

Abstract
Previous studies have focused considerable attention on the effects of estrogen on excitatory synaptic input to hippocampal CA1 pyramidal cells. Estrogen increases the density of dendritic spines and synapses on CA1 pyramidal cells and increases the sensitivity of these cells to excitatory synaptic input. Little is known, however, about the effects of estrogen on inhibitory synaptic input to CA1 pyramidal cells. We have used immunohistochemistry for glutamic acid decarboxylase and whole-cell voltage-clamp recording of IPSCs and EPSCs at multiple time points after estrogen treatment to (1) investigate estrogen regulation of synaptic inhibition in CA1 and (2) evaluate how estrogen affects the interaction between inhibitory and excitatory input to CA1 pyramidal cells. We find that estrogen transiently suppresses GABAA-mediated inhibition of CA1 pyramidal cells at a time point before changes in excitatory input to these cells occur. This finding is consistent with the suggestion that transient disinhibition of CA1 pyramidal cells is involved in estrogen-induced dendritic spine formation. We have also found that at a later time after estrogen, inhibition of CA1 pyramidal cells recovers in parallel with enhancement of NMDA-mediated excitatory input. The concurrent enhancement of GABAA and NMDA-mediated input to CA1 pyramidal cells restores a balance of excitatory and inhibitory input to these cells and increases the potential dynamic range of CA1 pyramidal cell responses to synaptic input.
Keywords: GABA, glutamic acid decarboxylase, IPSCs, AMPA, NMDA, seizures
Previous studies have shown that estrogen induces structural and functional changes in excitatory input to hippocampal CA1 pyramidal cells in adult female rats. Estrogen increases the density of dendritic spines (Gould et al., 1990; Woolley and McEwen, 1993; Woolley et al., 1997; McEwen et al., 1999) and spine synapses (Woolley and McEwen, 1992; Woolley et al., 1996; Leranth et al., 2000) on CA1 pyramidal cells. These structural changes in excitatory input are paralleled by increases in synaptic excitability of CA1 pyramidal cells, particularly synaptic input mediated by the NMDA subtype of the glutamate receptor (Weiland, 1992; Wong and Moss, 1992; Gazzaley et al., 1996; Woolley et al., 1997).
Although considerable attention has been focused on the effects of estrogen on excitatory synaptic input to CA1 pyramidal cells, little is known about the effects of estrogen on inhibitory input to these cells. Interestingly, studies of the effects of estrogen on dendritic spine density on cultured hippocampal neurons have shown that estrogen increases spine density via an activity-dependent mechanism that requires transient suppression of GABAergic inhibitory synaptic transmission (Murphy et al., 1998). Several parallels between estrogen regulation of spine density in vitro and in vivosuggest that similar mechanisms may be involved. Both effects require several days of estrogen exposure (Woolley and McEwen, 1993; Murphy and Segal, 1996), and both are blocked by NMDA receptor antagonists (Woolley and McEwen, 1994; Murphy and Segal, 1996) and antagonists of classical estrogen receptors (Murphy and Segal, 1996; McEwen et al., 1999). The possibility that changes in GABAergic neurotransmission are involved in estrogen regulation of spine density in the hippocampusin vivo is supported by the observation that most hippocampal neurons that express classical estrogen receptors are GABAergic interneurons [in the dorsal hippocampus, where spine changes occur (Weiland et al., 1997; Hart and Woolley, 2000)].
The goals of the current study were to (1) determine whether estrogen regulates GABAergic synaptic transmission in vivo as it doesin vitro and (2) determine the relationship between inhibitory and excitatory synaptic transmission in the CA1 region at two time points within the estrogen treatment protocol known to regulate dendritic spines. One time point was chosen to be before estrogen-induced spine density changes occur, and one time point was after estrogen-induced differences in spine density are established [based on previous studies (Woolley and McEwen, 1993)]. We used immunohistochemistry for glutamic acid decarboxylase (GAD), the rate-limiting enzyme in GABA synthesis, and whole-cell voltage-clamp recording of IPSCs and EPSCs in CA1 pyramidal cells to show the following. (1) Estrogen transiently decreases GABAergic inhibition of these cells. This disinhibition results in enhancement of excitatory synaptic input at a time before spine changes occur. (2) At a later time, when spine density is increased in estrogen-treated animals, GABAergic inhibition is also increased. The counteracting effects of estrogen on inhibitory and excitatory input at the later time point restore a balance of excitatory and inhibitory input to CA1 pyramidal cells and also potentially increase the dynamic range of the responses of these cells to synaptic input.
MATERIALS AND METHODS
Animals and hormone treatment. Adult female Sprague Dawley rats (180–220 gm) were housed on a 12 hr light/dark cycle with food and water available ad libitum. All rats were bilaterally ovariectomized (OVX) under methoxyflurane anesthesia using aseptic procedures. On the third only or third and fourth days after surgery, rats were injected subcutaneously either with 10 μg of 17β-estradiol benzoate in 100 μl of sesame oil (E) or with 100 μl of sesame oil alone (O) and allowed to survive for various times after injection.
The densities of GAD65- and GAD67-immunoreactive cells were quantified at the following time points (Fig.1A) (n= 6 for all groups): 3 d OVX (3DO; before any injections), 2 or 24 hr after the first O injection (21O and 241O) or E injection (21E and 241E), 48 hr after the second O injection (482O), and 2 and 48 hr after the second E injection (22E and 482E). Surgeries and treatment times were staggered so that all animals were perfused together. IPSCs and EPSCs in CA1 pyramidal cells were recorded in slices from animals killed at the 3DO, 241O, 241E, 482O, and 482E time points (Fig.1A).
Fig. 1.
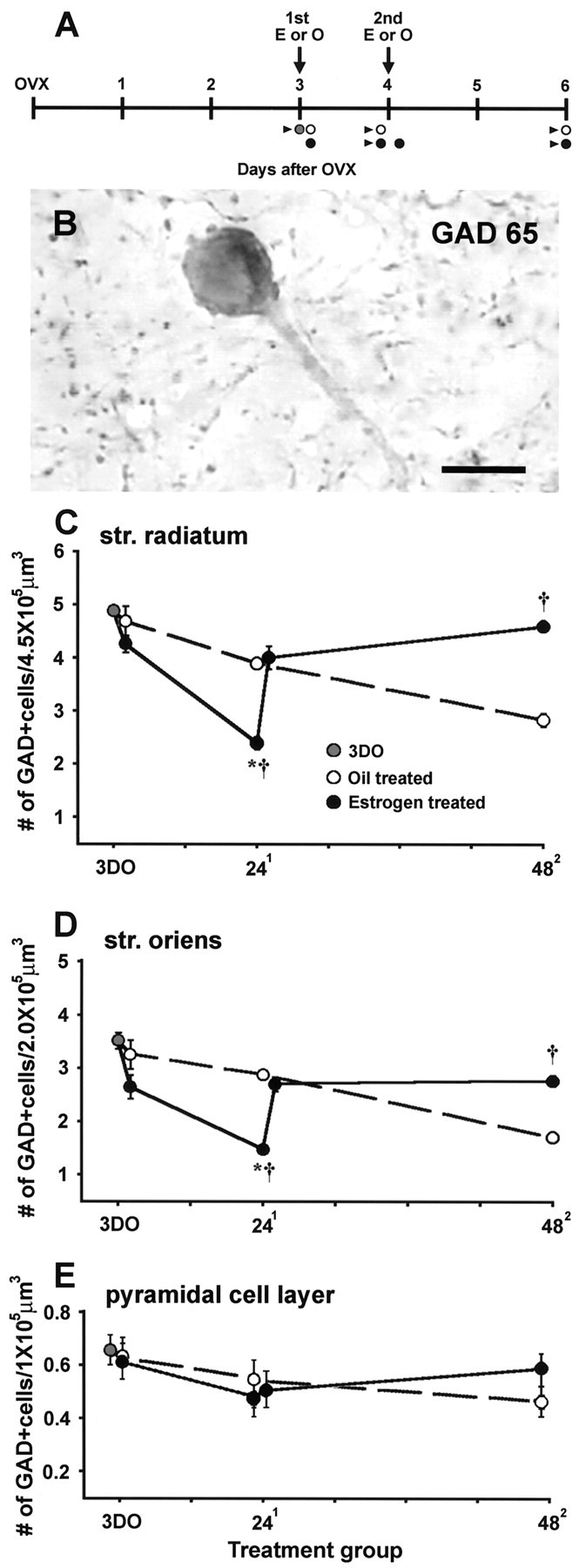
Estrogen regulates GAD65 immunoreactivity in CA1.A, Treatment schedule used in all studies. All rats were OVX and received O (white circles) or E (black circles) injections on the third only or third and fourth days after OVX. For immunohistochemistry, six animals were perfused at each of the following time points: 3 d OVX (before any injections; gray circle), 2 or 24 hr after the first O or E injection, 48 hr after the second O injection, and 2 or 48 hr after the second E injection. Electrophysiological analyses were performed on a variable number of animals at the following time points (arrowheads): 3DO, 241O, 241E, 482O, and 482E. B, Representative GAD65-immunoreactive cell in the CA1 str. radiatum. Scale bar, 10 μm. C–E, Time course of changes in the density of GAD65-immunoreactive cells in the CA1 str. radiatum (C), str. oriens (D), and pyramidal cell layer (E) in O- and E-treated animals, compared with 3DO animals. In str. radiatum and str. oriens, GAD65 immunoreactivity declines gradually in O-treated controls, but in E-treated animals, GAD65 decreases sharply at 241E and then recovers by 482E. Asterisksindicate a significant difference from 3DO, and crossesindicate a significant difference from the O-treated control at the same time point (p < 0.05). In the pyramidal cell layer, GAD65 immunoreactivity follows a similar pattern, but differences reflect only a statistical trend (p < 0.1). Note that y-axes in C–E are different.
Immunohistochemistry. All rats were deeply anesthetized with Nembutal (80 mg/kg) and transcardially perfused with cold 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. After perfusion, brains were removed, blocked, and post-fixed overnight in the same solution at 4°C. The brains were rinsed with 0.1 m PB, cryoprotected in 30% sucrose, and coronally sectioned through the dorsal hippocampus (50 μm) using a freezing microtome. Sections were stained immunohistochemically using the avidin–biotin–peroxidase method described below. Sections from each brain were processed for GAD65 (monoclonal to rat GAD 65 kDa isoform; Chemicon, Temecula, CA) or GAD67 (polyclonal to rat GAD 67 kDa isoform; Chemicon).
