

Abstract
During development of neuronal circuits, presynaptic and postsynaptic functions are adjusted in concert, to optimize interneuronal signaling. We have investigated whether activation of glutamate receptors affects presynaptic function during synapse formation, when constitutive synaptic vesicle recycling is downregulated. Using primary cultures of hippocampal neurons as a model system, we have found that chronic exposure to both NMDA and non-NMDA glutamate receptor blockers during synaptogenesis produces an increase in miniature EPSC (mEPSC) frequency, with no significant changes in mEPSC amplitude or in the number of synapses. Enhanced synaptic vesicle recycling, selectively in glutamatergic nerve terminals, was confirmed by the increased uptake of antibodies directed against the lumenal domain of synaptotagmin. No increased uptake was detected in neuronal cultures grown in the chronic presence of TTX, speaking against an indirect effect caused by decreased electrical activity. Enhanced mEPSC frequency correlated with a reduction of synaptophysin-synaptobrevin–vesicle-associated membrane protein 2 (VAMP2) complexes detectable by immunoprecipitation. Intracellular perfusion with a peptide that inhibits the binding of synaptophysin to synaptobrevin–VAMP2 induced a remarkable increase of mEPSC frequency in control but not in glutamate receptor blocker-treated neurons. These findings suggest that activation of glutamate receptors plays a role in the downregulation of the basal rate of synaptic vesicle recycling that accompanies synapse formation. They also suggest that one of the mechanisms through which this downregulation is achieved is an increased interaction of synaptophysin with synaptobrevin–VAMP2.
Keywords: hippocampal neurons, glutamate receptors, synaptic vesicle recycling, presynaptic release, synaptobrevin–VAMP2, synaptophysin
Activity-dependent regulation of synaptic properties has been shown to play a central role in modulating synaptic function in the mature nervous system. In the case of glutamatergic synapses in the mammalian CNS, a large body of evidence indicates that synaptic activity affects postsynaptic properties, for instance by controlling spine density and by regulating the synaptic localization of glutamate receptors. Glutamate released during spontaneous activity and acting on AMPA receptors has been shown to exert a trophic effect on spines (McKinney et al., 1999a), whereas synapse inactivation has been found to produce a compensatory enhancement of postsynaptic spine density (Kirov and Harris, 1999). Furthermore, synaptic activity operates a continuous remodeling of postsynaptic densities, regulates PSD-95 cluster dynamics (Okabe et al., 1999), and affects the redistribution of glutamate receptors toward or away from synaptic sites (Rao and Craig, 1997; O'Brien et al., 1998; Liao et al., 1999; Lissin et al., 1999). Finally, an increase in miniature EPSC (mEPSC) frequency has been demonstrated in hippocampal slice cultures chronically treated with NMDA receptor blockers, possibly as a consequence of presynaptic sprouting (McKinney et al., 1999b). Altogether, these data have indicated that activation of glutamate receptors influences the functional properties of the mature CNS synapses.
It is not as clear whether similar mechanisms also operate during neuronal development and synaptogenesis. Formation and maturation of synapses are complex processes, which require both the recruitment at sites of contact of proteins and organelles already present in the presynaptic and postsynaptic elements as well as a change in gene expression (Passafaro and Sheng, 1999; Sanes and Lichtman, 1999;Verderio et al., 1999a; Craig and Lichtman, 2000; Garner et al., 2000). A powerful experimental system to investigate the mechanisms of synaptogenesis is represented by primary cultures of hippocampal neurons. In these neurons synaptic vesicles are already present at very early developmental stages, before axons have made a contact with postsynaptic cells. At these stages, basal synaptic vesicle exocytosis occurs at high rate from the entire distal axonal arbor. Synaptogenesis coincides with a downregulation of this basal rate (Matteoli et al., 1992; Kraszewski et al., 1995; Coco et al., 1998; Verderio et al., 1999a) and with the coalescence of preassembled packages of synaptic vesicles into the larger clusters characteristic of mature synapses (Matteoli et al., 1992; Kraszewski et al., 1995; Ahmari et al., 2000). These changes are likely to depend, at least partially, on retrograde signals mediated by the activation of the postsynaptic side of the newly formed synaptic contact (Verderio et al., 1999b). We have now investigated the possible role of glutamate receptor activation in mediating the regulation of synaptic vesicle recycling. We demonstrate that neurons chronically exposed to glutamate receptor blockers during synaptogenesis are characterized by a higher basal rate of synaptic vesicle exocytosis. Furthermore, we provide evidence for a role of a decreased synaptophysin-synaptobrevin–vesicle-associated membrane protein 2 (VAMP2) interaction in this effect.
MATERIALS AND METHODS
Hippocampal cell cultures. Primary neuronal cultures were prepared from the hippocampi of 18-d-old fetal rats as previously described (Bartlett and Banker, 1984; Matteoli et al., 1992). APV (100 μm) and CNQX (20 μm) or TTX (1 μm) were added to the medium after 24 hr. Glutamate receptor inhibitors were removed immediately before electrophysiological recordings. Medium containing freshly prepared inhibitors was substituted every other day. A modification of the method of Furshpan et al. (1976) was used to grow single neurons on small islands of substrate, consisting in a fine mist of poly-l-lysine sprayed on glass coverslips (Verderio et al., 1999b).
Electrophysiology. Whole-cell patch-clamp recordings were obtained from 14- to 20-d-old neurons with an Axopatch 200B amplifier and pClamp software (Axon Instruments, Foster City, CA). Recordings were performed in the voltage-clamp mode. Currents were sampled at 2 kHz and filtered at 2–5 kHz. External solution [Krebs'—Ringer's–HEPES (KRH)] had the following composition (in mm): 125 NaCl, 5 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2 CaCl2, 6 glucose, and 25 HEPES–NaOH, pH 7.4. mEPSCs were recorded in the presence of 1 μm tetrodotoxin (TTX). Recording pipettes were fabricated from capillary glass using a two stage puller (Narishige, Tokyo, Japan) and had tip resistances of 3–5 MΩ when filled with the intracellular solution of the following composition (in mm): 130 K-gluconate, 10 KCl, 1 EGTA, 10 HEPES, 2 MgCl2, 4 MgATP, and 0.3 Tris-GTP. With such an intracellular solution, the chloride equilibrium potential was calculated to be approximately −63 mV. Voltage-clamp recordings were performed with a holding potential of −60 mV, thus avoiding contaminating GABAA-mediated responses. Recordings were performed at room temperature. Off-line analysis of mEPSCs used Axograph Software (Axon Instruments). Events had to exceed a threshold of two to three times the SD of the baseline noise. In a set of experiments a peptide corresponding to 1–32 N-terminal sequence of VAMP2 (SATAATVPPAAPAGEGGPPAPPPNLTSNRRL) was introduced into single neurons forming autaptic contacts by diffusion from the pipette. The concentration of the peptide in the pipette was 160 μm. Exchange times for small peptides was estimated to be 1–3 min based on test reagents in previous studies (Rosenmund et al., 1994). mEPSC activity was recorded for up to 30 min. Series resistance (80–90%) and the cell capacitance were compensated and continuously monitored during recording.
