

Abstract
Regulation of neuronal NMDA receptors (NMDARs) by group I metabotropic glutamate receptors (mGluRs) is known to play a critical role in synaptic transmission. The molecular mechanisms underlying mGluR1-mediated potentiation of NMDARs are as yet unclear. The present study shows that in Xenopus oocytes expressing recombinant receptors, activation of mGluR1 potentiates NMDA channel activity by recruitment of new channels to the plasma membrane via regulated exocytosis. Activation of mGluR1α induced (1) an increase in channel number times channel open probability, with no change in mean open time, unitary conductance, or reversal potential; (2) an increase in charge transfer in the presence of NMDA and the open channel blocker MK-801, indicating an increased number of functional NMDARs in the cell membrane; and (3) increased NR1 surface expression, as indicated by cell surface Western blots and immunofluorescence. Botulinum neurotoxin A or expression of a dominant negative mutant of synaptosomal associated protein of 25 kDa molelcular mass (SNAP-25) greatly reduced mGluR1α-mediated potentiation, indicating that receptor trafficking occurs via a SNAP-25-mediated form of soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor-dependent exocytosis. Because group I mGluRs are localized to the perisynaptic region in juxtaposition to synaptic NMDARs at glutamatergic synapses in the hippocampus, mGluR-mediated insertion of NMDARs may play a role in synaptic transmission and plasticity, including long-term potentiation.
Keywords: metabotropic glutamate receptors; NMDA receptors; receptor trafficking; channel gating; protein kinase C; (1S,3R)-1-amino-cyclopentane-1,3-dicarboxylic acid; ACPD
Activation of metabotropic glutamate receptors (mGluRs) modulates NMDA receptor (NMDAR) activity and is implicated in synaptic transmission and activity-dependent synaptic plasticity (Pin and Duvoisin, 1995; Conn and Pin, 1997; Nicoletti et al., 1999). mGluRs are encoded by a gene family of eight members; further diversity is generated by alternative RNA splicing of the receptor C-terminal domains. By sequence homology, agonist selectivity, intracellular signaling mechanisms, and differential targeting in neurons, mGluRs can be divided into three classes. Group I mGluRs (mGluR1 and mGluR5) couple to diverse intracellular signaling transduction pathways. Group I mGluRs can couple positively to phospholipase C, activation of which leads to stimulation of protein kinase C (PKC) and release of intracellular Ca2+ (Conn and Pin, 1997), or to adenylyl cyclase, activation of which stimulates cAMP formation (Aramori and Nakanishi, 1992; Joly et al., 1995). Group I mGluRs couple negatively to NMDARs via a G-protein-dependent, membrane-delimited signaling pathway in cortical neurons (Yu et al., 1997) and depolarize CA3 neurons via a G-protein-independent, Src-dependent signaling cascade (Heuss et al., 1999).
Immunocytochemical studies reveal strikingly different distributions of group I mGluRs within the hippocampus. Although mGluR1 is localized primarily to somatostatin-positive GABAergic interneurons, mGluR5 is highly expressed in pyramidal neurons (Shigemoto et al., 1997). At excitatory glutamatergic synapses, mGluR1 and mGluR5 are concentrated in the perisynaptic zone at the periphery of postsynaptic densities (PSDs; Baude et al., 1993; Lujan et al., 1996). Localization of group I mGluRs in close proximity to NMDARs and AMPA receptors (AMPARs), which are concentrated at PSDs, and to critical signaling proteins is consistent with a modulatory role in glutamatergic transmission.
Activation of group I mGluRs induces potentiation of NMDA EPSCs in hippocampal (Aniksztejn et al., 1992; Challiss et al., 1994), striatal (Pisani et al., 1997), and subthalamic nucleus (Awad et al., 2000) slices and potentiation of NMDA currents in lamprey motoneurons (Krieger et al., 2000) and frog spinal cord neurons (Holohean et al., 1999). However, the molecular mechanisms underlying potentiation of NMDARs by group I mGluRs are as yet unclear. Mice with null mutations of either mGluR1 (Aiba et al., 1994a,b; Conquet et al., 1994; Bordi, 1996) or mGluR5 (Lu et al., 1997) display reductions of hippocampal long-term potentiation (LTP) and abnormalities of motor coordination and associative learning. Metabotropic glutamate receptor signaling is also implicated in neurodegenerative diseases (Conn and Pin, 1997;Nicoletti et al., 1999), cortical development (Kaczmarek et al., 1997), and addiction (Wolf, 1998).
Activation of PKC induces rapid delivery of NMDARs to the cell surface of Xenopus oocytes and hippocampal neurons (Lan et al., 2001). The present study was undertaken to examine the hypothesis that potentiation of NMDARs by mGluR1 occurs at least in part by receptor trafficking. We used Xenopus oocytes to express a homogeneous population of receptors of known subtype in a geometrically simple system. Moreover, the molecular machinery for protein trafficking is highly conserved from yeast to mammals (Bennett and Scheller, 1993). Experiments involving patch-clamp recording, charge transfer measurements, cell surface Western blots, and immunofluorescence reveal that activation of mGluR1α promotes delivery of new NMDA channels to the plasma membrane by regulated exocytosis, a mechanism that accounts entirely for potentiation of NMDAR activity. These studies provide molecular insight into synaptic function of group I mGluRs and define a novel mechanism for potentiation of NMDAR function.
MATERIALS AND METHODS
Expression constructs. Rat NMDAR subunit NR1-4b (NR1100; Zukin and Bennett, 1995) cDNA was cloned in this laboratory (Durand et al., 1992); rat NR1-1a (NR1011) and mGluR1α cDNAs were gifts from Dr. S. Nakanishi (Kyoto University, Kyoto, Japan); mouse ε1, ε2, and ε3 (corresponding to rat NR2A, NR2B, and NR2C, respectively) cDNAs were gifts from Dr. M. Mishina (Niigata University, Tokyo, Japan). cDNAs were subcloned into the pBluescript SK(−) vector (Stratagene, La Jolla, CA) for oocyte expression. Synaptosomal associated protein of 25 kDa molecular mass (SNAP-25) and SNAP-25(Δ20) cDNAs were gifts from Dr. R. Y. Tsien (Howard Hughes Medical Institute, University of California, San Diego, CA). Capped mRNAs were synthesized as runoff transcripts from linearized plasmid cDNAs with T3 or T7 polymerase (mMessage mMachine transcription kit; Ambion, Austin, TX; 2 hr at 37°C). The concentration and integrity of mRNAs were assessed after staining with ethidium bromide by direct comparison of sample mRNAs with an RNA standard ladder (Life Technologies, Gaithersburg, MD).
Electrophysiology of recombinant NMDA receptors expressed inXenopus oocytes. Selected stage V and VI oocytes from adult female Xenopus laevis (Xenopus I, Ann Arbor, MI) were injected with a mixture of in vitrotranscribed mRNAs (20 ng of mRNA/cell; NR1:NR2:mGluR1α ratio, 1:2:1) using a Nanoject injector (Drummond Scientific, Broomall, PA) as described previously (Zheng et al., 1997). Oocytes were maintained at 18°C in culture buffer (in mm: 103 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 5 HEPES, pH 7.5).
Whole-cell recording. Whole-cell currents were recorded from oocytes (2–6 d after injection) at ambient temperature using a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA) by two-microelectrode voltage clamp, filtered at 20 Hz, and digitized on-line at 100 Hz (Zheng et al., 1997). Currents were elicited by bath application of NMDA (300 μm with 10 μmglycine) at a holding potential of −60 mV. Oocytes were perfused in Mg2+-free, normal frog Ringer's solution consisting of (in mm): 116 NaCl, 2.0 KCl, 1.0 CaCl2, and 10 HEPES, pH 7.2 (Zheng et al., 1997). Because Ca2+ influx through the NMDAR can cause Ca2+ amplification (Zheng et al., 1997), Ca2+ inactivation (Legendre et al., 1993), and activation of chloride channels endogenous to the oocyte (Leonard and Kelso, 1990) and thereby affect measurements of PKC potentiation, all experiments except those illustrated in Figures 1-3were performed in Ringer's solution in which CaCl2 was replaced by BaCl2. The pipette resistance ranged from 0.5 to 2 MΩ with an internal solution consisting of (in mm): 3000 KCl and 10 HEPES, pH 7.2. For mGluR1α activation, oocytes were incubated in (1S,3R)-1-amino-cyclopentane-1,3-dicarboxylic acid (ACPD; 100 μm, 2 min; Sigma, St. Louis, MO) and washed. ACPD potentiation is defined as the ratio of the NMDA-elicited current measured after ACPD application to that measured before ACPD application. For direct PKC activation, oocytes were incubated in 100 nm12-O-tetradecanoylphorbol 13-acetate (TPA; Sigma) for 10 min. TPA potentiation is defined as the ratio of the NMDA-elicited current measured after TPA application to that measured before TPA application.
Fig. 1.