Freshly cut sections were rinsed in PB and incubated in 1% sodium borohydride for 10 min, rinsed, and then incubated in H2O2 (0.5% for 30 min, 1.0% for 1 hr, and 0.5% for 30 min). After rinsing in Tris buffer (TB), pH 7.4, sections were incubated for 1 hr in a nonspecific blocking solution containing 5% normal serum, 3% bovine serum albumin (BSA), and 0.3% dimethylsulfoxide (DMSO) in 0.5 mTris-buffered saline (TBS). Sections were then rinsed and incubated in the primary antibody or antisera (2 μg/ml for GAD65 or 1:10,000 for GAD67) solution containing 1% normal serum, 3% BSA, and 0.3% DMSO for 48 hr at 4°C in 0.5 m TBS. Some sections from each brain were incubated without the primary to determine nonspecific secondary antibody staining.
After primary incubation, sections were rinsed thoroughly with 0.1m TBS and incubated in biotinylated secondary antibody (1:400; anti-mouse IgG for GAD65 or anti-rabbit IgG for GAD67) solution containing 1% normal serum, 2% BSA, and 0.3% DMSO in 0.1m TBS for 3 hr. The sections were rinsed with 0.1m TBS and incubated in avidin–biotin HRP complex (1:500; Vector Elite Kit) for 3 hr. Next, the sections were rinsed and preincubated in TB, pH 7.6, containing 0.025% 3,3′-diaminobenzidine for 20 min followed by addition of 0.01% H2O2 for an additional 20 min. Finally, the sections were rinsed, mounted onto subbed slides, dried, dehydrated in graded ethanols, cleared in xylene, and coverslipped under Permount.
Quantification of GAD65- and GAD67-labeled cells. Tissue sections for quantitative analysis of GAD-immunoreactive cells were coded, and the code was not broken until analysis was complete. Unbiased estimates of the density of GAD65- and GAD67-labeled cells were obtained using the optical disector principle and random systematic sampling (Gundersen et al., 1988). For both left and right sides of each brain, 10 sectors (184 × 246 μm) were randomly chosen for each layer of the CA1 region. Four nonadjacent tissue sections were analyzed for each brain. The starting point for cell counting was set at 5 μm below the surface of the section and stepped down five times at 2 μm per step for a total of 10 μm. Labeled cells that were sharply in focus and inside the counting frame or that intersected the upper horizontal and right vertical were counted at each step; cells that intersected the left vertical and lower horizontal of the counting frame were not counted. Labeled cells were visualized with a 50× oil-immersion lens on an Olympus BX60 microscope (Olympus Optical, Tokyo, Japan) with a Dage DC330 camera (Dage MTI, Inc., Michigan City, IN) and Image-Pro Plus software (Media-Cybernetics, Silver Spring, MD). The density of labeled cells was calculated by dividing the sum of all cells counted by the volume of all disectors counted. Means were calculated for each animal, and the data were analyzed statistically using ANOVA with Tukey post hoc comparisons.
Slice preparation and maintenance. On each recording day, two animals (one control and one estrogen-treated) were coded, and the code was not broken until all data analysis for those animals was complete. All rats used for electrophysiological recordings were anesthetized with Nembutal (80 mg/kg) and transcardially perfused with ice-cold oxygenated (95% O2/5% CO2) artificial CSF (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 25 dextrose, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, and 2 CaCl2, pH 7.5. The brain was quickly removed and cooled in ice-cold oxygenated ACSF. By use of a vibroslicer, 300-μm-thick transverse hippocampal slices were cut into a bath of ice-cold oxygenated ACSF. Slices were transferred to a holding chamber where they remained submerged in oxygenated ACSF at 35°C for 30 min. The slices then remained in oxygenated ACSF at room temperature (∼24°C) until used for recording.
Whole-cell voltage-clamp recording. Slices were transferred to a recording chamber mounted on a Zeiss Axioskop (Oberkochen, Germany) where they were submerged in oxygenated ACSF maintained at 35 ± 1°C. Neurons in the slice were visualized using infrared differential interference videomicroscopy (Hamamatsu, Hamamatsu City, Japan). Somatic whole-cell voltage-clamp recordings were obtained from CA1 pyramidal neurons using patch electrodes made from thick-walled borosilicate glass (Garner Glass, Claremont, CA) pulled on a P-97 micropipette puller (Sutter Instrument Company, Novato, CA) with an open tip resistance of 3–5 MΩ in ACSF. Series resistance (average, 12 ± 4.1 MΩ) was compensated (70%), and a recording was terminated if a significant increase occurred. Data were collected with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) and acquired and analyzed using Igor Pro software (WaveMetrics, Inc., Lake Oswego, OR).
Synaptically evoked IPSCs and miniature IPSCs (mIPSCs) were recorded using a pipette solution containing (in mm): 140 CsCl, 2 MgCl2, 10 HEPES, 2 EGTA, 2 MgATP, 0.3 NaGTP, and 10 Na2-creatinine phosphate, with 0.10% biocytin, pH 7.2–7.3. Synaptically evoked IPSCs were recorded in ACSF containing 5 μm kynurenic acid to block glutamate receptor-mediated synaptic transmission. A stimulating electrode (patch pipette with chlorided silver wire) was placed in the CA1 pyramidal cell layer 50–100 μm away from the recording electrode. Stimulus–response curves were generated at a holding potential of −70 mV by varying stimulus intensity from the minimum current necessary to evoke a postsynaptic response to the current that produced a maximal response; stimuli were delivered at 0.1 Hz. Synaptically evoked IPSCs were all blocked by the GABAA antagonists 10 μm bicuculline or 2 μm SR-95531 (SR). The stimulus–response protocol was repeated at least three times per cell, and the average of peak IPSC amplitude at each stimulus intensity was calculated for each cell. IPSC rise and decay kinetics was obtained from maximal currents, and data were averaged per cell. Stimulus–response curves were analyzed statistically with repeated measures ANOVA. IPSC rise time and time to 50% decay for cells from O and E animals were analyzed statistically with Student's ttest.
Miniature IPSCs were recorded from the same cells as synaptically evoked IPSCs after addition of 5 μm TTX to the bath. Data were collected from the first 565 mIPSCs per cell. Like synaptically evoked IPSCs, mIPSCs were blocked by 10 μm bicuculline or 2 μm SR. The frequency of mIPSCs and mean mIPSC amplitude and decay time were analyzed statistically using ANOVA followed by Tukey post hoc comparisons. Miniature IPSC amplitude and decay time histograms were analyzed statistically using the Kolmogorov–Smirnov test. Statistical association between mIPSC amplitude and decay time was analyzed by linear regression.
EPSCs and mixed postsynaptic currents evoked by str. radiatum stimulation were recorded using a pipette solution containing (in mm): 130 Cs-gluconate, 2 CsCl, 2MgCl2, 20 HEPES,10 Na2-creatinine phosphate, 2 EGTA, 2 MgATP, and 0.3 NaGTP, with 0.10% biocytin, pH 7.3. Before recordings began, a cut was made between the CA3 and CA1 regions to prevent retrograde activation of CA3 pyramidal cells. A stimulating electrode was placed in the str. radiatum 150–250 μm laterally from the soma of the recorded cell, midway between the pyramidal cell layer and the str. lacunosum. At least three sets of minimum-to-maximum stimulus–response curves were generated for each cell from mixed postsynaptic currents as well as from isolated AMPA- and NMDA-mediated EPSCs. AMPA-mediated currents were blocked with 30 μm CNQX, and NMDA-mediated currents were blocked with 10 μm APV. Means for total charge transfer were calculated at each stimulus intensity for each cell and were analyzed statistically using repeated measures ANOVA. Decay times were fit with biexponentials and also were analyzed by comparing the mean time to 50% decay of currents evoked at the stimulus intensity that produced a half-maximal postsynaptic response using Student's t test.
RESULTS
Time course of changes in GAD65 and GAD67 immunoreactivity after estrogen
Quantitative analysis of GAD65- and GAD67-immunoreactive cells revealed a transient estrogen-induced decrease in GAD65 immunoreactivity in both the str. radiatum and str. oriens of CA1. Similar changes were observed in the pyramidal cell layer, albeit to a much lesser extent. In control animals, the density of GAD65-labeled cells (Fig. 1B) in both dendritic layers decreased gradually from the baseline at 3DO to the end of the treatment period at 482O (Fig. 1C,D,dashed lines) (p < 0.05). In estrogen-treated animals, the density of GAD65-labeled cells was decreased slightly at 21, substantially decreased by the 241 time point (Fig.1C,D, solid lines) (p < 0.05 from 3DO and 241O), but recovered at the 22 and 482time points so that GAD65 labeling at 482E was significantly greater than that at 482O (p < 0.05) and not different from that at 3DO (p > 0.1). Counts of GAD65-immunoreactive cells in the pyramidal cell layer also showed a similar pattern of changes, but these differences represented only a statistical trend (Fig. 1E) (p < 0.1).
In contrast to GAD65 labeling, estrogen had no effect on the density of GAD67-labeled cells at any time point after OVX or estrogen treatment (p > 0.1; data not shown). Because GAD65 and GAD67 are generally coexpressed in the same neurons (Houser and Esclapez, 1994; Sloviter et al., 1996; Stone et al., 1999), the lack of effect on GAD67 suggests that GAD65 immunoreactivity is suppressed in the same cells in which GAD67 is unchanged.
Changes in GAD65 immunoreactivity could result from estrogen-induced differences in GAD65 expression and/or some other change in GAD65 that alters its antigenicity. Our subsequent analyses of synaptic currents in CA1 pyramidal cells suggest the former, i.e., changes in expression, because differences in GAD65 immunoreactivity are paralleled by differences in inhibitory synaptic function (see below). Although the reversal of GAD suppression between 241E and 22E is quite rapid, it is possible that even this change represents an increased GAD expression because estrogen is known to very rapidly activate neuronal protein synthetic machinery (Jones et al., 1988, 1990) and GAD expression has been shown previously to be rapidly modulated by other manipulations (Bowers et al., 1998; Szabo et al., 2000; Churchill et al. 2001).
