

Abstract
In addition to actions mediated by changes in gene expression, steroids can directly modulate several transmitter-gated and voltage-gated ion channels. Despite numerous studies showing that steroids enhance or reduce ion channel activity, the site(s) that mediates steroid recognition is not known. To identify the regions in which steroids bind and affect ion channel activity, we have taken advantage of the observation that human α4β2 neuronal nicotinic receptors are potentiated by an estrogen steroid, 17β-estradiol, whereas a rat α4β2 receptor is not. Mutations indicate that a sequence (AGMI) at the end of the C terminus of the human α4 subunit forms a binding site required for 17β-estradiol potentiation. In contrast, ethynyl β-estradiol (an oral contraceptive) potentiates both human and rat α4β2 receptors. A single tryptophan in the C terminus of both the rat and the human α4 subunit is sufficient for potentiation by ethynyl β-estradiol, probably through a π–π interaction. Mutation of this tryptophan eliminates the ability of ethynyl β-estradiol to potentiate rat receptors. However, in human receptors it was necessary to mutate both the AGMI sequence and the tryptophan to eliminate potentiation by ethynyl β-estradiol. The findings that β-estradiol requires the AGMI sequence but that a single C-terminal tryptophan is sufficient for potentiation by ethynyl β-estradiol indicate that the C terminus forms a binding site for these steroids. The binding site(s) for block appears to differ from those involved in potentiation because the C-terminal sequence does not affect block by steroids such as progesterone, and progesterone does not competitively inhibit potentiation.
Keywords: nicotinic receptors, estrogen, steroids, potentiation, binding site, ligand-gated ion channel
The physiological actions of steroids are typically associated with initiation of gene transcription after binding to cytosolic steroid receptors. However, steroids can also have immediate effects that require a more direct mechanism of action, including effects on neuronal excitability by “neuroactive” steroids (Paul and Purdy, 1992; Rupprecht and Holsboer, 1999). Rapid modulation of neuronal excitability by neuroactive steroids is particularly interesting because some of these steroids are produced in the brain (so-called neurosteroids), either de novo from cholesterol or by enzyme-mediated modification of another steroid (Robel and Baulieu, 1995). Localized production and the ability to alter ion channel kinetics suggest the possibility of selective actions of neurosteroids in specific brain regions. For example, aromatase P450, the enzyme that produces estradiol, is found both presynaptically and postsynaptically (Balthazart and Ball, 1998), and RNA encoding aromatase P450 is found in the hippocampus and the temporal and frontal neocortex of the adult human brain (Stoffel-Wagner et al., 1999). Estrogen has been shown to influence various ion channels (McEwen and Alves, 1999; Valverde et al., 1999); therefore, local estrogen synthesis could affect activity at both sides of the synapse.
Although it is thought that steroids can bind to ion channels, efforts to locate a specific binding site have been unsuccessful. Here, we show that 17β-estradiol, an estrogenic steroid, directly potentiates responses from the human α4β2 neuronal nicotinic receptor, and that the C terminus of the α4 subunit is likely to form the binding site necessary for potentiation.
The α4β2 receptor represents a major part of the total brain content of nicotinic receptors (Flores et al., 1992), accounts for 80–90% of the high-affinity nicotine binding sites in the brain (Marubio et al., 1999), and is diminished in the brains of patients with Alzheimer's disease (Sugaya et al., 1990; Wevers et al., 1999). Nicotinic receptors are involved in a variety of neurological disorders (Kuryatov et al., 1997; Lindstrom, 1997; Paterson and Nordberg, 2000), and increasing evidence suggests that presynaptic nicotinic receptors have an important physiological role in modulating the release of many neurotransmitters (McGehee et al., 1995; Gray et al., 1996; Coggan et al., 1997; Wonnacott, 1997; Paterson and Nordberg, 2000). Therefore, modulation of nicotinic receptors by estrogen or other compounds is likely to influence synaptic signaling in several pathways.
We previously established that 17β-estradiol, βE2, inhibits a receptor formed from a rat α4β2 neuronal nicotinic receptor (Paradiso et al., 2000), in contrast to the ability of βE2 to potentiate human α4β2 receptors (Buisson et al., 1998). We explored this apparent contradiction to determine the difference between block and potentiation and to identify the portions of the protein that are responsible for potentiation.
MATERIALS AND METHODS
Synthesis and expression of cDNA encoding chimeric and mutated subunits. cDNA constructs for human α4 (Kuryatov et al., 1997) and β2 (Anand and Lindstrom, 1990) subunits were kindly provided by Dr. Jon Lindstrom (University of Pennsylvania, Philadelphia, PA) in pSP64 and were transferred to pcDNA3 (Invitrogen, Carlsbad, CA) for use. Rat α4 and β2 subunits were kindly provided by Dr. J. Patrick (Baylor College of Medicine, Houston, TX) (Sabey et al., 1999). Because of the high homology between the rat and human subunits, identical restriction sites could be found for the production of chimeras. Three sets of chimeric α4 subunits were made; for each set, one chimera was made with the rat sequence from the N terminus to the chimeric join, whereas the other chimera was made with the human sequence at the N terminus. The chimera pairs were made at the following amino acid residues (numbered for the mature protein): X1, 131/132 (AatII); X2, 296/297 (Alw44I); and X3, rat 268/human 268 or human 267/rat 269 (ScaI). The human α4 subunit was excised with HindIII and BamHI, whereas rat α4 was excised with HindIII andXbaI; next, the subunits were digested separately with the specific restriction endonuclease, and fragments were purified by gel electrophoresis. pcDNA3 was digested with HindIII andXbaI or BamHI (as appropriate), and the opened vector was purified. The two appropriate subunit fragments from the subunits were then mixed with the opened pcDNA3 and ligated.
All point mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). For reasons that are not clear to us, it was more efficient to mutate the rat α4 construct in pcDNA3. Accordingly, many of the mutations in the C-terminal tail region were generated this way, after it had been shown that other regions of the subunit did not affect potentiation (see Results). All chimeras and point mutations were sequenced through the entire translated region to confirm that the appropriate changes had been introduced, and that no additional mutations were generated in the sequence.