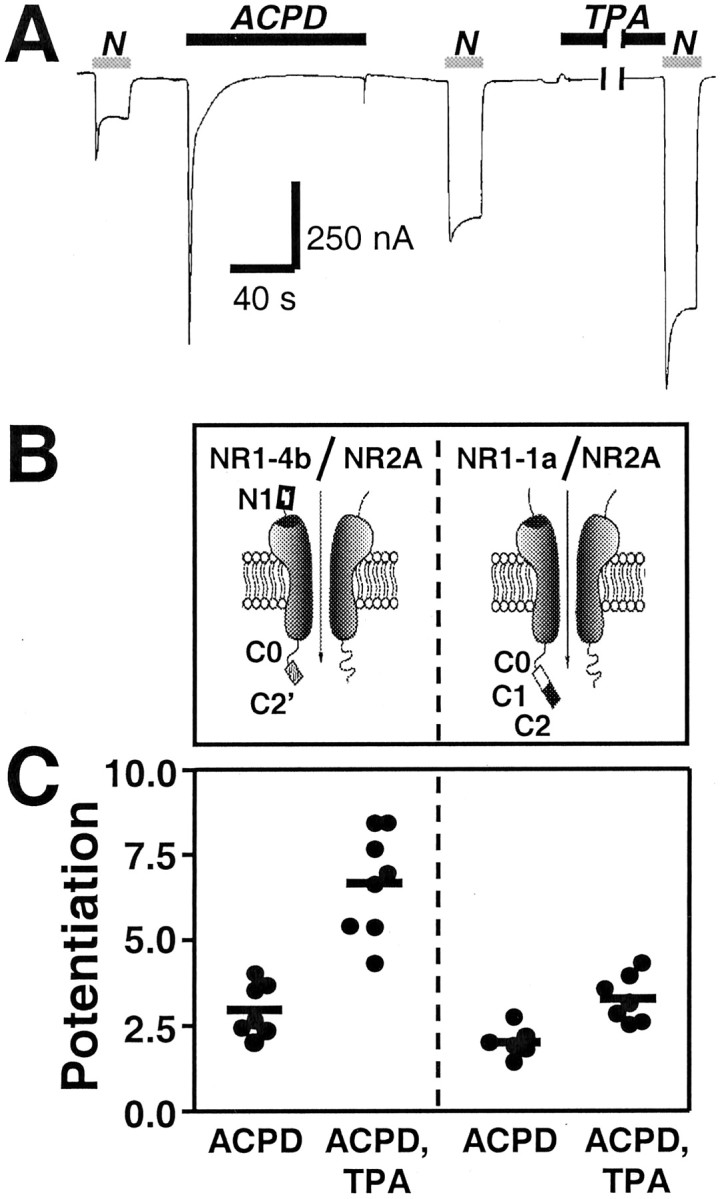
Activation of mGluR1α potentiates NMDA whole-cell currents; alternative splicing of the NR1 subunit has little effect on ACPD potentiation. A, Typical sequence showing NMDA-activated whole-cell currents recorded in Ca2+Ringer's solution at Vh = −60 mV from oocytes expressing mGluR1α and NR1-4b/NR2A receptors before and after application of ACPD (100 μm, 2 min), followed by application of TPA (100 nm, 10 min). NMDA, 300 μm; glycine, 10 μm. ACPD elicited a large inward current that decayed rapidly to near baseline within 30 sec, ascribable to Cl− efflux through Ca2+-activated Cl− channels endogenous to the oocyte. ACPD significantly potentiated NMDA responses. Application of the phorbol ester TPA to oocytes after ACPD treatment further potentiated NMDA responses. B, Schematic representation of the NR1 splice variants. N1,C1, C2, and C2′ are alternatively spliced cassettes; C0 is the region between the fourth transmembrane domain and the first splice site in the C terminal (Zheng et al., 1999). C, Summary of several experiments illustrating that ACPD potentiation did not differ significantly for NR1-4b/NR2A versus NR1-1a/NR2A receptors. In contrast, potentiation by TPA (applied to oocytes after ACPD) was significantly greater for NR1-4b/NR2A receptors compared with NR1-1a/NR2A receptors (p < 0.01, one-way ANOVA followed by Bonferroni's t test). Each data point is from a single experiment; horizontal bars represent the means.
Fig. 2.
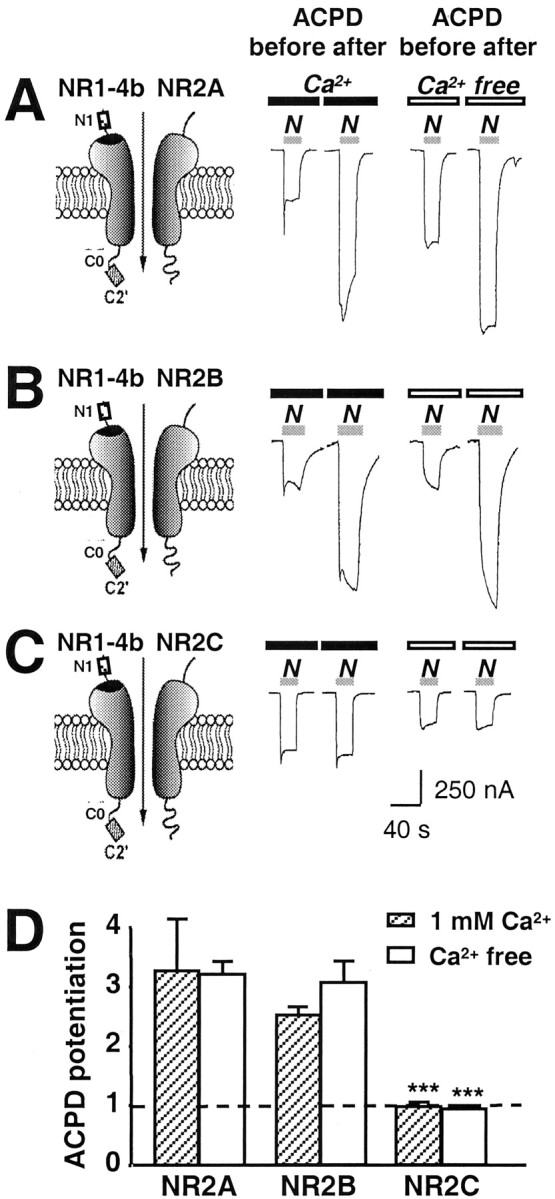
The NR2 subunit alters ACPD potentiation. NMDA-activated whole-cell currents were recorded in Ca2+ or Ca2+-free Ringer's solution from oocytes expressing mGluR1α and NMDARs.A–C, ACPD (100 μm, 2 min) potentiated NMDA currents in oocytes expressing NR1-4b/NR2A or NR1-4b/NR2B receptors; no potentiation was observed for NR1-4b/NR2C receptors.N1, C0, and C2′ are defined in the legend to Figure 1. D, Summary of three experiments illustrating ∼3.33-fold potentiation by ACPD of NR1-4b/NR2A and 2.57-fold for NR1-4b/NR2B receptors in Ca2+Ringer's solution but little or no potentiation of NR1-4b/NR2C receptors.
Fig. 3.

ACPD potentiates npo.A, B, NMDA-activated single channels recorded from outside-out patches excised from oocytes expressing recombinant NR1-4b/NR2A and mGluR1α receptors; patches were excised before (A) and after (B) application of ACPD (100 μm, 2 min). NMDA, 100 μm; glycine, 10 μm. The main unitary conductance was 44 ± 1 pS at Vh = −60 mV. C, ACPD treatment potentiated thenpo from 0.050 ± 0.006 recorded from patches excised before ACPD (n = 5) to 0.204 ± 0.034 recorded from different patches excised from the same oocytes after ACPD treatment (n = 5; p< 0.001). D, Single-channel current–voltage relationships. ACPD did not alter the main unitary conductance of NMDA-activated channels (44 ± 1 pS before ACPD;n = 5; vs 44 ± 1 pS after ACPD;n = 5). ACPD treatment did not affect the reversal potential (Erev = 0 mV before ACPD;n = 5; and 0 mV after ACPD; n = 5). E, F, Histograms of mean open time durations for NMDA-activated channels before and after application of ACPD to the oocyte. Mean open times were determined by a single exponential fit to the event lists generated by single opening and closing events. ACPD did not alter the mean open time.
Single-channel recording. Single-channel currents were recorded from outside-out patches excised from devitellinized oocytes (2–7 d after injection) as described previously (Araneda et al., 1999). Pipette resistance ranged from 7 to 12 MΩ with an internal solution consisting of (in mm): 115 CsCl, 5.0 EGTA, and 10 HEPES, pH 7.2. External solutions were as described for whole-cell recording to permit direct comparisons between NMDA-elicited whole-cell and single-channel currents. NMDA (100 μm with 10 μm glycine) solution was delivered to the patch from a multibarrel array fed by gravity.
Single-channel current amplitudes were determined from means of Gaussian fits to all-point amplitude histograms. The number of active channels times channel open probability (npo) was calculated as the total channel open time divided by recording time. Open time durations were calculated from single-channel openings above baseline but may include events from more than one channel in the same patch. All data are presented as mean ± SEM for 4–12 experiments performed with different oocytes. Statistical significance was assessed by the Student's t test (SigmaPlot 3.0) and ANOVA (CLR ANOVA 1.3).
Surface Western blot analysis. Control and ACPD-treatedXenopus oocytes expressing mGluR1α and NR1-4b/NR2A receptors were screened for NMDA currents in the range of 100–300 nA and ACPD potentiation of ∼2–3. Oocytes were incubated in external recording solution in the absence or presence of ACPD (100 μm, 2 min) and washed twice, and surface proteins were biotinylated with the membrane-impermeant reagent sulfosuccinimidyl 2-(biotinamido) ethyl-1,3′-dithiopropionate (sulfo-NHS-SS-biotin, 1.5 mg/ml, 30 min at 4°C; Pierce, Rockford, IL) according to the method of Chen et al. (1999). Cell extracts were prepared as described by Hollmann et al. (1994). To isolate biotinylated surface proteins from nonsurface proteins, cell extracts were incubated with Neutravidin-linked beads (Pierce) for 2 hr at 4°C, centrifuged, and washed. Bound proteins were eluted from beads by incubation with SDS-PAGE gel loading buffer containing DTT (which releases the proteins from the biotin moiety) and subjected to gel electrophoresis together with aliquots of total protein.