Synaptic inhibition at the 241 time point
Synaptically evoked IPSCs
Estrogen-induced differences in GAD65 immunoreactivity were paralleled by functional differences in GABAAinhibition of CA1 pyramidal cells. Isolated IPSCs were recorded using a CsCl-based internal solution so that the GABAA-mediated IPSC was an inward current. Series of synaptically evoked IPSCs in cells from 3DO, 241O (Fig.2A), and 241E (Fig. 2B) animals were used to generate stimulus–response curves (Fig. 2C). Comparison of these stimulus–response curves showed that evoked IPSC amplitude was significantly reduced in cells from 241E animals compared with those from 241O and 3DO animals (p < 0.05).
Fig. 2.
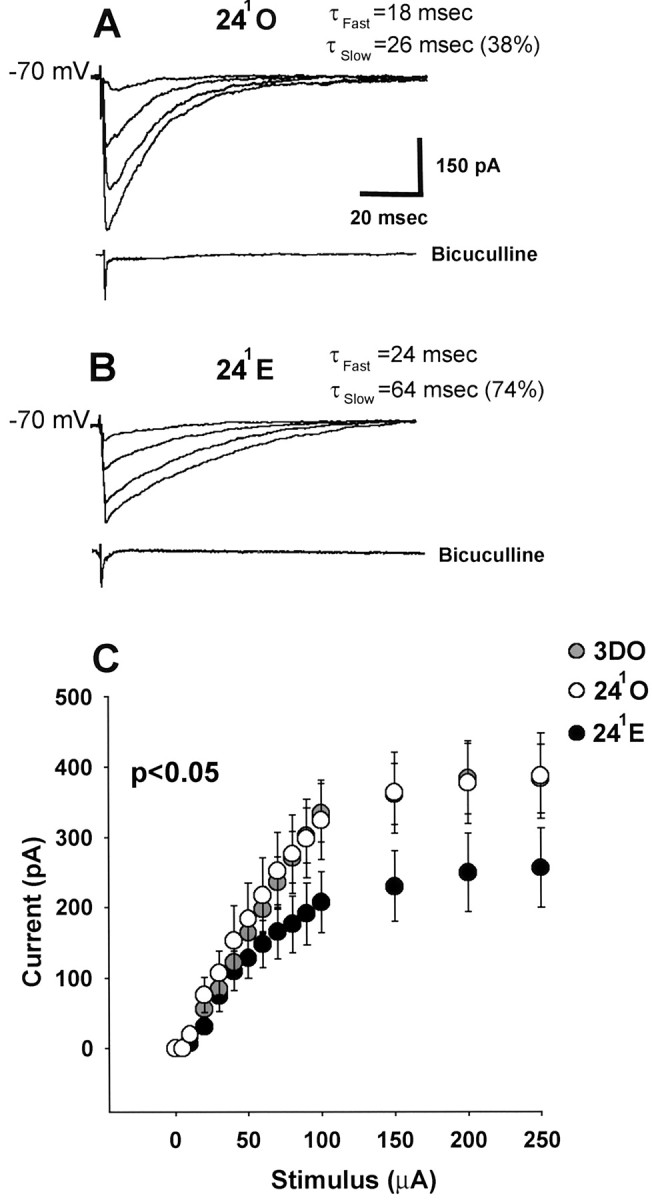
Estrogen reduces amplitude and increases decay time of synaptically evoked IPSCs in CA1 pyramidal cells at the 241 time point. Recordings were made with a CsCl internal solution. A, Representative individualtraces of IPSCs evoked in a 241O cell using 50, 100, 150, and 250 μA stimulating currents.B, Representative individual traces of IPSCs evoked in a 241E cell using the same stimulus intensities as in A. Evoked currents are blocked by bicuculline. τdecay fast and τdecay slowvalues in A and B apply specifically to the cells shown. C, Averaged stimulus–response curves for 3DO (gray circles; n = 12 cells from 6 animals), 241O (white circles; n = 18 cells from 10 animals), and 241E (black circles;n = 22 cells from 10 animals) groups. Peak IPSC amplitudes are significantly reduced in 241E cells compared with 241O and 3DO cells (p < 0.05).
In addition to decreasing evoked IPSC amplitude, estrogen also prolonged the decay of evoked IPSCs at the 241 time point (Fig. 2Avs B). Synaptically evoked IPSCs were best described by two time constants, τdecay fast and τdecay slow (Pearce, 1993), each of which was greater in the 241E than in the 241O or 3DO groups (Table1; τdecay fast,p < 0.05; τdecay slow,p < 0.01). Additionally, at 241E, τdecay slowaccounted for an average of 71 ± 4.5% of the synaptic current compared with only 38 ± 2.9% at 241O (Table 1; p < 0.01). We also analyzed IPSC decay kinetics on the basis of time to 50% decay, which was significantly greater in 241E compared with 241O cells (Table 1; p < 0.01). In contrast to decay times, rise times of synaptically evoked IPSCs were unchanged at the 241 time point (Table 1; p > 0.1).
Table 1.
Parameters of synaptically evoked and miniature IPSCs
| Treatment group | |||||
|---|---|---|---|---|---|
| 3DO | 241O | 241E | 482O | 482E | |
| Evoked IPSCs | |||||
| τrise(msec) | 0.9 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.0 ± 0.1 |
| τdecay fast (msec) | 11.3 ± 3.2 | 11.9 ± 2.9 | 16.1 ± 2.4*,† | 12.7 ± 3.1 | 10.9 ± 4.2 |
| τdecay slow(msec) | 31.5 ± 6.1 | 30.9 ± 5.2 | 43.8 ± 5.61-160,1-164 | 32.7 ± 6.7 | 29.3 ± 5.9 |
| τslow relative amplitude (%) | 40 ± 3.1 | 38 ± 2.9 | 71 ± 4.51-160,1-164 | 42 ± 3.3 | 39 ± 2.8 |
| Time to 50% decay (msec) | 14.7 ± 1.9 | 13.1 ± 1.6 | 22.1 ± 2.11-160,1-164 | 12.6 ± 1.3 | 11.2 ± 1.1 |
| Max amplitude (pA) | 383.2 ± 49.5 | 390.8 ± 49.9 | 242.8 ± 48.1*,† | 175.0 ± 20.3 | 391.7 ± 43.11-164 |
| Miniature IPSCs | |||||
| Frequency (Hz) | 12.5 ± 0.4 | 11.6 ± 0.8 | 8.4 ± 0.7*,† | 7.1 ± 0.9 | 15.5 ± 0.81-164 |
| τrise(msec) | 0.69 ± 0.1 | 0.59 ± 0.1 | 0.64 ± 0.1 | 0.60 ± 0.1 | 0.56 ± 0.1 |
| Decay time (msec) | 7.4 ± 1.1 | 7.2 ± 1.0 | 9.8 ± 1.6 | 7.4 ± 2.1 | 7.4 ± 1.1 |
| Amplitude (pA) | 14.5 ± 2.9 | 14.1 ± 2.7 | 19.6 ± 4.1 | 17.4 ± 3.4 | 15.3 ± 2.9 |
Data are means ± SEM. Units are indicated in parentheses.
indicates a significant difference from 3DO (p < 0.05).
F1-160: indicates a significant difference from 3DO (p < 0.01).
indicates a significant difference from the oil-treated control at the corresponding time point (p < 0.05).
F1-164: indicates a significant difference from the oil-treated control at the corresponding time point (p < 0.01).
There are at least two possible sources of the estrogen-induced increase in decay time of synaptically evoked IPSCs. First, prolonged IPSC decay could be caused by estrogen-induced changes in the subunit composition of postsynaptic GABAA receptors (Smith et al., 1998). Second, estrogen could alter the relative contribution to the IPSC of somatic versus dendritic GABAA synapses, which have been shown to produce synaptic currents with varying decay times. Pearce (1993) demonstrated two anatomically and functionally distinct subpopulations of GABAA synapses on CA1 pyramidal cells: somatic GABA inputs that produce synaptic currents with predominantly fast rise and decay times and dendritic GABA inputs that produce slowly rising and decaying synaptic currents. If estrogen alters the balance of somatic versus dendritic GABA input in favor of dendritic inputs, this might account for the prolongation of synaptically evoked IPSCs at 241E. However, two observations make this explanation insufficient to account for the prolonged IPSC decay that we observed. First, we detected no difference in IPSC rise times between 241O and 241E cells, which is inconsistent with the slow GABAA currents described by Pearce. Second, slow GABAA currents arise primarily from distal dendritic inputs and so are not likely to contribute substantially to postsynaptic currents evoked by stimulation in the pyramidal cell layer. Together, these observations favor an alteration in postsynaptic GABAA receptor subunit composition as a source of prolonged GABAA IPSCs at 241E.
Miniature IPSCs
To determine whether the reduced amplitude of synaptically evoked IPSCs in 241E cells was paralleled by a decrease in either the frequency and/or amplitude of individual synaptic events, we recorded GABAA-mediated mIPSCs in TTX. Analysis of mIPSCs in 3DO, 241O (Fig.3A), and 241E (Fig. 3B) cells showed a significant decrease in mIPSC frequency in 241E cells compared with those in both 3DO and 241O cells (Fig. 3C, Table1; p < 0.05). In contrast to mIPSC frequency, the mean amplitude of mIPSCs was not significantly affected by estrogen treatment, although there was a trend toward larger currents in the 241E cells (Fig.4, Table 1; p < 0.1). Comparison of mIPSC amplitude histograms for 3DO (Fig.4A), 241O (Fig.4B), and 241E (Fig.4C) cells showed that the distribution was skewed toward larger currents in the 241E group (Fig.4D) (p < 0.01), accounting for the statistical trend toward greater mean mIPSC amplitude at 241E. Together, these results indicate that the reduction in amplitude of synaptically evoked IPSCs at 241E is not caused by a decrease in the amplitude of individual GABAA-mediated synaptic currents but may result from a decrease in the number of functional GABAA synapses or the probability of GABA release.
Fig. 3.
