

Abstract
GABAA receptor-mediated responses manifest as either hyperpolarization or depolarization according to the intracellular Cl− concentration ([Cl−]i). Here, we report a novel functional interaction between the Na–K–Cl cotransporter (NKCC) and GABAA receptor actions on glutamatergic presynaptic nerve terminals projecting to ventromedial hypothalamic (VMH) neurons. The activation of presynaptic GABAA receptors depolarizes the presynaptic nerve terminals and facilitates spontaneous glutamate release by activating TTX-sensitive Na+ channels and high-threshold Ca2+ channels. This depolarizing action of GABA was caused by an outwardly directed Cl− driving force for GABAA receptors; that is, the [Cl−]i of glutamatergic nerve terminals was higher than that predicted for a passive distribution. The higher [Cl−]i was generated by bumetanide-sensitive NKCCs and was responsible for the GABA-induced presynaptic depolarization. Thus, GABAAreceptor-mediated modulation of spontaneous glutamatergic transmission may contribute to the development and regulation of VMH function as well as to the excitability of VMH neurons themselves.
Keywords: sEPSCs, presynaptic GABAA receptors, intraterminal Cl− concentration, NKCC, mechanical dissociation, VMH, GABA-induced depolarization
GABA is the primary inhibitory neurotransmitter throughout the mammalian CNS. The activation of ionotropic GABAA receptors inhibits neuronal excitability by increasing the Cl− conductance of the membrane. In adult neurons, this typically results in postsynaptic hyperpolarization, although in developing neurons GABAA receptor activation causes a depolarization because of the high intracellular Cl− concentration ([Cl−]i) (Obrietan and van den Pol, 1995; Chen et al., 1996). The high [Cl−]i results from inwardly directed Cl− transporters such as the Na–K–Cl cotransporter (NKCC) (Plotkin et al., 1997;Clayton et al., 1998; Kakazu et al., 1999). This GABA-induced depolarization can elevate the intracellular Ca2+ concentration ([Ca2+]i) via the activation of voltage-dependent Ca2+channels (VDCCs) (Leinekugel et al., 1995; Obrietan and van den Pol, 1995; Owens et al., 1996) and may contribute to several aspects of CNS development, such as the rate and direction of neuritic growth (Mattson and Kater, 1987; Obrietan and van den Pol, 1996) and gene expression (Vaccarino et al., 1992; Bading et al., 1993).
Activation of presynaptic GABAA receptors in sensory afferent neurons inhibits neurotransmitter release despite its depolarizing action (Levy, 1977; Rudomin and Schmidt, 1999). This primary afferent depolarization (PAD) is thought to block the conduction of action potentials (APs) in the presynaptic terminals both by membrane shunting and by the inactivation of Na+ channels (Segev, 1990; Graham and Redman, 1994; Cattaert and El Manira, 1999), causing a reduction of monosynaptic EPSPs in target neurons (Cattaert et al., 1992). The activation of presynaptic GABAA receptors inhibits both electrically evoked and high K+-evoked release of neurotransmitter or hormone from nerve terminals (Dyball and Shaw, 1978; Nicoll and Alger, 1979; Pickles, 1979; Tachibana and Kaneko, 1987; Saridaki et al., 1989; Rudomin, 1990; Zhang and Jackson, 1995).
In immature rat hypothalamic neurons, GABA evokes postsynaptic depolarization because of the high [Cl−]i, suggesting a possible excitatory role of GABAergic transmission during development (Chen et al., 1996). However, it is still unknown whether presynaptic GABAA receptor activation can increase the probability of neurotransmitter release and whether the presynaptic nerve terminals have the higher [Cl−]i that may be responsible for GABA-induced presynaptic depolarization. In the present study, we have investigated this hypothesis directly using mechanically dissociated rat ventromedial hypothalamic (VMH) neurons, taking care to preserve the attached glutamatergic nerve terminals [the “synaptic bouton” preparation (Rhee et al., 1999)]. This preparation allows us to focus selectively on presynaptic GABAA receptors and to study the effects of their activation on glutamatergic transmission.
MATERIALS AND METHODS
Preparation. Wistar rats (12–15 d old) were decapitated under pentobarbital anesthesia (50 mg/kg, i.p.). The brain was quickly removed and transversely sliced at a thickness of 370 μm by use of a microslicer (VT1000S; Leica, Nussloch, Germany). Slices were kept in the control incubation medium (see below) saturated with 95% O2 and 5% CO2 at room temperature (21–24°C) for at least 1 hr before the mechanical dissociation. For dissociation, slices were transferred into a 35 mm culture dish (Primaria 3801; Becton Dickinson, Rutherford, NJ), and the region of the VMH was identified under a binocular microscope (SMZ-1; Nikon, Tokyo, Japan). Details of the mechanical dissociation have been described previously (Rhee et al., 1999). Briefly, mechanical dissociation was accomplished using a custom-built vibration device and a fire-polished glass pipette oscillating at ∼3–5 Hz (0.1–0.2 mm). The tip of the fire-polished glass pipette was lightly placed on the surface of the VMH region with a micromanipulator. The tip of glass pipette was vibrated horizontally for ∼2 min. Slices were removed, and the mechanically dissociated neurons were allowed to settle for 15 min and adhere to the bottom of the dish. Such neurons undergoing dissociation retained short portions of their proximal dendrites.