Cell surface immunolabeling of oocytes. Xenopusoocytes expressing NR1-4b/NR2A receptors with mGluR1α were labeled with monoclonal antibody 54.1 directed to the extracellular loop of the NR1 subunit (Gazzaley et al., 1996). Oocytes were screened for NMDA currents in the range of 100–300 nA and ACPD potentiation of approximately twofold to threefold. Oocytes were incubated in 20 ml of external recording solution in the absence or presence of ACPD (100 μm, 2 min), devitellinized, and fixed in 4% paraformaldehyde and 2% sucrose (1 hr at room temperature). Oocytes were then incubated with NR1 antibody (10 μg/ml, 4°C overnight), followed by biotinylated horse anti-mouse IgG (Vector Laboratories, Burlingame, CA; 10 μg/ml, 1.5 hr at room temperature) and FITC-conjugated avidin (Vector Laboratories; 10 μg/ml, 1.5 hr at room temperature). Oocytes were rinsed, placed in a chamber 1.5 mm deep with a coverslip forming the bottom (MatTek, Ashford, MA), and covered with ProLong mounting medium (Molecular Probes, Eugene, OR) to reduce fluorescence quenching. Cross-sectional and tangential images of oocytes were viewed by a Bio-Rad MRC 600 Kr/Ar laser scanning confocal microscope and acquired using COMOS software (Bio-Rad, Hercules, CA). FITC fluorescence was measured (excitation wavelength, 488 nm; emission, 515 nm) with a 40× objective lens (aperture, 1.3); laser intensity, photomultiplier gain, and pinhole aperture were kept constant.
RESULTS
Activation of mGluR1α potentiates NMDA responses
To examine the interaction between group I mGluRs and NMDARs, we coexpressed mGluR1α with NR1-4b/NR2A receptors in Xenopusoocytes and examined the effect of mGluR activation on NMDA responses by whole-cell recording under voltage clamp. NR1-4b/NR2A and -B have the highest cell surface expression (Okabe et al., 1999) and highest degree of PKC potentiation (Zheng et al., 1999). Application of the mGluR agonist ACPD (100 μm) in Ca2+-free Ringer's solution to the oocyte elicited a large inward current (∼750 nA) that decayed to near baseline in <30 sec (Fig.1A). The ACPD-elicited current is ascribable to Cl− efflux through Ca2+-activated Cl− channels endogenous to the oocyte (Saugstad et al., 1996). It is well documented that activation of mGluR1 can lead to an increase of [Ca2+]i by activation of phospholipase C and release of Ca2+ from inositol triphosphate (IP3)-sensitive intracellular stores (Pin and Duvoisin, 1995). Hence, the ACPD-evoked current indicates effective expression of mGluR1α and targeting of functional receptors to the cell surface of the oocyte.
To examine the effect of mGluR1α activation on NMDAR responses, we recorded whole-cell currents elicited by NMDA (300 μmwith 10 μm glycine) before and after bath application of ACPD (100 μm, 2 min) to the oocyte. Treatment with ACPD significantly increased NMDA-elicited responses (Fig.1A; compare first and second NMDA responses). In Ca2+ Ringer's solution, ACPD potentiation (defined as the ratio of NMDA current amplitude measured after ACPD to that measured before ACPD) was 2.97 ± 0.30 (n = 7) (Fig. 1C).
To investigate the effect of alternative splicing of the NR1 subunit on mGluR1-mediated potentiation of NMDAR responses, we coexpressed mGluR1α with either NR1-1a/NR2A (NR1011/NR2A) or NR1-4b/NR2A (NR1100/NR2A) receptors inXenopus oocytes (Fig. 1B). NR1-1a/NR2A and -B have the lowest cell surface expression (Okabe et al., 1999) and lowest degree of PKC potentiation (Zheng et al., 1999). Application of ACPD potentiated NMDA responses of NR1-4b/NR2A receptors to 2.97 ± 0.30 times the control response in Ca2+Ringer's solution (n = 7) (Fig.1A,C). ACPD potentiated responses of NR1-1a/NR2A receptors to 2.03 ± 0.18 times the control response in Ca2+ Ringer's solution (n= 7) (Fig. 1C). Differences in ACPD-induced potentiation between splice variants did not reach statistical significance in either Ca2+- containing or Ca2+-free (Ba2+) Ringer's solution (one-way ANOVA followed by Bonferroni test, p > 0.05 for comparisons between each receptor pair). The smaller magnitude of the mGluR1α potentiation may have obscured an effect of NR1 splicing similar to that observed for PKC potentiation (Zheng et al., 1999).
To investigate the effects of the NR2 subunit on ACPD-induced potentiation, we coexpressed mGluR1α with NR1-4b/NR2A, NR1-4b/NR2B, or NR1-4b/NR2C receptors in Xenopus oocytes. The mGluR1 agonist ACPD (100 μM) significantly potentiated NMDA-induced currents of NR1-4b/NR2A and NR1-4b/NR2B receptors (n = 3) (Fig.2A,B). Potentiation was to 3.33 ± 0.88 (n = 3) (Fig.1A) and 2.57 ± 0.14 (n = 3) times the control responses in Ca2+Ringer's solution, respectively (Fig. 2D). The degree of ACPD potentiation did not differ significantly for NR1-4b/NR2A versus NR1-4b/NR2B receptors. As observed for TPA potentiation (Mori et al., 1993), ACPD did not potentiate responses of NR1-4b/NR2C receptors (n = 3) (Fig. 2C); potentiation was to 1.02 ± 0.04 times the control response in Ca2+ Ringer's solution (n= 3) (Fig. 2D). Similar results were observed for the three receptor subtypes in Ba2+ Ringer's solution. Thus, the NR2 subunit composition of NMDAR affected the degree of ACPD potentiation.
To rule out the possibility that ACPD directly activates NMDA receptors (the mGluR agonist 2-(2′,3′)-dicarboxycyclopropylglycine at high concentrations may act on the NMDA receptor as an agonist) (Opitz et al., 1994; Yu et al., 1997), we applied a high concentration of ACPD (100 μm) to oocytes expressing NR1-4b/NR2A receptors in the absence of mGluR1α. Under these conditions, ACPD elicited no response and did not potentiate NMDA currents (data not illustrated), indicating that the Cl− response was mGluR-dependent and that ACPD did not activate or potentiate NMDA channels directly.
ACPD increases npo but not conductance or open time
To examine effects of mGluR1 activation on NMDA channel conductance and gating, we recorded channel activity in outside-out patches excised from oocytes expressing mGluR1α and NR1-4b/NR2A receptors before and after ACPD treatment. We compared patches excised before and after ACPD treatment, because patch formation inhibits exocytosis, presumably by deformation of the membrane and disruption of association with cytoplasmic elements (Lan et al., 2001)
In control patches, NMDA (10 μm) activated single channels with a conductance of γ = 44 ± 1 pS at −60 mV, which did not vary with voltage (n = 5) (Fig.3A,D). Occasional transitions from the main conductance state to sublevels (total dwell time of <1% of that of the main state; data not shown) were excluded from the analysis. NMDA channel activity in patches excised after application of ACPD (100 μm, 2 min) was markedly potentiated (Fig. 3A,B). ACPD increasednpo by approximately fourfold from 0.050 ± 0.006 before ACPD (n = 5) to 0.209 ± 0.027 after ACPD (n = 5; p < 0.001) (Fig. 3C).
mGluR1 activation did not alter NMDA single-channel conductance (n = 5) (Fig. 3A,B) or reversal potential (Erev ∼ 0), as evidenced by comparison of current–voltage relationships (n = 5) (Fig. 3D). Moreover, ACPD did not significantly change the mean duration of openings (Fig. 3E,F). The distribution of open times was fit by a single exponential, consistent with the presence of a single open state (Fig. 3E,F) (τ = 6.35 ± 0.26 msec before ACPD; n = 5; vs τ = 6.08 ± 0.29 msec after ACPD; n = 5). Potentiation of npo without change in γ was also induced by the mGluR agonist (S)-3,5-dihydroxyphenylglycine (100 μm; data not shown).
mGluR1 activation induces an increase in the number of active NMDA channels
The results reported thus far indicate that ACPD increases the number of active channels in the cell membrane times the open probability. To distinguish between effects of mGluR1 activation on the number of functional channels in the membrane, n, and channel open probability, po, we recorded NMDA whole-cell currents in the presence of MK-801 (5 μm) from control (Fig.4B, left trace) and ACPD-treated (Fig. 4B, right trace) oocytes by a modification of the method of Jahr (1992) as adapted by Rosenmund et al. (1995). This method takes advantage of the essentially irreversible block of NMDA-elicited currents by the open-channel blocker MK-801. To determine the number of channels in the whole oocyte, N, we calculated the cumulative charge transfer, Q, which is the integral of NMDA-elicited current during the time required for complete block by MK-801. The number of channels, N, can be calculated from Q as follows:
where tbl is the time constant for MK-801 block (tbl = 1/kbl [MK-801], andkbl = 2.5 × 107m/sec; Jahr, 1992). From the single-channel recordings (Fig. 3), ACPD does not change single-channel conductance, and we would not expected it to change tbl. The channel number,N, for control and ACPD-treated oocytes was normalized to the NMDA-elicited whole-cell current, which corrects for any differences in levels of expression within and between the two groups. For control oocytes, which exhibited control currents in the range of 100–300 nA, the mean number of channels per 100 nA was 6.8 ± 0.8 × 105, and the mean channel density for 100 nA was 0.02 μm−2, assuming a surface area for the oocyte of 3 × 107 μm2(Zampighi et al., 1999). ACPD increased N ∼1.6-fold (NACPD/Ncontrol= 1.6; p < 0.01). This analysis indicates that mGluR1 activation increases the number of functional NMDA channels per cell.