Nicotinic receptors were expressed in Xenopus oocytes by injecting the nuclei of defolliculated oocytes using the blind method described by Colman (1984). cDNA constructs encoding an α4 and a β2 subunit at a concentration of 200 ng/μl, mixed at a 1:1 ratio, were used for all experiments. Nuclei were microinjected with 13.6 nl of cDNA-containing solution into the nucleus of the oocyte. Oocytes were incubated at 18°C for 2–4 d before biophysical recordings.
Two-electrode voltage-clamp recordings. A two-electrode voltage clamp (Warner Instruments, Hamden, CT) was used to record currents from oocytes. Both voltage and current electrodes had resistances of ∼1 MΩ and were filled with 3 mKCl; silver chloride pellets were used for the bath and virtual ground. The oocyte recording chamber had a volume of ∼0.1 ml and was continuously perfused with solution (either bath or test) at a rate of ∼7 ml/min. Solutions were switched by hand, using Teflon rotary valves (Rheodyne, Rohnert Park, CA). Because of the lipophilic nature of the steroids used in our experiments, glass syringes, Teflon tubing, and Teflon valves were used in the perfusion apparatus.
To prevent the activation of Ca2+-activated chloride currents found in oocytes, Ca2+ was replaced with Ba2+ (Briggs et al., 1995). The following external solution was therefore used (in mm): 96 NaCl, 2 KCl, 1.8 BaCl2, 1 MgCl2, and 10 HEPES, pH 7.3. Acetylcholine (ACh) chloride (Sigma, St. Louis, MO) was prepared as a 1 m stock solution in ion exchange-purified distilled water and stored in aliquots at −20°C. Steroids were purchased from Sigma or Steraloids (Newport, RI) or were synthesized by the laboratory of Dr. Doug Covey (Washington University, St. Louis, MO). Stock solutions were prepared as 10 mm steroid dissolved in DMSO and stored at room temperature in the dark. Working solutions were made up on the day of the experiment in external solution. The standard test concentration of 15 μm steroid resulted in a solution containing 0.15% DMSO, but control experiments with DMSO alone indicated that this concentration had no effect on ACh responses (data not shown) (Paradiso et al., 2000).
Data were filtered at 20 Hz using an eight-pole Bessel filter (Frequency Devices, Haverhill, MA), digitized at 50 Hz using a Digidata 1200 (Axon Instruments, Union City, CA), and analyzed with a PC clone computer using pClamp6 and pClamp8 software (Axon Instruments). The response was measured as the difference between the average response in a 1–2 sec window including the peak of the response and the average baseline current in an ∼2 sec window before the onset of response. The effect of steroids was determined by taking the ratio of the response in the presence of steroid to the average of control responses measured before and after the test application. One complicating factor was that the amount of potentiation produced by our standard test applications varied between batches of oocytes, with mean values from 1.6-fold to 5.9-fold increases in response in the presence of βE2 on different batches. A single-factor ANOVA test gave a p value of 0.001 that the difference between batches was random. We do not have an explanation for the variability. To compensate for the variability, in some cases (as noted in Fig. 5) we compared the amount of potentiation within a given batch of oocytes, in addition to tests on pooled data.
Fig. 5.

Mutations in the C-terminal tail of the α4 subunit affect potentiation by βE2. Oocytes were injected with an α4 construct and wild-type β2 subunit. The first column shows the C-terminal amino acid sequence for each construct with the first entry (r, rat;h, human) indicating the origin of the sequence before the C terminus of the subunit. The sequence follows the predicted end of the M4 membrane spanning region, with the proline pair (PP) aligned vertically. The critical four residues are shown in bold, and the locations of mutations areboxed. The next column briefly describes each construct, followed by the n value. The bar graph to the right shows the mean ratio of the response to 1 μm ACh plus 15 μm βE2 to the response to 1 μm ACh alone in the same oocyte. The vertical lines at 1 and 3.85 indicate the average response from rat (no potentiation) and human α4β2 receptors. Error bars indicate the SEM. *indicates a p < 0.05 that the difference between the ratio for each construct and the ratio for human wild-type receptor, obtained from the same batch of oocytes (see Materials and Methods), arises by chance (evaluated using the t test assuming unequal variances); (*) is the same as *, except data from these three constructs were compared with the pooled data from all human wild-type receptors because wild-type responses were not determined from the same batch of oocytes.
Data analysis and statistical tests were done using Excel (Microsoft, Seattle, WA) and Sigma Plot (SPSS, Chicago, IL).
RESULTS
Potentiation of human α4β2 receptors
Coapplication of 15 μm βE2 with 1 μmACh potentiated (3.9-fold ± 1.2 SD; n = 84) the response of human α4β2 nicotinic receptors expressed inXenopus oocytes (Fig.1A) but did not potentiate responses from a rat α4β2 receptor (Fig.1B) (1.02 ± 0.14 SD; n = 36). Potentiation was assessed from the peak response to coapplication of ACh and steroid, compared with the peak response to ACh alone. It should be noted that two forms of the rat α4 subunit are expressed in the rat brain, α4-1 and α4-2 (Deneris et al., 1988). The forms differ only at the extreme C terminus, and the mRNAs for the two forms are present at approximately equal levels in rat brain. For simplicity, we refer to rat α4-1 subunits as rat α4 in this paper.
Fig. 1.

βE2 potentiates responses from human α4β2 nicotinic receptors expressed in Xenopus oocytes but not responses from rat α4β2 receptors. A, The responses of an oocyte injected with human wild-type α4 plus β2 subunit cDNA to an application of 1 μm ACh and then to 1 μm ACh coapplied with 15 μm βE2 and finally the recovery response to 1 μm ACh alone (∼2 min after the co-application). Preapplication of βE2 did not increase the amount of potentiation (data not shown). B, Similar responses of an oocyte injected with rat wild-type α4 plus β2 subunit cDNA.
As seen for other studies of potentiation of ligand-gated ion channels, the amount of potentiation of the human α4β2 receptor is greater at lower agonist concentrations (Fig.2A,B). Accordingly, the experiments reported below were performed using 1 μm ACh because this represents a submaximal agonist concentration (Fig. 2A). The lack of potentiation for the rat α4β2 receptor did not result from major differences in activation of rat or human receptors, because 1 μm ACh is a low concentration for both types of receptors (Fig. 2A). The concentration of 15 μm βE2 used in Figure 1Aprovides close to maximal potentiation (Fig. 2C).