For the slice preparation, the brain was quickly removed and transversely sliced at a thickness of 250 μm by use of a microslicer (VT1000S; Leica). The slices were kept in a cold low-Na+ medium (see below) for at least 1 hr. Thereafter the slices were transferred into a recording chamber, and the VMH was identified under an upright microscope (Axioscope;Zeiss). Bath solution was perfused at 8–10 ml/min.
All experiments conformed to the guiding principles for the care and use of animals approved by the Council of the Physiological Society of Japan, and all efforts were made to minimize the number of animals and any suffering.
Electrical measurements. Most of the electrical measurements were performed using the conventional whole-cell patch-clamp recording mode at holding potentials (VH values) of −57 to −63 mV, except where indicated. Membrane voltage was controlled and currents were recorded by the use of a patch-clamp amplifier (CEZ-2300; Nihon Kohden, Tokyo, Japan). Patch pipettes were made from borosilicate capillary glass (1.5 mm outer diameter; 0.9 mm inner diameter; G-1.5; Narishige, Tokyo, Japan) in two stages on a vertical pipette puller (PB-7; Narishige). The resistance of the recording pipettes filled with internal solution was 5–6 MΩ. Electrode capacitance and liquid junction potential were compensated for, but series resistance was not. Neurons were visualized under phase contrast on an inverted microscope (Diapot; Nikon). Current and voltage were continuously monitored on an oscilloscope (VC-6023; Hitachi) and a pen recorder (RECTI-HORIT-8K; Sanei, Tokyo, Japan) and recorded on a digital-audio tape recorder (RD-120TE; TEAC). Membrane currents were filtered at 1 kHz (E-3201A Decade Filter; NF Electronic Instruments, Tokyo, Japan), digitized at 4 kHz, and stored on a computer equipped with pCLAMP 8.0 (Axon Instruments). All experiments were performed at room temperature (21–24°C), except for the slice experiments that were performed at 30–33°C.
To record evoked EPSCs (eEPSCs) in the slice preparation, a glass stimulation pipette (∼5 μm diameter), filled with the incubation medium, was positioned around the amygdala region. Brief (100 μsec) voltage pulses were applied by the pipette at a stimulation frequency of 0.1 Hz using the PULSE software (HEKA). Data were filtered at 3 kHz and digitized at 10 kHz.
Data analysis. Spontaneous EPSCs (sEPSCs) were counted and analyzed using the MiniAnalysis program (Synaptosoft). Spontaneous events were initially detected automatically by use of an amplitude threshold of 3 pA at VH values of −57 to −63 mV and then visually accepted or rejected on the basis of the rise and decay times. Events with brief rise times (0.5–1.5 msec) and with decay times that were well fitted by a single-exponential function were selected for analysis. The amplitudes and interevent intervals of large numbers of sEPSCs obtained from a single neuron were examined by constructing all-point cumulative probability distributions and compared using the Kolmogorov–Smirnov (K–S) test with StatView software (SAS Institute, Inc.). Values of p < 0.05 were considered significant. Averaged sEPSC frequency and amplitude were normalized to the control conditions and are provided as means ± SEM. Differences in sEPSC amplitude and frequency were tested with Student's paired two-tailed t test using their absolute values. Values of p < 0.05 were considered significant.
Solutions. The incubation medium consisted of (in mm) 124 NaCl, 5 KCl, 1.2 KH2PO4, 24 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, and 10 glucose saturated with 95% O2 and 5% CO2. The pH was ∼7.45. The low-Na+ medium consisted of (in mm) 230 sucrose, 2.5 KCl, 1.25 Na2HPO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 30 glucose. The standard external solution consisted of (in mm) 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES. Ca2+-free external solution consisted of (in mm) 150 NaCl, 5 KCl, 5 MgCl2, 2 EGTA, 10 glucose, and 10 HEPES. These external solutions were adjusted to pH 7.4 with Tris base. The ionic composition of the internal (patch pipette) solution was (in mm) 145 Cs-methanesulfonate, 5 tetraethylammonium-Cl, 5 CsCl, 2 EGTA, and 10 HEPES with pH adjusted to 7.2 with Tris base. In some experiments, as indicated below, 4 mm Mg-ATP was added to this pipette solution.
Drugs. Drugs used in the present study were tetrodotoxin (TTX), bicuculline, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX),cis-aminocrotonic acid (CACA), EGTA, GABA, muscimol, bumetanide, baclofen, nicardipine, and Mg-ATP (all from Sigma, St. Louis, MO); GYKI52466 from Research Biochemicals (Natick, MA); ω-conotoxin GVIA (ω-CgTx) and ω-agatoxine IVA (ω-AgTx) from the Peptide Institute (Osaka, Japan); and furosemide from Tokyo Kasei (Tokyo, Japan). CGP55845A was a kind gift from Dr. B. Willi. CNQX, bicuculline, and bumetanide were dissolved in dimethylsulfoxide at 10 mm as a stock solution. All drug-containing solutions were applied using the “Y-tube system” that results in solution exchange within ∼20 msec (Akaike and Harata, 1994).