Fig. 4.
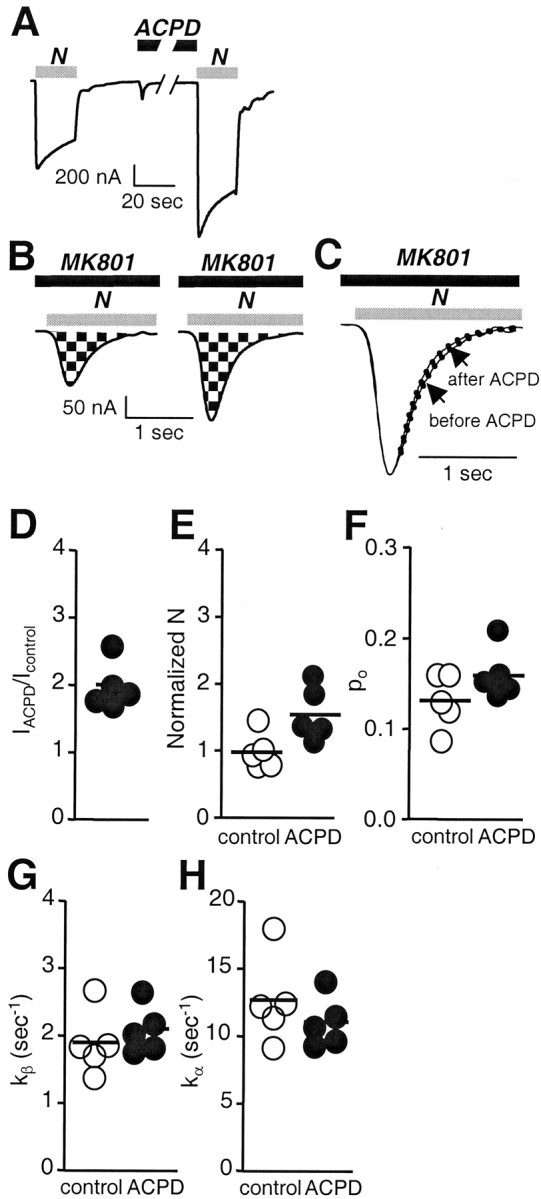
mGluR1α activation increases NMDA channel number and modestly (but not significantly) increases open probability.A, ACPD (100 μm, 2 min), bath-applied, potentiated whole-cell currents elicited by application of NMDA (1 mm with 50 μm glycine). To avoid contributions by Ca2+ inactivation, Ca2+ amplification to NMDA responses, or both, recording was in Ba2+ Ringer's solution as the extracellular solution. Vh = −60 mV.B, ACPD increased the number of functional NMDA channels expressed at the cell surface (N). Currents were elicited by application of NMDA (1 mm NMDA with 50 μm glycine) in the continuous presence of the open channel blocker MK-801 (5 μm) from control (left) and ACPD-treated (right) oocytes at a holding potential of −60 mV. The NMDA inward current increased to a peak value, after which it decayed exponentially as MK-801 entered and blocked NMDA channels as they opened. The cumulative charge transfer, Q, which is the total current flow during the time interval for complete block by MK-801, was obtained by integration of the current trace over time. The larger integrated current in ACPD-treated oocytes indicated an increased number of functional channels per cell. C, Agonist-evoked currents inB were normalized to the same peak amplitude and used for kinetic analysis. The more rapid decay of the NMDA current in ACPD-treated oocytes versus control in this pair of oocytes indicates an increased rate of channel opening, kβ, but the difference was not significant in the pooled data.D–H, Quantization of data in A–C.D, Potentiation of NMDA whole-cell current,IACPD/Icontrol, was 2.2 ± 0.2 times control (p < 0.001; n = 5). E, The increase in channel number, N, was significant (p < 0.01), andNACPD/Ncontrol= 1.6 (n = 5). F, The open probabilities were not significantly different (po, control = 0.13 ± 0.01; po, ACPD = 0.16 ± 0.01;po, control/po, ACPD = 1.2; n = 5). G, Opening rates, kβ, for control (1.9 ± 0.2/sec) and ACPD-treated oocytes (2.1 ± 0.2/sec;p < 0.01). H, Closing rates,kα, for control (12.7 ± 1.5/sec) and ACPD-treated oocytes (11.1 ± 0.8/sec).
The data in Figure 4B also enabled us to analyze the effect of mGluR1 activation on NMDA channel gating. We first calculated channel open probability in control and ACPD-treated oocytes from the NMDA-elicited whole cell current, I, the single-channel current, i (which is not changed by mGluR1 activation) (Fig.3D), and the number of channels per cell, N, as follows:
from which po, ACPD/po, control = 1.2. The ACPD-induced increase in channel open probability was not significant.
As an independent measure of the effect of mGluR1 activation on channel gating, we analyzed the decay of NMDA-elicited current in the presence of MK-801 from the data in Figure 4B normalized to the same peak amplitude (Fig. 4C). Provided thatkbl [MK-801] ≫kα andkβ (the closing and opening rates, respectively), the decay can be described by a single exponential with a rate constant, kβ, for channel opening. Activation of mGluR1 did not significantly changekβ (1.9 ± 0.2/sec for control oocytes vs 2.1 ± 0.2/sec for ACPD-treated oocytes) (Fig.4G). From measurements ofpo andkβ and the relationpo =kβ/(kα+ kβ), we calculated values ofkα = 12.7 ± 1.5/sec in control oocytes versus 11.1 ± 0.8/sec in ACPD-treated oocytes (Fig.4H). These values ofkα are substantially less than the value of ∼167/sec predicted from the ∼6 msec mean open time of single channels. The apparent discrepancy arises because thepo equation as applied to macroscopic current assumes an equilibrium between unliganded and bursting states and thus will yield a kα value corresponding to termination of bursting rather than of individual openings. Moreover, kα calculated in this manner will be overestimated in that the probability that a receptor is liganded is greater than the probability that it is open,po, calculated from Nand I.
mGluR1 activation delivers NMDA channels to the cell membrane via exocytosis
The results reported thus far indicate that mGluR1 activation induces an increase in the number of active channels at the cell surface but does not distinguish between insertion of new NMDA channels and unmasking of silent channels. To distinguish between these possibilities, we performed two additional experiments. First, we examined the effects of loading oocytes with the light chains of type A botulinum neurotoxin (BoNT A), which is known to inactivate SNAP-25 and to prevent SNAP-25-dependent exocytosis (Montecucco and Schiavo, 1995). Treatment of oocytes with BoNT A reduced the degree of mGluR1-mediated potentiation of NMDA-elicited currents by ∼50% (Fig.5A–C). A mixture of type A, B, and E BoNTs reduced ACPD potentiation by ∼60% (Fig.5C). No significant change in the resting potential, input resistance, and basal NMDA response was caused by BoNTs (data not shown). BoNT A also reduces TPA potentiation by ∼50% (Lan et al., 2001). No effects on mGluR1-mediated potentiation were observed when only the vehicle (10 mm DTT) was injected. Presumably, toxin treatment attenuated the number of new channels inserted by mGluR1 activation.
Fig. 5.
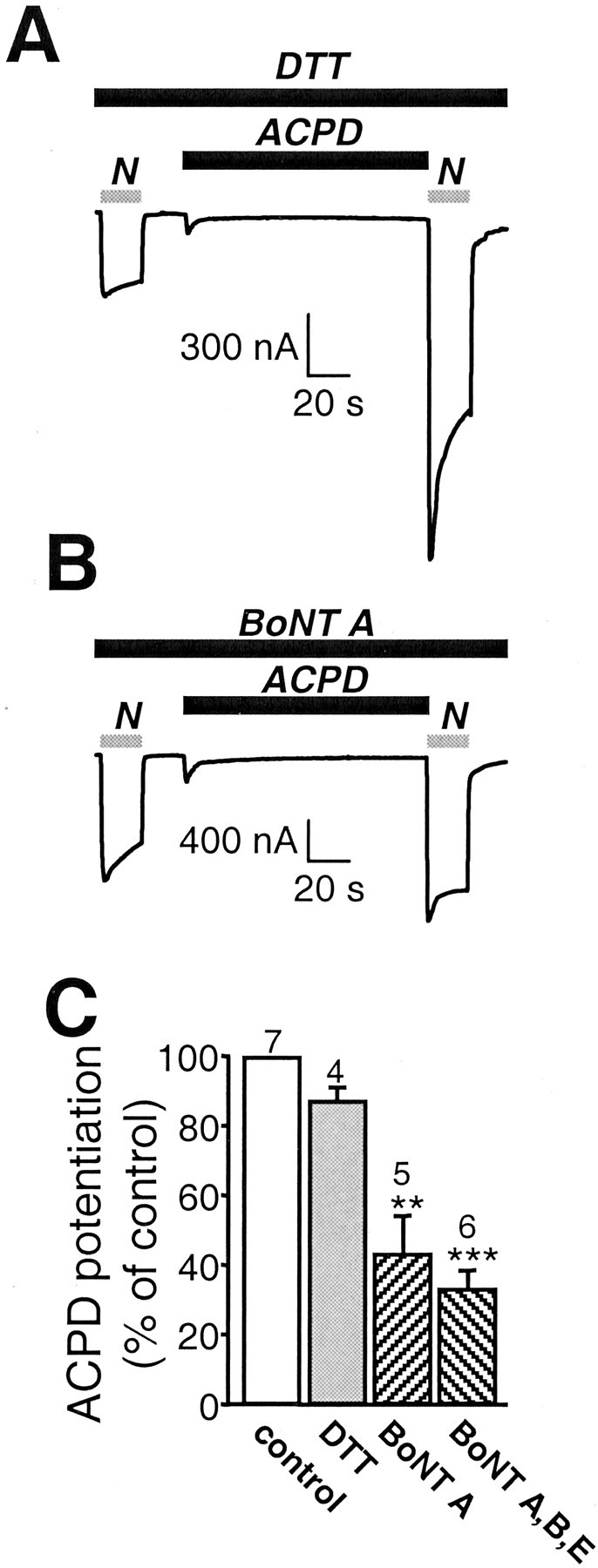
mGluR1α promotes delivery of NMDA channels to the cell membrane via exocytosis. A–C, Microinjection of the light chain of botulinum toxin type A BoNT (50 ng), an enzyme known to cleave SNAP-25, into oocytes 5 hr before recording reduced ACPD potentiation of NMDA-elicited currents by ∼50%. A, B, Representative NMDA-elicited whole-cell currents in an oocyte loaded with 50 nl of DTT (5 mm) in Aand 50 ng of BoNT A and DTT in B before and after ACPD application. BoNT A reduced potentiation but not basal NMDA-elicited currents or ACPD-elicited Cl− currents.C, Quantification of A andB. A mixture of type A, B, and E BoNTs reduced ACPD potentiation by 60%.
