

Abstract
Although adenosine is an important neuromodulator in the CNS, its role in modulating sympathetic outflow at the level of the spinal cord has not been studied. Because very little is known about adenosine A1 receptors (A1Rs) in the spinal cord, we determined their location and role with particular reference to the control of sympathetic preganglionic activity and interneuronal activity in the rat. High levels of immunoreactivity for A1Rs were observed throughout the spinal cord. Immunostaining was dense in the intermediolateral cell column (IML) and intercalated nucleus, regions containing retrogradely labeled sympathetic preganglionic neurons (SPNs). Electron microscopy revealed A1R immunoreactivity (A1R-IR) within presynaptic terminals and (to a lesser extent) postsynaptic structures in the IML, as well as the luminal membrane of endothelial cells lining capillaries. Using double-labeling techniques, some presynaptic terminals were observed to synapse onto SPNs. To investigate the effects of activating these A1Rs, visualized whole-cell patch-clamp recordings were made from electrophysiologically and morphologically identified SPNs and interneurons. Applications of the A1R agonist cyclopentyladenosine (CPA) reduced the amplitude of EPSPs elicited by stimulation of the lateral funiculus, an effect blocked by the A1R antagonist 8-cyclopentyl-1,3-dipropylxanthine. These effects were attributable to adenosine acting at a presynaptic site because CPA application increased the paired-pulse ratio. CPA did not affect evoked IPSPs. These data show that activating A1Rs reduces fast excitatory, but not inhibitory, transmission onto SPNs and interneurons in the IML and that A1Rs may play a protective role on neurons involved in the control of sympathetic outflow.
Keywords: sympathetic, adenosine A1 receptors, blood vessel, excitatory amino acid transmission, spinal cord, electron microscopy
Adenosine, generated partly by the extracellular catabolism of ATP through the ectonucleotidase cascade (Lee et al., 1981; Cunha et al., 1992), is an important neuromodulator in the CNS, acting on G-protein-linked cell surface receptors classified as A1, A2, and A3. A major role for adenosine acting at A1 receptors (A1Rs) may be neuroprotective because release of adenosine during hypoxic or ischemic episodes inhibits neurotransmission to counteract possible excitotoxicity (Fowler, 1989;Rudolphi et al., 1992; Katchman and Hershkowitz, 1993). On A1Rs, adenosine exerts its action presynaptically and postsynaptically to elicit mainly inhibitory effects. Presynaptic A1Rs reduce neurotransmitter release via activation of G-proteins that inhibit adenylate cyclase activity (Stiles, 1992). A1R-mediated reductions in release of both glutamate and GABA have been reported in the hypothalamus, substantia nigra reticulata, and the periaqueductal gray regions (Shen and Johnson, 1997; Bagley et al., 1999; Oliet and Poulain, 1999). However, in the hippocampus, although depression of EPSCs during hypoxia was elicited by A1R activation, depression of IPSCs was attributable to another mechanism (Katchman and Hershkowitz, 1993). Postsynaptic A1Rs seem to be pharmacologically identical to those located presynaptically (Thompson et al., 1992), and activation causes hyperpolarization particularly through opening of potassium channels (Trussell and Jackson, 1987; Gerber et al., 1989).
Because adenosine acting on A1Rs may play a crucial role in neuroprotection during stressful events, it seems important to know its exact role in areas of the CNS involved in autonomic functions. Studies indicate roles for the A1Rs in synaptic transmission in the brainstem (Thomas and Spyer, 1999), but little is known of the spinal cord. Ligand binding studies using radioactive A1-selective agonists pinpointed intense binding in the dorsal and ventral horn of the lumbar spinal cord (Goodman and Synder, 1982;Geiger et al., 1984; Choca et al., 1987). Moreover, hybridization studies localizing mRNA for the A1Rs showed concentrated labeling in ventral horn neurons and moderate hybridization throughout the spinal gray matter, although the white matter was unlabeled (Reppert et al., 1991). However, little mention is made of regions involved in sympathetic control, such as the intermediolateral cell column (IML).
Despite a lack of evidence for A1R localization in the IML, there is a sign that A1Rs play a role in determining sympathetic outflow. Intrathecal injections of an A1R agonist decreased blood pressure and heart rate, although it was not determined whether this was a result of sympathoinhibition (Koh et al., 1996). Furthermore, such intrathecal applications cannot pinpoint sites of action of drugs that may include receptors on blood vessels, as well as those located presynaptically and postsynaptically on neurons.
We therefore studied the localization of the A1Rs at the light and electron microscopic level using immunohistochemistry, concentrating on thoracic spinal cord, which contains neurons involved in cardiovascular control. When these studies revealed a striking density of A1Rs in the IML and extensive vascular and neuronal localization in the spinal cord, electrophysiological experiments were performed to examine the role of A1Rs in modulating the activity of neurons contributing to sympathetic outflow.
MATERIALS AND METHODS
Immunohistochemistry. Male rats (150–200 gm;n = 10) were deeply anesthetized by intraperitoneal Sagatal (60 mg/kg) and transcardially perfused with fixative containing 4% paraformaldehyde and 0.1–0.5% glutaraldehyde in 0.1m phosphate buffer (PB), pH 7.4. All experiments were performed under a UK Home Office License and in accordance with the regulations of the UK Animals (Scientific Procedures) Act of 1986. Efforts were made to minimize animal suffering and to use only the minimum number of animals required. Spinal cords were removed at the thoracic level and post-fixed in perfusing fixative for 4 hr. The tissue was then cut into ∼1 cm sections, embedded in gelatin (10%), fixed (0.5% glutaraldehyde), sectioned at 50 μm on a vibrating microtome (Leica, Nussloch, Germany), and collected into PBS, pH 7.2. For electron microscopy, sections were cryoprotected by incubation in 10% sucrose in 0.1m PB for 10 min, followed by 20% sucrose in 0.1m PB for 20 min and then freeze-thawed twice in liquid nitrogen to permeabilize the membranes. For light microscopy, the tissue was permeabilized using 0.1% Triton X-100 included with the primary antibody solution.
Sections were immersed in primary antibody in PBS for 12–48 hr at 4°C.We used various primary antisera directed against different parts of the A1Rs so that similar patterns of labeling would indicate specific localization of the receptor. The antiserum used for the majority of the labeling in this study was raised in rabbit against a unique intracellular sequence of the rat A1R, residues 310–323, and was diluted 1:200–1:400. This antibody has been shown previously to specifically recognize the adenosine A1 receptor in Xenopusoocytes expressing the A1 receptor but not in untransfected cells, as well as in rat CNS by Western blotting (Smith et al., 2001). Another antibody raised in rabbit against a similar sequence (amino acids 304–326; 1:100; Sigma, Poole, UK) has been reported previously to specifically recognize the A1 receptor by Western blotting in human brain and in Chinese hamster ovary cells stably expressing the A1 receptor but not in untransfected cells (Schindler et al., 2001). A primary antibody raised in goat against a peptide mapping near the C terminus of the adenosine A1 receptor was diluted at 1:200–1:800 (Santa Cruz Biotechnology, Santa Cruz, CA). Finally, another antibody raised in rabbit and generated against an extracellular sequence of the A1R (amino acids 163–176; Affinity BioReagents, Exeter, UK) was used at a dilution of 1:200. The specificity of this antibody to the A1 receptor has been shown by Western blotting in the CNS, as well as in A1 receptor-transfectedXenopus oocytes (Smith et al., 2001). For additional controls, sections were incubated with PBS in place of primary antiserum or with the goat primary antiserum that had been preabsorbed with the peptide antigen for 1 hr before use (1 μg of peptide for 1 μg of antibody).
For light and electron microscopy, sections were washed three times for 10 min each in PBS, placed into biotinylated secondary antibodies to rabbit IgG or goat IgG as appropriate, diluted 1:200 in PBS (Vector Laboratories, Peterborough, UK) for 5 hr at 4°C, washed three times for 10 min each in PBS, and then put into Vectastain Elite ABC reagent (Vector Laboratories) for 18–20 hr at 4°C. These sections were then washed in Tris HCl buffer, pH7.4, and incubated in diaminobenzidine (DAB) solution (5 mg in 10 ml of Tris buffer with 0.01% H2O2) for 10 min. Some sections were put on subbed slides for light microscopy only. Other sections were washed in 0.1 m PB for 10 min and post-fixed in 0.5% osmium tetroxide (in 0.1 mPB) for 45 min. After washing in 0.1 m PB, the sections were then dehydrated through a series of ethanols, followed by two 10 min washes in propylene oxide (Fisher Scientific, Loughborough, UK). The sections were then immersed in Durcupan ACM resin (Fluka, Neu-Ulm, Germany) for 12–20 hr, mounted on glass slides, and placed in an oven at 60°C for 48 hr to polymerize the resin. Slides were subsequently examined at the light microscope level, and images were obtained via an integrating analog CCD camera (JVC KYF 55B) attached to an Acquis image capture system (Synoptics, Cambridge, UK) and adjusted for brightness–contrast–intensity and color balance using Corel PhotoPaint 9 before printing.
When areas with suitable staining for electron microscopy were selected, the coverslip was removed, and the relevant area was cut out and glued to the flat surface of a resin block. After trimming of the block, serial ultrathin sections (70 nm) were cut using a Leica Ultra Cut S ultramicrotome and collected on Formvar-coated 1 mm slot grids. The sections were then stained with lead citrate before viewing on a Phillips CM10 transmission electron microscope. Negatives were digitized using an Umax Astra 2200 scanner and manipulated in Corel Draw 9.