Exo-endocytotic assay. An exo-endocytotic assay to monitor SV recycling was performed using rabbit polyclonal antibodies directed against the intravesicular domain of rat synaptotagmin I [Syt-ecto antibodies (Abs)], applied for 3, 5, or 25 min, as previously described (Matteoli et al., 1992; Kraszewski et al., 1995). Incubations with the antibody were performed in KRH or in KRH containing 50 mm KCl, always in the presence of APV (100 μm) and CNQX (20 μm). After fixation and staining (Matteoli et al., 1992), cells were photographed with Kodak TMAX 400 film on a Zeiss Axiophot microscope equipped with epifluorescence microscopy or acquired with a Bio-Rad (Hercules, CA) MRC-1024 confocal microscope equipped with LaserSharp 3.2 software. Acquired images were processed and quantitatively analyzed with NIH Image software from National Institutes of Health (Bethesda, MD), as previously described (Coco et al., 1998; Verderio et al., 1999b).
Immunoblotting. Total homogenates from rat brains and cell extracts from cultured hippocampal neurons were subjected to SDS-PAGE electrophoresis, Western blotting, and immunostaining as described (Coco et al., 1997; Verderio et al., 1999b). Immunoreactive bands were visualized either with enhanced chemiluminescence (Amersham Pharmacia Biotech, Milan, Italy) or by iodinated protein A. Quantitation of the signal was performed by NIH Image software from the National Institutes of Health.
Immunoprecipitation. Neuronal cell pellets were dissolved in 1 ml of extraction buffer containing (in mm): KCl 140, EDTA 2, HEPES–KOH 20, pH 7.3, and 1% (v/v) Triton X-100. Extraction was performed for 1 hr at 4°C followed by a centrifugation for 3 min at 700 × g. We added ∼10 μg of IgG of the monoclonal antibodies against synaptobrevin–VAMP2 or polyclonal antibodies against synaptophysin overnight to 25 μl of G-Sepharose suspension (Amersham Pharmacia Biotech, Piscataway, NJ) or A-Agarose suspension (Pierce, Rockford, IL). The beads were collected by centrifugation at 200 × g for 1 min and added to 200 μl of extraction supernatant. Incubation was performed for 1 hr at 4°C. Beads were then collected by centrifugation at 200 ×g for 1 min, washed three times in extraction buffer, and analyzed by SDS-PAGE and Western blotting. The supernatant of the immunoprecipitation was analyzed in parallel (Becher et al., 1999b).
Antibodies. Rabbit polyclonal antibodies directed against the intravesicular domain of rat synaptotagmin I (Syt-ecto Abs) were generated as previously described (Matteoli et al., 1992) using a synthetic peptide corresponding to the residue 1–19 of the protein. Antibodies against SV2 and GAD were kind gift of Drs. K. Buckley (Harvard University, Boston, MA) and M. Solimena (Yale University, New Haven, CT), respectively. Antibodies against synaptobrevin–VAMP2, synaptophysin (monoclonal C7.1–4 and polyclonal G95), and synaptotagmin I were a kind gift of Dr. R. Jahn (Gottingen, Germany). Monoclonal antibodies against synaptophysin were purchased from Boehringer Mannheim (Indianapolis, IN). Antibodies against β-tubulin were from Sigma (Milan, Italy). Anti-rabbit rhodamine-conjugated antibodies were purchased from Boehringer Mannheim. Anti-mouse fluorescein-conjugated antibodies were from Jackson ImmunoResearch (West Grove, PA).
Statistical analysis. Results are presented as means ± SE. Data were statistically compared using the Student's ttest. Differences were considered significant if p < 0.05 and are indicated by an asterisk in all figures, whereas those atp < 0.01 are indicated by double asterisks.
RESULTS
Chronic blockade of glutamate receptors enhances mEPSC frequency
Primary cultures of embryonic hippocampal neurons were maintained in the chronic presence of glutamate receptor antagonists (APV, 100 μm; CNQX, 20 μm). Despite of the block of glutamate receptor activation, neurons were able to differentiate and to form a synaptic network. Basal synaptic activity, in the form of mEPSCs, was monitored after 14–20 d in culture, after removal of glutamate antagonists, in KRH and in the presence of 1 μmTTX. Figure 1 shows that chronic blockade of glutamate receptors markedly increases mEPSC frequency (control, 2.47 ± 0.38 Hz, n = 38; APV–CNQX-treated, 4.88 ± 0.63 Hz, n = 37; mean ± SE;p < 0.01) (Fig. 1C) with no significant changes in mEPSC amplitude (Fig. 1D). A similar increase in mEPSC frequency was recorded from single hippocampal neurons grown on poly-l-lysine microislands in control conditions or in the chronic presence of glutamate receptor blockers (mEPSC frequency, control, 2.07 ± 1.4 Hz,n = 17; APV–CNQX-treated, 4.54 ± 1.95 Hz,n = 18; mean ± SE; p < 0.01; mEPSC amplitude, control, 16.8 ± 1.9 pA, n = 17; APV–CNQX-treated, 17.7 ± 0.6 pA, n = 18; mean ± SE; p > 0.1).
Fig. 1.

Differential effect of glutamate receptor chronic blockade on mEPSC frequency and amplitude. A,Representative recordings from 15-d-old hippocampal neurons, maintained in control medium or in the presence of 100 μm APV and 20 μm CNQX. B, Distribution of mEPSC frequencies in control and APV–CNQX-treated cells. C, D, Analysis of the average mEPSC frequency (C) and cumulative amplitude distribution (D) in control and treated cells reveals the specific effect of glutamatergic chronic blockade on the frequency, but not the amplitude, of mEPSCs.