RESULTS
Spontaneous EPSCs
Hypothalamic neurons receive strong GABAergic inputs (Decavel and van den Pol, 1990a,b). To distinguish functional GABAA receptors on glutamatergic presynaptic nerve terminals projecting to VMH neurons from those on the postsynaptic membrane, we used two experimental strategies. First, the VH of VMH neurons was adjusted to the reversal potential of GABA-induced currents (EGABA) to minimize the change of postsynaptic currents resulting from the activation of postsynaptic GABAA receptors by exogenous GABA application. Second, ATP was excluded from the pipette solution to induce a selective and rapid rundown of the postsynaptic GABAA response (Shirasaki et al., 1992; Akaike, 1995; Harata et al., 1997).
Exogenous GABA (5 μm) induced large outward currents in all VMH neurons when held at −40 mV and when applied shortly after membrane rupture (Fig.1Aa). The GABA responses obtained with pipettes that contained no ATP gradually declined by 80% within 15 min after the membrane rupture, whereas this substantial decrease in peak current was not observed when the pipettes contained 4 mm Mg-ATP (Fig.1Ab). The GABA-induced postsynaptic currents (Fig.1B) reversed polarity at a VHof −61.1 ± 0.3 mV (n = 4;EGABA). This compares with the theoretical Cl− equilibrium potential (ECl) of −69.9 mV calculated from the Nernst equation using extracellular and intracellular Cl− concentrations of 161 mm[Cl−]o and 10 mm[Cl−]i, respectively. This difference may be partially explained by the small permeability of the GABAA receptors to MeSO3− (aP[MeSO3−]/P[Cl−] ratio of 0.01 will shift the predictedEGABA to −66.5 mV) [see also Chen et al. (1996)], and we conducted no further study to elucidate any other source of this difference. Thus in all of the following experiments, the VH for each neuron was finely adjusted to the experimentally measured postsynapticEGABA, and each neuron was well dialyzed with ATP-free pipette solution.
Fig. 1.

Experimental protocols and glutamatergic sEPSCs.A, The effect of intracellular ATP ([ATP]i) on the postsynaptic GABA response recorded from VMH neurons. a, GABA (5 μm)-induced currents in neurons perfused with pipette solution with or without 4 mm ATP. VH was −40 mV. GABA was applied (drug application indicated by thehorizontal bars) at 4 min intervals at the indicated times after the membrane rupture. b, Time courses of GABA responses with a pipette solution with or without ATP. Mean current amplitudes were normalized to the initial response obtained within the first 1 min after membrane rupture (n = 4). B, a, Postsynaptic responses induced by 5 μm GABA at various VHvalues (−80 to −40 mV). The pipette solution contained 4 mm Mg-ATP. b, The mean GABA current–voltage (I–V) relationship. All current amplitudes were normalized to that obtained at a VH of −40 mV (n = 4). C, A typicaltrace of sEPSCs in the presence and absence of 10 μm CNQX. VH was adjusted toEGABA. D, a,Traces of sEPSCs recorded at various VHvalues. b, The corresponding mean I–Vrelationship. In b, each point is the mean of four neurons, and all recordings were performed in the presence of 10 μm bicuculline.
In neurons held under these conditions, the recorded spontaneous postsynaptic currents were completely and reversibly blocked by adding 10 μm CNQX (n = 4) (Fig. 1C) and 3 μm GYKI52466, a specific antagonist of the AMPA receptor, but were not affected by adding either 10 μm bicuculline or 10 μmAP-5 (data not shown). Furthermore, these sEPSCs clearly reversed close to 0 mV (Fig. 1D). These results are consistent with the sEPSCs being generated by currents passing through AMPA receptor channels.
GABAergic modulation of sEPSCs
To examine whether the activation of GABAA receptor influences glutamatergic transmission, GABA was reapplied to the neurons 30–40 min after membrane rupture and after the postsynaptic response had been minimized. GABA increased the sEPSC frequency to 144.7 ± 18.8% of the control (p < 0.05;n = 10) (Fig.2Ba, inset). The cumulative distribution of sEPSC frequency was significantly shifted to the left by GABA, indicating an increase in sEPSC frequency, whereas that of sEPSC amplitude was not changed (Fig.2Bb). The results suggest that GABA acts presynaptically to facilitate glutamate release at these synapses. However, if both GABAA and GABAB receptors are localized on the same glutamatergic nerve terminals, the application of GABA could activate both receptor types at the same time. Functional GABAB receptors do indeed appear to exist on the glutamatergic nerve terminals because sEPSC frequency was greatly reduced to 41.12 ± 7.43% of the control by adding 30 μm baclofen, a selective GABAB receptor agonist (p< 0.01; n = 5) (Fig. 2C,D). Mean current amplitude was unaffected. Application of 10 μmCGP55845A, a selective GABAB receptor antagonist, did not affect glutamatergic transmission (p > 0.2; n = 13) (Fig. 2C,D). These results suggest that the activation of GABABreceptors attenuates the action of GABAA receptor on sEPSC frequency.
Fig. 2.