For fluorescence light microscopy, sections incubated in rabbit primary antibody were detected by incubation for 4 hr in Cy3-conjugated donkey anti-rabbit IgG (1:1000; Stratech, Luton, UK). Goat primary antibodies were detected by biotinylated anti-goat (1:200; Vector Laboratories), followed by streptavidin–Alexa488(1:1000; Molecular Probes, Rijnsburger-Weg, The Netherlands). Sections were dried onto gelatinized slides at 4°C, covered with Vectamount (Vector Laboratories), and stored at 4°C. Cy3 was visualized with a custom Cy3 filter set and Alexa488 with a standard FITC filter set, and digital images were acquired as above.
Retrograde labeling of sympathetic preganglionic neurons and immunohistochemistry. To retrogradely label the complete population of sympathetic preganglionic neurons (SPNs), male rats (150 gm; n = 3) were injected intraperitoneally with 0.1 ml of 1% Fluorogold (Fluorochrome Inc., Englewood, NJ) 7 d before immunohistochemistry. The rats were then deeply anesthetized with pentobarbitone sodium (60 mg/kg, i.p.) and perfused transcardially with 0.9% NaCl, followed by fixative containing 4% paraformaldehyde. Sections were washed in PBS, and the A1R immunostaining was performed for fluorescence detection as described above. Fluorogold was visualized with UV illumination and immunostaining with either Cy3 or FITC filters as appropriate.
SPNs with defined ganglionic projections were retrogradely labeled from the superior cervical ganglion (n = 5) or the adrenal gland (n = 3) in 150–200 gm rats under halothane anesthesia (5% in O2) by the injection of 5–10 μl of 1% cholera toxin B chain (CTB) (List Biologic, Campbell, CA) in saline. After 3–7 d of recovery, the rats were anesthetized with intraperitoneal Sagatal (60 mg/kg) and perfused transcardially with 4% paraformaldehyde and 0.025–0.1% glutaraldehyde as described above. Sections were cut on the vibrating microtome at 50 μm and freeze-thawed in liquid nitrogen as described above. Retrogradely transported CTB was detected before the A1R immunohistochemistry in two ways, both requiring incubation in goat anti-CTB (1:10,000; List Biologic) for 12–24 hr at 4°C. One approach was a preembedding gold procedure performed on tissue that had been perfused with low glutaraldehyde to facilitate penetration of gold particles into the tissue. Sections were transferred from primary antibody into secondary antibodies recognizing goat IgG conjugated to 1 nm gold particles (Amersham Pharmacia Biotech, Little Chalfont, UK) diluted 1:200 in pH 7.6 Tris-HCl buffer containing 1% fish gelatin and 1% goat serum for 18–20 hr at 4°C. After thoroughly rinsing the sections (four times for 10 min each) in PBS, fixing in 2% glutaraldehyde in 0.1 m PB for 2 min, and washing in distilled deionized water (four times for 10 min each), the gold particles were silver enhanced for 5–10 min using an IntenSE silver enhancement kit (Amersham Pharmacia Biotech). The other method used biotinylated anti-goat IgG after primary antibody incubations, detected with Vector ABC kit (Vector Laboratories), and visualized with the tetramethylbenzidine (TMB), pH6.0, method (Marfurt et al., 1988) to provide a crystalline reaction product. In both cases, sections were then incubated in a rabbit primary antibody against the A1R, which was visualized using the DAB method described above. Sections were then osmicated and processed for light and electron microscopy as described above.
Electrophysiology. Rats aged 10–15 d were anesthetized with urethane (2 gm/kg, i.p.). The thoracic spinal cord was exposed, and the dorsal and ventral roots were cut to isolate the cord. The upper and middle thoracic spinal cord was removed and submerged in ice-cold sucrose artificial CSF (aCSF) containing (in mm): 217 sucrose, 26 NaHCO3, 3 KCl, 2 MgSO4, 2.5 NaH2PO4, 1 CaCl2, and 10 glucose (equilibrated with 95%O2–5%CO2). The dorsal and ventral roots were cut, and the dura mater was removed. The pia mater was carefully teased away from the spinal cord, and the clean spinal cord was immersed in warm agar that was placed on ice for rapid setting. Slices (250 μm thick) of the embedded spinal cord were cut on a Vibroslice and placed into the recording chamber or a holding chamber for later use. The sections were submerged in aCSF (in mm: 124 NaCl, 26 NaHCO3, 3 KCl, 2 MgSO4, 2.5 NaH2PO4, 2 CaCl2, and 10 glucose) and superfused at a rate of 3–5 ml/min. All experiments were performed at room temperature. A visualized patch-clamp recording set-up was used with an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan). The IML was located at 10× magnification, and the cells were visualized at 60× magnification for recording. Both SPNs and interneurons within and around the IML were targeted for these experiments, identified both electrophysiologically and anatomically (at the end of recording).
Whole-cell patch-clamp recordings were obtained from neurons after achieving tight (>5 GΩ) resistance seals. Recordings were made in current-clamp mode using an Axopatch 1D (Axon Instruments, Foster City, CA). Patch electrodes (tip diameter of 3 μm; resistance of 4–6 MΩ) were filled with (in mm): 130 K-gluconate, 10 KCl, 11 EGTA, 2 MgCl2, 1 CaCl2, 10 HEPES, 5 Na2ATP, and 0.3 Na2GTP, pH 7.2 (295 mOsm). Neurobiotin (0.5%) was included in the patch solution and diffused into the neuron during recording. For some experiments, the filling solution was composed of (in mm): 110 Cs2SO4, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, 5 tetraethylammonium (TEA), and 5 Na2ATP.
Neurons were first characterized by applying hyperpolarizing and depolarizing current pulses (1 sec duration). Hyperpolarizing current pulses (−10 to −150 pA) produced voltage responses that were characteristically different for the two types of neuron studied. The input resistance of the neuron could also be calculated from the response to a hyperpolarizing current pulse. Depolarizing current pulses brought the neurons to threshold for firing, and the shapes of the action potentials were examined to further differentiate the neurons.
The lateral funiculus (lf), which contains the descending fibers originating in the areas of the brainstem involved in sympathetic control, was stimulated using a bipolar stimulating electrode placed just below the surface of the slice. The lf was stimulated at two times the threshold for response using an isolated stimulator (model DS2A; Digitimer, Hertfordshire, UK). Single-pulse stimulation was used except for the series of experiments looking at paired-pulse ratios when a second stimulus was applied 100–300 msec after the first. The response in control solution was always an EPSP, but on blocking this EPSP with selective antagonists, an IPSP could also be obtained at more depolarized potentials using the same stimulation parameters. The input resistance of the neuron was also tested just before, during, and after application of drugs.
Drugs were applied in the superfusing solution at a rate of 3–5 ml/min, and the concentration given is the final concentration in the bath. Drugs used were the A1R agonist cyclopentyladenosine (CPA) and the A1R antagonists 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (dissolved in dimethylsulfoxide) and 8-cyclopentyl-1,3-dimethylxanthine (CPT) (dissolved in 0.1N NaOH). To block the EPSPs, the excitatory amino acid antagonists 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX) (dissolved in 0.1N NaOH) or 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide disodium (NBQX), and d(−)-2-amino-5-phosphopentanoic acid (AP-5) were applied. To block synaptic activity, tetrodotoxin (TTX), DNQX, AP-5, strychnine, and bicuculline were applied. All drugs were obtained from Sigma/RBI (Poole, UK) and dissolved in water unless otherwise stated.
Data analysis. The voltage responses to current pulses were plotted to reveal the presence of any currents specific to either type of neuron. The action potential duration, amplitude, and afterhyperpolarization (AHP) were measured for each neuron because interneurons display action potentials of significantly shorter durations and smaller AHPs (S. Deuchars et al., 2000).
The EPSP (and IPSP) amplitude was measured as the peak change from the holding potential and averaged over 10–20 consecutive sweeps for control and drug responses. Plotting the drug response as a mean percentage of the control response in each case showed the effects of the drugs on the EPSP. The effects of the drugs were tested statistically using the paired Student's t test, and differences were considered significant when p < 0.05. Any effects of the drugs on resting potential and input resistance were also noted. The responses of a neuron to paired-pulse stimulation of the lf were given as the ratio of the second response to the first response. Paired-pulse ratios were then calculated in control medium and in CPA and compared to determine whether there was a significant change.
Histology. After recordings of up to 4 hr, electrodes were very slowly removed from neurons, and the slice was immersed in fixative containing 0.5% glutaraldehyde–4% paraformaldehyde for up to 18 hr. Slices were embedded in gelatin and immersed in the same fixative. Sections (50 μm thick) were cut on a vibrating microtome, freeze-thawed twice by immersing in liquid nitrogen, and incubated in extravidin–peroxidase (1:250; Sigma) for 24–48 hr, which was then visualized with DAB (Sigma). Sections were then processed for light microscopy or for light and electron microscopy as described previously (Deuchars and Thomson, 1995) and reconstructed using a drawing tube attached to a light microscope. This allowed anatomical confirmation of the type of neuron recorded.
RESULTS
Immunohistochemistry
Specificity of antibodies
The specificity of many of the antibodies has been reported previously (see Materials and Methods) (Schindler et al., 2001; Smith et al., 2001). In addition, no staining was observed in control sections that had no primary antibody present or in which the primary antibody was preadsorbed against the control peptide (only available with the goat antibody from Santa Cruz Biotechnology), indicating that the antibodies recognized the appropriate sequence in the tissue. Furthermore, the pattern of immunostaining was identical with all antibodies tested (Figs. 1,2) and was also consistent with previous reports of A1 receptor localization using in situhybridization (Reppert et al., 1991) or autoradiography (Goodman and Synder, 1982; Geiger et al., 1984; Choca et al., 1987), and so we are confident that our procedures specifically localized the adenosine A1 receptor.