Elevations in mEPSC frequency could result from an increase in the number of synapses formed and/or from an increase in the presynaptic rate of release. To distinguish between these two possibilities, the number of synapses per micrometer of neurite was evaluated in control neurons (Fig.2A) and in neurons grown in the presence of APV and CNQX (Fig. 2B). Synaptic contacts were revealed by immunofluorescence staining for the synaptic vesicle protein SV2, which specifically labels synaptic contacts in hippocampal cultures (Matteoli et al., 1992; Coco et al., 1997). Figure 2C shows the lack of any significant difference in the number of synapses per micrometer of neurite between control and APV–CNQX-treated neurons (number of synapses per micrometer of neurite, control, 0.183 ± 0.02; number of cells = 19; APV–CNQX, 0.188 ± 0.01; number of cells = 24; p > 0.1).
Fig. 2.
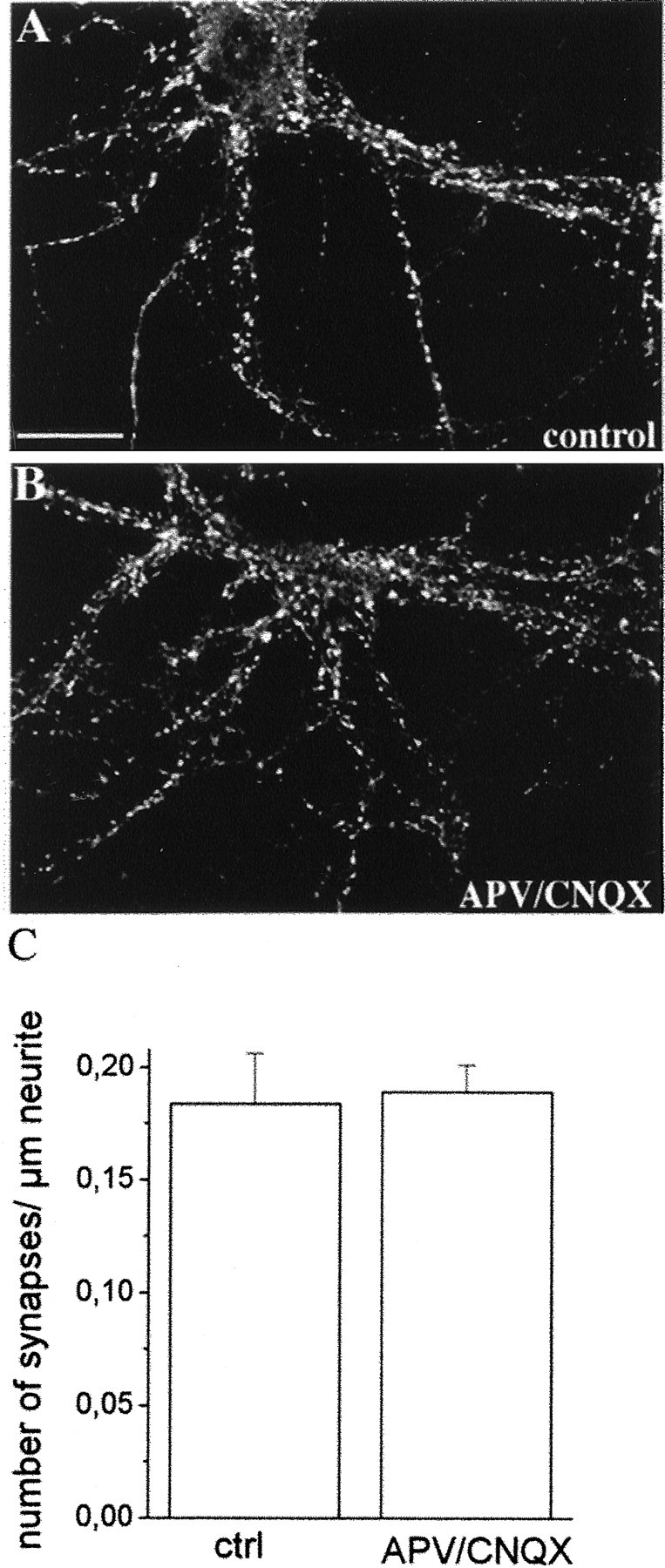
Glutamatergic block does not affect synapse number. Immunofluorescence stainings of control (A) and APV–CNQX-treated (B) cultures with antibodies directed against the synaptic vesicle protein SV2. Immunofluorescent puncta represent sites of synaptic contacts. Scale bar, 20.8 μm. C, Histogram showing the quantitative analysis of the number of synapses present for micrometer of neurite length. No significant difference is detectable between control and APV–CNQX-treated neurons.