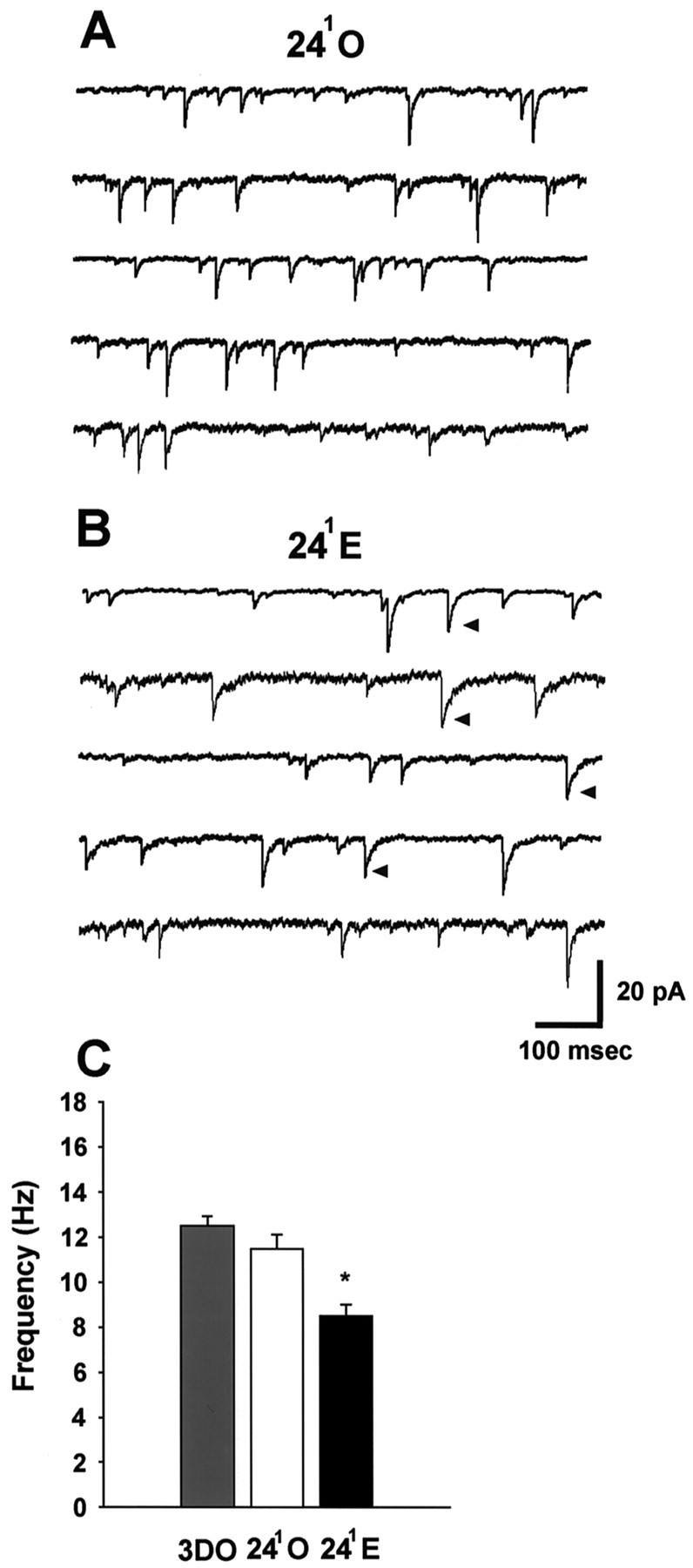
Estrogen reduces the frequency of mIPSCs in CA1 pyramidal cells at the 241 time point.A, Representative traces from five different 241O cells showing mIPSCs.B, Representative traces from five different 241E cells showing mIPSCs.Arrowheads indicate representative mIPSCs with a prolonged decay time in 241E cells.C, Bar graph of mIPSC frequency in 3DO (gray bar; n = 12 cells from 6 animals), 241O (white bar;n = 18 cells from 10 animals), and 241E (black bar;n = 22 cells from 10 animals) groups. Data are from the same cells shown in Figure 2C. Theasterisk indicates a significant difference from 3DO and 241O (p < 0.05).
Fig. 4.
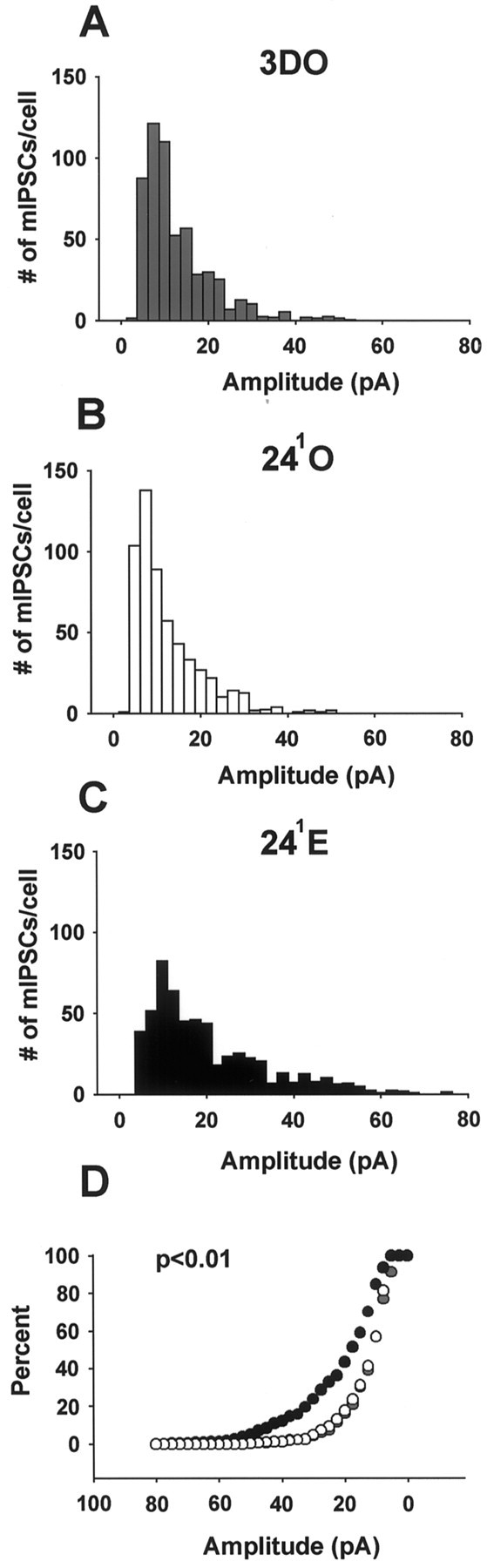
Estrogen alters mIPSC amplitude distributions at the 241 time point. A–C, mIPSC amplitude histograms for the 3DO (A), 241O (B), and 241E (C) groups. Note the shift toward larger-amplitude mIPSCs in the 241E group. D, Cumulative histogram of mIPSC amplitudes for the 3DO (gray circles), 241O (white circles), and 241E (black circles) groups. Note the leftwardshift in mIPSC amplitude in the 241E group (p < 0.01). Data are from the same cells shown in Figure 3. Mean mIPSC amplitudes are not significantly different and are shown in Table 1.
In agreement with the prolonged decay of synaptically evoked IPSCs in 241E cells, a subpopulation of mIPSCs in the same cells also showed prolonged decay times compared with 3DO (Fig. 5A) and 241O (Fig. 5B) cells. Similar to the data for mIPSC amplitude, there was a statistical trend toward increased mean mIPSC decay time at 241E (Table 1; p < 0.1), which reflected a bimodal distribution of mIPSC decay times for this group (Fig. 5C). A significant proportion of mIPSCs in the 241E group was prolonged compared with mIPSCs in 3DO or 241O (Fig. 5D) (p < 0.01); clearly bimodal decay time histograms were observed for 20 of 22 cells at 241E. In contrast to decay time, mIPSC rise time was not affected by estrogen (Table 1; p > 0.1). Interestingly, regression analysis of the first 50 mIPSCs per cell showed a weak but statistically significant correlation between mIPSC amplitude and decay time (r = 0.33;p < 0.01; data not shown). Thus, the larger-amplitude mIPSCs were a subset of the mIPSCs with prolonged decay times.
Fig. 5.
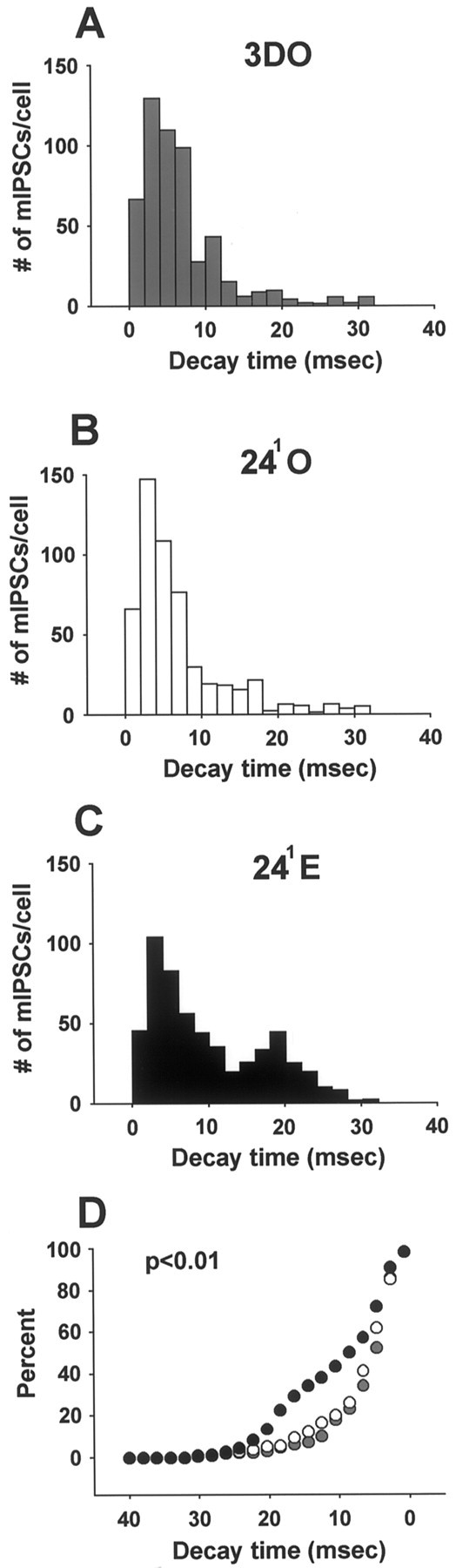
Estrogen alters mIPSC decay time distributions at the 241 time point. A–C, mIPSC decay time histograms for the 3DO (A), 241O (B), and 241E (C) groups. Note the subpopulation of mIPSCs with longer decay times in the 241E group. D, Cumulative histogram of mIPSC decay times for the 3DO (gray circles), 241O (white circles), and 241E (black circles) groups. Note theleftward shift in mIPSC decay time in the 241E group (p < 0.01). Data are from the same cells shown in Figures 3 and 4. Mean mIPSC decay times are not significantly different and are shown in Table 1.