Fig. 1.
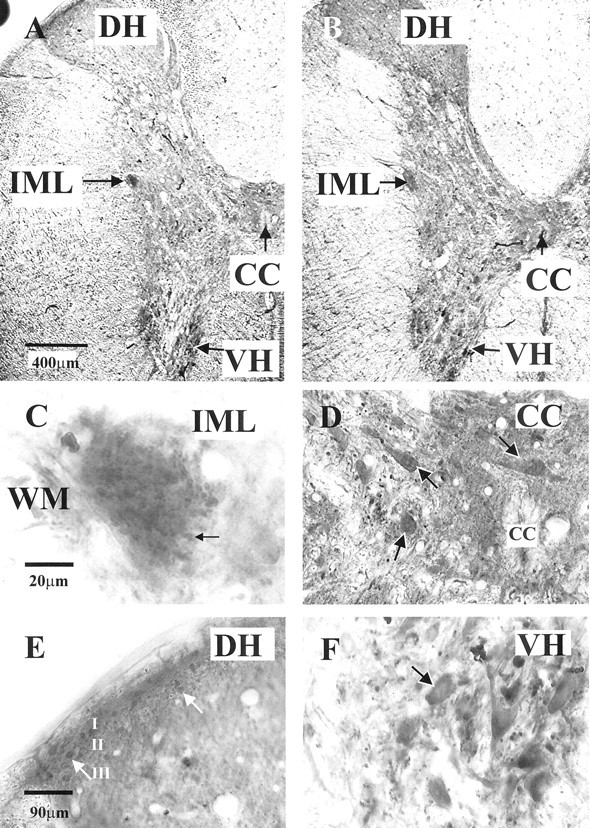
Adenosine A1 receptor immunoreactivity in the thoracic spinal cord. A, Low magnification of staining obtained with the A1 receptor antibody raised in rabbit (gift from Dr. Mike Yates, Leeds University) in the thoracic spinal cord. Immunoreactivity was visualized with diaminobenzidine. Labeling was observed throughout the spinal cord. Even at low magnification, the IML is clearly seen as heavily stained. Staining is also dense around the central canal (CC), the dorsal horn (DH), and the ventral horn (VH). B, Low magnification of staining obtained with the A1 receptor antibody raised in goat (Santa Cruz Biotechnology), prepared identically to the material inA, except for the primary antibody. Note that the pattern of staining is almost identical, with the IML and ventral horn being particularly prominent. C, High magnification of the IML. Staining was a compact collection of punctate structures covering the IML adjacent to the white matter (WM). The arrow indicates one such punctate structure. D, In the vicinity of the central canal, labeling could be observed in the somata and dendrites of neurons (arrows), as well as presumptive fibers.E, In the dorsal horn, staining of fibers was dense in lamina II, and labeled neuronal somata could also be observed in laminas II and III (arrows). F, In the ventral horn, labeled fibers, somata, and dendrites of large neurons (arrow) were observed.
Fig. 2.
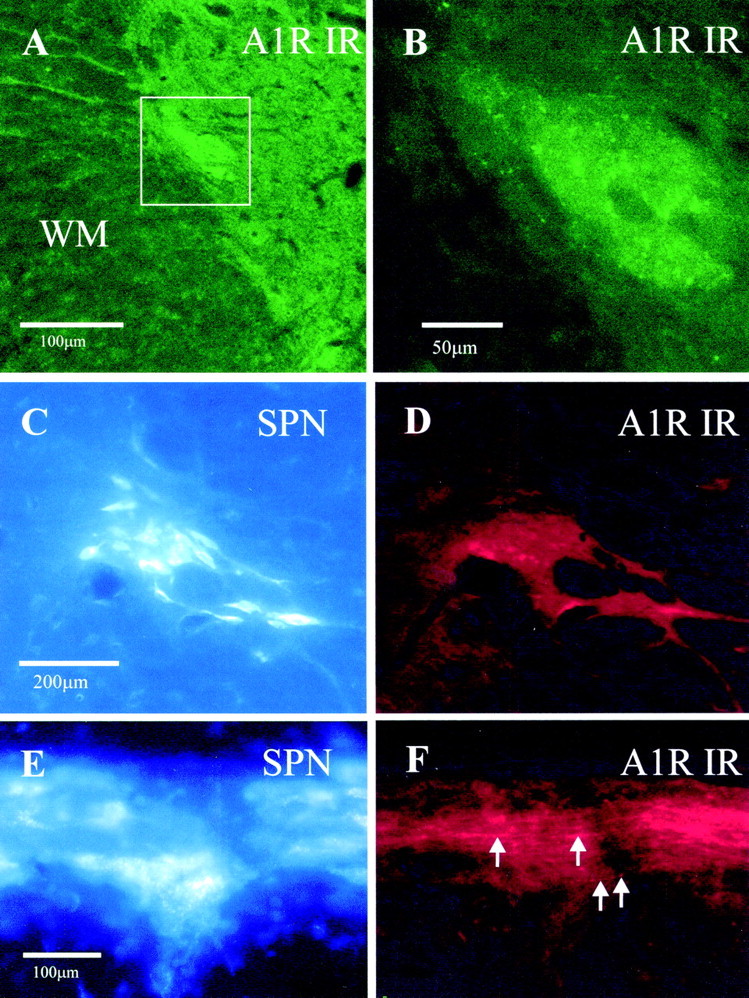
Fluorescence images indicating that adenosine A1 receptor immunoreactivity in the IML is dense in the region of retrogradely labeled SPNs. A, Low magnification of adenosine A1R-IR detected with the antibody raised in goat and visualized with Alexa488 viewed through a FITC filter set. The IML contains dense labeling and so stands out from surrounding structures. The boxed area surrounding the IML is shown at higher magnification in B.B, Higher magnification of the boxed areain A. At this magnification, it is clear that the staining in the IML has a punctate appearance. C, SPNs retrogradely labeled in the IML by an intraperitoneal injection of Fluorogold and visualized by UV illumination in a coronal section. SPNs appear blue–white under these conditions and contain Fluorogold in cell bodies and dendrites. The same section is shown inD. D, A1R-IR in the same section asC, detected with Cy3-conjugated secondary antibodies and viewed through a Cy3 filter set so that immunoreactivity for the adenosine A1R appears red. The A1R-IR makes the IML stand out as brighter red than the surrounding neuropil, and this bright staining persists into the lateral funiculus.E, SPNs retrogradely labeled in the IML by an intraperitoneal injection of Fluorogold and visualized by UV illumination in a longitudinal section. The same section is shown inF. F, A1R-IR in the same area of the section as E but viewed through a Cy3 filter set so that immunoreactivity for the adenosine A1 receptor appearsred. The A1 immunoreactivity runs in fibers along the IML (arrows).
Light microscopy
Adenosine A1 receptor immunostaining was observed throughout the spinal cord (Fig. 1). In the ventral horn, strong staining was apparent in somata of presumptive motor neurons, as well as fibers (Fig.1A,B,F). In the dorsal horn, staining was strongest in lamina II in both fibers and cell bodies (Fig.1A,B,E). Labeled neurons and fibers were also apparent in the vicinity of the central canal (Figs. 1A,B,D). The IML was evident even at low magnifications as a region of dense staining (Figs. 1A,B, 2A). The predominant staining was punctate in nature (Figs. 1C,2B). When SPNs were retrogradely labeled by intraperitoneal injections of Fluorogold, then this dense region of A1R-IR precisely overlaid the retrogradely labeled SPNs (Fig.2C–F). In longitudinal sections, the labeled fibers appeared to run in a ladder-like pattern similar to that described for the distribution of SPNs (Petras and Cummings, 1972) and to follow the spread of SPNs at the rungs of the ladder.
Electron microscopy
Ultrastructural examination of the IML always revealed A1R-IR highly targeted to the luminal membrane of endothelial cells of blood vessels (Fig. 3A), in myelinated fibers in the lateral funiculus (n = 30) (Fig. 3C) and in presynaptic terminals (n = 100) onto dendritic shafts (n = 85) (Fig.3B,E,F), spines (n = 1), and somata (n = 2) (Fig.4E). In accordance with the receptors being incorporated into the membrane, reaction product was often observed adjacent to the membrane (Figs.3B,D,4A–C,E,F). In addition, some cytoplasmic reaction product was observed (Fig.4D), presumably reflecting A1Rs that are being trafficked to the membrane or have been internalized during agonist induced desensitization (Saura et al., 1998). Some postsynaptic targets of A1R-IR terminals were identified as SPNs by the presence of crystalline reaction product when retrogradely transported CTB was visualized with TMB (n = 30) (Fig.3E,F). In other cases, SPNs were identified by the presence of silver intensified gold particles when CTB was localized with the preembedding immunogold approach (n = 30) (Fig. 4A–F). In these cases, the labeled structures were followed in serial sections to verify the presence of several gold particles and so confirm that labeling was specific (Fig. 4A,B). Where A1R-immunoreactive terminals synapsed onto unlabeled postsynaptic structures, it was impossible to say whether the postsynaptic cell is an interneuron or part of an SPN in which there is no reaction product; however, the electrophysiological experiments do suggest that interneurons are also likely targets of these A1R-immunoreactive terminals (see below). A1R-IR was also detected in the soma of retrogradely labeled SPNs in which it was associated with endoplasmic reticulum but not with the synaptic specialization (Fig.4E).