Enhanced mEPSC frequency reflects an increased rate of synaptic vesicle exo-endocytotic recycling
To determine whether the change in mEPSC frequency reflected an enhanced basal rate of synaptic vesicle exocytosis, independent of postsynaptic effects, we investigated whether an increase in this rate could be detected in neurons grown in the continual presence of the glutamate receptor blockers APV–CNQX. At this aim, we used an optical method to measure synaptic vesicle exocytosis and recycling: the immunocytochemical assay based on antibodies directed against the intravesicular domain of the synaptic vesicle protein synaptotagmin I (Syt-ecto Abs). These antibodies are internalized in the lumen of synaptic vesicles after their exo-endocytosis (Matteoli et al., 1992;Kraszewski et al., 1995; Malgaroli et al., 1995; Coco et al., 1998;Verderio et al., 1999b), and their uptake closely reflects levels of vesicle recycling. Mature cultures, grown in control medium (Fig.3A–C) or in the presence of glutamate receptor blockers (Fig. 3D–F), were exposed to Syt-ecto Abs for 5 min in the same extracellular solution used for electrophysiology, in the presence of glutamate receptor blockers and in the absence of external stimuli. They were then fixed, permeabilized, stained for the internalized antibodies, and counterstained for the synaptic vesicle protein SV2 to reveal the entire synaptic population. Figure 3A–C (see also details in Fig. 3G,H) shows that, in control neurons, only few synaptic contacts were labeled by internalized Syt-ecto Abs (Fig.3B,G) above threshold levels (Mundigl et al., 1995). However, when neurons were grown in the presence of APV and CNQX, a high percentage of synapses were clearly positive for internalized Syt-ecto Abs (Fig. 3E,M). The percentages of labeled synapses in control and treated cultures were found to be 24.55 ± 2.00 in control cultures (number of examined synapses = 69,834) and 43.46 ± 2.2 in APV–CNQX-treated cultures (number of examined synapses = 59,323; p < 0.001) (Fig.3Q). An increase in the number of labeled synaptic contacts was detected when control and treated cultures were incubated for 25 min in the presence of Syt-ecto Abs. However, even in this case, the number of synaptic contacts labeled above threshold was found to be significantly higher in cultures treated with glutamate receptor antagonists [control, percentages of labeled synapses = 44.83 ± 3.3; number of examined synapses = 43,939; APV–CNQX-treated, percentages of labeled synapses = 55.89 ± 2.8, number of examined synapses 33,253; p < 0.05 (Fig. 3Q)]. When neurons were stimulated by a brief application of 55 mm KCl (Fig.3I,L,O,P), virtually all synaptic contacts were found to be labeled by the internalized Syt-ecto Abs (Fig. 3I,O) both in control (Fig. 3I) and treated (Fig. 3O) cultures (control, percentages of labeled synapses = 95.6 ± 3.5, number of examined synapses = 14,869; APV–CNQX-treated, percentages of labeled synapses = 93.4 ± 3.2; number of examined synapses = 12,597; p > 0.1) (Fig.3Q). No difference in the number of labeled synapses could be detected between control cultures and cultures grown in the chronic presence of TTX (data not shown). These data demonstrate that synapses formed in vitro in the presence of glutamate receptor blockers have a higher rate of basal synaptic vesicle exo-endocytosis.
Fig. 3.

Chronic treatment with glutamatergic blockers upregulates synaptic vesicle recycling. We incubated 15-d-old neurons from control (A–C,G–L) or APV–CNQX-treated (D–F, M–P) cultures for 5 min (A–H, M, and N, histogram in Q) or 25 min (histogram inQ) in the presence of Syt-ecto Abs in KRH containing glutamate receptor blockers. In a set of experiments, incubation was performed for 3 min in the presence of 55 mm KCl (I,L, O,P, histogram in Q). After this incubation, neurons were washed, fixed, detergent-permeabilized, reacted with fluorescein-conjugated goat anti-rabbit IgGs (B, E, G,I, M,O), and counterstained with antibodies against SV2 followed by rhodamine-conjugated goat anti-mouse IgGs (A,D, H, L, N,P). Puncta of immunoreactivity represent presynaptic nerve terminals that outline perikarya and dendrites. Syt-ecto Abs are internalized only at few synaptic contacts in control cultures (B, G) and in most synaptic contacts in APV–CNQX-treated neurons (E, M).C,F, Fluorescein- and rhodamine-merged images. Incubation in the presence of KCl results in Syt-ecto Ab internalization in virtually all synaptic contacts, both in control (I) and treated (O) cultures. Scale bars: A–C, 20.8 μm;D–F, 25 μm; G–P, 3.5 μm.Q, Histogram showing the quantitative evaluation of Syt-ecto-positive synapses in control and in APV–CNQX-treated neurons.
The effect of glutamate antagonists is selective for glutamatergic synapses
In primary cultures of hippocampal neurons, a low percentage of the total neuronal population is represented by interneurons that use GABA as neurotransmitter (Benson et al., 1994). We investigated therefore whether APV–CNQX treatment affected synaptic vesicle recycling selectively in glutamatergic or also in GABAergic nerve terminals. Control (Fig.4A–C, details in Fig.4G,H) and APV–CNQX-treated (Fig.4D–F, details in Fig. 4I,L) neurons were exposed in basal conditions to Syt-ecto Abs for 5 min, and then stained, after fixation, both for the internalized antibodies and for the GABA-synthesizing enzyme glutamic acid decarboxylase (GAD). This enzyme is a marker of GABAergic nerve terminals (Fig.4A,D,G,I). Interestingly, although only ∼25% of all synapses are labeled for Syt-ecto Abs above threshold under this condition (see above), nearly all GAD-positive synapses were also positive for Syt-ecto Abs (Fig.4A,C,G,H). Thus, even in control conditions, these inhibitory synapses are characterized by a high rate of synaptic vesicle exocytosis. In neurons grown in APV–CNQX-containing medium, a large number of synapses showed high rate of recycling (Fig.4E,L). The number of such recycling synapses largely exceeded GABAergic synapses (Fig.4D–F,I,L), suggesting a massive effect of APV–CNQX on the rate of synaptic vesicle exo-endocytosis at glutamatergic boutons (Fig. 4M) (percentage of Syt-ecto-positive synapses, control, 124 ± 20.7; number of cells = 15; APV–CNQX, 304.7 ± 40, number of cells = 17; values normalized to GAD-positive synapses; p < 0.001). To further establish whether APV–CNQX altered the extent of synaptic vesicle recycling occurring at GABAergic terminals, the intensity of the immunocytochemical labeling for internalized Syt-ecto Abs was normalized to the intensity of GAD immunostaining in control and APV–CNQX-treated synapses. Figure 4N shows that APV–CNQX treatment did not significantly affect the ratio between Syt-ecto Abs and GAD immunocytochemical signals (Syt-ecto Abs/GAD mean density, control, 1.05 ± 0.03; n = 31; APV–CNQX, 1.15 ± 0.05, n = 33, p > 0.1). Furthermore, no significant differences in GAD expression were detected in control versus treated neurons (data not shown). Altogether these data suggest that the functional block of glutamatergic receptors specifically affects the extent of synaptic vesicle recycling at glutamatergic, but not at GABAergic, nerve terminals.
Fig. 4.