Analysis of mIPSCs corroborated the interpretation that prolonged IPSC decay was caused by something other than an enhancement of the slow dendritic GABAA inputs described by Pearce (1993). Because mIPSCs with prolonged decay were frequent in our somatic recordings and distal dendritic inputs should be only infrequently represented in somatic recordings, it is unlikely that these inputs underlie the mIPSCs with prolonged decay. In addition, as with evoked IPSCs, mIPSC rise time was not affected by estrogen. Thus, these data point toward changes in postsynaptic GABAA receptor subunit composition as a more likely explanation for prolonged IPSC decay. Also, because we observed a distinct subpopulation of mIPSCs with prolonged decay times rather than a uniform increase in mIPSC decay time, this indicates that the GABA synapses altered at 241E are a subset of all GABA synapses detectable in somatic recordings.
Postsynaptic currents evoked by str. radiatum stimulation
IPSCs evoked by pyramidal cell layer stimulation and mIPSCs primarily reflect somatic and proximal dendritic GABA inputs, whereas we observed the greatest effects of estrogen on GAD65 immunoreactivity in the str. radiatum and str. oriens, which contain GABA neurons that provide primarily dendritic inputs. To investigate how estrogen affects the interaction between EPSCs and mixed somatic and dendritic GABAA IPSCs at the 241 time point, we recorded postsynaptic currents elicited by stimulation of the principal excitatory pathway into the CA1 region, the Schaffer collateral axons in the str. radiatum. Recordings were made in normal ACSF followed by addition of a GABAA receptor antagonist (bicuculline or SR). Mixed currents evoked by str. radiatum stimulation were recorded at −70 mV holding potential with a Cs-gluconate internal solution so that glutamate receptor-mediated EPSCs were inward currents and the GABAA-mediated IPSC was an outward current. In agreement with a reduction in disynaptic inhibition at 241E, recordings in normal ACSF showed that mixed postsynaptic currents in cells from the 241E group were prolonged compared with those in 241O cells (Fig.6A). The mean time to 50% decay of synaptically evoked currents was 42% greater in 241E cells than in 241O cells (Fig. 6C) (p < 0.01). This difference in decay time was very likely caused by the difference in GABAA-mediated inhibition because it was eliminated by addition of bicuculline or SR (Fig.6B,D). Recording at a holding potential of +40 mV in the presence of a GABAA antagonist and CNQX showed no effect of estrogen on NMDA-mediated currents at the 241 time point. The averaged peak amplitude of NMDA-mediated EPSCs was 56.3 ± 7.1 pA in the 241E group versus 57.1 ± 5.9 pA in the 241O group (p> 0.1; data not shown).
Fig. 6.

Reduced inhibition of CA1 pyramidal cells at the 241E time point prolongs postsynaptic currents evoked by str. radiatum stimulation. Recordings were made with a Cs-gluconate internal solution. A, Representative individual traces of str. radiatum-evoked postsynaptic currents in a 241O and a 241E cell in normal ACSF. B, Representative individualtraces of str. radiatum-evoked postsynaptic currents in the same 241O and 241E cells after addition of SR-95531 to the bath. C, Bar graph of averaged decay times in normal ACSF for 241O (white bar; n = 12 cells from 6 animals) and 241E (black bar;n = 12 cells from 6 animals) groups. Thedouble asterisks indicate a significant difference from 241O (p < 0.01).D, Bar graph of averaged decay times in ACSF plus a GABAA antagonist (bicuculline or SR-95531) for the same 241O and 241E cells shown inC. In the presence of the GABAA blocker, decay times are not different for the 241O and 241E groups.
Synaptic inhibition at the 482 time point
Synaptically evoked IPSCs
Similar to experiments at the 241time point, the amplitude of synaptically evoked IPSCs in CA1 pyramidal cells at the 482 time point also paralleled GAD65 immunoreactivity. Note that at this later time point, GAD65 staining is greater in 482E than in 482O animals (Fig. 1C–E). As with experiments at the 241 time point, isolated IPSCs were recorded using a CsCl-based internal solution so that the GABAA-mediated IPSC was an inward current. Analysis of stimulus–response curves for synaptically evoked IPSCs in 3DO, 482O (Fig.7A), and 482E (Fig. 7B) cells showed that peak IPSC amplitude was greater in cells from 482E than from 482O animals (Fig. 7C) (p < 0.01). The reversal of the O versus E relationship at 482 compared with 241 is caused partly by the decrease in IPSC amplitude in 482O cells and partly by the recovery of IPSC amplitude in 482E cells. In cells from 482E animals, evoked IPSC amplitudes had recovered to values that were no longer different from that in 3DO cells (Fig. 7C) (p> 0.1).
Fig. 7.
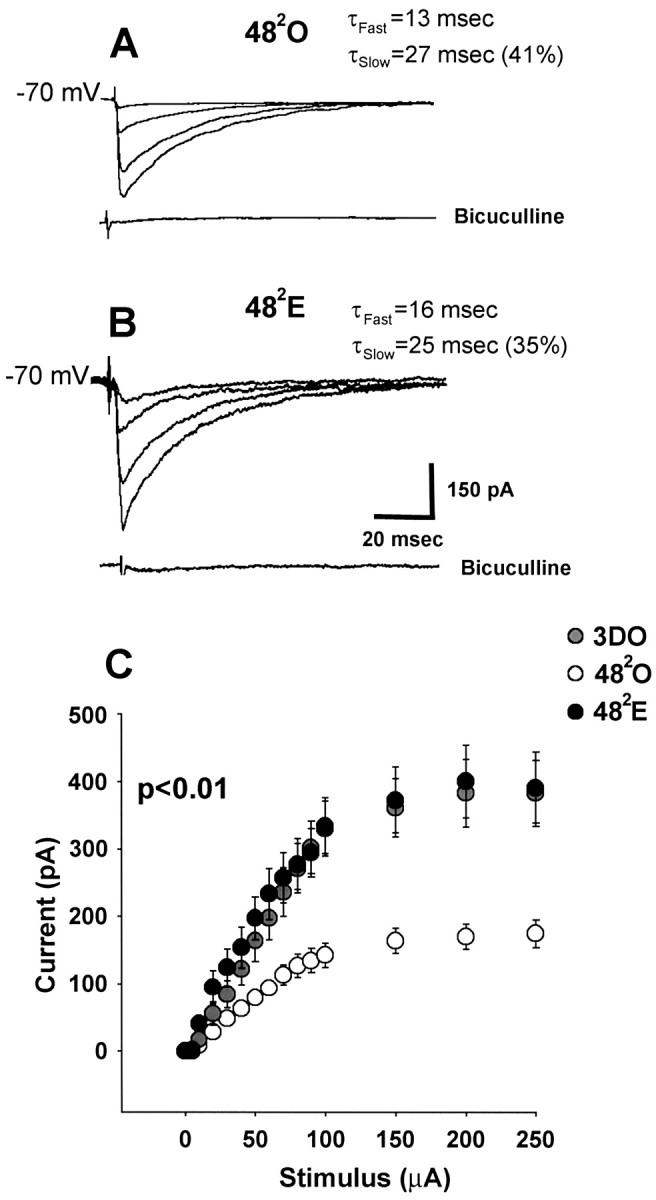
In estrogen-treated animals at the 482 time point, synaptically evoked IPSC amplitude has recovered to baseline values and is greater than that in oil-treated controls. Recordings were made with a CsCl internal solution. A, Representative individualtraces of IPSCs evoked in a 482O cell using 50, 100, 150, and 250 μA stimulating currents.B, Representative individual traces of IPSCs evoked in a 482E cell using the same stimulus intensities as in A. Evoked currents are blocked by bicuculline. τdecay fast and τdecay slowvalues in A and B apply specifically to the cells shown. C, Averaged stimulus–response curves for 3DO (gray circles; n = 12 cells from 6 animals), 482O (white circles; n = 20 cells from 10 animals), and 482E (black circles;n = 22 cells from 10 animals) groups. There is no difference in peak IPSC amplitude between 482E and 3DO groups (p > 0.1), whereas IPSC amplitude is significantly reduced in the 482O group compared with both 482E and 3DO groups (p < 0.01).
In contrast to 241, the decay kinetics of evoked IPSCs at the 482 time point was not different between 3DO, 482O, and 482E. There were no differences detected in τdecay fast, τdecay slow, or time to 50% decay of evoked IPSCs (Table 1;p > 0.1). Thus, the prolonged decay time of synaptically evoked IPSCs seen in 241E cells was no longer evident at the 482E time point. Like the data at 241, there were also no differences in rise times of synaptically evoked IPSCs at 482 (Table 1; p > 0.1).
Miniature IPSCs
Consistent with the pattern established at the 241 time point, the frequency of mIPSCs at the 482 time point paralleled GAD65 staining and the amplitude of synaptically evoked IPSCs. Analysis of mIPSCs in 3DO, 482O (Fig.8A), and 482E (Fig. 8B) cells showed that mIPSC frequency in cells from the 482E group had recovered to values that were no longer different from those of the baseline 3DO group (Fig.8C, Table 1; p > 0.1) but were significantly higher than those in the 482O group (Fig. 8C, Table 1;p < 0.01). In contrast to results of mIPSC analysis at the 241 time point, we detected no evidence of a difference in mIPSC amplitude or kinetics (means or histograms) between 3DO, 482O, and 482E (Table 1; p > 0.1). Thus the subpopulations of larger and prolonged mIPSCs seen in 241E cells were no longer apparent at the 482E time point.
Fig. 8.