Fig. 3.
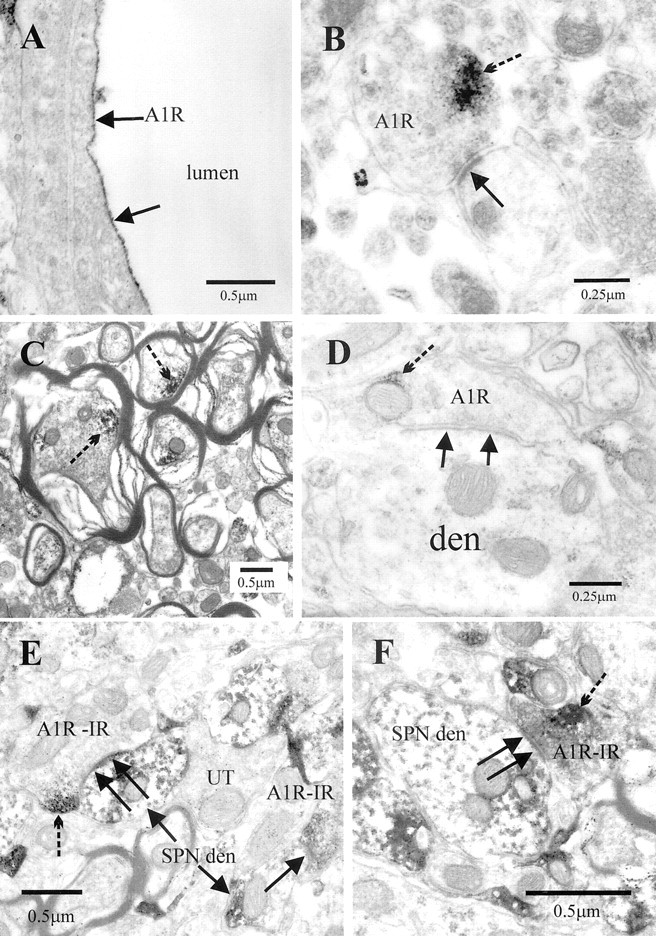
Electron microscopic localization of the adenosine A1R-IR in the IML. A, A capillary in which A1R-IR is highly targeted to the luminal membrane of endothelial cells of blood vessels (arrows). B, A presynaptic terminal in the IML containing immunoreactivity for the A1R adjacent to the membrane (broken arrow indicates immunoreaction product). This terminal forms an asymmetric-type synaptic contact (arrow) with a dendritic structure.C, A1R-IR (broken arrows) was detected in numerous myelinated fibers in the lateral funiculus. D, A presynaptic terminal in the IML containing A1R-IR adjacent to the membrane (broken arrow indicates immunoreaction product). This terminal forms an asymmetric-type synaptic contact (arrows) with a dendritic structure (den). E, F, A1R-IR terminals formed synaptic contacts (arrows) with structures identified as SPN dendrites (SPN den) by the presence of crystalline reaction product (arrows) as a result of retrograde labeling with cholera toxin B chain, which was visualized with the TMB method. Note that the A1R-IR (broken arrows) is adjacent to the plasma membrane but some distance from the synaptic face.UT, unlabeled terminal.
Fig. 4.
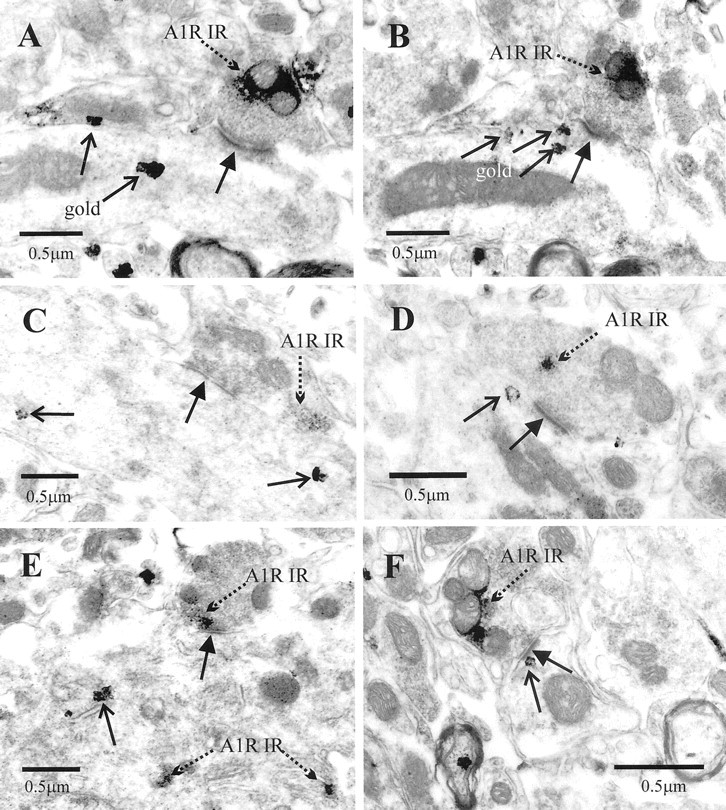
Adenosine A1R-IR terminals form synaptic contacts with structures identified as SPNs by retrograde labeling.A, An A1R-IR terminal (broken arrow) forms an asymmetric-type synaptic contact (arrow) with a dendritic structure that contains silver-intensified gold particles (open arrows), indicating that it is a retrogradely labeled structure. The same terminal is shown inB. Note that the reaction product is adjacent to the membrane of the terminal but some distance from the active zone.B, The same terminal shown in A but serially several sections on, confirming that the dendrite is retrogradely labeled. The section has been tilted slightly with the goniometer to visualize the synaptic specialization. The silver grains in this section (open arrows) are spatially separate to those in A, indicating that these are different particles and not the result of intensification of the initial gold particles throughout the dendrite. C, D,F, Synaptic terminals (A1R-IR) containing A1 receptor immunoreactivity form synaptic contacts (arrows) with dendrites identified as retrogradely labeled SPNs by the silver-intensified gold particles (open arrows). Immunoreactivity is both adjacent to the membrane of labeled terminals (C, F) and within the cytoplasm (D). E, An A1R-IR terminal forms a synaptic contact (arrow) with the soma of an SPN. The soma contains not only silver-intensified gold particles (open arrows) but also A1R-IR associated with the endoplasmic reticulum (broken arrow).
Electrophysiology
These studies were performed on both SPNs and interneurons within the IML of the thoracic spinal cord because it is likely that both of these neurons could be targeted by synapses containing adenosine A1 receptors. The two groups of neurons were distinguished by their very different electrophysiological and morphological characteristics. From the voltage responses to hyperpolarizing current pulses, it was noted that SPNs showed a delayed return to the holding potential at the end of a current pulse, indicative of activation of anIA (Fig.5A). In contrast, interneurons within the IML are characterized by a sag in the voltage response to a hyperpolarizing current pulse, suggesting activation of anIH (Fig. 5B). The interneuronal action potentials are significantly shorter in duration than those of SPNs (Fig. 5C) and have a complex but smaller afterhyperpolarization. Neurons were filled with Neurobiotin during the recording procedure, and 10 interneurons and nine SPNs could be recovered and reconstructed at the end of the experiment. SPNs had axons that coursed ventrally straight out of the IML to the ventral horn in which they exited. Interneuronal axonal arborization, in contrast, was extensive, and the axons coursed both dorsally and ventrally and showed varicosities within the IML, which could be attributable to sites of synaptic contact. Because identification of the SPNs and interneurons was easily determined from electrophysiology and could be confirmed with anatomy, the responses of both types of neurons to applications of A1R agonists and antagonists therefore could be compared.
Fig. 5.

CPA reduces the amplitude of EPSPs elicited in both SPNs and interneurons. A, On the leftare the voltage responses of an SPN to hyperpolarizing (averages of 3 sweeps) and depolarizing (single sweep) current pulses. Thetraces show a delayed return to resting potential at the end of the hyperpolarizing current pulses, indicative of activation of an IA (arrow). The action potential duration was 7.2 msec, and the afterhyperpolarization was quite simple with two components. On the right are average traces (of 10 sweeps) of the EPSP elicited by lf stimulation in control solution, CPA (100 nm), and then after switching back to standard aCSF. The EPSP amplitude was decreased by CPA application. B, The left shows the voltage responses of an interneuron to the same hyperpolarizing (averages of 3 sweeps) and depolarizing (single sweep) current pulses. At hyperpolarized potentials, a sag in the voltage response was observed, suggesting that an IH was activated. The action potential duration was 3.2 msec, and the AHP shows a distinct fast and slower phase. On the right are average traces (of 10 sweeps) of the EPSP elicited in the interneuron by lf stimulation. The EPSP amplitudes in control aCSF are not significantly different from those elicited in the SPN. In addition, the effects of CPA on the interneuronal EPSP are similar to those observed in the SPN. C, Comparisons of the duration of the interneuronal and SPN action potentials. D, Pooled data showing that the effects of CPA on the EPSPs elicited in SPNs are not significantly different from those in interneurons.