Blockade of glutamate receptors affect glutamatergic, but not GABAergic, presynaptic nerve terminals.A–F, Immunofluorescence images of control (A–C) and APV–CNQX-treated (D–F) 15-d-old hippocampal neurons incubated for 5 min in the presence of Syt-ecto Abs (B,E) and counterstained with antibodies against the synthetic enzyme GAD (A, D). In control neurons, the few synapses labeled by Syt-ecto Abs are generally GAD positive (A, B, see also merged image inC). In APV–CNQX-treated neurons, the synapses positive for Syt-ecto Abs (E) largely exceed GABAergic terminals (D) (see merged image inF). G–L, Details of control (G, H) and APV–CNQX-treated (I, L) neurons stained for Syt-ecto Abs (H, L) and for GAD (G,I). Scale bar: A–F, 30.7 μm;G–L, 10.2 μm. M, Histogram showing the quantitative analysis of the percentages of Syt-ecto Ab-positive synapses in control and APV–CNQX-treated neurons, normalized to the number of GAD-positive synapses. N, Histogram showing that the amount of internalized Syt-ecto Abs is not significantly different in GABAergic terminals of control and treated neurons. Syt-ecto Ab intensity values are normalized to GAD immunoreactivity.
The stimulatory effect of glutamate receptor blockers on mEPSC frequency correlates with a decreased level of synaptophysin-synaptobrevin–VAMP2 complex
We have previously shown that formation of synaptic contacts among hippocampal neurons in vitro correlates with a change in the basal rate of synaptic vesicle exo-endocytosis. Before the formation of synaptic contacts, basal synaptic vesicle exocytosis occurs at high rate from the entire distal axonal arbor. Formation of synaptic contacts coincides with a downregulation of this basal rate (Kraszewski et al., 1995; Coco et al., 1998; Verderio et al., 1999a) and with the coalescence of packages of synaptic vesicles into the larger clusters characteristic of mature synapses (Matteoli et al., 1992; Kraszewski et al., 1995; Ahmari et al., 2000). This change was found to be dependent on the presence of the postsynaptic site, as strikingly demonstrated by the analysis of single neurons grown on poly-l-lysine-coated microislands. The axons of these neurons form autaptic contacts (Fig.5A–C) but often extend additional branches that lack a postsynaptic target, thus allowing the simultaneous analysis in a same cell of synaptic vesicle exocytosis in compartments facing or not a postsynaptic target (Verderio et al., 1999b). Incubation of these neurons with Syt-ecto Abs under conditions that produce labeling of isolated cells but no detectable labeling of mature autapses (5 min in basal conditions) (Fig. 5C, small arrowheads) does result in an intense labeling of isolated axonal branches (Fig. 5C, large arrowhead). The higher rate of synaptic vesicle recycling observed at glutamatergic synapses after chronic exposure to glutamate receptor antagonists seems to recapitulate immature stages of neuronal development. We tested therefore whether the synaptic vesicle recycling process in drug-treated synapses had some properties characteristic of the “immature” nonsynaptic release.
Fig. 5.

Basal rate of exo-endocytosis is downregulated after contact with a postsynaptic cell. A–C, Synaptic vesicle exo-endocytosis occurs at different basal rates, in distinct compartments of a same 14-d-old neuron grown in a microisland, depending on the contact with the postsynaptic target. An efficient internalization of Syt-ecto Abs takes place in basal conditions in the isolated axon (C, large arrowhead) but not at autaptic contacts (C, small arrowheads) of the same neuron, shown as a bright field in A. B, Double immunolabeling of the same neuron for the synaptic vesicle protein SV2. Scale bar, 18.75 μm.
It is well established that synaptophysin can form a complex with synaptobrevin–VAMP2, and this complex is mutually exclusive with the association of synaptobrevin–VAMP2 with other solubleN-ethylmaleimide-sensitive factor attached protein (SNAP) receptors (SNAREs) in the fusion complex (Edelman et al., 1995;Washbourne et al., 1995). The property of synaptophysin to associate with synaptobrevin–VAMP2 was reported to be regulated during development and to occur only after formation of synaptic contacts (Becher et al., 1999a,b). We therefore investigated whether treatment with glutamate receptor blockers altered the level of the synaptophysin-synaptobrevin–VAMP2 complex. Immunoprecipitations with antibodies directed against synaptobrevin–VAMP2 (Fig.6A) or synaptophysin (data not shown) were performed from extracts of 12- to 14-d-old control cultures or from cultures chronically treated with glutamate receptor blockers. Western blot analysis of the immunoprecipitates revealed that the presence of complexes was drastically reduced (62% average reduction) in neurons grown in the continual presence of the drugs. No reduction in the complex formation was detected in cultures chronically treated with TTX (Fig. 6A).
Fig. 6.
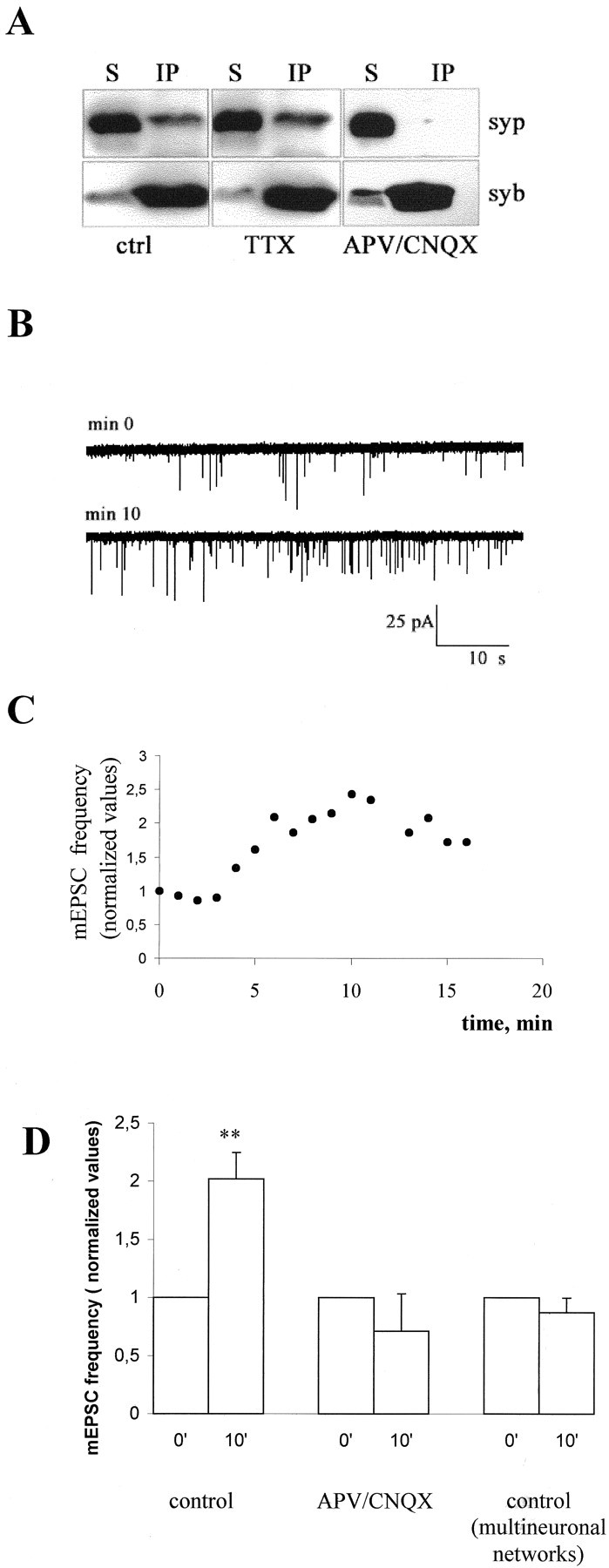