Presynaptic GABAA and GABAB receptors on the glutamatergic nerve terminals.A, A typical trace of sEPSCs observed before, during, and after the application of 5 μm GABA.B, Cumulative distributions of interevent intervals (a; p < 0.01; GABA vs control; K–S test) and amplitudes (b;p = 0.216; K–S test) in the same neuron shown in A (438 and 258 events for control and GABA, respectively).Insets, The mean sEPSC frequency (a) and amplitude (b) from 11 neurons. All columns were normalized to the respective control. C, Typical traces of sEPSCs observed before, during, and after the application of 30 μm baclofen (top) or 10 μmCGP55845A (bottom) in the same neuron. D, Cumulative distributions of interevent intervals (a) and current amplitudes (b) recorded from the same neuron shown inC (560 events for control, 128 events for baclofen, and 538 events for CGP55845A). Baclofen significantly shifted sEPSC frequency (p < 0.001), but CGP55845A did not (p = 0.492). Insets, The mean sEPSC frequency (a) and amplitude (b) from five neurons. All columnswere normalized to the control (dotted lines). *p < 0.05; **p < 0.01. These definitions of * and ** are applied to all subsequent figures.Amp., Amplitude; Bac, baclofen;CGP, CGP55845A; Cont, control;Freq., frequency; ns, not significant.
To isolate the GABAA receptor actions, GABA was applied in the presence of GABAB blockade (10 μm CGP55845A). Under these conditions, GABA facilitated sEPSC frequency to 225.1 ± 25.7% of the control (p < 0.01; n = 13) (Fig.3A,Ba, inset) without affecting the mean current amplitude. This was a significantly greater facilitation than that observed in the absence of CGP55845A (p < 0.05, Student's unpaired ttest). All facilitation was blocked by adding 10 μm bicuculline, a selective GABAA receptor antagonist (n = 8) (Fig. 3C). Taken together, these results suggest that both functional GABAA and GABABreceptors are located on the glutamatergic nerve terminals projecting to VMH neurons and that the activation of these two receptors could modulate the glutamatergic transmission in a competitive manner.
Fig. 3.
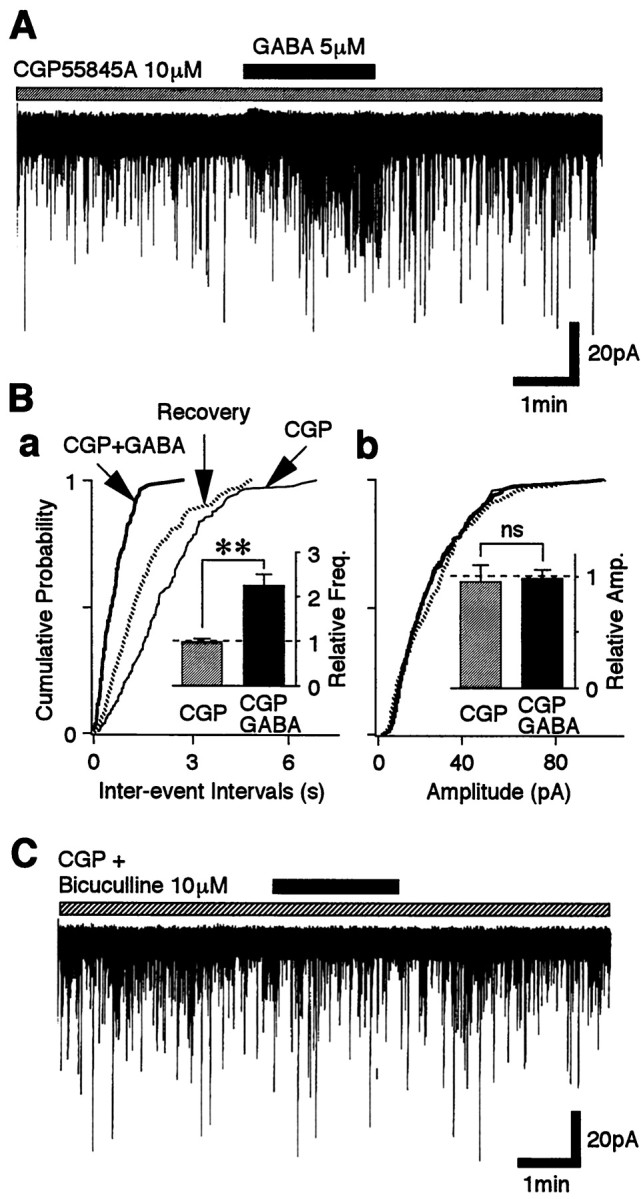
GABA facilitates glutamate release via the GABAA receptor. A, A typicaltrace of sEPSCs observed before, during, and after the application of 5 μm GABA in the presence of 10 μm CGP55845A. B, Cumulative distributions of interevent intervals (a; p < 0.001) and current amplitudes (b;p = 0.615) for sEPSCs recorded from the same neuron shown in A (437 and 462 events for CGP55845A and GABA, respectively). Insets, The mean sEPSC frequency (a) and amplitude (b) from 14 neurons. All columns were normalized to the control (dotted lines). C, A typicaltrace of sEPSCs observed before, during, and after the application of 5 μm GABA in the presence of both 10 μm CGP55845A and 10 μm bicuculline.