Fig. 2.
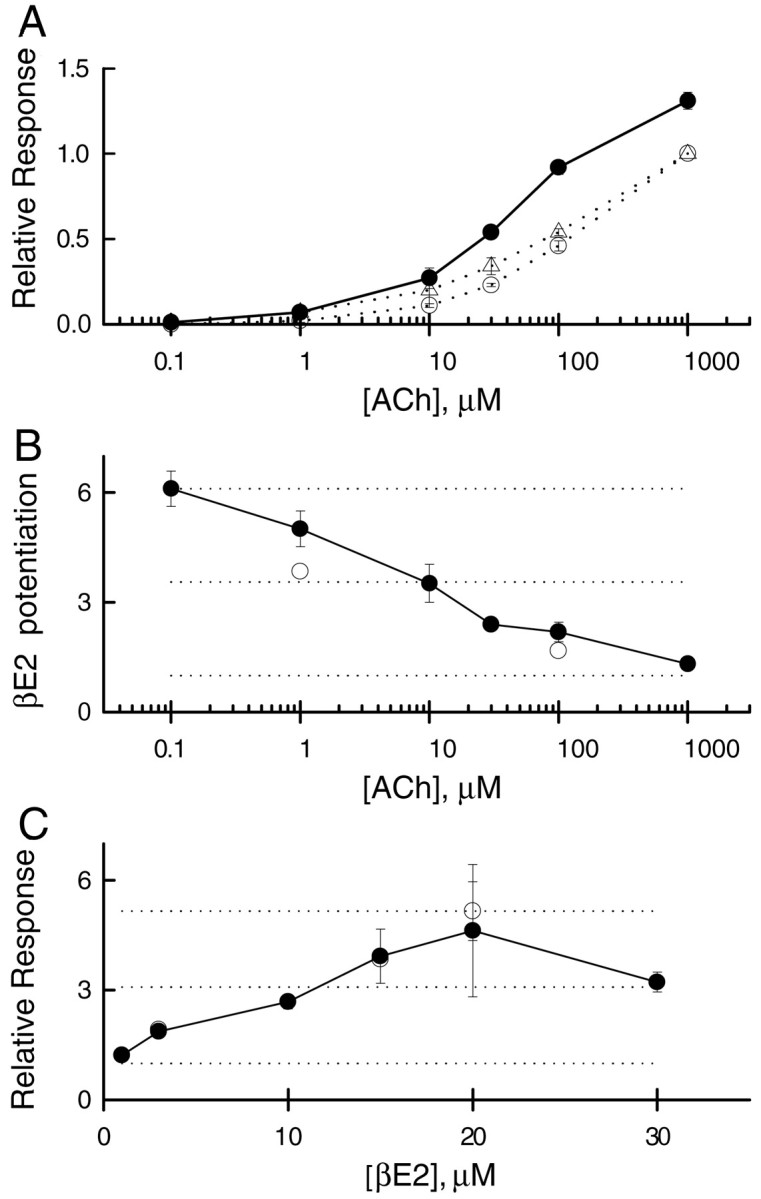
Concentration–response data for ACh and βE2.A, The relative response to various concentrations of ACh, normalized to the response to 1 mm, in the absence of βE2. Human α4β2, open circles; rat α4β2,open triangles. Coapplication of 15 μmβE2 enhances the response from the human receptor (filled circles). Lines simply connect the points, whereas points show mean ± 1 SEM for three to six oocytes (derived from two batches).B, Potentiation of the human wild-type receptor response produced by coapplication of 15 μm βE2 plus various concentrations of ACh indicates that potentiation is greater at lower concentrations of ACh. The dotted lines show values of 1 (no potentiation), half the observed maximal potentiation, and maximal observed potentiation. Filled circles show data derived from oocytes tested at multiple concentrations (3 oocytes at each point), whereas open circles show pooled data from all oocytes tested (36 oocytes at 100 μm ACh and 84 oocytes at 1 μm ACh). C, The potentiation produced by various concentrations of βE2 coapplied with 1 μmACh indicate an EC50 value of ∼10 μm for βE2. The apparent decrease between 20 μm and 30 μm βE2 is not statistically significant, although it could reflect mixed potentiation and block by βE2 at high concentrations. Filled circles show data derived from oocytes tested at multiple concentrations (3 oocytes at 3 μm, 2 oocytes at 20 μm, and 6 oocytes at other concentrations), whereas open circles show pooled data from all oocytes tested (13 oocytes at 3 μm, 84 oocytes at 15 μm, and 4 oocytes at 20 μm). The two symbols overlap at 3 and 15 μm βE2.
Potentiation and inhibition by steroids are separate and independent processes
In addition to causing potentiation, steroids have been shown to inhibit the responses of neuronal nicotinic α4β2 receptors (Bertrand et al., 1991; Valera et al., 1992; Sabey et al., 1999;Paradiso et al., 2000). Indeed, human α4β2 receptors expressed in oocytes are inhibited by some steroids (Fig.3, Table1). Accordingly, we examined the question of whether potentiation and inhibition might be related processes; for example, inhibition could result from the action of an “inverse agonist” that bound to the same site as potentiating steroids. Our observations indicate that potentiation is distinct from inhibition.
Fig. 3.

The onset and offset of potentiation are rapid. The response of a single oocyte to a long application of 1 μm ACh shows a slow decline reflecting desensitization (light trace). When a long application is made but the solution is switched from 1 μm ACh to 1 μmACh plus 15 μm βE2 during the application, the response is rapidly potentiated, and on return to 1 μm ACh alone rapidly returns to the same level as the response to 1 μmACh alone. In contrast, when the solution is switched to one containing 1 μm ACh plus 15 μm PROG, the response is rapidly blocked but recovers extremely slowly on return to 1 μm ACh alone. The responses were taken sequentially, with ACh alone first, followed by ACh plus βE2 and then ACh plus PROG. The holding current has been subtracted but the traces have not been normalized. There apparently was a small increase in response amplitude between the initial control application and the test applications.Bars indicate onset and duration of drug application.