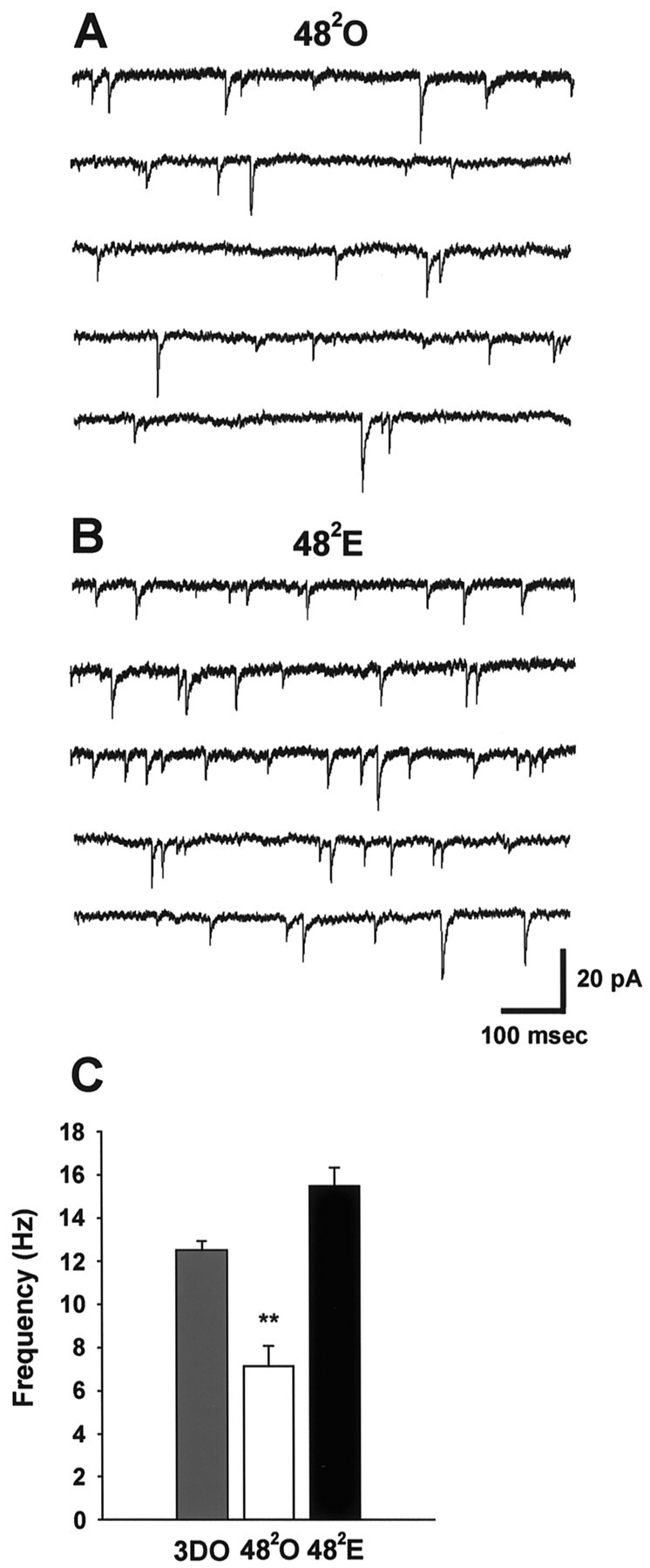
At the 482 time point, the mIPSC frequency in estrogen-treated animals has recovered to baseline values and is greater than that in oil-treated controls.A, Representative traces from five different 482O cells showing mIPSCs.B, Representative traces from five different 482E cells showing mIPSCs.C, Bar graph of mIPSC frequency in 3DO (gray bar; n = 12 cells from 6 animals), 482O (white bar;n = 20 cells from 10 animals), and 482E (black bar;n = 22 cells from 10 animals) groups. Data are from the same cells shown in Figure 7. The double asterisksindicate a significant difference from 3DO and 482E (p < 0.01).
Postsynaptic currents evoked by st. radiatum stimulation
Similar to our analysis of the 241time point, we also used str. radiatum stimulation to investigate the interaction between GABAA-mediated IPSCs and glutamate receptor-mediated EPSCs at the 482 time point. These recordings were made using a Cs-gluconate internal solution. In this case, mixed synaptic currents evoked at a holding potential of −70 mV in normal ACSF were not different in amplitude or time course between CA1 pyramidal cells in 482O and 482E groups (Fig.9A). Addition of a GABAA antagonist (SR) revealed a small, but statistically significant, difference in the time to 50% decay of the EPSC (Fig. 9B) (482E, 6% greater than that of 482O;p < 0.05). We used subtraction of postsynaptic currents recorded in the presence of SR from currents recorded in normal ACSF to reveal the GABAA-mediated component of total postsynaptic current elicited by str. radiatum stimulation (Fig. 9B, inset). In agreement with larger evoked IPSCs in cells from 482E animals, the amplitude of SR-sensitive current was 36% greater in the 482E than in the 482O group (Fig. 9C) (p < 0.05). Interestingly, addition of APV eliminated the small difference in time to 50% decay of EPSCs at −70 mV (Fig. 9D), suggesting that estrogen enhancement of the NMDA-mediated component of the EPSC might account for the difference in EPSC decay time.
Fig. 9.
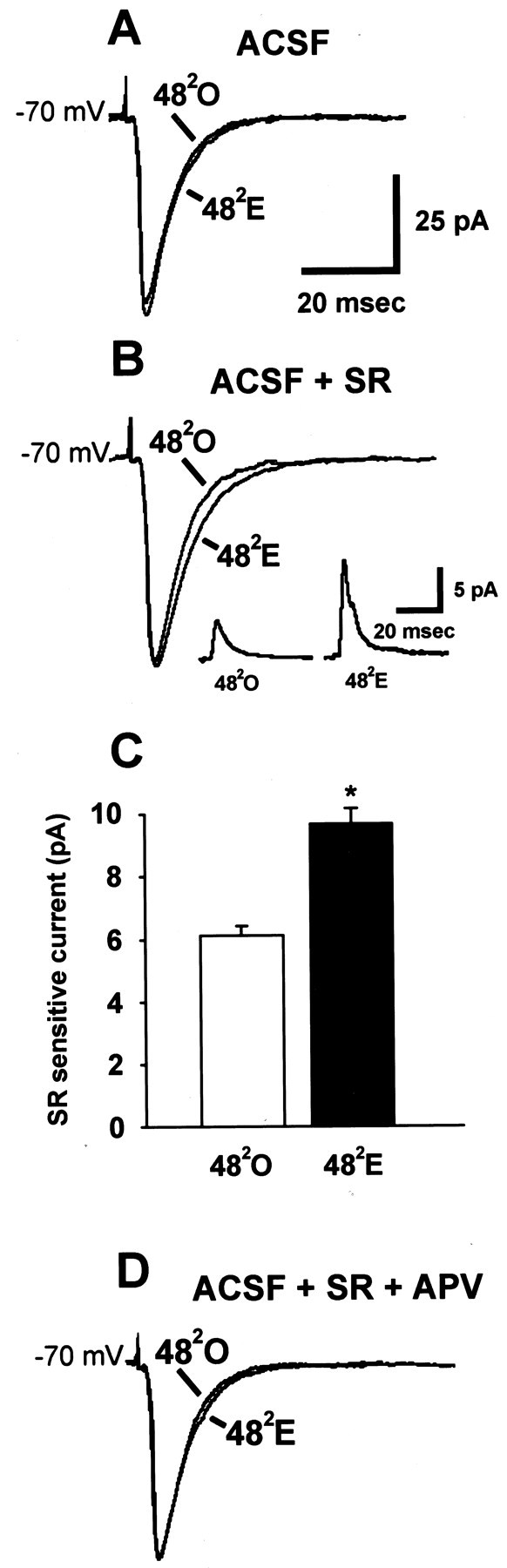
At the 482 time point, GABAA receptor-mediated currents are enhanced in estrogen-treated animals, but mixed postsynaptic currents (EPSC + IPSC) are similar to those in oil-treated controls. Recordings were made with a Cs-gluconate internal solution. A, Representative individual traces of str. radiatum-evoked postsynaptic currents in a 482O and a 482E cell in normal ACSF. B, Representative individualtraces of str. radiatum-evoked postsynaptic currents in the same 482O and 482E cells after addition of SR-95531. Inset, SR-sensitive (GABAA-mediated) currents obtained by subtraction of the normal ACSF currents shown in A from the ACSF + SR-95531 currents shown in B. C, Averaged amplitudes of SR-sensitive postsynaptic currents evoked by str. radiatum stimulation in the 482O (white bar; n = 16 cells from 8 animals) and 482E (black bar;n = 16 cells from 9 animals) groups. Theasterisk indicates a significant difference from 482O (p < 0.05).D, Representative individual traces of str. radiatum-evoked postsynaptic currents in the same 482O and 482E cells shown inA and B after addition of APV and SR-95531.
This observation suggested that the effect of the enhanced GABAA-mediated IPSC on the total postsynaptic current (EPSC + IPSC) evoked by str. radiatum stimulation in 482E cells might be masked by concurrent enhancement of NMDA EPSCs. A previous analysis based on current-clamp recording with sharp electrodes (Woolley et al., 1997) showed a steeper stimulus–response relationship in 482E than in 482O cells when initial slopes of NMDA-mediated EPSPs were plotted versus stimulus intensity. AMPA-mediated EPSPs were not directly evaluated in this previous study, but baseline synaptic responses were unaffected by estrogen. These results suggested that the sensitivity of CA1 pyramidal cells to NMDA, but not non-NMDA, glutamate receptor-mediated synaptic input is enhanced by estrogen. To determine directly whether NMDA- and/or AMPA-mediated currents are enhanced by estrogen at the 482 time point, we recorded AMPA (Fig.10A) and NMDA (Fig.10C) EPSCs at a holding potential of +40 mV to relieve Mg2+ block of the NMDA receptor. Comparison of stimulus–response curves for isolated AMPA (Fig.10B) and NMDA (Fig. 10D) currents showed that estrogen treatment enhanced total charge transfer of NMDA- but not AMPA-mediated EPSCs. There was no effect of estrogen on the stimulus–response relationship for AMPA-mediated currents (Fig.10B) (p > 0.1), whereas NMDA currents were significantly greater in 482E compared with 482O cells (Fig. 10D) (p < 0.01). Total charge transfer of maximal NMDA-mediated EPSCs was 26% greater at 482E than at 482O (Fig. 10D) (p < 0.01). Thus, direct analysis of EPSCs confirmed the effect of estrogen to enhance the synaptic sensitivity of CA1 pyramidal cells to NMDA-mediated input (Woolley et al., 1997). The apparent lack of effect of estrogen on total synaptic currents evoked by str. radiatum stimulation at a holding potential near rest (−70 mV) (Fig. 9A) is likely caused by balanced, opposing enhancements of the NMDA EPSC and GABAA IPSC.
Fig. 10.