To confirm that GABA facilitates sEPSC frequency via GABAA receptors, the GABAAreceptor agonist muscimol was tested. Muscimol (0.5 μm) greatly increased sEPSC frequency to 264.0 ± 26.7% of the control (p < 0.01; n = 10) without affecting mean sEPSC current amplitude (Fig.4Aa,Ab). Muscimol at 5 μm did not cause any further facilitation of sEPSC frequency (264.1 ± 29.0% of the control), whereas 50 μm muscimol actually caused a reduced facilitation of sEPSC frequency (178.6 ± 21.7% of the control). To evaluate this concentration dependence further, the time course of muscimol-induced sEPSC facilitation was analyzed further (Fig.4Ac). Low concentrations of muscimol persistently facilitated sEPSCs throughout the application period, whereas high concentrations of muscimol only transiently increased sEPSC frequency. The muscimol action on sEPSC frequency was easily reproduced after repeated applications (Fig. 4B). The kinetics of sEPSCs, including their rise times and the decay time constants, was unaltered by muscimol (the 10 and 90% rise times being 0.87 ± 0.05 and 0.85 ± 0.15 msec; the time constants of the decay being 3.19 ± 0.24 and 3.21 ± 0.15 msec for control and muscimol, respectively; n = 11). Furthermore, CACA (1 or 10 μm), a selective GABAC receptor agonist, did not alter sEPSC frequency and amplitude (n = 4; data not shown). Therefore, because of its specificity of action on GABAA receptors, muscimol, rather than GABA, was used in the following experiments.
Fig. 4.
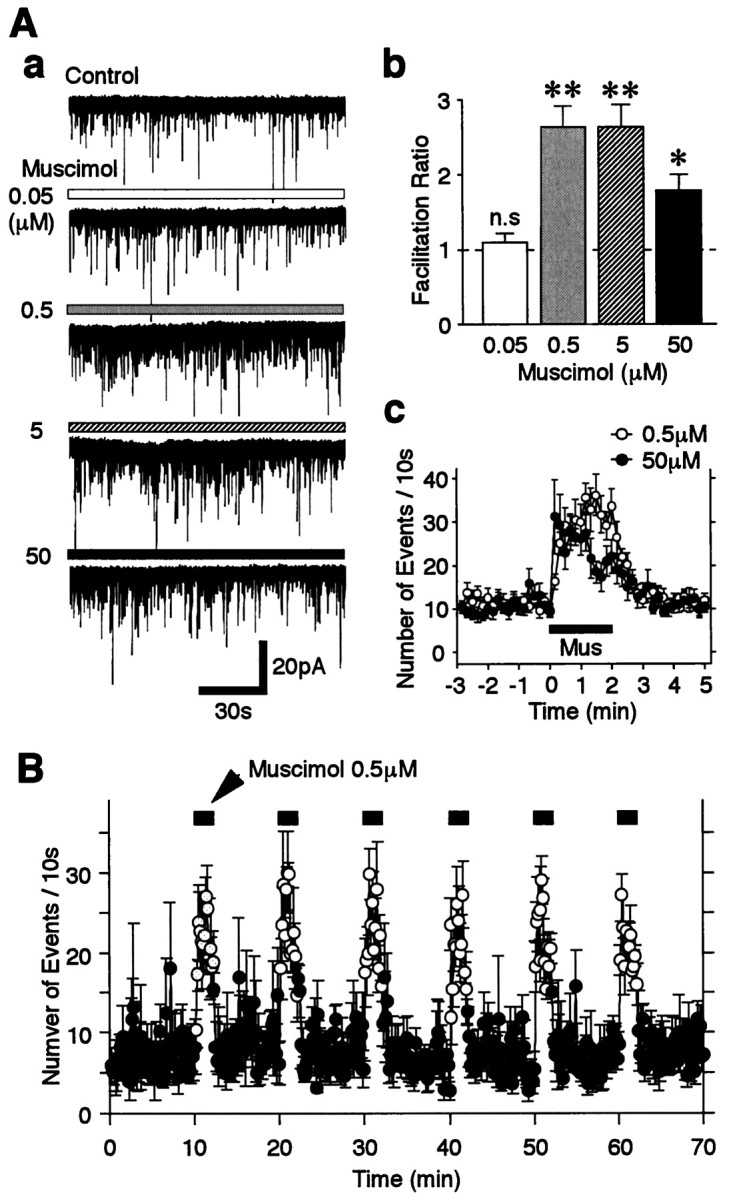
Concentration–response relationship of muscimol.A, a, Typical traces of sEPSCs in the absence of (Control) and in the presence of four different muscimol concentrations as indicated. b, Mean ratio of sEPSC frequency facilitation from 8 to 10 neurons. Allcolumns were normalized to the control (dotted line). Note that the facilitation induced by 50 μm muscimol was less than that induced by 0.5 μm muscimol. c, Time course of sEPSC frequency before, during, and after the application of 0.5 and 50 μm muscimol. The number of events in every 10 sec period was summed and plotted. Each point is the mean ± SEM from 10 and 8 neurons for 0.5 and 50 μm muscimol, respectively. Note that 0.5 μm muscimol has a persistent effect during the period of application, whereas 50 μm muscimol-induced sEPSC facilitation was rapidly attenuated during the period of application. B, The time course of sEPSC frequency during six consecutive applications of 0.5 μm muscimol. The number of events in every 10 sec period (open circles, presence of muscimol; closed circles, absence of muscimol) was summed and plotted. Each point is the mean ± SEM from five neurons. Mus, Muscimol;n.s., not significant.