The A1R agonist CPA decreased the EPSPs elicited by lateral funiculus stimulation in both SPNs and interneurons
We chose to stimulate the lateral funiculus because this contains descending axons from areas of the brainstem that make monosynaptic connections onto SPNs, whereas dorsal horn input onto SPNs is polysynaptic and thus more difficult to analyze (Dembowsky et al., 1985). Lateral funiculus stimulation elicited responses in all SPNs and interneurons tested. These consisted of a fast EPSP in control solution, which occurred at a constant latency and could follow high-frequency stimulation (up to 50 Hz). These EPSPs were therefore considered to be of a monosynaptic nature and were not significantly different in amplitude or latency for the two groups of neurons. These EPSPs could be abolished by applications of the excitatory amino acid receptor antagonists CNQX (20 μm) or NBQX (20 μm) and AP-5 (50 μm), showing that these responses are elicited by release of glutamate (n = 6) (Fig. 6C). This was expected because, in previous work, stimulation of the rostral ventrolateral medulla (RVLM) elicited monosynaptic EPSPs in SPNs that were mediated by activation of the excitatory amino acid receptors, indicating that glutamate is the major neurotransmitter in this pathway (Deuchars et al., 1995). The effects of bath applications of 100 nm CPA, which is selective for A1Rs, were determined on both SPNs and interneurons. CPA decreased the amplitude of the EPSP elicited in all SPNs tested from 10.5 ± 1.2 (mean ± SEM) to 5.6 ± 0.7 mV, a decrease of 44.5 ± 5% (n = 13; measured at −70 mV) (Fig.5A,D). The effect of this drug on the EPSPs elicited in interneurons was almost identical with CPA decreasing the amplitude of these EPSPs from 9.0 ± 0.9 to 5.3 ± 0.8 mV, a decrease of 44.1 ± 4.5% (n= 11) (Fig. 5B,D). Not surprisingly, these decreases in amplitude were not significantly different for the two groups of neurons, and so all neurons were considered together for the rest of the analysis. When CPA application ceased, the drug washed off slowly, and the EPSP amplitude returned to near control values (Fig. 6).
Fig. 6.
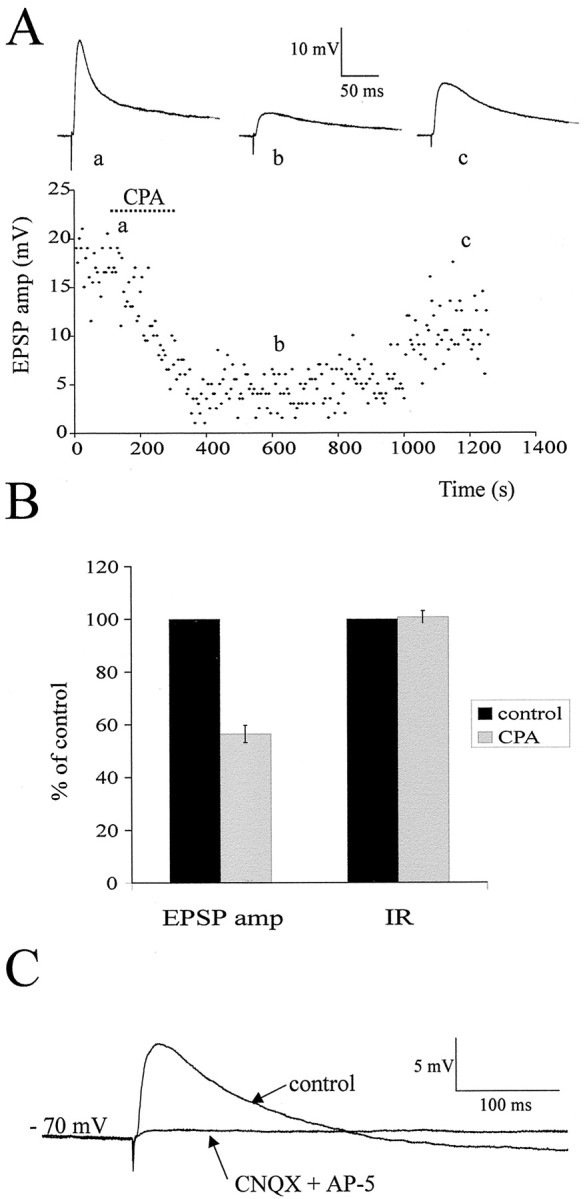
Time course of action of CPA and pharmacology of the EPSP. A, Graph of the amplitude of the EPSP elicited by lf stimulation in an SPN over time with sample EPSPs shown at thetop for each of the conditions (average of 10 consecutive sweeps). It can be clearly seen that CPA application caused a decrease in EPSP amplitude that slowly recovered as the CPA was washed off. B, Pooled data from 26 neurons showing the effect of CPA on EPSP amplitude and input resistance. CPA caused a significant decrease in EPSP amplitude but had no significant effect on input resistance. C, In another SPN, the EPSP was abolished by application of the excitatory amino acid receptor antagonists CNQX (20 μm) and AP-5 (50 μm), indicating that the EPSP is mediated by activation of these receptors postsynaptically.
The effects of CPA could be antagonized by the A1R antagonist DPCPX
The A1R antagonist DPCPX (200–500 nm) was applied to neurons to selectively block the reduction observed with CPA. DPCPX was first applied during application of CPA in which the EPSPs had been reduced from 9.1 ± 1.3 to 5.8 ± 1.1 mV, a decrease of 39.4 ± 6.0% (n = 9). DPCPX antagonized the effects of CPA, and the EPSP amplitude returned to 8.8 ± 1.0 mV without effect on holding potential or input resistance (Fig.7).
Fig. 7.
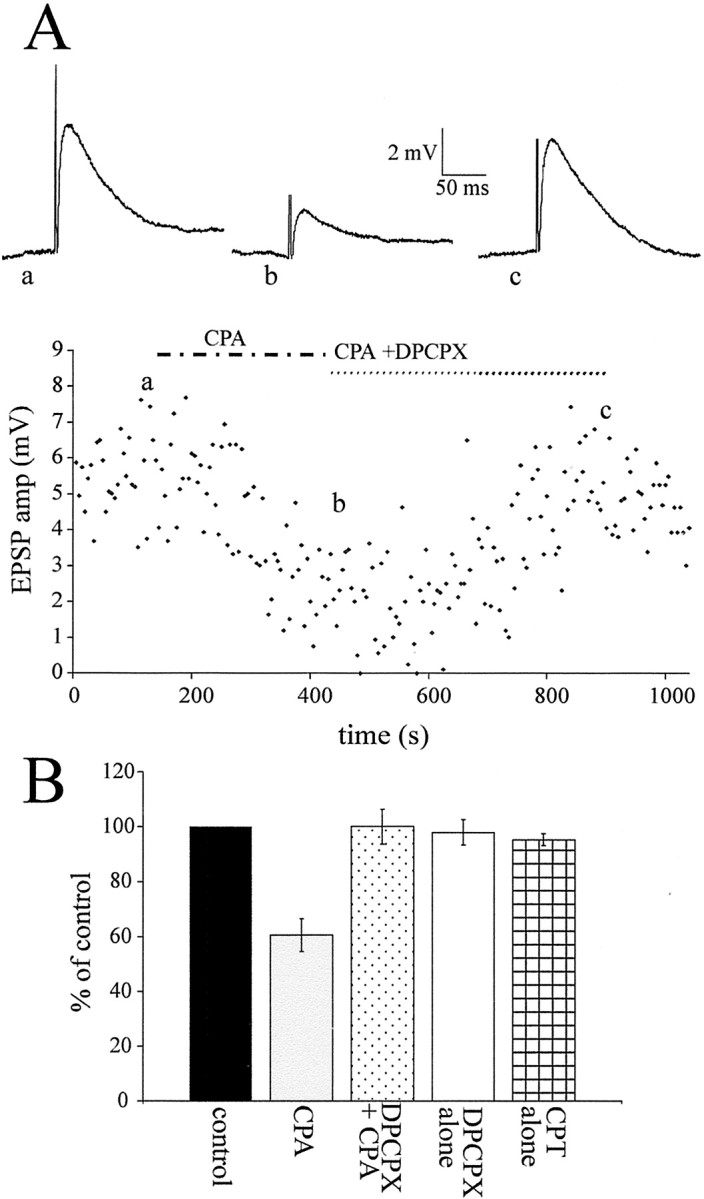
DPCPX antagonized the effects of CPA.A, Graph of the EPSP amplitude elicited by lf stimulation against time in an interneuron with traces of the EPSP above (averages of 10 sweeps). CPA (100 nm) decreased the EPSP amplitude, an effect that was antagonized by application of DPCPX (300 nm), the A1R antagonist, together with CPA.B, Pooled data showing the effect of CPA on the EPSP and the antagonism by DPCPX, which restored the EPSP to the control amplitude. Both DPCPX and CPT (10 μm), another A1R antagonist, had no effect on EPSP amplitude when applied alone.
DPCPX (500 nm) and another A1R antagonist, CPT (10 μm), were applied alone to determine whether there was an ongoing release of adenosine, which tonically reduced the EPSP amplitude. Neither drug had an effect on holding potential or the evoked EPSP in which the amplitude was unchanged in DPCPX [10.6 ± 1.4 to 10.5 ± 1.7 mV (n = 5)] and CPT [8.9 ± 0.9 to 8.6 ± 0.8 mV (n = 8)] (Fig.7B). Thus, in our recording conditions, there is no tonically active adenosine.
The effects observed with CPA are attributable to a presynaptic action
To determine the sites of the A1R mediating the effects of CPA, the relative amplitude of the synaptic response to two stimuli applied to the lf at short intervals was determined in control aCSF and in CPA. If a change in paired-pulse ratio is observed with CPA, this is probably attributable to activation of A1R presynaptically, which then decreases the probability of neurotransmitter release.
