML, Nonneman A, editors. Toxin-induced models of neurological disorders. Plenum; New York: 1994. pp. 139–151. [Google Scholar]
- 62.Thornburg JE, Moore KE. Supersensitivity to dopamine agonists following unilateral, 6-hydroxydopamine-induced striatal lesions in mice. J Pharmacol Exp Ther. 1975;192:42–49. [PubMed] [Google Scholar]
- 63.Todd RD, Carl J, Harmon S, O'Malley KL, Perlmutter JS. Dynamic changes in striatal dopamine D2 and D3 receptor protein and mRNA in response to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) denervation in baboons. J Neurosci. 1996;16:7776–7782. doi: 10.1523/JNEUROSCI.16-23-07776.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uhl GR, Walther D, Mash D, Faucheux B, Javoy-Agid F. Dopamine transporter messenger RNA in Parkinson's disease and control substantia nigra neurons. Ann Neurol. 1994;35:494–498. doi: 10.1002/ana.410350421. [DOI] [PubMed] [Google Scholar]
- 65.Vingerhoets FJG, Snow BJ, Lee CS, Schulzer M, Mak E, Calne DB. Longitudinal fluorodopa positron emission tomographic studies of the evolution of idiopathic Parkinsonism. Ann Neurol. 1994;36:759–764. doi: 10.1002/ana.410360512. [DOI] [PubMed] [Google Scholar]
- 66.Walaas SI, Aswaad SW, Greengard P. A dopamine-and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. Nature. 1984;301:69–71. doi: 10.1038/301069a0. [DOI] [PubMed] [Google Scholar]
- 67.Zigmond MJ, Stricker EM. Animal models of Parkinsonism using selective neurotoxins: clinical and basic implications. Int Rev Neurobiol. 1989;31:1–79. doi: 10.1016/s0074-7742(08)60277-9. [DOI] [PubMed] [Google Scholar]
- 68.Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- 69.Zoli M, Fuxe LF. Wiring and volume transmission in the central nervous system: the concept of closed and open synapses. Prog Neurobiol. 1996;49:363–380. doi: 10.1016/0301-0082(96)00020-2. [DOI] [PubMed] [Google Scholar]