Table 1.
Progesterone block of the human α4β2 receptor in the absence and presence of β-estradiol indicates a lack of competition
| No β-estradiol | +3 μm βE2 | +15 μm βE2 | |
|---|---|---|---|
| 3 μmPROG | 0.37 ± 0.13 (n = 7) | 0.22 ± 0.05 (n = 4) | 0.25 ± 0.03 (n = 4) |
| 15 μm PROG | 0.10 ± 0.04 (n = 4) | 0.08 ± 0.04 (n = 7) | 0.05 ± 0.01 (n = 3) |
βE2 refers to 17β-estradiol. PROG (3 or 15 μm) was pre-applied for 20 sec before a test pulse containing 1 μm ACh plus the concentration of PROG that was pre-applied plus 0, 3, or 15 μm βE2. The table shows the relative response in the presence of PROG (3 or 15 μm) to the response in the absence of PROG: the first data column is normalized to 1 μm ACh (no βE2); the second column is normalized to 1 μm ACh plus 3 μm βE2; and the third column is normalized to 1 μm ACh plus 15 μm βE2. The SD follows and the n value is shown in parentheses.
We used progesterone (PROG) as an example of an inhibitory steroid. One difference between inhibition and potentiation is that inhibition reversed relatively slowly (Fig. 3) (Paradiso et al., 2000). We also found that the requirements for the steroid structures that produce potentiation (discussed in next section) differ from those that produce inhibition (Paradiso et al., 2000). Finally, pharmacological experiments indicate that inhibition and potentiation are independent processes. We examined this by coapplying an inhibiting steroid (PROG) with a potentiating steroid (βE2) and determining whether inhibition was reduced by potentiation (Table 1). If the presence of the potentiating steroid reduced inhibition, this would suggest that the two mechanisms shared some parts of a pathway (perhaps as early as a binding site). For the competition experiments, 3 μm or 15 μm PROG was preapplied for 20 sec to ensure that the inhibiting steroid had ample time to interact with its binding site.
For either concentration of PROG there was no significant difference between the amounts of block among the different βE2 concentrations (Table 1) (p > 0.06 for all comparisons). In an additional experiment, 3 μm PROG and 15 μm βE2 were preapplied together for 20 sec to allow interactions between steroids to reach equilibrium. The response was then tested with 1 μm ACh plus 3 μm PROG and 15 μmβE2, and the test response was reduced to 0.22 ± 0.06 (four cells). The reduction did not differ from that seen when only 3 μm PROG was preapplied (Table 1). Accordingly, the actions of steroids to potentiate and to block responses of the human α4β2 receptor do not show competition and so are unlikely to involve binding to the same or overlapping portions of the receptor.
However, block is a confounding factor in the analysis of potentiation. In particular, the presence of block might be significant in comparisons among steroids of differing structure because it is likely that steroids that potentiate can also block to some extent. Hence, stronger block might be observed as weaker potentiation, or vice versa.
Features of steroid structure important for potentiation
We first examined the ability of several steroids and some estrogen receptor agonists to potentiate human α4β2 nicotinic receptors. Some important basic features of steroid structure are shown in Figure 4A, including the ring nomenclature, with the A ring highlighted to indicate its importance, and arrows marking the critical carbon 3 and 17 positions. The standard concentration of steroid adopted was 15 μm, because this concentration produced close to maximal potentiation for βE2 (Fig. 2C). In addition, these steroids can precipitate out of solution at concentrations of >20 μm; thus 15 μmrepresents a reasonable test concentration while still being low enough to minimize issues of steroid solubility.
Fig. 4.

Steroids tested for their ability to potentiate the human α4β2 nicotinic receptor. The structures of the steroids are identified by the abbreviations in the text, and values indicate the ratio of the response to coapplication of 1 μm ACh and 15 μm of the test compound to the response to 1 μm ACh alone in the same oocyte (±1 SD, with the number of oocytes tested in parentheses). The stereochemistry at all optically active centers is shown for βE2 andent-βE2; only partial stereochemistry for the other steroids is shown, but they are identical to that for βE2 at the additional centers. A, The structure in the top left corner shows the steroid ring backbone with the rings labeled A–D. The A ring is inbold, and arrows point to carbons 3 and 17 to indicate the importance of these positions. Although βE2 potentiates responses, its mirror image (ent-βE2) does not. B, Steroids containing a saturated A ring (3β17βED) or lacking a hydroxyl group at carbon 3 (3MeβE2 andmestranol) do not potentiate. C, Absence of a hydroxyl group at carbon 17 (E0,E1) or addition of a hydroxyl group at carbon 16 (E3) reduces potentiation. D, Potentiation occurs with an α-hydroxyl at carbon 17 (αE2), additional unsaturation in the B ring (Δ7 βE2), or an ethynyl group added to carbon 17 (ethynyl βE2).
The mirror image of βE2 (ent-βE2) (Fig.4A) was ineffective at potentiating; therefore, the steroid interacts with a site that recognizes the stereochemistry of the steroid. Enantioselectivity indicates that βE2 does not act by some bulk effect on the membrane, because the physical properties of enantiomers are identical, suggesting that βE2 is binding to a protein.
Strong potentiation required an unsaturated A ring and a free hydroxyl group at the 3 and 17 position (Fig. 4A). An A ring-saturated analog of βE2 (3β17βED) and the 3-methyl ether of βE2 (3MeβE2) were ineffective at potentiating (Fig.4B), as was the 3-methyl ether of 17α-vinyl βE2 (mestranol). Removal of the 17-hydroxyl group (E0) or replacement with a keto group (E1) produced ineffective compounds, as did the addition of a third hydroxyl group (E3) at carbon 16 (Fig. 4C). Because our experiments do not separate affinity from efficacy, it is possible that some of the structural requirements for potentiation reflect occupancy, whereas others reflect the ability to produce potentiation.