Second, we examined the ability of a dominant negative mutant of SNAP-25 to block mGluR1-mediated potentiation. SNAP-25(Δ20), a truncation mutant lacking the C-terminal 20 amino acids, corresponds to yeast sec9-Δ17, a dominant negative mutant of the yeast SNAP-25 homolog (Yao et al., 1999). Mutant and wild-type SNAP-25 were expressed in oocytes by injection of their cRNAs 48 hr after injection of NMDAR cRNAs, and ACPD potentiation was measured ∼20 hr later. ACPD-induced potentiation of NMDA currents in control oocytes (2.5 ± 0.3;n = 3) (Fig.6A,D) and in oocytes expressing full-length SNAP-25 (2.9 ± 0.2; n = 4) (Fig. 6B,D) was greater than in oocytes expressing SNAP-25(Δ20) (1.5 ± 0.2; n = 5;p < 0.01) (Fig. 6C,D). Expression of SNAP-25(Δ20) did not affect the NMDA-elicited current or endogenous Ca2+-activated Cl− currents. These findings indicate that NMDA channels inserted at the cell surface by mGluR1α activation are delivered, at least in part, by SNAP-25-mediated solubleN-ethylmaleimide sensitive fusion protein attachment protein receptor (SNARE)-dependent exocytosis.
Fig. 6.
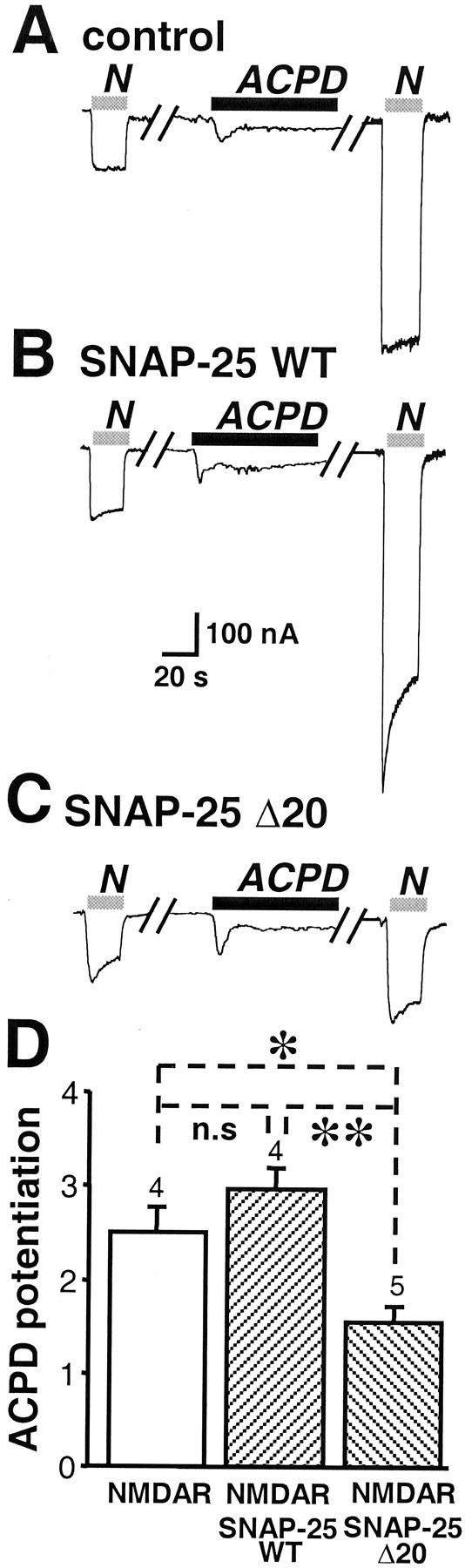
A dominant negative mutant of SNAP-25 reduces ACPD potentiation of NMDARs. A, ACPD potentiation of NMDA-elicited currents in oocytes expressing mGluR1α and NR1-4b/NR2A receptors as in Figure 2A. B, Coexpression of wild-type SNAP-25 with NMDARs had no effect on ACPD potentiation. C, Coexpression of SNAP-25(Δ20), a dominant negative mutant of SNAP-25, with NMDARs markedly reduced ACPD potentiation. D, Quantification of the effects of wild-type and mutant SNAP-25 on ACPD potentiation.
Although the data indicate that ACPD increases the number of functional NMDA channels at the cell surface via a SNARE-dependent mechanism, they do not exclude a contribution from reduction in internalization (or another mechanism of silencing). As an estimate of the rate of constitutive exocytosis in the oocyte, we measured reappearance of functional channels at the cell surface after quasi-irreversible block by MK-801. MK-801 (1 μm) blocked completely the NMDA-elicited current (Fig.7A, first response). A test application of NMDA at 3 min elicited a small response, indicative of a small number of newly inserted channels, which were opened by the agonist (Fig. 7A, second response). The small size of the test response is indicative of a relatively slow rate of constitutive exocytosis and is consistent with the half-time of ∼40 min reported for recovery of the acetylcholine response after irreversible block of nicotinic acetylcholine receptors expressed in Xenopusoocytes (Akabas et al., 1992). Because under steady-state conditions, the rate of internalization equals the rate of exocytosis, the rate of internalization is slow (Fig. 7A), and a reduction in the rate of internalization (or of silencing) could account for, at most, a very small fraction of the potentiation by ACPD.
Fig. 7.
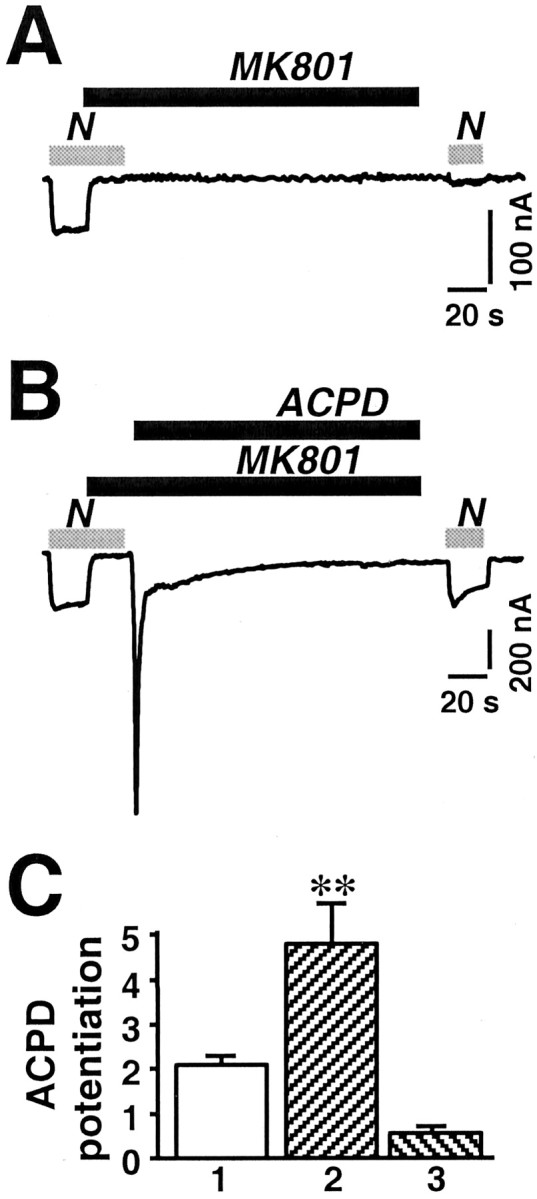
mGluR1α potentiation increases the rate of exocytosis of NMDA channels in the cell membrane. Whole-cell recordings were obtained from Xenopus oocytes expressing NR1-4b/NR2A receptors in Ca2+-free Ringer's solution. The open channel blocker MK-801 (1 μm) was used to estimate the rate of delivery of functional channels to the cell surface in control and ACPD-treated oocytes. NMDA, 300 μm; glycine, 10 μm. A, Application of MK-801 (1 μm) in the presence of agonist completely blocked the NMDA response. Three minutes after block and washout of NMDA, MK-801 was removed; then a test application of NMDA elicited a very small response (∼10% of control), attributable to either recovery of a small number of channels from block or insertion of new channels. B, After complete block of the NMDA response by MK-801 (1 μm), ACPD (100 μm) was applied for 2 min in the continuous presence of MK-801. After washout of ACPD and MK-801, the peak of the NMDA-induced response was slightly smaller than that of the control response. The greater decay of this response is ascribable to residual MK-801. C, Quantitation of data in A and B. Test responses were normalized to initial currents. Bar 1,IACPD/Icontrol, where IACPD is the NMDA-elicited current after ACPD. In this batch of oocytes. ACPD-induced potentiation was to ∼2 times the control response. Bar 2,IACPD, MK-801/IMK-801, whereIACPD, MK-801 is the current after block of the control response by MK-801 and potentiation by ACPD, andIMK-801 is the current after block of the control response by MK-801 followed by 3 min recovery. In the presence of MK-801, ACPD potentiation was to 4.5 times the recovered response observed after block by MK-801 in A, a value that could not be accounted for by twofold potentiation of the recovered response.Bar 3, IACPD, MK-801/Icontrol. The potentiation measured as the ratio of the potentiated response after MK-801 block to the control response was somewhat smaller than the potentiated response without MK-801 minus the control response.