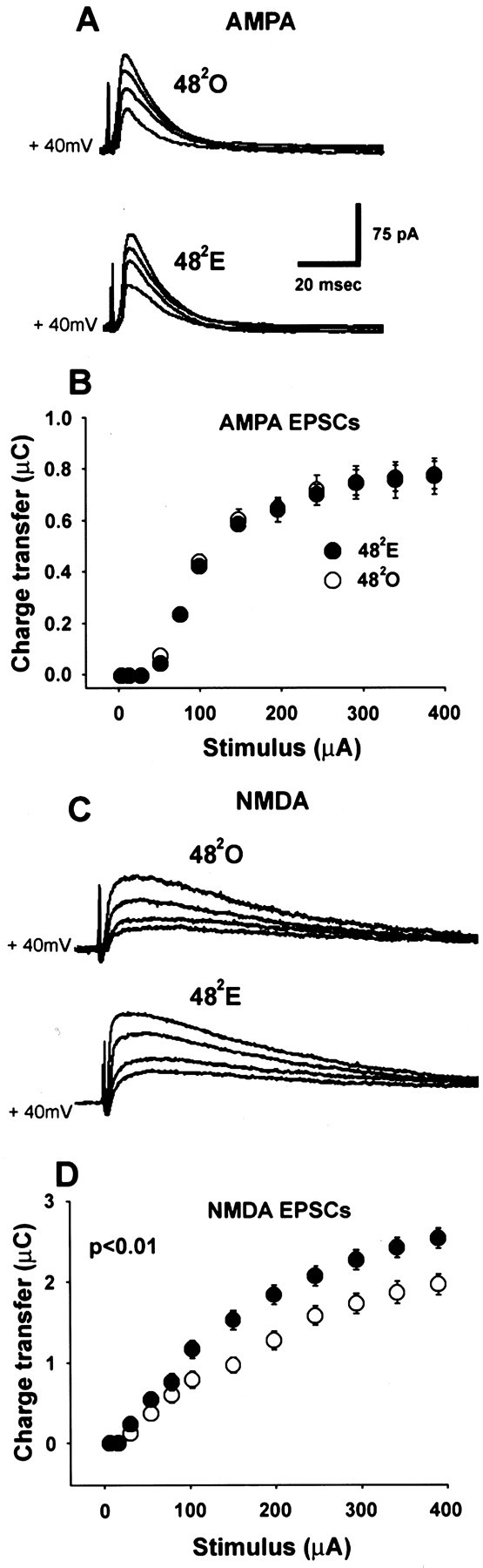
Estrogen enhances NMDA- but not AMPA-mediated EPSCs at the 482 time point. A, Representative individual traces of str. radiatum-evoked postsynaptic currents in a 482O (upper traces) and a 482E (lower traces) cell in ACSF containing APV + SR-95531 (AMPA-mediated EPSCs). Postsynaptic currents were evoked using 50, 150, 250 and 400 μA stimulating currents. B, Stimulus–response curves generated from isolated AMPA receptor-mediated EPSCs for 482O (white circles;n = 16 cells from 9 animals) and 482E (black circles;n = 16 cells from 9 animals) groups.C, Representative individual traces of str. radiatum-evoked postsynaptic currents in the same 482O (upper traces) and 482E (lower traces) cells inA but in ACSF containing CNQX + SR-95531 (NMDA-mediated EPSCs). Postsynaptic currents were evoked using 50, 150, 250 and 400 μA stimulating current. D, Stimulus–response curves generated from isolated NMDA receptor-mediated EPSCs for 482O (white circles; same cells shown in B) and 482E (black circles; same cells shown in B) groups. NMDA-mediated charge transfer is significantly greater in 482E than in 482O cells (p < 0.01).
DISCUSSION
We have used immunohistochemical and electrophysiological analyses to demonstrate that estrogen regulates a dynamic balance between excitatory and inhibitory synaptic input to hippocampal CA1 pyramidal cells. Ovariectomy results in a gradual decline in GAD65 immunoreactivity. Superimposed on this gradual decline is a phasic effect of estrogen, in which GAD65 immunoreactivity is further suppressed 24 hr after a single estrogen treatment (241) but recovers by 48 hr after a second estrogen treatment (482).
Whole-cell voltage-clamp recordings reveal that the effects of estrogen on GAD65 are paralleled by changes in functional inhibition of CA1 pyramidal cells. Reduced inhibition at 241is reflected by lower-amplitude synaptically evoked IPSCs and reduced mIPSC frequency. Because mean mIPSC amplitude is not reduced by estrogen (if anything, it is increased), these data suggest that estrogen decreases the number of functional GABAergic synapses on CA1 pyramidal cells and/or decreases the probability of transmitter release at GABAergic synapses at the 241E time point. Because there is no concomitant effect of estrogen on CA1 pyramidal cell EPSCs at this time point, reduced disynaptic inhibition prolongs the total postsynaptic response to excitatory input.
In estrogen-treated animals at the 482time point, GAD65 immunoreactivity, synaptically evoked IPSC amplitude, and mIPSC frequency are restored to original values. Together, the recovery of inhibition in estrogen-treated animals and the gradual decrease in inhibition in ovariectomized controls result in greater inhibition in estrogen-treated than in control animals at this later time point. Additional analysis revealed that, at 482, estrogen also enhances NMDA-mediated EPSCs in parallel with enhancement of the GABAA-mediated IPSC, restoring a balance of excitatory and inhibitory input to these cells.
GAD65 versus GAD67
We found an effect of estrogen on the 65 kDa but not the 67 kDa isoform of GAD. The significance of this difference is difficult to predict because functional differences between the two GAD isoforms are not well understood (Erlander and Tobin, 1991; Soghomonian and Martin, 1998). Although GAD65 and GAD67 are encoded by different genes (Erlander et al., 1991), they are generally coexpressed (Houser and Esclapez, 1994; Sloviter et al., 1996; Stone et al., 1999). GAD65 tends to be concentrated in neuronal membranes, particularly in axonal varicosities, but is also expressed in the cell body (Erlander et al., 1991; Kaufman et al., 1991) of GABAergic neurons. GAD67 is more highly expressed in cell bodies than is GAD65 but is also found to a lesser extent in axons (Gonzales et al., 1991). Both GAD isoforms require a cofactor, pyridoxal-phosphate, to be active (Martin et al., 1991). Interestingly, because a large fraction of GAD that exists in the “apo” form (i.e., not bound to cofactor) is GAD65, it has been proposed that GAD65 preferentially responds to rapid changes in demand for GABA (Soghomonian and Martin, 1998). Thus estrogen effects on GAD65 may reflect differences in the capacity of CA1 interneurons for activity-dependent changes in GABA synthesis.
Estrogen-sensitive interneurons
In dorsal CA1, at least one form of the nuclear estrogen receptor (ERα) is expressed primarily in GABAergic interneurons. The greatest concentration of ERα-immunoreactive GAD cells is found at the border between str. radiatum and str. lacunosum-moleculare (Weiland et al., 1997; Hart and Woolley, 2000); however, only a small subset [<30% (Hart and Woolley, 2000)] of all GAD neurons in this region express ERα. Thus, although one interneuron may contact over 1000 pyramidal cells (Freund and Buzsaki, 1996), it seems unlikely that this small subpopulation of interneurons could mediate the highly consistent effects of estrogen on inhibition via direct interneuron–pyramidal cell connections. However, some interneurons at the radiatum–lacunosum border also project to other interneurons (Kunkel et al., 1988; Lacaille and Schwartzkroin, 1988) and so are well positioned to mediate multiplicative effects of estrogen via connections with interneurons that then project to pyramidal cells. Indeed, some targets of border interneurons are interneurons in the pyramidal cell layer (Banks et al., 2000). One speculative possibility is that estrogen modulates somatic inhibition of pyramidal cells indirectly via border interneuron to pyramidal cell layer interneuron interactions.
The role of disinhibition in dendritic spine formation
There is a rich literature describing activity-dependent maintenance or formation of dendritic spines (for review, seeHarris, 1999; Smart and Halpain, 2000). Two lines of reasoning support the suggestion that disinhibition of CA1 pyramidal cells at the 241E time point is involved, at least in part, in estrogen-induced dendritic spine/synapse formation. First, as would be required of a causal factor, the disinhibition at 241 occurs before spine/synapse changes occur (Woolley and McEwen, 1993). Second, the estrogen-induced changes in GAD immunoreactivity and functional inhibition of CA1 pyramidal cells that we observed very closely parallel the findings of Murphy et al. (1998) who demonstrated that estrogen-induced dendritic spine formation on cultured hippocampal neurons is caused by a transient reduction in GABA-mediated neurotransmission.
However, although our data are, in part, consistent with the possibility that disinhibition is involved in estrogen-induced dendritic spine formation, disinhibition alone cannot completely account for spine formation in vivo. If disinhibition were sufficient to induce new spines, spine density would be increased in ovariectomized, oil-treated animals, which showed a gradual reduction in inhibition between the 3DO and 482O time points. However, previous studies have shown that spine density is low at the 482O time point (Gould et al., 1990; Woolley and McEwen, 1993; Woolley et al., 1997), or even with longer periods of ovariectomy (Woolley and McEwen, 1993). At least two factors distinguish disinhibition of CA1 pyramidal cells observed at the 241E versus 482O time points; these factors may be related to different consequences of disinhibition at 241E versus 482O for spine formation. First, although evoked IPSC amplitude and mIPSC frequency were similarly low in 241E and 482O animals, inhibitory currents at 241E had several features not seen in any other group. Both synaptically evoked IPSCs and a substantial subpopulation of mIPSCs were significantly prolonged in 241E cells. In addition, mIPSC amplitudes were shifted toward larger values at 241E. These features suggest that inhibition at 241E is functionally distinct from that seen at 482O. Second, the disinhibition at 241E results from a sharp decline in measures of GABAergic synaptic transmission as opposed to the gradual decline observed at 482O. The consequences of sharp versus gradual disinhibition for dendritic spine formation are currently unknown, but this difference in timing could be important in the regulation of spine density. A third factor that should be considered in the mechanism of estrogen regulation of spine density in vivo is the possibility that extrahippocampal afferents interact with disinhibition of CA1 pyramidal cells to regulate spine density. In agreement with this suggestion,Leranth et al. (2000) have shown recently that removal of subcortical input to the hippocampus via fimbria/fornix transection blocks the effect of estrogen to increase dendritic spine synapse density in CA1in vivo. It is conceivable that the timing of interactions with subcortical inputs is effective at the 241 but not the 482 time point.