Consistent with the structural features required for potentiation discussed above, the steroids shown in Figure 4Dpotentiated responses. A diastereoisomer of βE2, 17α-estradiol (αE2) potentiated responses almost as well as βE2, indicating that the presence of the 17-hydroxyl group is critical, whereas the stereochemistry is less important (Fig. 4D). Two other steroids were at least as effective at potentiation as βE2: 17α-vinyl βE2 (ethynyl βE2) and a steroid with a partially unsaturated B ring (Δ7 βE2). These three steroids (Fig.4D) contain the same critical structural features of βE2: an unsaturated A ring and free hydroxyl groups at the 3 and 17 position.
We believe that the steroids directly interact with the nicotinic receptor and are not acting through estrogen receptors. Although the human α4β2 nicotinic receptor and the nuclear estrogen receptors share requirements for steroid structure (Anstead et al., 1997; Kuiper et al., 1997; Wiese et al., 1997), there are some clear differences between agonists for the nicotinic receptor and the estrogen receptor. Specifically, for human estrogen receptors, αE2 is much worse at binding or activating than is βE2 and E0, whereas E1 and E3 are comparable with αE2 in affinity and activity at human estrogen receptors (Kuiper et al., 1997; Wiese et al., 1997). In addition, two nonsteroidal agonists for the estrogen receptor inhibited responses from the α4β2 receptor: diethyl stilbestrol (0.45 ± 0.12 SD;n = 4) and kampferol (0.86 ± 0.09 SD;n = 3). Accordingly, although there are similarities in the structure–activity relationship for potentiation of the nicotinic receptor and activation of the estrogen receptor, there are clear differences that indicate structurally distinct estradiol binding sites for the α4β2 nicotinic receptor and the estrogen receptors. The differences in agonist selectivity combined with the speed of the response allow us to conclude that the potentiation of the α4β2 receptor is not mediated by steroids acting through an estrogen receptor.
The C-terminal tail of the human α4 subunit is required for potentiation by estradiol
The observation that the human α4β2 receptor is potentiated by βE2 and that the rat α4β2 receptor is not provided the opportunity to define the portions of the receptor that are required for potentiation. We first determined whether the species of origin for the α4 or the β2 subunit was critical, by expressing all four subunit combinations. For this work, we used a standard test application of 1 μm ACh plus 15 μm βE2 and measured the ratio of the response in the presence of steroid to the response in the absence of steroid. Both receptors containing the human α4 subunit were potentiated (human α4 plus human β2, 1.58 ± 0.36 SD; n = 11) (human α4 plus rat β2, 2.08 ± 0.34 SD; n = 6), whereas receptors containing the rat α4 subunit were not (rat α4 plus human β2, 0.78 ± 0.09 SD; n = 3) (rat α4 plus rat β2, 0.77 ± 0.04 SD; n = 4). Accordingly, our attention focused on the differences between human and rat α4 subunits.
We produced three pairs of chimeric subunits between the human and rat α4 subunits, with joining points located in the N-terminal extracellular region (in the cysteine–cysteine loop), at the C-terminal end of the M3 membrane spanning region, and in the middle of the main cytoplasmic loop. In each case, potentiation mapped to the C-terminal portion of the human α4 subunit (data not shown). We noted that the rat and human α4 subunit differ at the extreme C-terminal region, in the extracellular “tail” that follows the M4 membrane spanning region. Accordingly, we constructed subunits in which only the last two (rat, PPWLA AC) or three (human, PPWLA GMI) residues were exchanged. This small region was sufficient to transfer potentiation to the rat α4 subunit and remove it from the human α4 subunit (Fig.5A). It is interesting to note that there is no evidence that any other nonhomologous portions of the α4 subunits have a significant role in producing potentiation, because the potentiation observed was the same regardless of the nature of the subunit preceding the tail (Fig. 5A). We examined the possibility that the terminal cysteine of the rat α4 subunit acted to prevent potentiation, for example by disulfide bond formation (DiPaola et al., 1989). However, deletion of cysteine or replacement with isoleucine or serine did not confer potentiation (Fig.5B).
To further determine that block and potentiation are separate, we also tested the ability of PROG to block responses from the subunits with exchanged C-terminal tails. As shown in Table2, we found that the exchange had little or no effect on block, confirming that the sites involved in potentiation and block are distinct. None of the differences were significant (p > 0.2 for all pairwise comparisons). It therefore appeared that the human α4 C terminus did not affect block, but instead contained a sequence of amino acids that conferred potentiation.
Table 2.
The C terminus is not involved in steroidal inhibition
| Human WT | Rat WT | Human with Rat CT | Rat with Human CT | |
|---|---|---|---|---|
| α4 CT sequence | h-PPWLAGMI | r-PPWLAAC | h-PPWLAAC | r-PPWLAGMI |
| 1 μm ACh plus 15 μmPROG | 0.16 ± 0.10 (n = 4) | 0.08 ± 0.02 (n = 5) | 0.12 ± 0.11 (n = 4) | 0.13 ± 0.18 (n = 3) |
CT, C terminus; WT, wild type. The italic letter appearing before the CT sequence indicates the species origin for the subunit (h, human; r, rat). Inhibition by steroids co-applied with agonist results in a reduced peak response followed by an additional rapid reduction in response to a steady-state level of block. Inhibition is indicated by the ratio of the steady-state response to 1 μm ACh plus 15 μm PROG versus the peak response from 1 μm ACh alone. The SD and the n value are also indicated.
Specific residues in the human α4 C terminus are required for potentiation
Mutation of residues in the human α4 tail sequence revealed that the nature of the final four residues (AGMI) was critical, whereas the preceding two residues (WL) were less important for potentiation by βE2 (Fig. 5C). Deletion of the ultimate isoleucine removed potentiation, whereas substitution with methionine reduced it. Similarly, alterations of the methionine, glycine, or alanine reduced potentiation.
In contrast, mutation of leucine to alanine and mutation of tryptophan to leucine or alanine did not reduce βE2 potentiation (Fig.5C). These observations indicate that these residues are unlikely to interact strongly with βE2.