As a further test of ACPD-induced recruitment of new channels to the cell surface, we blocked NMDA currents with MK-801 as in Figure7A and then applied ACPD in the presence of MK-801 (Fig.7B). In these oocytes, a test application of NMDA elicited a much larger response, ∼4.5 times that observed in control oocytes at 3 min after MK-801 block (Fig. 7A) (p< 0.01) and approximately the same size as the control response (p < 0.001) (Fig. 7C). If ACPD potentiation were attributable entirely to insertion of new channel molecules (Fig. 4), and constitutive insertion were negligible, the amplitude of the test NMDA current after ACPD application in the presence of MK-801 (IACPD, MK-801) would equal the amplitude of the test NMDA current after ACPD alone (IACPD) minus the amplitude of the initial control current (Icontrol).IACPD, MK-801 was slightly smaller than the predicted value, possibly an effect of residual MK-801 (Fig.5D, bars 1, 3). These findings demonstrate that ACPD causes a rapid increase in the number of functional NMDARs in the cell surface.
ACPD increases surface expression of NMDARs
The electrophysiological experiments presented thus far argue for mGluR1-mediated increase in the number of active channels via exocytosis but do not provide a measure of the total number of channel molecules expressed at the surface. To examine the effect of mGluR1α on the surface expression of NMDARs, we performed cell surface Western blots by a modification of the method of Chen et al. (1999). Control and ACPD-treated oocytes expressing mGluR1α and NR1/NR2A receptors were surface-labeled with sulfo-NHS-SS-biotin, and surface proteins were separated from intracellular proteins by reaction with Neutravidin beads and centrifugation. Samples of surface and total cell protein were treated with DTT (which releases the biotin moiety from surface proteins) and subjected to electrophoresis. Membranes were probed with antibody 54.1, a monoclonal antibody directed to the extracellular loop of the NR1 subunit (Gazzaley et al., 1996) (Fig.8A). Analysis of band densities indicated an increase in surface NR1 expression to 1.7 ± 0.2 times control (n = 4; p < 0.01) (Fig. 8B,D), with no change in total cell NR1 protein (Fig. 8C).
Fig. 8.
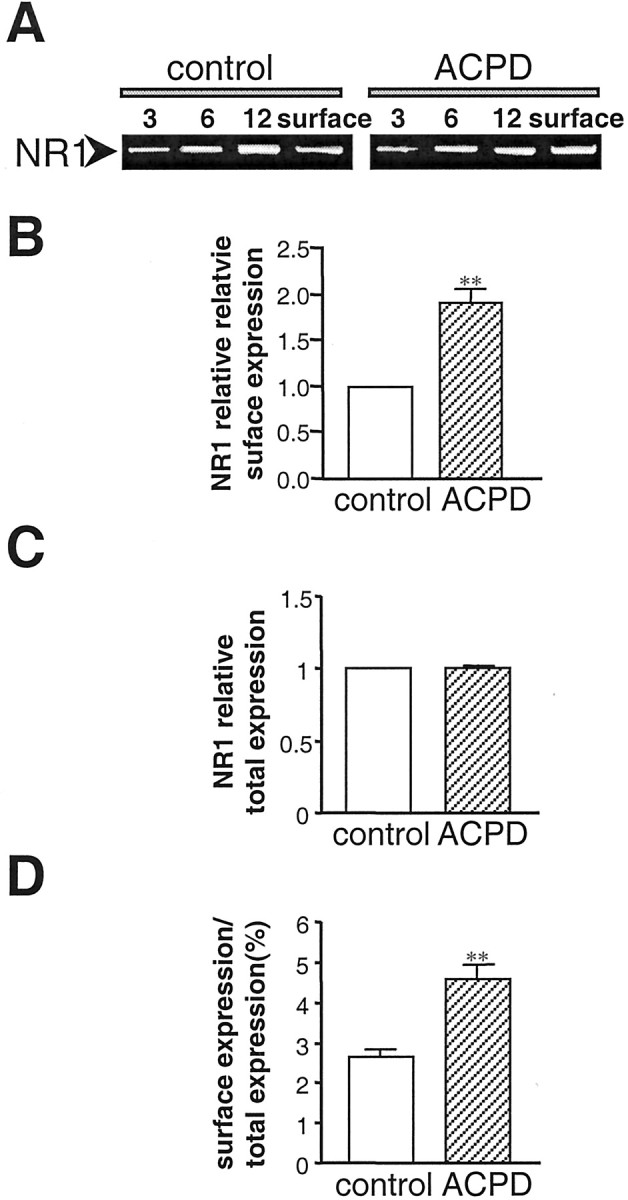
mGluR1α increases NMDAR surface expression. NR1 surface and total cell expression in control and ACPD-treated oocytes is shown, as assessed by Western blot analysis of surface proteins isolated by biotinylation. A, Representative Western blot of surface protein from oocytes expressing NR1-4b/NR2A with mGluR1α receptors probed with anti-NR1 antibody 54.1. 3, 6, 12, Micrograms of protein in samples of total cell extract before Neutravidin bead extraction loaded on each lane;surface, aliquot of Neutravidin bead-isolated receptors.B–D, Quantitative analysis of the effects of ACPD on surface expression (B), total cell protein (C), and fractional surface expression (D) of NMDARs. Surface expression was increased to 1.7 ± 0.2 times control (n = 4;p < 0.01). Total cell NR1 was not changed. The proportion of NR1 expressed at the cell surface increased from 2.6 to 4.5%.
In addition, we assessed the effect of mGluR1α activation on receptor surface expression by immunofluorescence. Control and ACPD-treated nonpermeabilized oocytes expressing NR1-4b/NR2A and mGluR1α receptors were reacted with antibody 54.1, followed by a biotinylated secondary antibody and fluorescein-conjugated avidin. Optical sections through the oocyte were viewed by confocal microscopy and captured by COMOS software. Control oocytes expressing NMDARs exhibited clear immunofluorescence, which appeared concentrated at the external surface (Fig. 9A,C). ACPD dramatically increased NR1 surface immunolabeling (Fig. 9B,D). Little or no immunofluorescence was observed for oocytes reacted with the FITC-tagged secondary antibody in the absence of the primary antibody (Fig. 9E) or for water-injected oocytes labeled with the N-terminal antibody (Fig. 9F). These findings indicate specificity of the immunofluorescence labeling. ACPD did not increase the resting conductance of the oocytes after the Ca2+-activated Cl− current subsided. Moreover, control experiments indicate that TPA treatment does not increase the permeability of the oocyte membrane to the antibody (Lan et al., 2001). We therefore infer that ACPD is unlikely to affect the permeability of the oocyte membrane.
Fig. 9.
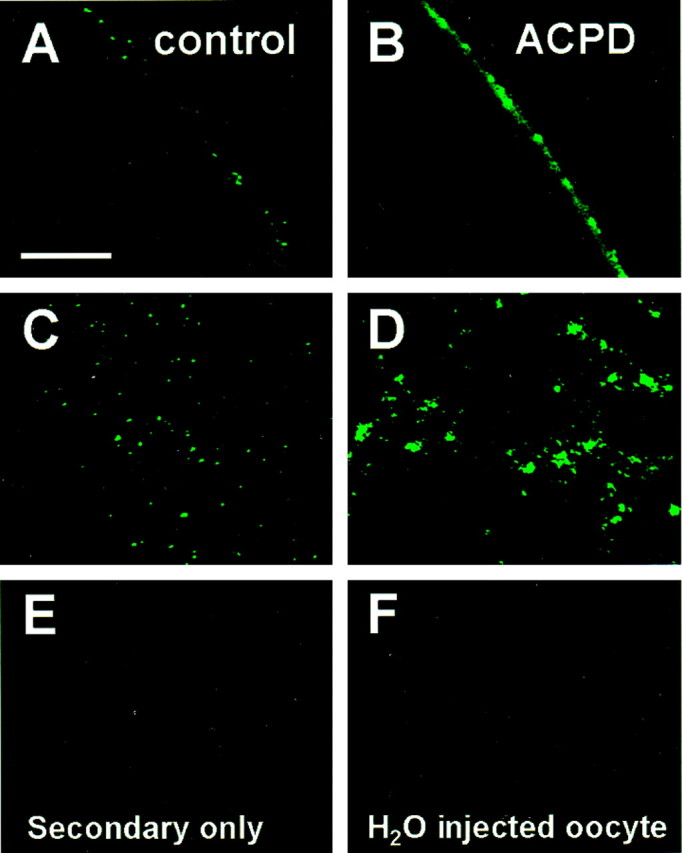
mGluR1α increases NR1 surface immunofluorescence. Oocytes expressing NR1100/NR2A receptors were incubated in external recording solution in the presence or absence of ACPD (100 μm, 10 min) and subjected to immunocytochemistry. Immediately after incubation, intact oocytes were devitellinized mechanically, transferred to glass coverslips, and fixed with paraformaldehyde (4%) and sucrose (2%). To label surface NMDARs, fixed oocytes were incubated with NR1 antibody 54.1, followed by FITC-conjugated secondary antibody. NMDARs on the oocyte surface were then visualized using confocal microscopy. Surface fluorescence was expressed as the mean intensity of fluorescence per unit area. Shown are representative oocytes expressing NR1-4b/NR2A receptors from control (A, B) and ACPD (C, D) treatment groups labeled by the extracellular epitope antibody. Control oocytes labeled by a secondary antibody in the absence of primary antibody (E) and water-injected oocytes labeled as inA–D showed negligible fluorescence (F).