In control solution, the paired-pulse ratio was 0.88 ± 0.11 (n = 8). Because this ratio is <1, the control response is a paired-pulse depression in the control solution likely attributable to a high probability of neurotransmitter release with the first stimulus, causing depletion of synaptic vesicles so that there is a smaller response to the second stimulus. When CPA was applied, the paired-pulse ratio increased to 1.26 ± 0.15, a significant change from the control ratio of 0.88 ± 0.11. Thus, there was a switch from paired-pulse depression to paired-pulse facilitation observed with CPA (n = 8) (Fig.8A). This is because, in the presence of CPA, the first EPSP is much smaller, as discussed above. This means that there are more synaptic vesicles remaining in the presynaptic cleft that can be released with the second stimulus, so paired-pulse depression does not occur. In addition, because fewer vesicles were released with the first stimulus, there was an elevated level of intracellular calcium; thus, the second stimulus can release more vesicles and the EPSP is larger. So now we see paired-pulse facilitation, and these data indicate a presynaptic site of action of adenosine.
Fig. 8.

Effects of CPA are attributable to a presynaptic action. A, Responses of an SPN to twin pulse stimulation of the lf (200 msec apart) in control solution showed paired-pulse depression (i.e., the response to the second pulse was smaller than the first). In CPA, the second response is the same size as the first reduced response, with the ratio now being 1:1. On theright are the pooled data showing the paired-pulse ratio increases in the presence of CPA, indicative of a presynaptic site of action for adenosine. B, Effects of applications of CPA and DPCPX plus CPA in a neuron recorded using a patch solution in which cesium sulfate is the main component. CPA decreased the EPSP amplitude without effect on the membrane potential. This reduction was antagonized by DPCPX. C, In synaptic block medium containing TTX, DNQX, AP-5, strychnine, and bicuculline, action potentials and the response to lf stimulation were blocked. Hyperpolarizing current pulses were then applied to check input resistance, and CPA was applied. There was no change in membrane potential or input resistance with application of CPA in this recording medium.
In addition, experiments were performed using a patch pipette solution with the main component being cesium sulfate. TEA, the potassium channel blocker, was also included in the solution. Inclusion of these two nonselective potassium channel-blocking agents in the patch solution has been shown to dramatically reduce postsynaptic effects of adenosine attributable to opening of potassium channels (Li and Perl, 1994). Under these recording conditions, the effects of CPA on the EPSP amplitude were identical to those observed previously, i.e., a decrease in amplitude of the evoked EPSP from 8.9 ± 1.5 to 4.8 ± 1.0 mV, a decrease of 42.9 ± 12% (n = 5) (Fig.8B). In addition, DPCPX also antagonized these effects of CPA (n = 3) (Fig. 8B). This suggests that the effects observed were not attributable to a change in postsynaptic potassium currents.
CPA had no significant effect on the input resistance of the neurons, measured as the change in voltage to a hyperpolarizing current pulse at a holding potential of −70 mV (Fig. 6B). Furthermore, in these recording conditions (whole-cell current clamp), CPA did not cause the SPNs or interneurons to hyperpolarize. To further test for postsynaptic sites of action, the effects of CPA on the holding potential and input resistance were also tested in synaptic block medium containing TTX (0.6 μm), DNQX (20 μm), AP-5 (50 μm), bicuculline (5 μm), and strychnine (2 μm) immediately after establishing whole-cell configuration. In this medium, CPA had no effect on the membrane potential or input resistance of the neurons tested (n= 6) (Fig. 8C). These data therefore indicate that, in young rats, the major site of action of the A1Rs is presynaptic, in accordance with the immunohistochemical localization of the A1Rs in adult tissue described above.
Fast IPSPs elicited by lf stimulation are not affected by applications of CPA
Stimulating the lateral funiculus activates both excitatory and inhibitory pathways that descend from the RVLM (Deuchars et al., 1997); thus, we were interested to see whether activation of A1Rs also affects inhibitory inputs onto these neurons. In control conditions, the response observed with lf stimulation was an EPSP that was always reduced in amplitude with CPA. To determine whether IPSPs were underlying these EPSPs, the excitatory amino acid receptor antagonists DNQX or NBQX and AP-5 were applied to eight neurons. In five of these neurons, an IPSP was revealed, whereas in the other three neurons, no additional postsynaptic potential was elicited. These fast IPSPs had a very short constant latency and could follow high-frequency stimulation. In addition, the IPSP was still robust in the presence of the excitatory amino acid antagonists that have been shown previously to block polysynaptic IPSPs elicited by RVLM stimulation (Deuchars et al., 1997). These data indicate that the IPSP was elicited at least in part by activation of a monosynaptic pathway. In the presence of these excitatory amino acid antagonists, CPA had no effect on the IPSP amplitude because, in control conditions, the amplitude was −4.9 ± 0.7 mV and, in CPA, the amplitude was −4.9 ± 0.8 mV (Fig.9). Thus, there was no significant difference in the amplitudes of this fast IPSP in control and CPA solutions. On two occasions, after washout of CPA, the GABAA receptor antagonist bicuculline was superfused onto the slice, which blocked the IPSP completely, whereas strychnine had no effect (data not shown). This indicates that the fast IPSPs elicited by lf stimulation are attributable to activation of GABAA receptors. Thus, the A1 receptors are located presynaptically on excitatory and not inhibitory fibers in the lf and have a selective effect on excitatory transmission.
Fig. 9.
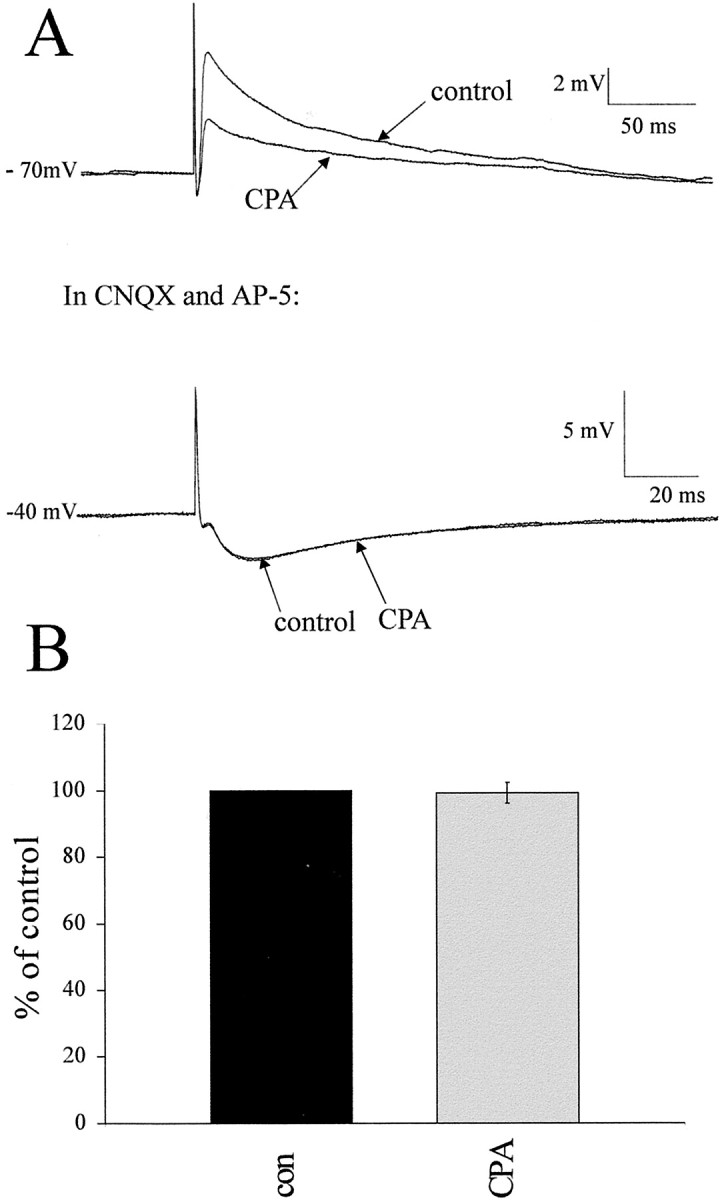
Fast IPSPs elicited by lf stimulation are not affected by CPA. A, Reponses of an interneuron to lf stimulation in control and CPA-containing aCSF. CPA significantly reduced the EPSP amplitude. The EPSP was then antagonized by application of CNQX and AP-5 to reveal an underlying IPSP (seen here at −40 mV), which increased in amplitude as the membrane was depolarized. This IPSP was not affected by CPA application at the same concentration (100 nm) as that which reduced the EPSP. B, Pooled data from five neurons in which an IPSP was revealed. In all five cases, the IPSP was not affected by CPA, suggesting a selective site of action of the adenosine.
DISCUSSION
These studies indicate that the A1R is located on excitatory terminals in the intermediolateral cell column that innervates both SPNs and interneurons. These interneurons may play a role in sympathetic control because all interneurons in this region have activity correlated to sympathetic activity (Chau et al., 2000). Activation of these presynaptic A1Rs reduces release of an excitatory amino acid (probably glutamate) from these excitatory terminals but not of GABA from inhibitory terminals. Previous studies on the distribution of A1Rs in the spinal cord failed to report any degree of binding in regions involved in sympathetic control. Thus, our studies are extremely pertinent because the degree of immunoreactivity observed here and the effects of the A1R agonists indicate a crucial role for adenosine in the control of sympathetic activity.