Impairment in the formation of the synaptophysin-synaptobrevin–VAMP2 complex is detectable in APV–CNQX-treated neurons and is responsible for the increase in mEPSC frequency. A, Triton X-100 extracts of control and APV–CNQX-treated 12-d-old neurons were immunoprecipitated using the monoclonal antibody against synaptobrevin–VAMP2 (syb). Immunoprecipitates (IP) and their corresponding supernatants (S) were analyzed using polyclonal antibodies against synaptophysin (syp). Note that synaptophysin is efficiently immunoprecipitated from control cultures and is almost completely detectable in the supernatant of APV–CNQX- treated cultures. B, Representative recordings from a single 12-d-old hippocampal neuron forming autaptic contacts, intracellularly perfused via the patch pipette with a peptide corresponding to the 32-residue-long N-terminal segment of synaptobrevin–VAMP2, which inhibits complex formation. C, Time course of the increase in the frequency of mEPSCs recorded from the same neuron as inB. D, Histogram showing the increase in mEPSC frequency occurring in control neurons 10–11 min after the beginning of recordings (p < 0.002). No effect is detectable in either single neurons maintained in APV–CNQX (p > 0.1) or in neurons grown in polyneural networks (p > 0.1). Values are normalized to the first or second minute of recording.
The perturbation of synaptophysin-synaptobrevin–VAMP2 interaction increases mEPSC frequency in control but not in glutamate receptor blocker-treated neurons
A 32-residue-long N-terminal segment of synaptobrevin–VAMP2 was previously shown to inhibit complex formation in vitro(Washbourne et al., 1995). To investigate whether the formation of the synaptophysin-synaptobrevin–VAMP2 complex plays a role in downregulating the constitutive fusion rate of synaptic vesicles, mEPSCs were recorded from single hippocampal neurons growing in microislands and forming autaptic contacts, during the intracellular perfusion of this peptide via the patch pipette. The peptide was found to cause a remarkable increase in the mEPSC frequency recorded from control but not from APV–CNQX-treated neurons (Fig.6B–D). Ten or eleven minutes after the beginning of recording, the frequency of mEPSCs was found to be 2.02 ± 0.22 in control cells (values normalized to the first or second minute of recording; n = 6; p < 0.002), and 0.71 ± 0.31 in APV–CNQX-treated cells (n = 5;p > 0.1) (Fig. 6D). No change in mEPSC frequency was recorded from neurons in multineuronal networks, which receive synaptic inputs from non injected neurons, excluding therefore a postsynaptic site of action for the peptide (mEPSC frequency in multineuronal networks 10 or 11 min after the beginning of recording, 0.87 ± 0.12; values normalized to the first or second minute of recording, n = 5;p > 0.1) (Fig. 6D). No increase in mEPSC frequency was detected in either control or APV–CNQX-treated neurons intracellularly perfused with a scrambled peptide or with the peptide corresponding to the 1–32 N-terminal residues of synaptobrevin–VAMP1 (data not shown). Although other mechanisms of action of the peptide cannot be ruled out, these findings strongly support the hypothesis that the interaction between synaptobrevin–VAMP2 and synaptophysin has a role in the regulation of synaptic vesicle exocytosis and that a decrease of this complex plays a role in the effect of chronic treatment with glutamate receptor blockers.
DISCUSSION
We have investigated the role of glutamate receptor activation on presynaptic function in primary cultures of hippocampal neurons. We show that the chronic blockade of NMDA and non-NMDA glutamate receptors during synaptogenesis produces an increase in mEPSC frequency, with no significant changes in mEPSC amplitude, both in multineuronal networks and in single neurons grown in microislands. Furthermore, we demonstrate that the increase in mEPSC frequency, produced by the long-term treatment of hippocampal cultures with glutamate receptor antagonists, occurs in the absence of significant changes in the number of synaptic sites. The increase in mEPSC frequency appears instead to correlate with an increase in the basal rate of synaptic vesicle exo-endocytotic recycling, specifically occurring at glutamatergic terminals. A similar increase in mEPSC frequency, but not amplitude, has been previously demonstrated in hippocampal slice cultures as a consequence of the chronic treatment with NMDA receptor blockers. In the latter experimental model, however, antagonists were applied after synapses had already established, and the observed effect was suggested to result from massive sprouting and neosynaptogenesis (McKinney et al., 1999b). Interestingly, short time exposure (2–3 d) of cortical or spinal cultures to glutamate receptor blockers after neurons have already established synaptic contacts results in an enhancement of mEPSC amplitude (O'Brien et al., 1998; Turrigiano et al., 1998). This suggests that blocking of synaptic activity may result in distinct functional changes, depending on the specific time window in which such a blockade occurs (Gomperts et al., 2000). In our experiments, the increase in mEPSC frequency, recorded from neurons chronically treated with glutamate blockers, is not accompanied by differences in the amplitude of mEPSCs. The lack of effect on mEPSC amplitude is in contrast with some previously published work, reporting a decrease in mEPSC amplitude after chronic glutamate receptor blockade in hippocampal postnatal neurons in culture (Gomperts et al., 2000). This discrepancy may be explained by differences between culture preparations (embryonic vs postnatal cultures) or other experimental variables (presence vs absence of glial cells, which are known to influence both the number and the properties of newly formed synapses;Pfrieger and Barres, 1997; Verderio et al., 1999c; Ullian et al., 2001).