Not only the nature but the position of the AGMI residues is critical. For convenience, we will speak as though the two prolines at the end of the M4 region define a fixed position with respect to the membrane and the rest of the receptor. Moving the AGMI sequence out from the prolines by inserting an alanine or moving it in toward the prolines by deleting the tryptophan removed potentiation (Fig. 5D). These observations demonstrate that the presence of AGMI as the terminal sequence, in and of itself, is not sufficient to confer potentiation. However, the relative position of AGMI with respect to the prolines is not the only critical feature, because extension of the tail by adding one or two residues after the isoleucine also reduced or removed potentiation (Fig. 5D). Both prolines were mutated to glycine, with the thought that increased flexibility might result in a structure that retains potentiation. However, this mutation also reduced potentiation (Fig. 5D), perhaps because the rigid bend introduced by the prolines is required for correct positioning of the tail. These observations are consistent with the idea that the tail occupies a relatively confined volume and must be able to adopt a particular conformation to allow recognition of βE2. It is possible that reduced potentiation found with the relatively small changes of alanine to glycine or glycine to alanine (Fig. 5C) also reflects a tight binding pocket requiring a specific conformation.
The C-terminal tail sequence acts as a steroid binding site
We think that the tail is most likely to be involved in recognition of the steroid. The data summarized in Figure 5 do not rule out the possibility that the AGMI sequence is required for conformational changes involved in transducing the binding of βE2 into the observed potentiation. However, an additional observation provides strong evidence that C-terminal residues interact directly with the steroid. We found that ethynyl βE2 potentiates rat α4β2 receptors (Fig. 6), which is unique among the steroids we have tested (Paradiso et al., 2000). For the rat α4β2 receptor, this steroid has an EC50 value that is between 3 and 10 μm (data not shown), and 15 μm yields ∼75% of the potentiation seen with 30 μm (the highest concentration tested).
Fig. 6.

Mutations in the C terminus of the α4 subunit differentially affect the ability of steroids to potentiate responses to ACh. The horizontal axis indicates the sequence of the C terminus of the α4 subunit, injected with the wild-type β2 subunit. The bars indicate the response to 1 μm ACh and 15 μm steroid (βE2 or ethynyl βE2) compared with 1 μm ACh alone. Error bars indicate the SEM, with the n value indicated above each bar. Thedotted line at the value of 1.0 indicates no potentiation.
We reasoned that the unsaturated ethynyl moiety (C≡C) might interact with the aromatic group in the tryptophan side chain in the C-terminal tail. Accordingly, we mutated the tryptophan to leucine to remove the π electrons while retaining the hydrophobic nature of the side chain. The mutation was made in both the human (WLAGMI to LLAGMI) and rat tail (WLAAC to LLAAC). We then compared the abilities of βE2 and ethynyl βE2 to potentiate receptors containing the constructs. As shown in Figure 6, receptors containing α4 subunits that had a tail with sequences of either WLAGMI or LLAGMI were potentiated by both βE2 and ethynyl βE2, as expected based on our previous results, because the AGMI sequence is present. In contrast, a tail sequence of WLAAC supported potentiation only by ethynyl βE2 but not βE2. Finally, LLAAC did not support potentiation by either steroid, consistent with it lacking both the tryptophan and the AGMI sequence.
The absence of steroid potentiation in the LLAAC construct could also be explained by an increase in steroid block. In fact, application of βE2 to receptors containing the W to L mutation produced a 26% reduction in the peak response (Fig. 6). To further address this, we tested the ability of progesterone to block the LLAAC construct and compared this with block of wild-type rat receptors. Progesterone was coapplied with ACh to match the protocol for application of potentiating steroids. At 3 μm, progesterone reduced the peak response of rat α4β2 receptors by 43% (±0.08 SD;n = 6) and that of LLAAC-containing receptors by 45% (±0.09 SD; n = 4). At 15 μm, however, progesterone reduced the peak response of rat α4β2 receptors by 63% (±0.04 SD; n = 6) and that of LLAAC-containing receptors by 76% (±0.06 SD; n = 4). Although there is a small increase in the ability of high concentrations of progesterone to block the LLAAC receptors, this small difference is not enough to account for the complete loss of potentiation by ethynyl estradiol.
These results from Figure 6 demonstrate that the AGMI sequence is not absolutely required for potentiation, because ethynyl βE2 can potentiate a receptor with the WLAAC tail sequence. Similarly, the tryptophan residue (W) is not required for potentiation, because both βE2 and ethynyl βE2 can potentiate receptors with the sequence LLAGMI. Our interpretation of these results is that both βE2 and ethynyl βE2 interact with the AGMI sequence. However, the vinyl group of ethynyl βE2 can also interact with the tryptophan. Accordingly, to remove binding of ethynyl βE2 it was necessary to remove both sites of interaction from the protein.
On the basis of these data, it appears that the steroid rings and/or the ethynyl group bind to the α4 C terminus, leaving the two hydroxyl groups of the steroid free to hydrogen bond to amino acids elsewhere in the protein. The data indicate that the C terminus acts as a binding domain, although additional regions of the receptor are likely necessary for the conformational changes required for potentiation.
DISCUSSION
The binding site involved in steroid potentiation differs from that for inhibition
17β-Estradiol and some structurally similar steroids potentiate responses of human α4β2 receptors, whereas many other steroids inhibit responses. Our work indicates that steroids must bind to different regions of the receptor to have these two divergent actions. In pharmacological experiments, progesterone produces the same level of block for control responses and for responses potentiated by 3 or 15 μm βE2, indicating that PROG and βE2 must bind to nonoverlapping regions of the receptor to produce block or potentiation. In addition, mutations of the α4 subunit have shown that the regions of the receptor that are important for potentiation do not affect block. Hence, although block and potentiation both involve binding of steroids, the C-terminal amino acids that are essential for potentiation are not necessary for block. Previous studies of GABAA (Zaman et al., 1992) and NMDA (Park-Chung et al., 1997) receptors have also found that inhibition and potentiation by steroids are independent processes.
Steroid recognition by the C terminus of the α4 subunit
Our observations were made using a physiological assay, and potentiation reflects both steroid binding and the ensuing conformational changes that result in potentiation. Our data indicate that the C terminus of the α4 subunit is involved in steroid recognition and that the specific residues have less of an effect on the subsequent conformational changes. The most compelling evidence for this conclusion is the finding that the tryptophan in the C terminus has a specific role in recognizing the 17α ethynyl moiety (C≡C) in ethynyl βE2, probably through a π–π interaction between the benzene ring of the tryptophan and the ethynyl group. We think that ethynyl βE2 interacts with two portions of the C terminus, both the tryptophan and the terminal tetrapeptide AGMI, because the presence of either allows potentiation by ethynyl βE2. 17β-Estradiol, in contrast, interacts only with the AGMI peptide.