Another interesting observation was an abundance of A1R immunoreactivity in the luminal membrane of endothelial cells in the spinal cord. We believe that this is the first localization in both the peripheral and central vasculature. This is also relevant to our studies because Koh et al. (1996, 1998) used an intrathecal method to determine the effects of A1R activation on cardiovascular variables. Therefore, their observations may be attributable to activation of receptors located on the blood vessels that may cause nitric oxide-dependant relaxation (Marshall, 2000), possibly influencing neuronal activity. This necessitates the in vitro approach to determine the exact role of adenosine in modulating synaptic transmission in the spinal cord, and our studies show clearly that activation of A1Rs reduces excitatory neurotransmitter release from presynaptic terminals onto SPNs and interneurons.
Sources of adenosine
It is well established that adenosine plays an important role in modulating synaptic transmission throughout the CNS, but where does this adenosine come from? Adenosine may be released from vesicles within the presynaptic terminal itself or may be a result of the breakdown of ATP released from the presynaptic neurons (Cunha, 2001). An additional alternative could be that adenosine is formed from the breakdown of ATP intracellularly during periods of hypoxia or ischemia. In other autonomic regions, a major source of adenosine may be attributable to the extracellular metabolism of ATP. For example, St. Lambert et al. (1997) showed in the brainstem that at least some effects of adenosine were attributable to breakdown of ATP extracellularly but also that there was release of adenosine itself. Because there is very little endogenous activation of A1Rs during our experiments, it would be difficult to try and dissect out the sources of adenosine under these recording conditions. However, because SPNs are excited by activation of the P2X7 receptor, whose endogenous ligand is ATP (J. Deuchars et al., 2000), it is likely, in accordance with other CNS regions, that a probable source of the adenosine is from extracellular catabolism of ATP.
Is there a tonic effect of adenosine?
Because applications of DPCPX and CPT had no effect on EPSPs elicited by lf stimulation or resting membrane potential, basal levels of adenosine are not sufficient to activate A1Rs under our experimental conditions. Oliet and Poulain (1999) also saw no effect of CPT on EPSPs evoked with single stimuli; however, stimulating the hypothalamus with a train of stimuli caused a depression in the response over time that was CPT sensitive. We stimulated the lf with similar trains of stimuli at a rate of 1 Hz (which is slow enough to prevent paired-pulse depression occurring); however, no reduction in EPSP amplitude was observed (S. Deuchars, unpublished observations). One possibility is that tonically released adenosine is broken down or taken up by terminals or glial cells too fast for effects to be observed in conditions of the experiments. This may therefore merit further investigation using either inhibitors of adenosine metabolism or uptake, such as in the dorsal horn (Ackley et al., 2000) in which adenosine antagonists alone also have little effect.
Is there a postsynaptic effect of adenosine?
At the electron microscopic level, both presynaptic and, to a lesser degree, postsynaptic A1Rs were located in the IML. However, electrophysiological data did not reveal postsynaptic A1Rs. The majority of A1R-mediated postsynaptic effects are attributable to potassium channels opening, which causes hyperpolarization (Trussell and Jackson, 1987; Gerber et al., 1989). The potassium reversal potential in our experiments was −96.8 mV, far removed from the membrane potential at which the effects of CPA were studied (−40 to −70 mV). A1R opening of potassium channels involves activation of G-proteins; thus, the whole-cell patch-clamp technique may wash out vital second messengers for observation of this postsynaptic response. However, other reports observed postsynaptic hyperpolarization using similar whole-cell patch-clamp techniques (Li and Perl, 1994; Herlenius and Lagercrantz, 1999). Interestingly, at the electron microscopic level, A1Rs were associated with the endoplasmic reticulum rather than the somatic membrane, suggesting that A1Rs are synthesized in the postsynaptic neuron and then transported to the terminals. In support of this, studies report adenosine-mediated decreases in synaptic transmission in sympathetic ganglia (Alkadhi et al., 1984; Hogan et al., 1998). Therefore, combining our anatomical and physiological data, it seems likely that the major effect of the A1Rs in the IML is presynaptic.
Source of the presynaptic terminals
The location of A1Rs in myelinated axons in the lf and the fact that stimulating the lf elicited EPSPs that were reduced in amplitude by CPA suggests that the A1Rs may be located on presynaptic terminals of descending fibers from higher centers. SPNs and interneurons are innervated by direct inputs from supraspinal regions, including the RVLM, the A5 region of the pons, raphe nuclei, paraventricular nucleus, and lateral hypothalamus (Strack et al., 1989). In situhybridization reveals strong neuronal expression of the A1R in the RVLM (Reppert et al., 1991), suggesting that these neurons produce the A1R and may transport it to their terminals in the IML.
Inhibitory transmission was not affected by activation of A1Rs
Interestingly, CPA affected EPSPs but not IPSPs, suggesting that there are no A1Rs in inhibitory terminals onto SPNs and interneurons. There is a monosynaptic inhibitory pathway from the RVLM region of the brainstem onto SPNs (Deuchars et al., 1997) distinct from the excitatory bulbospinal pathway originating in that region. Thus, it is feasible that A1Rs are located exclusively on excitatory terminals within the IML. There is evidence that A1R agonists reduce release of inhibitory and excitatory transmitters in the hypothalamus, substantia nigra reticulata, and the periaqueductal gray regions (Shen and Johnson, 1997; Bagley et al., 1999; Oliet and Poulain, 1999). However, in CA1 neurons, DPCPX failed to block depression of IPSCs induced by hypoxia (Katchman and Hershkowitz, 1993), showing a selective site of action of adenosine similar to that observed here.
What is the role of adenosine in the control of sympathetic outflow?
As described above, adenosine may play an important role in modulating synaptic transmission. Furthermore, adenosine acting at A1Rs has a neuroprotective role in the CNS because ischemic episodes or hypoxia results in adenosine production, which inhibits the release of excitatory amino acids to prevent excitotoxicity (Fowler, 1989; Simpson et al., 1992; Katchman and Hershkowitz, 1993; Sweeney, 1997).
In the autonomic nervous system, adenosine has long been known to have profound effects at the level of the end organ and will cause vasodilatation in skeletal and cardiac muscle to increase oxygen supply during hypoxic episodes (Marshall, 2000). In addition, there is a role for adenosine in the control of neuronal activity in areas of the brainstem crucial for cardiovascular regulation (Thomas and Spyer, 1999). However, at the level of the spinal cord, little was known about the role that it plays in the control of neurons involved in sympathetic control. Although intrathecal studies suggest that adenosine in the spinal cord modulates blood pressure and heart rate (Koh et al., 1996), these may be attributable to effects at the blood vessels rather than on neuronal activity. This study therefore is the first to show the role for adenosine in the control of neurons influencing sympathetic outflow from the spinal cord.
In accordance with the many previous studies in the brain (see above), this study shows that activation of A1Rs reduces release of glutamate from presynaptic terminals in the IML without effect on inhibitory transmission. Because excessive release of glutamate is an important mechanism underlying the excitotoxic effects of hypoxia and ischemia (Hara et al., 1993; Schroder et al., 1999) and adenosine is produced under such conditions (Rudolphi et al., 1992), our data are consistent with a neuroprotective role for adenosine in the IML. This is important because sympathetic outflow at the level of our recordings in the thoracic spinal cord is mainly to the heart and blood vessels in which abnormal activity could lead to circulatory problems.
We have established that adenosine A1 receptors are located on excitatory presynaptic terminals innervating neurons in the IML involved in controlling sympathetic outflow. Activation of these receptors reduces the release of transmitter from these excitatory terminals. We therefore conclude that adenosine acting on A1 receptors can play an important role in determining the level of activity of the sympathetic nervous system at the level of the spinal cord.
Footnotes
We thank the British Heart Foundation and Wellcome Trust for their generous support, Brenda Frater for recovery of filled neurons, and Dr. Ida Llewellyn-Smith for help with recovery of filled neurons
Correspondence should be addressed to Dr. Susan A. Deuchars, School of Biomedical Sciences, Worsley Building, University of Leeds, Leeds, LS2 9NQ, UK. E-mail: S.A.Deuchars@leeds.ac.uk.