The increase in presynaptic rate of exo-endocytotic recycling, induced by the chronic blockade of glutamate receptors may be produced by changes in the regulation of the synaptic vesicle release machinery and one of such changes may be (or may be reflected by) a modified interaction of synaptophysin with synaptobrevin–VAMP2. It has been reported previously that the formation of the complex between synaptophysin and synaptobrevin–VAMP2 occurs only at mature synapses and not in developing neurons (Becher et al., 1999a,b). This complex is mutually exclusive with the interaction of synaptobrevin–VAMP2 with syntaxin and SNAP25 and is therefore thought to play an inhibitory role on the formation of the SNARE fusion complex (Calakos and Scheller, 1994; Edelman et al., 1995; Washbourne et al., 1995). We now demonstrate that the functional block of glutamate receptors results in a downregulation of the synaptophysin-synaptobrevin–VAMP2 complex. This downregulation, in turn, may play a role in the increase in spontaneous release from the presynaptic terminal, as indicated by our demonstration that the intracellular injection of a peptide that strongly inhibits complex formation in vitro (Washbourne et al., 1995) enhances spontaneous (this study) and evoked (E. Pravettoni, S. Armano, and M. Matteoli, unpublished observations) neurotransmitter release in single neurons forming autaptic contacts. The same peptide did not produce any detectable increase in mEPSC frequency either in single neurons chronically grown in the presence of APV–CNQX, where complex formation is already impaired, or in control polyneuronal networks, thus excluding a postsynaptic site of action for the peptide. The developmentally regulated property of synaptophysin to associate with synaptobrevin–VAMP2, correlates with a change in its antigenic characteristics during neuronal development both in situ and in culture (S. Coco, U. Schenk, P. De Camilli, and M. Matteoli, unpublished observations). Possibly, the two changes are closely linked to each other and may reflect a post-translational modification of synaptophysin (Becher et al., 1999a). In agreement with this possibility, treatment with glutamate receptor blockers prevents a shift in the antigenic properties of synaptophysin, which occur during neuronal development in situ and in culture (Coco, Schenk, De Camilli, and Matteoli, unpublished observations). In view of our data, the lack of synaptic transmission defects in synaptophysin knock-out mice (McMahon et al., 1996) is quite puzzling. However, chronic adjustments and/or redundancy with other proteins could explain this discrepancy.
It remains unclear how glutamate receptor blockade generates a signal that is then transduced into presynaptic changes. Enhanced mEPSC frequency is unlikely to be explained by a reduced neuronal spiking caused by blockade of excitatory innervation, because neither impairment of synaptophysin-synaptobrevin–VAMP2 complex formation nor increase in synaptic vesicle recycling were detectable in neurons chronically grown in the presence of TTX. Although we cannot exclude a possible involvement of presynaptic glutamate receptors, it is notable that APV–CNQX treatment specifically affects glutamatergic, but not GABAergic terminals, in spite of the known presence of glutamate receptors on inhibitory terminals (Satake et al., 2000). This finding, together with the property of axons to switch from a “high basal rate” to a “low basal rate” after contact with a postsynaptic cell (this study, Kraszewski et al., 1995; Coco et al., 1998), supports the view that the effects of APV–CNQX may be attributable, at least in part, to a postsynaptic block that in turn impairs a negative feedback signaling to the presynapse, preventing therefore a full maturation of the presynaptic compartment. Such signals could, for example, be mediated by neurotrophic factors that act on presynaptic receptors. Interestingly, inhibitory GABAergic presynaptic terminals are characterized by a higher rate of synaptic vesicle exo-endocytosis than excitatory terminals (this study; Burke and Rudomin, 1977). These terminals do not appear to downregulate synaptic vesicle recycling when synapses form (our unpublished observations). One could therefore hypothesize the existence of a developmentally regulated tuning of presynaptic release that operates selectively at excitatory synapses and that can be partially prevented when postsynaptic excitatory receptors are blocked.
A role of postsynaptic cells in regulating presynaptic transmitter release has been clearly demonstrated at Drosophila and mammalian neuromuscular junctions. In Drosophilaloss-of-function mutants of DGluRII, a muscle-specific glutamate receptor, the decreased postsynaptic sensitivity is compensated for by an increase in transmitter release from the neuron. Such increase occurs in the absence of any sprouting of the presynaptic arborization (Petersen et al., 1997). Interestingly, similar changes in presynaptic transmitter release have been documented in neuregulin knock-out mice and in animal models of myasthenia gravis (Cull-Candy et al., 1980;Plomp et al., 1992; Sandrock et al., 1997). Together, these data have suggested the existence, at the neuromuscular junction, of a muscle-to-motoneuron retrograde signaling that modulates presynaptic release (Davis and Goodman, 1998; Davis et al., 1998). Our data support the existence of similar mechanisms in CNS neurons during development and synaptogenesis. In these immature neurons, the efficiency of interneuronal signaling is in the middle of a dynamic range, which can be tuned, either postsynaptically or presynaptically, depending on the demand of the target cells for receiving an appropriate amount of neurotransmitter. Functional block of glutamate receptors could therefore mimic an earlier stage of development, in which neurons, characterized by a reduced receptor responsivity, require an increased release of transmitter from presynaptic terminals.
Footnotes
This work was supported by European Community Grant QLGR3-CT-2000-01343 (coordinator E. Scarfone), by Telethon Italy Grant 1042 (M.M.), and by National Institutes of Health Grant NS 36251 (P.D.C.). We thank Roberto Gramignoli for help in some experiments. We thank Drs. R. Jahn (Gottingen, Germany) and M. Solimena (Yale University) for the generous gift of antibodies. We also thank Prof. C. Montecucco (University of Padova), Prof. F. Clementi (University of Milan), and Prof. F. Valtorta (DIBIT, Milan) for discussion.
A.B. and S.C. contributed equally to this paper.