Dynamic balance of excitatory and inhibitory synaptic input to CA1 pyramidal cells
Functional analysis of GABAA- and NMDA-mediated synaptic input to CA1 pyramidal cells shows that both are enhanced in estrogen-treated compared with control animals at the 482 time point. This (482) is the same time point used in previous studies that demonstrated estrogen-induced increases in CA1 dendritic spine and synapse density (Gould et al., 1990; Woolley and McEwen, 1992, 1993; Woolley et al., 1997) and enhancement of excitatory synaptic input to CA1 pyramidal cells (Wong and Moss, 1992; Woolley et al., 1997). Previous studies suggested that synaptic sensitivity to NMDA-mediated input was increased in parallel with dendritic spine/synapse numbers (Woolley et al., 1997). Here, direct analysis of NMDA and AMPA EPSCs at 482 confirmed that NMDA currents are increased by estrogen, leading to a balance in enhancement of NMDA-mediated excitatory input and GABAA-mediated inhibitory input to CA1 pyramidal cells. Although this balance results in little net effect of estrogen on normal synaptic transmission, it might also be expected to increase the dynamic range of the responses of CA1 pyramidal cells to synaptic input. Enhancement of NMDA currents may underlie estrogen facilitation of long-term potentiation in CA1 (Cordoba-Montoya and Carrer, 1997) and hippocampus-dependent seizure susceptibility (Terasawa and Timiras, 1968; Buterbaugh and Hudson, 1991), circumstances that involve substantial NMDA receptor activation.
Our results may also help to explain a puzzling dichotomy in the effects of estrogen on the susceptibility of female rats to kainic acid-induced seizures, which depend in part on hippocampal activity (Ben-Ari et al., 1981; Lothman and Collins, 1981). Woolley (2000) found that a slightly lower proportion of 482E than 482O rats developed behavioral seizures when treated with kainic acid; however, when seizures were initiated, they progressed more rapidly and were more severe in 482E than in 482O animals. It is conceivable that the slight protective effect of estrogen on seizure initiation is caused by enhanced synaptic inhibition of hippocampal pyramidal cells. However, after the barrier of greater inhibition in 482E is overcome, enhanced sensitivity to excitatory input makes hippocampal neurons more prone to developing and propagating synchronous discharge associated with seizure activity.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS37324 and by the Alfred P. Sloan Foundation. C.N.R. was supported by National Institutes of Health Training Grant GM08061. We thank Drs. Nelson Spruston and Indira Raman for help with experimental setup and for their critical reading of this manuscript.
Correspondence should be addressed to Dr. Catherine S. Woolley, Department of Neurobiology and Physiology, Northwestern University, 2153 North Campus Drive, Evanston, IL 60208. E-mail:cwoolley@northwestern.edu.
REFERENCES
- 1.Banks MI, White JA, Pearce RA. Interactions between distinct GABAA circuits in hippocampus. Neuron. 2000;25:449–457. doi: 10.1016/s0896-6273(00)80907-1. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Tremblay E, Riche D, Ghilini G, Naquet R. Electrographic, clinical and pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping using the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience. 1981;6:1361–1391. doi: 10.1016/0306-4522(81)90193-7. [DOI] [PubMed] [Google Scholar]
- 3.Bowers G, Cullinan WE, Herman JP. Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci. 1998;18:5938–5947. doi: 10.1523/JNEUROSCI.18-15-05938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buterbaugh GG, Hudson GM. Estradiol replacement to female rats facilitates dorsal hippocampal but not ventral hippocampal kindled seizure acquisition. Exp Neurol. 1991;111:55–64. doi: 10.1016/0014-4886(91)90050-m. [DOI] [PubMed] [Google Scholar]
- 5.Churchill L, Taishi P, Guan Z, Chen L, Fang J, Krueger JM. Sleep modifies glutamate decarboxylase mRNA within the barrel cortex of rats after a mystacial whisker trim. Sleep. 2001;24:261–266. doi: 10.1093/sleep/24.3.261. [DOI] [PubMed] [Google Scholar]
- 6.Cordoba-Montoya DA, Carrer HF. Estrogen facilitates induction of long-term potentiation in the hippocampus of awake rats. Brain Res. 1997;778:430–438. doi: 10.1016/s0006-8993(97)01206-7. [DOI] [PubMed] [Google Scholar]
- 7.Erlander MG, Tobin AJ. The structural and functional heterogeneity of glutamic acid decarboxylase: a review. Neurochem Res. 1991;16:215–226. doi: 10.1007/BF00966084. [DOI] [PubMed] [Google Scholar]
- 8.Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 9.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 10.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzales C, Kaufman DL, Tobin AJ, Chesselet MF. Distribution of glutamic acid decarboxylase (Mr 67,000) in basal ganglia of the rat: an immunohistochemical study with a selective cDNA-generated polyclonal antibody. J Neurocytol. 1991;20:953–961. doi: 10.1007/BF01187913. [DOI] [PubMed] [Google Scholar]
- 12.Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gundersen HJG, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 14.Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- 15.Hart SA, Woolley CS. Colocalization of ER-α and GAD immunoreactivity in the CA1 region of the adult female rat hippocampus. Soc Neurosci Abstr. 2000;26:1148. [Google Scholar]
- 16.Houser CR, Esclapez M. Localization of mRNAs encoding two forms of glutamic acid decarboxylase in the rat hippocampal formation. Hippocampus. 1994;4:530–545. doi: 10.1002/hipo.450040503. [DOI] [PubMed] [Google Scholar]
- 17.Jones KJ, McEwen BS, Pfaff DW. Quantitative assessment of early and discontinuous estradiol-induced effects on ventromedial hypothalamic and preoptic area proteins in female rat brain. Neuroendocrinology. 1988;48:561–568. doi: 10.1159/000125063. [DOI] [PubMed] [Google Scholar]
- 18.Jones KJ, Harrington CA, Chikaraishi DM, Pfaff DW. Steroid hormone regulation of ribosomal RNA in rat hypothalamus: early detection using in situ hybridization and precursor–product ribosomal DNA probes. J Neurosci. 1990;10:1513–1521. doi: 10.1523/JNEUROSCI.10-05-01513.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunkel DD, Lacaille JC, Schwartzkroin PA. Ultrastructure of stratum lacunosum-moleculare interneurons of hippocampal CA1 region. Synapse. 1988;2:382–394. doi: 10.1002/syn.890020405. [DOI] [PubMed] [Google Scholar]
- 21.Lacaille JC, Schwartzkroin PA. Stratum lacunosum-moleculare interneurons of hippocampal CA1 region. II. Intrasomatic and intradendritic recordings of local circuit synaptic interactions. J Neurosci. 1988;8:1411–1424. doi: 10.1523/JNEUROSCI.08-04-01411.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leranth C, Shanabrough M, Horvath TL. Hormonal regulation of hippocampal spine density involves subcortical mediation. Neuroscience. 2000;101:349–356. doi: 10.1016/s0306-4522(00)00369-9. [DOI] [PubMed] [Google Scholar]
- 23.Lothman EW, Collins RC. Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalography and neuropathological correlates. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- 24.Martin DL, Martin SB, Wu SJ, Espina N. Cofactor interactions and the regulation of glutamate decarboxylase activity. Neurochem Res. 1991;16:243–249. doi: 10.1007/BF00966087. [DOI] [PubMed] [Google Scholar]
- 25.McEwen BS, Tanapat P, Weiland NG. Inhibition of dendritic spine induction on hippocampal CA1 pyramidal neurons by a nonsteroidal estrogen antagonist in female rats. Endocrinology. 1999;140:1044–1047. doi: 10.1210/endo.140.3.6570. [DOI] [PubMed] [Google Scholar]
- 26.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- 29.Sloviter RS, Dichter MA, Rachinsky TL, Dean E, Goodman JH, Sollas AL, Martin DL. Basal expression and induction of glutamate decarboxylase and GABA in excitatory granule cells of the rat and monkey hippocampal dentate gyrus. J Comp Neurol. 1996;373:593–618. doi: 10.1002/(SICI)1096-9861(19960930)373:4<593::AID-CNE8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 30.Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–554. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 31.Smith SS, Gong QH, Hsu FC, Markowitz RS, ffrench-Mullen JM, Li X. GABA(A) receptor alpha4 subunit suppression prevents withdrawal properties of an endogenous steroid. Nature. 1998;392:926–930. doi: 10.1038/31948. [DOI] [PubMed] [Google Scholar]
- 32.Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- 33.Stone DJ, Walsh J, Benes FM. Localization of cells preferentially expressing GAD67 with negligible GAD65 transcripts in the rat hippocampus. A double in situ hybridization study. Brain Res Mol Brain Res. 1999;71:201–209. doi: 10.1016/s0169-328x(99)00185-0. [DOI] [PubMed] [Google Scholar]
- 34.Szabo G, Kartarova Z, Hoertnagl B, Somogyi R, Sperk G. Differential regulation of adult and embryonic glutamate decarboxylases in rat dentate granule cells after kainate-induced limbic seizures. Neuroscience. 2000;100:287–295. doi: 10.1016/s0306-4522(00)00275-x. [DOI] [PubMed] [Google Scholar]
- 35.Terasawa E, Timiras PS. Electrical activity during the estrous cycle of the rat: cyclical changes in limbic structures. Endocrinology. 1968;83:207–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- 36.Weiland NG. Estradiol selectively regulates agonist binding sites on the N-methyl-d-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology. 1992;131:662–668. doi: 10.1210/endo.131.2.1353442. [DOI] [PubMed] [Google Scholar]
- 37.Weiland NG, Orikasa C, Hayashi S, McEwen BS. Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol. 1997;388:603–612. doi: 10.1002/(sici)1096-9861(19971201)388:4<603::aid-cne8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woolley CS. Estradiol facilitates kainic acid-induced, but not flurothyl-induced, behavioral seizure activity in adult female rats. Epilepsia. 2000;41:510–515. doi: 10.1111/j.1528-1157.2000.tb00203.x. [DOI] [PubMed] [Google Scholar]
- 40.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- 42.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via a N-methyl-d-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woolley CS, Wenzel HJ, Schwartzkroin PA. Estradiol increases the frequency of multiple synapse boutons in the hippocampal CA1 region of the adult female rat. J Comp Neurol. 1996;373:108–117. doi: 10.1002/(SICI)1096-9861(19960909)373:1<108::AID-CNE9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 44.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]