REFERENCES
- 1.Ackley MA, Baldwin SA, King AE. Inhibition of adenosine uptake attenuates glutamatergic synaptic transmission in lamina II of the rat spinal cord in vitro. Soc Neurosci Abstr. 2000;26:821.5. [Google Scholar]
- 2.Alkadhi KA, Brown TR, Sabouni MH. Inhibitory effect of adenosine on transmission in sympathetic ganglia. Naunyn Schmiedebergs Arch Pharmacol. 1984;328:16–19. doi: 10.1007/BF00496098. [DOI] [PubMed] [Google Scholar]
- 3.Bagley EE, Vaughan CW, Christie MJ. Inhibition by adenosine receptor agonists of synaptic transmission in rat periaqueductal grey neurons. J Physiol (Lond) 1999;516:219–225. doi: 10.1111/j.1469-7793.1999.219aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chau D, Johns DG, Schramm LP. Ongoing and stimulus-evoked activity of sympathetically correlated neurons in the intermediate zone and dorsal horn of acutely spinalized rats. J Neurophysiol. 2000;83:2699–2707. doi: 10.1152/jn.2000.83.5.2699. [DOI] [PubMed] [Google Scholar]
- 5.Choca JI, Proudfit HK, Green RD. Identification of A1 and A2 adenosine receptors in the rat spinal cord. J Pharmacol Exp Ther. 1987;242:905–910. [PubMed] [Google Scholar]
- 6.Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- 7.Cunha RA, Sebastiao AM, Ribeiro JA. Ecto-5′-nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat. J Neurochem. 1992;59:657–666. doi: 10.1111/j.1471-4159.1992.tb09420.x. [DOI] [PubMed] [Google Scholar]
- 8.Dembowsky K, Czachurski J, Seller H. An intracellular study of the synaptic input to sympathetic preganglionic neurones of the third thoracic segment of the cat. J Auton Nerv Syst. 1985;13:201–244. doi: 10.1016/0165-1838(85)90012-8. [DOI] [PubMed] [Google Scholar]
- 9.Deuchars J, Thomson AM. Innervation of burst firing spiny interneurons by pyramidal cells in deep layers of rat somatomotor cortex: paired intracellular recordings with biocytin filling. Neuroscience. 1995;69:739–755. doi: 10.1016/0306-4522(95)00288-t. [DOI] [PubMed] [Google Scholar]
- 10.Deuchars J, Atkinson L, Batten TFC, Deuchars SA. Evidence that the P2X7 receptor is targetted to presynaptic terminals in the brainstem and spinal cord of rats. J Physiol (Lond) 2000;526P:167P–167P. [Google Scholar]
- 11.Deuchars SA, Morrison SF, Gilbey MP. Medullary-evoked EPSPs in neonatal rat sympathetic preganglionic neurones in vitro. J Physiol (Lond) 1995;487:453–463. doi: 10.1113/jphysiol.1995.sp020892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deuchars SA, Spyer KM, Gilbey MP. Stimulation within the rostral ventrolateral medulla can evoke monosynaptic GABAergic IPSPs in sympathetic preganglionic neurons in vitro. J Neurophysiol. 1997;77:229–235. doi: 10.1152/jn.1997.77.1.229. [DOI] [PubMed] [Google Scholar]
- 13.Deuchars SA, Brooke RE, Deuchars J. Function of Kv3.1b channels in interneurons in the intermediolateral cell column of the rat. J Physiol (Lond) 2000;528P:109P. [Google Scholar]
- 14.Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- 15.Geiger JD, LaBella FS, Nagy JI. Characterization and localization of adenosine receptors in rat spinal cord. J Neurosci. 1984;4:2303–2310. doi: 10.1523/JNEUROSCI.04-09-02303.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerber U, Greene RW, Haas HL, Stevens DR. Characterization of inhibition mediated by adenosine in the hippocampus of the rat in vitro. J Physiol (Lond) 1989;417:567–578. doi: 10.1113/jphysiol.1989.sp017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodman RR, Synder SH. Autoradiographic localization of adenosine receptors in rat brain using [3H]cyclohexyladenosine. J Neurosci. 1982;2:1230–1241. doi: 10.1523/JNEUROSCI.02-09-01230.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara H, Sukamoto T, Kogure K. Mechanism and pathogenesis of ischemia-induced neuronal damage. Prog Neurobiol. 1993;40:645–670. doi: 10.1016/0301-0082(93)90009-h. [DOI] [PubMed] [Google Scholar]
- 19.Herlenius E, Lagercrantz H. Adenosinergic modulation of respiratory neurons in the neonatal rat brainstem in vitro. J Physiol (Lond) 1999;518:159–172. doi: 10.1111/j.1469-7793.1999.0159r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogan YH, Florent G, Hussain T, Alkadhi KA. Cyclic AMP antagonizes adenosine-induced inhibition of ganglionic transmission. Brain Res. 1998;787:242–247. doi: 10.1016/s0006-8993(97)01497-2. [DOI] [PubMed] [Google Scholar]
- 21.Katchman AN, Hershkowitz N. Adenosine antagonists prevent hypoxia-induced depression of excitatory but not inhibitory synaptic currents. Neurosci Lett. 1993;159:123–126. doi: 10.1016/0304-3940(93)90814-2. [DOI] [PubMed] [Google Scholar]
- 22.Koh HC, Shin IC, Hwang SJ, Paik DJ. Modification of cardiovascular response of adenosine A1 receptor agonist by cyclic AMP in the spinal cord of the rats. Neurosci Lett. 1996;219:195–198. doi: 10.1016/s0304-3940(96)13205-5. [DOI] [PubMed] [Google Scholar]
- 23.Koh HC, Shin IC, Hwang SJ, Kang JS, Lee CH, Ha JH, Paik DJ. Mediation of the cardiovascular response of adenosine A1 receptor through a GABA(B) receptor in the spinal cord of the rat. Neurosci Lett. 1998;243:81–84. doi: 10.1016/s0304-3940(98)00089-5. [DOI] [PubMed] [Google Scholar]
- 24.Lee KS, Schubert P, Emmert H, Kreutzberg GW. Effect of adenosine versus adenine nucleotides on evoked potentials in a rat hippocampal slice preparation. Neurosci Lett. 1981;23:309–314. doi: 10.1016/0304-3940(81)90016-1. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Perl ER. Adenosine inhibition of synaptic transmission in the substantia gelatinosa. J Neurophysiol. 1994;72:1611–1621. doi: 10.1152/jn.1994.72.4.1611. [DOI] [PubMed] [Google Scholar]
- 26.Marfurt CF, Turner DF, Adams CE. Stabilization of tetramethylbenzidine (TMB) reaction product at the electron microscopic level by ammonium molybdate. J Neurosci Methods. 1988;25:215–223. doi: 10.1016/0165-0270(88)90136-7. [DOI] [PubMed] [Google Scholar]
- 27.Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiol Scand. 2000;168:561–573. doi: 10.1046/j.1365-201x.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- 28.Oliet SH, Poulain DA. Adenosine-induced presynaptic inhibition of IPSCs and EPSCs in rat hypothalamic supraoptic nucleus neurons. J Physiol (Lond) 1999;520:815–825. doi: 10.1111/j.1469-7793.1999.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petras JM, Cummings JF. Autonomic neurons in the spinal cord of the Rhesus monkey: a correlation of the findings of cytoarchitectonics and sympathectomy with fiber degeneration following dorsal rhizotomy. J Comp Neurol. 1972;146:189–218. doi: 10.1002/cne.901460205. [DOI] [PubMed] [Google Scholar]
- 30.Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- 31.Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- 32.Saura CA, Mallol J, Canela EI, Lluis C, Franco R. Adenosine deaminase and A1 adenosine receptors internalize together following agonist-induced receptor desensitization. J Biol Chem. 1998;273:17610–17617. doi: 10.1074/jbc.273.28.17610. [DOI] [PubMed] [Google Scholar]
- 33.Schindler M, Harris CA, Hayes B, Papotti M, Humphrey PP. Immunohistochemical localization of adenosine A1 receptors in human brain regions. Neurosci Lett. 2001;297:211–215. doi: 10.1016/s0304-3940(00)01643-8. [DOI] [PubMed] [Google Scholar]
- 34.Schroder UH, Opitz T, Jager T, Sabelhaus CF, Breder J, Reymann KG. Protective effect of group I metabotropic glutamate receptor activation against hypoxic/hypoglycemic injury in rat hippocampal slices: timing and involvement of protein kinase C. Neuropharmacology. 1999;38:209–216. doi: 10.1016/s0028-3908(98)00180-4. [DOI] [PubMed] [Google Scholar]
- 35.Shen KZ, Johnson SW. Presynaptic GABAB and adenosine A1 receptors regulate synaptic transmission to rat substantia nigra reticulata neurons. J Physiol (Lond) 1997;505:153–163. doi: 10.1111/j.1469-7793.1997.153bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simpson RE, O'Regan MH, Perkins LM, Phillis JW. Excitatory transmitter amino acid release from the ischemic rat cerebral cortex: effects of adenosine receptor agonists and antagonists. J Neurochem. 1992;58:1683–1690. doi: 10.1111/j.1471-4159.1992.tb10041.x. [DOI] [PubMed] [Google Scholar]
- 37.Smith JA, Sivaprasadarao A, Munsey TS, Bowmer CJ, Yates MS. Immunolocalisation of adenosine A1 receptors in the rat kidney. Biochemical Pharmacology. 2001;61:237–244. doi: 10.1016/s0006-2952(00)00532-3. [DOI] [PubMed] [Google Scholar]
- 38.St. Lambert JH, Thomas T, Burnstock G, Spyer KM. A source of adenosine involved in cardiovascular responses to defense area stimulation. Am J Physiol. 1997;272:R195–R200. doi: 10.1152/ajpregu.1997.272.1.R195. [DOI] [PubMed] [Google Scholar]
- 39.Stiles GL. Adenosine receptors. J Biol Chem. 1992;267:6451–6454. [PubMed] [Google Scholar]
- 40.Strack AM, Sawyer WB, Hughes JH, Platt KB, Loewy AD. A general pattern of CNS innervation of the sympathetic outflow demonstrated by transneuronal pseudorabies viral infections. Brain Res. 1989;491:156–162. doi: 10.1016/0006-8993(89)90098-x. [DOI] [PubMed] [Google Scholar]
- 41.Sweeney MI. Neuroprotective effects of adenosine in cerebral ischemia: window of opportunity. Neurosci Biobehav Rev. 1997;21:207–217. doi: 10.1016/s0149-7634(96)00011-5. [DOI] [PubMed] [Google Scholar]
- 42.Thomas T, Spyer KM. A novel influence of adenosine on ongoing activity in rat rostral ventrolateral medulla. Neuroscience. 1999;88:1213–1223. doi: 10.1016/s0306-4522(98)00296-6. [DOI] [PubMed] [Google Scholar]
- 43.Thompson SM, Haas HL, Gahwiler BH. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol (Lond) 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trussell LO, Jackson MB. Dependence of an adenosine-activated potassium current on a GTP-binding protein in mammalian central neurons. J Neurosci. 1987;7:3306–3316. doi: 10.1523/JNEUROSCI.07-10-03306.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]