DISCUSSION
Modulation of ligand-gated channels by G-protein-linked receptors is likely to be an important mode of receptor regulation under physiological conditions. The present study demonstrates that mGluR1α-mediated potentiation of NMDARs in Xenopus oocytes is mediated by recruitment of new channel molecules to the cell surface via regulated exocytosis. We show that loading cells with BoNT A, which interferes with exocytosis by cleaving SNAP-25, markedly reduces ACPD potentiation of NMDAR responses. Moreover, expression of a dominant negative mutant of SNAP-25, SNAP-25(Δ20), nearly abolishes ACPD potentiation. Specificity of mGluR1α-stimulated insertion of NMDARs at the cell surface is indicated by the observation that NR1/NR2A and NR1/NR2B but not NR1/NR2C receptors exhibit ACPD-induced potentiation. The present study is the first to examine biophysical mechanisms at the single-channel level in a system in which activation of group I mGluRs induces potentiation of NMDAR activity. We show that activation of mGluR1α increases the number of functional NMDA channels at the cell surface, with no change in single-channel conductance, mean open duration, or open probability. Our study is also the first to demonstrate mGluR-mediated rapid trafficking of NMDARs in oocytes via a SNARE-dependent mechanism. A simple scenario is that activation of mGluR1α regulates membrane fusion events of NMDAR-containing vesicles by activation of endogenous protein kinases, which in turn phosphorylate a protein involved in receptor trafficking. One potential target is SNAP-25, which can be directly phosphorylated in an activity-dependent manner (Shimazaki et al., 1996; Genoud et al., 1999). The intracellular signaling pathway linking mGluR1 activation with an increase in regulated exocytosis is, however, unknown, and association of SNAP-25 or its binding partners with NMDARs remains to be established.
Recent studies by our laboratory show that activation of PKC induces rapid delivery of NMDARs to the cell surface of oocytes and hippocampal neurons and an increase in the NMDA channel opening rate (Lan et al., 2001). Several findings indicate that the molecular mechanisms underlying mGluR1-mediated potentiation of NMDARs differ from those of PKC potentiation. First, ACPD regulates receptor trafficking in the absence of a significant effect on channel gating (although the present data cannot exclude a proportionately smaller change in the channel opening rate). Second, whereas the degree of TPA potentiation is markedly affected by NR1 splicing (Zheng et al., 1999), the degree of ACPD potentiation is affected only modestly by NR1 splicing, an effect that was not statistically significant in the present study. Third, whereas PKC-induced potentiation of NMDAR activity is observed in oocytes and dissociated hippocampal neurons, ACPD potentiation is observed in oocytes and in hippocampal slices but not in dissociated neurons, suggesting that the intracellular signaling pathway linking mGluR activation to NMDAR potentiation requires preservation of neuronal circuitry (P. J. Conn, personal communication).
Finally, group I mGluRs couple to diverse intracellular signaling pathways and potentiate NMDA EPSCs and NMDA-elicited currents by PKC-independent signal transduction in some neuronal cell types and preparations and in expression systems. Electrophysiological studies of CA3 pyramidal neurons from rat hippocampal slice cultures indicate that synaptic activation of group I mGluRs (mGluR1) by mossy fiber stimulation evokes an EPSC by a G-protein-independent signaling pathway (Heuss et al., 1999). These studies show that an Src-family tyrosine kinase is an integral component in a signaling pathway that functionally links mGluR1 activation with a transient cationic conductance increase. Because NR1/NR2A NMDARs are functionally modified by tyrosine kinases such as Src (Wang and Salter, 1994), it is reasonable to propose that mGluR1 potentiates NMDA currents via activation of Src. Consistent with a role for tyrosine kinase-dependent modulation of NMDAR activity, we recently reported that insulin promotes rapid exocytosis of NMDARs in Xenopus oocytes (Skeberdis et al., 2001a). Alternatively, group I mGluRs might signal via a membrane-delimited signaling pathway, as has been observed for mGluR-mediated inhibition of NMDAR activity in embryonic mouse cortical neurons (Yu et al., 1997).
The role of PKC-mediated signaling cascades in mGluR potentiation of NMDARs is, at best, controversial. mGluR-mediated potentiation of NMDA EPSCs is reported to occur independently of PKC signaling in hippocampal (Harvey and Collingridge, 1993) and cerebellar slice preparations (Kinney and Slater, 1993), as is group I mGluR-mediated potentiation of NMDA currents in lamprey motoneurons (Krieger et al., 2000) and frog spinal cord neurons (Holohean et al., 1999). On the other hand, the intracellular pathway linking mGluRs to potentiation of NMDA responses appears to involve PKC in at least two preparations, although neither the mGluR nor NMDAR subtype or receptor class was identified in these preparations. PKC inhibitors block at least in part mGluR-mediated potentiation of NMDA EPSCs in hippocampal slice (Aniksztejn et al., 1992) and potentiation of NMDA currents in oocytes expressing rat brain mRNA (Kelso et al., 1992). Thus, multiple pathways might contribute to the group I mGluR-dependent regulation of NMDARs observed in the present study. This concept is supported by pharmacological studies; whereas mGluR-dependent potentiation of NMDARs is only partly blocked by selective PKC inhibitors, it is completely blocked by the broad-spectrum kinase inhibitor staurosporine (Skeberdis et al., 2001b).
mGluR potentiation is via regulated exocytosis
In the present study, ACPD potentiation of NMDA-elicited currents was inhibited by ∼50% injection of BoNT A and nearly completely abolished by expression of SNAP-25(Δ20) (Figs. 5, 6). These findings provide important new evidence for a mechanism involving regulated exocytosis of NMDARs to the cell surface of the oocyte. Inhibition of exocytosis by BoNT A is incomplete in a number of other systems, including exocytosis of Isoc channels (Yao et al., 1999) and NMDARs in Xenopus oocytes (Lan et al., 2001). The action of BoNT A on regulated exocytosis of NMDARs was relatively specific in that (1) ACPD-induced insertion of new channel molecules was not inhibited by BoNT B or E; and (2) constitutive expression of NMDARs and endogenous Ca2+-activated Cl− channels (Yao et al., 1999; our unpublished observations) and transfected epithelial Na+ channels (Yao et al., 1999) in oocytes is not reduced by BoNT A.
Structural basis of localization of group I mGluRs and NMDARs
Immunogold studies reveal a highly ordered arrangement of group I mGluRs and NMDARs at the postsynaptic membrane of CA1 synapses and indicate that the two classes of receptors are concentrated in microdomains in close proximity to one another (Baude et al., 1993;Lujan et al., 1996; Lujan et al., 1997; Shigemoto et al., 1997). Studies involving the yeast two-hybrid system and immunoprecipitation suggest that mGluR1α and mGluR5 may be cross-linked to NMDARs at synapses. The C-terminal tails of mGluR1 and mGluR5 contain a recognition sequence for binding by members of the Homer family of anchoring proteins (Tu et al., 1998). Homer proteins, in turn, link via Shank and PSD to each other and to IP3 receptors, NMDARs, and AMPARs (Tu et al., 1999). Thus, a scaffolding structure consisting of Homer and Shank may directly connect group I mGluRs to NMDARs, thereby providing strategic positioning for regulation of NMDARs by mGluRs at hippocampal synapses.
Physiological significance of mGluR1 potentiation of NMDARs
The physiological relevance of mGluR1 actions is underscored by observations that activation of group I mGluRs induces potentiation of NMDA EPSCs in rat hippocampal (Aniksztejn et al., 1992; Challiss et al., 1994), striatal (Pisani et al., 1997), and subthalamic nucleus (Awad et al., 2000) slices and potentiation of NMDA currents in lamprey motoneurons (Krieger et al., 2000) and frog spinal cord neurons (Holohean et al., 1999). Turnover and biotinylation studies of cerebellar granule neurons indicate a large pool of NR1 subunits (∼60% total NR1) in the cytoplasm of dendritic shafts and spines, where they are poised for rapid assembly with NR2 subunits and insertion at synaptic sites (Huh and Wenthold, 1999). Rapid trafficking of AMPARs between the plasma membrane and an intracellular compartment in response to synaptic activity (Nishimune et al., 1998; Osten et al., 1998; Song et al., 1998) and synaptic plasticity (Carroll et al., 1999) is thought to represent recycling.
Findings from the present study suggest a mechanism whereby group I mGluRs can augment the number of synaptic NMDARs, thereby modulating neuronal excitability and lowering the threshold for LTP and long-term depression. Moreover, activation of NMDARs potentiates mGluR5 activity (Challiss et al., 1994; Luthi et al., 1994; Alagarsamy et al., 1999); mechanisms implicated in this potentiation include a rise in intracellular Ca2+, activation of calcineurin, and inhibition of PKC (De Blasi et al., 2001). Thus, positive interactions between mGluRs and NMDARs may be reciprocal in nature and important to NMDAR-dependent LTP. The present study extends earlier studies by providing insight into the biophysical and molecular mechanisms underlying mGluR1-mediated regulation of NMDARs in a pure population of receptors. Insertion and retrieval of NMDARs to and from the plasma membrane are likely to be common and powerful mechanisms for regulating excitatory synaptic transmission.