Mechanism of GABAA receptor-mediated sEPSC facilitation
In immature neurons, either synaptically released or exogenously applied GABA induces membrane depolarization followed by increases in [Ca2+]i(Leinekugel et al., 1995; Obrietan and van den Pol, 1995). Alternatively, GABA-induced depolarization elicits increases in [Ca2+]i by activation of Ca2+ channels secondary to the activation of Na+ channels (Hales et al., 1994; Owens et al., 1996). Thus, it was tested whether GABAA receptor-mediated sEPSC facilitation depended on the activation of either voltage-dependent Na+ and/or Ca2+ channels.
Removal of extracellular Ca2+ greatly reduced sEPSC frequency to 53.1 ± 2.9% of the control (p < 0.01; n = 5) (Fig.5B) without affecting the distribution of current amplitudes. The results indicate that extracellular Ca2+ influxes markedly contribute to the generation of sEPSCs. In the Ca2+-free external solution, muscimol failed to facilitate sEPSC frequency (n = 5) (Fig.5A,B). Thus, muscimol-induced sEPSC facilitation seems to be dependent on the presence of extracellular Ca2+.
Fig. 5.
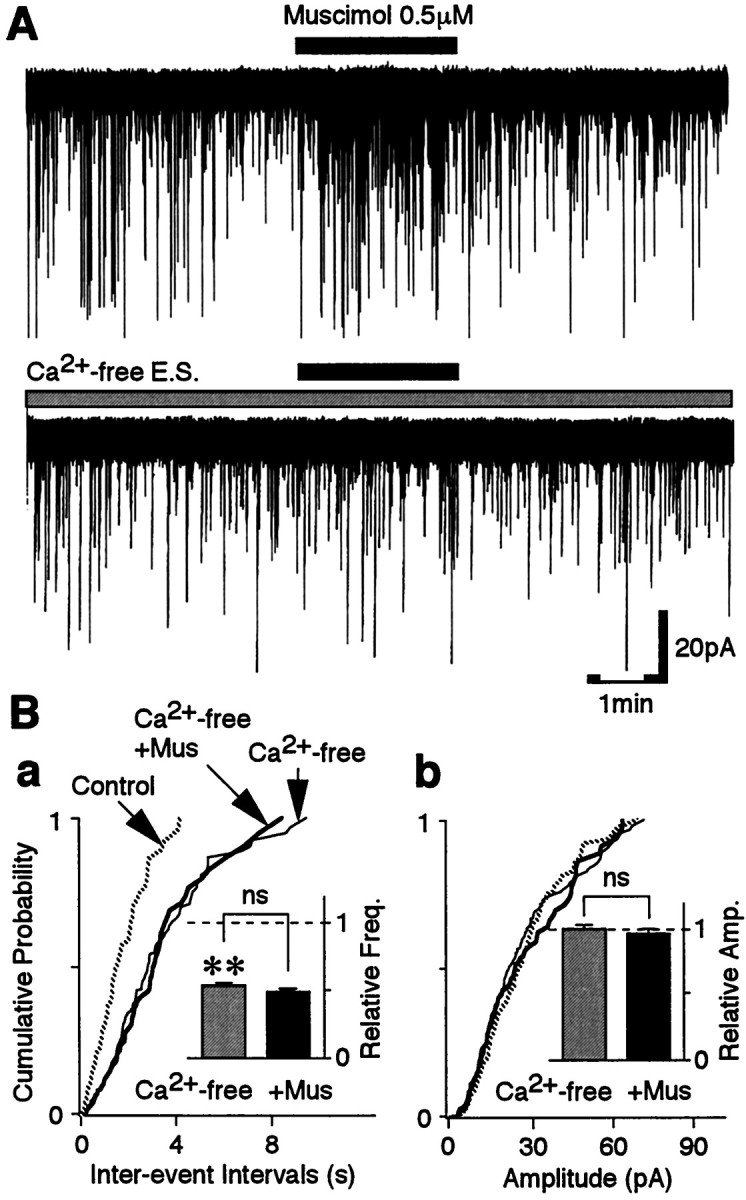
Muscimol-induced facilitation of sEPSC frequency is occluded in the Ca2+-free external solution.A, Typical traces of sEPSCs observed before, during, and after application of 0.5 μm muscimol in the control solution (top trace) and in the Ca2+-free external solution (E. S.; bottom trace). B, Cumulative distributions of interevent intervals (a;p = 0.399) and current amplitudes (b; p = 0.165) for sEPSCs recorded from the same neuron shown in A (398 events for the control, 206 events for Ca2+-free, and 75 events for muscimol). Insets, The mean sEPSC frequency (a) and amplitude (b) from five neurons. All columns were normalized to the control (dotted lines).