Because the two oxygen atoms in the βE2 molecule are only ∼1 nm apart (Anstead et al., 1997), it is possible that the C-terminal tail adopts a sufficiently extended conformation that allows it to provide much of the binding site for βE2. It is also possible that the α4 C-terminal carboxylate group forms a hydrogen bond to one of the hydroxyls of the steroid. However, the C-terminal tail cannot provide the entire binding site because it does not contain residues that specifically interact with both hydroxyl groups in βE2. In the estrogen receptor, a glutamate and a histidine interact with the 3- and 17-hydroxyls, respectively (Tanenbaum et al., 1998). It is thought that the interaction with the hydroxyl groups is critical for activation of estrogen receptors. In the α4β2 nicotinic receptor, the two hydroxyl groups appear to be essential for potentiation (Fig. 4), and it is possible that the hydroxyl groups are important in translating binding into potentiation.
All of the residues that we have identified as interacting with βE2 are hydrophobic. In the estrogen receptor, there are 15 hydrophobic interactions involved in stabilizing the association between βE2 and the receptor (Tanenbaum et al., 1998), and the free energy of binding is actually derived primarily from hydrophobic interactions (Anstead et al., 1997). The lower apparent affinity of the nicotinic receptor would suggest that there are fewer hydrophobic interactions between the receptor and βE2. However, the two receptors are similar in the sense that both apparently recognize steroids containing a rigid diol with a hydrophobic and appropriately shaped spacer region.
Estradiol is synthesized in the CNS
The high nanomolar to low micromolar levels of βE2 required for potentiation represent a high concentration relative to the concentration necessary to activate the α1 and β1 (0.1–1 nm) or β2 (10–100 nm) estrogen receptors (Petersen et al., 1998; Hanstein et al., 1999). However, the nongenomic actions of βE2 generally require nanomolar to micromolar concentrations (Moosmann and Behl, 1999; Valverde et al., 1999), and it has been proposed that local production by aromatase can result in high local concentrations of βE2 (Balthazart and Ball, 1998;Stoffel-Wagner et al., 1999). It is also possible that the relevant concentration of βE2 is the concentration found in the synaptic membrane, which would be increased by partitioning into the membrane. Therefore, the possibility exists that the local βE2 concentration might be significantly higher than the bulk CSF levels at sites at which the aromatase enzyme is located. Currently, the methods used to measure steroid levels are not capable of measuring local concentrations of steroid.
Steroids and ligand-gated ion channels
Transmitter-gated ion channels have been shown to be one target responsible for the rapid action of neuroactive steroids. Steroids can potentiate or inhibit the responses of several different receptors to transmitters, and in some cases a given steroid may potentiate one type of receptor while inhibiting another. Two well studied examples of steroid modulation of transmitter-gated ion channels are the potentiation of GABAA receptors (cf. Paul and Purdy, 1992) and the inhibition of NMDA receptors (Park-Chung et al., 1997). Although many studies have been made of the structural features of the steroid that enhance or reduce activity on a transmitter-gated channel, there has been no success to date in identifying the regions of any receptor that interact with steroids (Rick et al., 1998; Blanton et al., 1999). Studies of chimeric subunits have suggested that the ability of the anesthetic steroid alphaxalone to potentiate the GABAA receptor requires regions of the receptor located between the N terminus and the middle of the second transmembrane spanning region (Rick et al., 1998). In that study, however, it was not possible to distinguish between the parts of the receptor involved in steroid recognition and those regions required for conformational changes. The steroid promegestone inhibits the muscle-type nicotinic receptor found in Torpedo electric organ, and photolabeling experiments have shown that it interacts with residues in the M4 membrane spanning helix (Blanton et al., 1999). However, promegestone is an inhibitory steroid, and our data indicate that inhibition and potentiation of the α4β2 nicotinic receptor by steroids does not involve an overlapping binding site.
We show a direct interaction between the human α4β2 neuronal nicotinic receptor and β-estradiol by locating a specific region of the human α4β2 receptor that is necessary for potentiation of responses by estradiol steroids. This work represents the first example of a specific region required for steroidal effects on an ion channel, reveals an important role for the C terminus of ligand-gated ion channels, and suggests an additional mechanism for some of the nongenomic effects of estrogen.
Footnotes
This work was supported by National Institutes of Health Grants P01 GM47969 and R01 NS22356 (J.H.S.). J.H.S. is the Russell and Mary Shelden Professor of Anesthesiology. We thank Dr. Douglas F. Covey for providing steroids and for advice on their use and effects, G. Akk for comments during the studies, and J. Bracamontes for advice on molecular biology.
Correspondence should be addressed to Dr. Kenneth Paradiso, Department of Anesthesiology, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail:paradiso@morpheus.wustl.edu.