Footnotes
This work was supported by National Institutes of Health Grants NS 20752 and NS 31282 (R.S.Z.) and NS 07512 (M.V.L.B). M.V.L.B is the Sylvia and Robert S. Olnick Professor of Neuroscience. We thank Alice P. Wang for technical support. We acknowledge the Analytical Imaging Facility of the Albert Einstein College of Medicine (Michael Cammer, director).
Correspondence should be addressed to Dr. R. Suzanne Zukin, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: zukin@aecom.yu.edu.
REFERENCES
- 1.Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994a;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 2.Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994b;79:377–388. [PubMed] [Google Scholar]
- 3.Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 4.Alagarsamy S, Marino MJ, Rouse ST, Gereau RW4, Heinemann SF, Conn PJ. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat Neurosci. 1999;2:234–240. doi: 10.1038/6338. [DOI] [PubMed] [Google Scholar]
- 5.Aniksztejn L, Otani S, Ben-Ari Y. Quisqualate metabotropic receptors modulate NMDA currents and facilitate induction of long-term potentiation through protein kinase C. Eur J Neurosci. 1992;4:500–505. doi: 10.1111/j.1460-9568.1992.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 6.Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8:757–765. doi: 10.1016/0896-6273(92)90096-v. [DOI] [PubMed] [Google Scholar]
- 7.Araneda RC, Lan JY, Zheng X, Zukin RS, Bennett MV. Spermine and arcaine block and permeate N-methyl-d-aspartate receptor channels. Biophys J. 1999;76:2899–2911. doi: 10.1016/S0006-3495(99)77445-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- 10.Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bordi F. Reduced long-term potentiation in the dentate gyrus of mGlu1 receptor-mutant mice in vivo. Eur J Pharmacol. 1996;301:R15–R16. doi: 10.1016/0014-2999(96)00138-0. [DOI] [PubMed] [Google Scholar]
- 12.Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- 13.Challiss RAJ, Mistry R, Gray DW, Nahorski SR. Modulatory effects of NMDA on phosphoinositide responses evoked by the metabotropic glutamate receptor agonist 1S,3R-ACPD in neonatal rat cerebral cortex. Br J Pharmacol. 1994;112:231–239. doi: 10.1111/j.1476-5381.1994.tb13057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen N, Luo T, Raymond LA. Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 16.Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- 17.De Blasi A, Conn PJ, Pin J, Nicoletti F. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci. 2001;22:114–120. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- 18.Durand GM, Gregor P, Zheng X, Bennett MV, Uhl GR, Zukin RS. Cloning of an apparent splice variant of the rat N-methyl-d-aspartate receptor NMDAR1 with altered sensitivity to polyamines and activators of protein kinase C. Proc Natl Acad Sci USA. 1992;89:9359–9363. doi: 10.1073/pnas.89.19.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genoud S, Pralong W, Riederer BM, Eder L, Catsicas S, Muller D. Activity-dependent phosphorylation of SNAP-25 in hippocampal organotypic cultures. J Neurochem. 1999;72:1699–1706. doi: 10.1046/j.1471-4159.1999.721699.x. [DOI] [PubMed] [Google Scholar]
- 21.Harvey J, Collingridge G. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br J Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heuss C, Scanziani M, Gahwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci. 1999;2:1070–1077. doi: 10.1038/15996. [DOI] [PubMed] [Google Scholar]
- 23.Hollmann M, Maron C, Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 24.Holohean AM, Hackman JC, Davidoff RA. Mechanisms involved in the metabotropic glutamate receptor-enhancement of NMDA-mediated motoneurone responses in frog spinal cord. Br J Pharmacol. 1999;126:333–341. doi: 10.1038/sj.bjp.0702263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-d-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- 26.Jahr CE. High probability opening of NMDA receptor channels by L-glutamate. Science. 1992;255:470–472. doi: 10.1126/science.1346477. [DOI] [PubMed] [Google Scholar]
- 27.Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin JP. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaczmarek L, Kossut M, Skangiel-Kramska J. Glutamate receptors in cortical plasticity: molecular and cellular biology. Physiol Rev. 1997;77:217–255. doi: 10.1152/physrev.1997.77.1.217. [DOI] [PubMed] [Google Scholar]
- 29.Kelso SR, Nelson TE, Leonard JP. Protein kinase C-mediated enhancement of NMDA currents by metabotropic glutamate receptors in Xenopus oocytes. J Physiol (Lond) 1992;449:705–718. doi: 10.1113/jphysiol.1992.sp019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinney GA, Slater NT. Potentiation of NMDA receptor-mediated transmission in turtle cerebellar granule cells by activation of metabotropic glutamate receptors. J Neurophysiol. 1993;69:585–594. doi: 10.1152/jn.1993.69.2.585. [DOI] [PubMed] [Google Scholar]
- 31.Krieger P, Hellgren-Kotaleski J, Kettunen P, El Manira AJ. Interaction between metabotropic and ionotropic glutamate receptors regulates neuronal network activity. J Neurosci. 2000;20:5382–5391. doi: 10.1523/JNEUROSCI.20-14-05382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- 33.Legendre P, Rosenmund C, Westbrook GL. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leonard JP, Kelso SR. Apparent desensitization of NMDA responses in Xenopus oocytes involves calcium-dependent chloride current. Neuron. 1990;4:53–60. doi: 10.1016/0896-6273(90)90443-j. [DOI] [PubMed] [Google Scholar]
- 35.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 37.Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. 1997;13:219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 38.Luthi A, Gahwiler BH, Gerber U. Potentiation of a metabotropoic glutamatergic response following NMDA receptor activation in rat hippocampus. Pflügers Arch. 1994;427:197–202. doi: 10.1007/BF00585965. [DOI] [PubMed] [Google Scholar]
- 39.Montecucco C, Schiavo G. Structure and function of tetanus and botulinum neurotoxins. Q Rev Biophys. 1995;28:423–472. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 40.Mori H, Yamakura T, Masaki H, Mishina M. Involvement of the carboxyl-terminal region in modulation by TPA of the NMDA receptor channel. NeuroReport. 1993;4:519–522. doi: 10.1097/00001756-199305000-00014. [DOI] [PubMed] [Google Scholar]
- 41.Nicoletti F, Bruno V, Catania MV, Battaglia G, Copani A, Barbagallo G, Cena V, Sanchez-Prieto J, Spano PF, Pizzi M. Group-I metabotropic glutamate receptors: hypotheses to explain their dual role in neurotoxicity and neuroprotection. Neuropharmacology. 1999;38:1477–1484. doi: 10.1016/s0028-3908(99)00102-1. [DOI] [PubMed] [Google Scholar]
- 42.Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 43.Okabe S, Miwa A, Okado H. Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J Neurosci. 1999;19:7781–7792. doi: 10.1523/JNEUROSCI.19-18-07781.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Opitz T, Richter P, Reymann KG. The metabotropic glutamate receptor antagonist (+)-alpha-methyl-4-carboxyphenylglycine protects hippocampal CA1 neurons of the rat from in vitro hypoxia/hypoglycemia. Neuropharmacology. 1994;33:715–717. doi: 10.1016/0028-3908(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 45.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 46.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 47.Pisani A, Calabresi P, Centonze D, Bernardi G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenmund C, Feltz A, Westbrook GL. Synaptic NMDA receptor channels have a low open probability. J Neurosci. 1995;15:2788–2795. doi: 10.1523/JNEUROSCI.15-04-02788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 52.Skeberdis VA, Lan JY, Opitz T, Zheng X, Bennett MVL, Zukin RS. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology. 2001a;40:856–865. doi: 10.1016/s0028-3908(01)00005-3. [DOI] [PubMed] [Google Scholar]
- 53.Skeberdis VA, Lan J, Zheng X, Zukin RS, Bennett MV. Insulin promotes rapid delivery of N-methyl-d-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci USA. 2001b;98:3561–3566. doi: 10.1073/pnas.051634698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 55.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 56.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 57.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 58.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 59.Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- 60.Yu SP, Sensi SL, Canzoniero LM, Buisson A, Choi DW. Membrane-delimited modulation of NMDA currents by metabotropic glutamate receptor subtypes 1/5 in cultured mouse cortical neurons. J Physiol (Lond) 1997;499:721–732. doi: 10.1113/jphysiol.1997.sp021964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zampighi GA, Loo DD, Kreman M, Eskandari S, Wright EM. Functional and morphological correlates of connexin 50 expressed in Xenopus laevis oocytes. J Gen Physiol. 1999;113:507–524. doi: 10.1085/jgp.113.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng X, Zhang L, Wang AP, Bennett MV, Zukin RS. Ca2+ influx amplifies protein kinase C potentiation of recombinant NMDA receptors. J Neurosci. 1997;17:8676–8686. doi: 10.1523/JNEUROSCI.17-22-08676.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng X, Zhang L, Wang AP, Bennett MV, Zukin RS. Protein kinase C potentiation of N-methyl-d-aspartate receptor activity is not mediated by phosphorylation of N-methyl-d-aspartate receptor subunits. Proc Natl Acad Sci USA. 1999;96:15262–15267. doi: 10.1073/pnas.96.26.15262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zukin RS, Bennett MV. Alternatively spliced isoforms of the NMDARI receptor subunit. Trends Neurosci 18 1995. 306 313[erratum (1995) 18:441]. [DOI] [PubMed] [Google Scholar]