Because muscimol-induced sEPSC facilitation requires Ca2+ influxes from the extracellular site, we tested whether the activation of VDCCs is responsible for this Ca2+ influx. Cd2+ (100 μm), a general high-threshold VDCC blocker, significantly reduced sEPSC frequency to 59.15 ± 7.84% of the control (p < 0.05;n = 5; data not shown). In the presence of Cd2+, the facilitatory action of muscimol on sEPSC frequency was completely abolished (103.87 ± 11.24% of the Cd2+ condition; data not shown). However, because Cd2+ is known to block the GABAA response (Kumamoto and Murata, 1995;Fisher and Macdonald, 1998), we tested further the effect of a range of more specific VDCC antagonists, including ω-CgTx (N-type blocker), ω-AgTx (P/Q-type blocker), and nicardipine (L-type blocker), on the muscimol-induced sEPSC facilitation. Exposure of the mixture of these three antagonists (3 μmω-CgTx, 0.3 μm ω-AgTx, and 3 μm nicardipine) to VMH neurons decreased sEPSC frequency to 55.42 ± 6.46% of the control (p < 0.05; n = 5) without affecting the distribution of sEPSC current amplitudes (Fig.6B,C). In these conditions, muscimol again failed to facilitate sEPSC frequency (n = 5) (Fig. 6A,Ba,Ca).
Fig. 6.
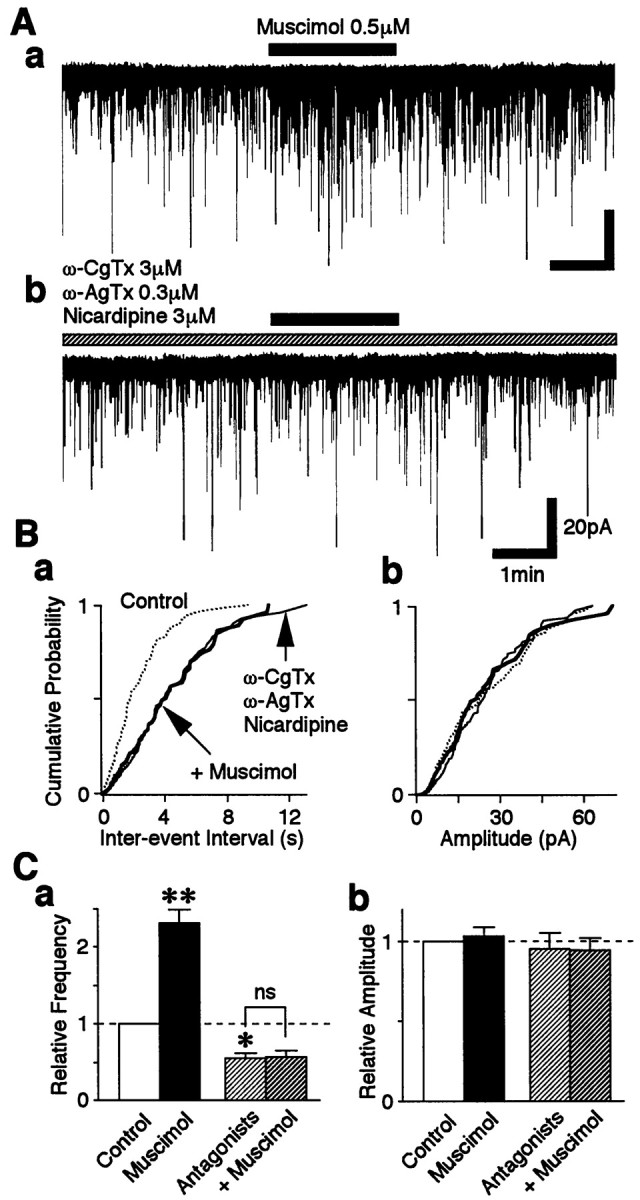
Muscimol-induced presynaptic depolarization activates high-threshold VDCCs. A, Typicaltraces of sEPSCs observed before, during, and after application of 0.5 μm muscimol in the control solution (a) and in the presence of 3 μmω-CgTx, 0.3 μm ω-AgTx, and 3 μmnicardipine (b). B, Cumulative distributions of interevent intervals (a;p = 0.399) and current amplitudes (b; p = 0.165) in the same neuron shown in A (436 events for the control, 233 events for VDCC antagonists, and 83 events for muscimol). C, The mean sEPSC frequency (a) and amplitude (b) of five neurons. All columnswere normalized to the control (dotted lines).
Next, we tested whether muscimol action on sEPSC frequency depends on a direct activation of VDCCs or is secondary to activation of voltage-dependent Na+ channels. In 12 of 14 neurons tested, 0.3 μm TTX completely abolished the facilitatory effect of 0.5 μm muscimol on sEPSC frequency (n = 12) (Fig.7A,B), suggesting that TTX-sensitive Na+ channels are responsible for the VDCC activation. In two neurons, however, muscimol somewhat increased sEPSC frequency despite the presence of TTX, although its facilitatory effect was reduced (data not shown). The variability of the TTX effect on muscimol-induced sEPSC facilitation did not seem to be related to the lower concentration of muscimol used because even the facilitatory effect of 50 μm muscimol was absent in the presence of 0.3 μm TTX (n = 4; data not shown). TTX itself decreased sEPSC frequency to 65.7 ± 6.1% of the control (p < 0.05; n = 12) (Fig.7B) without affecting the distribution of sEPSC current amplitudes, indicating that TTX-sensitive Na+ channels also contribute to the generation of sEPSCs.