REFERENCES
- 1.Anand R, Lindstrom J. Nucleotide sequence of the human nicotinic acetylcholine receptor β2 subunit gene. Nucleic Acids Res. 1990;18:4272. doi: 10.1093/nar/18.14.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 3.Balthazart J, Ball GF. New insights into the regulation and function of brain estrogen synthase (aromatase). Trends Neurosci. 1998;21:243–249. doi: 10.1016/s0166-2236(97)01221-6. [DOI] [PubMed] [Google Scholar]
- 4.Bertrand D, Valera S, Bertrand S, Ballivet M, Rungger D. Steroids inhibit nicotinic acetylcholine receptors. NeuroReport. 1991;2:277–280. doi: 10.1097/00001756-199105000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Blanton MP, Xie Y, Dangott LJ, Cohen JB. The steroid promegestone is a noncompetitive antagonist of the Torpedo nicotinic acetylcholine receptor that interacts with the lipid-protein interface. Mol Pharmacol. 1999;55:269–278. doi: 10.1124/mol.55.2.269. [DOI] [PubMed] [Google Scholar]
- 6.Briggs CA, McKenna DG, Piattonikaplan M. Human α7 nicotinic acetylcholine receptor responses to novel ligands. Neuropharmacology. 1995;34:583–590. doi: 10.1016/0028-3908(95)00028-5. [DOI] [PubMed] [Google Scholar]
- 7.Buisson B, Bertrand S, Bertrand D. Inhibition and potentiation of human nicotinic receptors by sex hormones. Soc Neurosci Abstr. 1998;24:530.19. [Google Scholar]
- 8.Coggan JS, Paysan J, Conroy WG, Berg DK. Direct recording of nicotinic responses in presynaptic nerve terminals. J Neurosci. 1997;17:5798–5806. doi: 10.1523/JNEUROSCI.17-15-05798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colman A. Expression of exogenous DNA in Xenopus oocytes. In: Hames BD, Higgins SJ, editors. Transcription and translation: a practical approach. Oxford; Washington, DC: 1984. pp. 49–69. [Google Scholar]
- 10.Deneris ES, Connolly J, Boulter J, Wada E, Wada K, Swanson LW, Patrick J, Heinemann S. Primary structure and expression of β2: a novel subunit of neuronal nicotinic acetylcholine receptors. Neuron. 1988;1:45–54. doi: 10.1016/0896-6273(88)90208-5. [DOI] [PubMed] [Google Scholar]
- 11.DiPaola M, Czajkowski C, Karlin A. The sidedness of the COOH terminus of the acetylcholine receptor δ subunit. J Biol Chem. 1989;264:15457–15463. [PubMed] [Google Scholar]
- 12.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 13.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 14.Hanstein B, Liu H, Yancisin MC, Brown M. Functional analysis of a novel estrogen receptor-β isoform. Mol Endocrinol. 1999;13:129–137. doi: 10.1210/mend.13.1.0234. [DOI] [PubMed] [Google Scholar]
- 15.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 16.Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- 18.Marubio LM, Arroyo-Jimenez MD, Cordero-Erausquin M, Lena C, Le Novere N, d'Exaerde AD, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 19.McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocr Rev. 1999;20:279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 20.McGehee DS, Heath MJS, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 21.Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paradiso K, Sabey K, Evers AS, Zorumski CF, Covey DF, Steinbach JH. Steroid inhibition of rat neuronal nicotinic α4β2 receptors expressed in HEK 293 cells. Mol Pharmacol. 2000;58:341–351. doi: 10.1124/mol.58.2.341. [DOI] [PubMed] [Google Scholar]
- 23.Park-Chung M, Wu FS, Purdy RH, Malayev AA, Gibbs TT, Farb DH. Distinct sites for inverse modulation of N-methyl-d-aspartate receptors by sulfated steroids. Mol Pharmacol. 1997;52:1113–1123. doi: 10.1124/mol.52.6.1113. [DOI] [PubMed] [Google Scholar]
- 24.Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- 25.Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- 26.Petersen DN, Tkalcevic GT, Koza-Taylor PH, Turi TG, Brown TA. Identification of estrogen receptor β2, a functional variant of estrogen receptor β expressed in normal rat tissues. Endocrinology. 1998;139:1082–1092. doi: 10.1210/endo.139.3.5840. [DOI] [PubMed] [Google Scholar]
- 27.Rick CE, Ye Q, Finn SE, Harrison NL. Neurosteroids act on the GABA-A receptor at sites on the N-terminal side of the middle of TM2. NeuroReport. 1998;9:379–383. doi: 10.1097/00001756-199802160-00004. [DOI] [PubMed] [Google Scholar]
- 28.Robel P, Baulieu EE. Neurosteroids: biosynthesis and function. Crit Rev Neurobiol. 1995;9:383–394. [PubMed] [Google Scholar]
- 29.Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22:410–416. doi: 10.1016/s0166-2236(99)01399-5. [DOI] [PubMed] [Google Scholar]
- 30.Sabey K, Paradiso K, Zhang J, Steinbach JH. Ligand binding and activation of rat nicotinic α4β2 receptors stably expressed in HEK293 cells. Mol Pharmacol. 1999;55:58–66. [PubMed] [Google Scholar]
- 31.Stoffel-Wagner B, Watzka M, Schramm J, Bidlingmaier F, Klingmuller D. Expression of CYP19 (aromatase) mRNA in different areas of the human brain. J Steroid Biochem Mol Biol. 1999;70:237–241. doi: 10.1016/s0960-0760(99)00114-4. [DOI] [PubMed] [Google Scholar]
- 32.Sugaya K, Giacobini E, Chiappinelli VA. Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer's disease. J Neurosci Res. 1990;27:349–359. doi: 10.1002/jnr.490270314. [DOI] [PubMed] [Google Scholar]
- 33.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci USA. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valera S, Ballivet M, Bertrand D. Progesterone modulates a neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1992;89:9949–9953. doi: 10.1073/pnas.89.20.9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, Mann GE, Vergara C, Latorre R. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the β subunit. Science. 1999;285:1929–1931. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- 36.Wevers A, Monteggia L, Nowacki S, Bloch W, Schutz U, Lindstrom J, Pereira EF, Eisenberg H, Giacobini E, de Vos RA, Steur EN, Maelicke A, Albuquerque EX, Schroder H. Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer's disease: histotopographical correlation with amyloid plaques and hyperphosphorylated-τ protein. Eur J Neurosci. 1999;11:2551–2565. doi: 10.1046/j.1460-9568.1999.00676.x. [DOI] [PubMed] [Google Scholar]
- 37.Wiese TE, Polin LA, Palomino E, Brooks SC. Induction of the estrogen specific mitogenic response of MCF-7 cells by selected analogues of estradiol-17β: a 3D QSAR study. J Med Chem. 1997;40:3659–3669. doi: 10.1021/jm9703294. [DOI] [PubMed] [Google Scholar]
- 38.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 39.Zaman SH, Shingai R, Harvey RJ, Darlison MG, Barnard EA. Effects of subunit types of the recombinant GABAA receptor on the response to a neurosteroid. Eur J Pharmacol. 1992;225:321–330. doi: 10.1016/0922-4106(92)90106-6. [DOI] [PubMed] [Google Scholar]