Correspondence should be addressed to Dr. Michela Matteoli, Consiglio Nazionale delle Ricerche Cellular and Molecular Pharmacology, and “B. Ceccarelli” Centers, Department of Medical Pharmacology, University of Milan, via Vanvitelli 32, 20129 Milan, Italy. E-mail:MichelaM@csfic.mi.cnr.it.
A. Bacci's present address: Department of Neurology and Neurological Sciences, Stanford University Medical Center, Stanford, CA 94035.
REFERENCES
- 1.Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett WP, Banker GA. An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. I. Cells which develop without intercellular contacts. J Neurosci. 1984;4:1944–1953. doi: 10.1523/JNEUROSCI.04-08-01944.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becher A, Drenckhahn A, Pahner I, Margittai M, Jahn R, Ahnert-Hilger G. The synaptophysin-synaptobrevin complex: a hallmark of synaptic vesicle maturation. J Neurosci. 1999a;19:1922–1931. doi: 10.1523/JNEUROSCI.19-06-01922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becher A, Drenckhahn A, Pahner I, Ahnert-Hilger G. The synaptophysin-synaptobrevin complex is developmentally upregulated in cultivated neurons but is absent in neuroendocrine cells. Eur J Cell Biol. 1999b;78:650–656. doi: 10.1016/S0171-9335(99)80050-8. [DOI] [PubMed] [Google Scholar]
- 5.Benson DL, Watkins FH, Stewaed O, Banker GA. Characterization of GABAergic neurons in hippocampal cell cultures. J Neurocytol. 1994;23:279–295. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- 6.Burke RE, Rudomin P. In: Handbook of physiology, Section 1, The nervous system (Brookhart JM, Mountcastle VM, Kandel ER, Geiger SR, eds), pp 877–944. Williams and Wilkins; Baltimore: 1977. [Google Scholar]
- 7.Calakos N, Scheller RH. Vesicle-associated membrane protein and synaptophysin are associated on the synaptic vesicle. J Biol Chem. 1994;269:24534–24537. [PubMed] [Google Scholar]
- 8.Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- 9.Coco S, Verderio C, De Camilli P, Matteoli M. Calcium dependence of synaptic vesicle recycling before and after synaptogenesis. J Neurochem. 1998;71:1987–1992. doi: 10.1046/j.1471-4159.1998.71051987.x. [DOI] [PubMed] [Google Scholar]
- 10.Craig AM, Lichtman JW. Synapse formation and maturation. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. John Hopkins University; Baltimore: 2000. pp. 571–612. [Google Scholar]
- 11.Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human endplates. J Physiol (Lond) 1980;299:621–638. doi: 10.1113/jphysiol.1980.sp013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis GW, Goodman CS. Genetic analysis of synaptic development and plasticity: homeostatic regulation of synaptic efficacy. Curr Opin Neurobiol. 1998;8:149–156. doi: 10.1016/s0959-4388(98)80018-4. [DOI] [PubMed] [Google Scholar]
- 13.Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- 14.Edelman L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci USA. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garner CC, Nash J, Huganir RL. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 2000;10:274–280. doi: 10.1016/s0962-8924(00)01783-9. [DOI] [PubMed] [Google Scholar]
- 17.Gomperts SN, Carroll R, Malenka RC, Nicoll RA. Distinct roles for ionotropic and metabotropic glutamate receptors in the maturation of excitatory synapses. J Neurosci. 2000;20:2229–2237. doi: 10.1523/JNEUROSCI.20-06-02229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- 19.Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao D, Zhang X, O'Brien RJ, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- 21.Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malgaroli A, Ting AE, Wendland B, Bergamaschi A, Villa A, Tsien RW, Scheller RH. Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science. 1995;268:1624–1628. doi: 10.1126/science.7777862. [DOI] [PubMed] [Google Scholar]
- 23.Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999a;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- 25.McKinney RA, Luthi A, Bandtlow CE, Gahwiler BH, Thompson SM. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Natl Acad Sci USA. 1999b;96:11631–11636. doi: 10.1073/pnas.96.20.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMahon HT, Bolshakov VY, Janz R, Hammer RE, Siegelbaum SA, Sudhof TC. Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc Natl Acad Sci USA. 1996;93:4760–4764. doi: 10.1073/pnas.93.10.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mundigl O, Verderio C, Kraszewski K, De Camilli P, Matteoli M. A radioimmunoassay to monitor synaptic activity in hippocampal neurons in vitro. Eur J Cell Biol. 1995;66:246–256. [PubMed] [Google Scholar]
- 28.O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 29.Okabe S, Kim HD, Miwa A, Kuriu T, Okado H. Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci. 1999;2:804–811. doi: 10.1038/12175. [DOI] [PubMed] [Google Scholar]
- 30.Passafaro M, Sheng M. Synaptogenesis: the MAP location of GABA receptors. Curr Biol. 1999;9:R261–263. doi: 10.1016/s0960-9822(99)80159-6. [DOI] [PubMed] [Google Scholar]
- 31.Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- 32.Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin treated rats. J Physiol (Lond) 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 34.Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 35.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 36.Sandrock AW, Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–604. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- 37.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 38.Satake S, Saitow F, Yamada J, Konishi S. Synaptic activation of AMPA receptors inhibits GABA release from cerebellar interneurons. Nat Neurosci. 2000;3:551–558. doi: 10.1038/75718. [DOI] [PubMed] [Google Scholar]
- 39.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 40.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–659. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 41.Verderio C, Coco S, Pravettoni E, Bacci A, Matteoli M. Synaptogenesis in hippocampal cultures. Cell Mol Life Sci. 1999a;55:1448–1462. doi: 10.1007/s000180050384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses, but not of synaptic vesicles in isolated axons. J Neurosci. 1999b;19:6723–6732. doi: 10.1523/JNEUROSCI.19-16-06723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verderio C, Bacci A, Coco S, Pravettoni E, Fumagalli G, Matteoli M. Astrocytes are required for the oscillatory activity in hippocampal neurons. Eur J Neurosci. 1999c;11:2793–2800. doi: 10.1046/j.1460-9568.1999.00697.x. [DOI] [PubMed] [Google Scholar]
- 44.Washbourne P, Schiavo G, Montecucco C. Vescicle-associated membrane protein-2 (synaptobrevin-2) forms a complex with synaptophysin. Biochem J. 1995;305:721–724. doi: 10.1042/bj3050721. [DOI] [PMC free article] [PubMed] [Google Scholar]