Fig. 7.
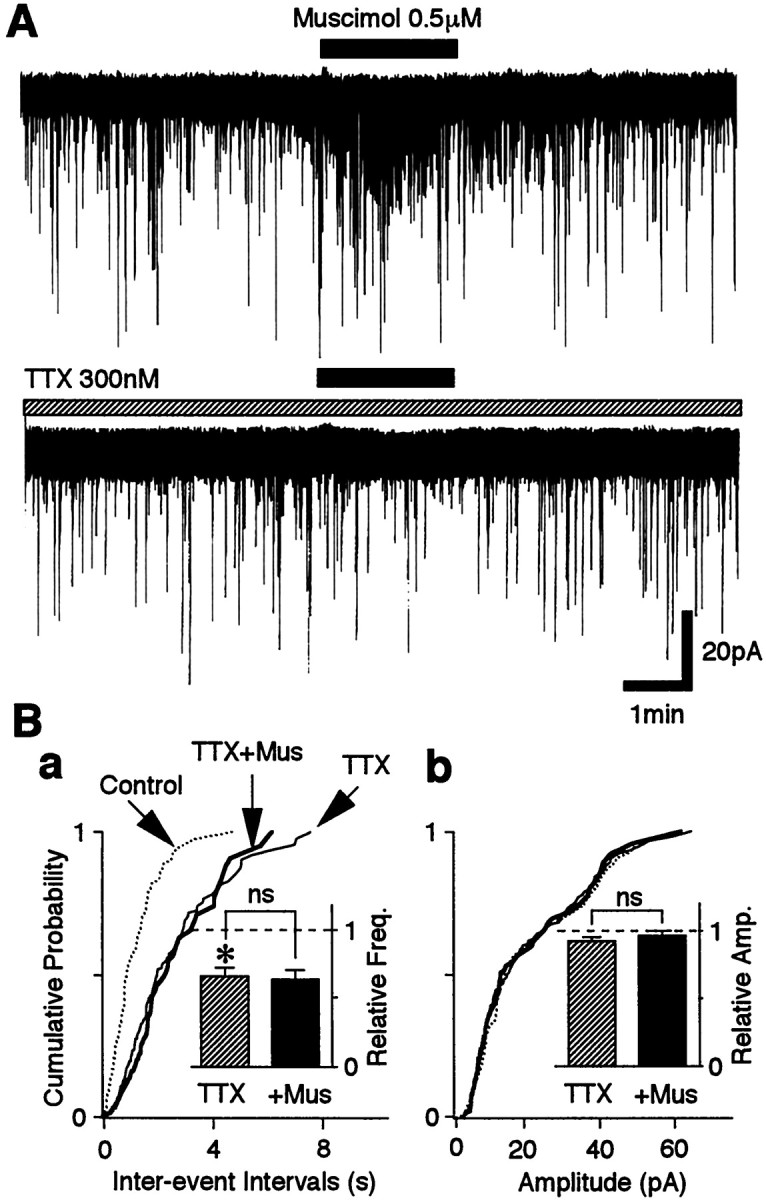
Muscimol-induced facilitation of sEPSC frequency is occluded in the presence of TTX. A, Typicaltraces of sEPSCs observed before, during, and after application of 0.5 μm muscimol in the control solution (top trace) and in the presence of 300 nmTTX (bottom trace). B, Cumulative distributions of interevent intervals (a;p = 0.282) and current amplitudes (b; p = 0.490) recorded from the same neuron shown in A (421 events for the control, 248 events for TTX, and 89 events for muscimol). Insets, The mean sEPSC frequency (a) and amplitude (b) from 12 neurons. All columnswere normalized to the control (dotted lines).
GABAA receptor-mediated presynaptic depolarization
Because muscimol-induced sEPSC facilitation depends on the activation of Na+ channels, it suggests that the activation of presynaptic GABAAreceptors depolarizes the glutamatergic nerve terminals. This indicates that the intraterminal [Cl−]i might be maintained higher than predicted for a passive distribution. If this hypothesis is true, the higher [Cl−]i might be established by some inwardly directed Cl−transport systems, such as the NKCC, the Cl−– HCO exchanger, and the Na+-dependent Cl−– HCO exchanger (Alvarez-Leefmans, 1990; Kaila, 1994; Plotkin et al., 1997;Clayton et al., 1998; Russell, 2000). In immature CNS neurons and sensory neurons, NKCC has a pivotal role in generating higher [Cl−]i and subsequent GABA-induced depolarization (Alvarez-Leefmans, 1990;Russell, 2000). To investigate the role of NKCC, we examined the effect of bumetanide, an NKCC blocker (Haas, 1989; Xu et al., 1994), on the muscimol-induced facilitation of sEPSC frequency. At low concentrations (∼10 μm), bumetanide is specific for NKCC (Russell, 2000).
As shown in Figure 4B, the facilitatory action of muscimol (0.5 μm) on sEPSC frequency was reproduced during repeated applications of muscimol. In contrast, in the continued presence of bumetanide (10 μm), the facilitatory effect of muscimol on sEPSC frequency was gradually attenuated (Fig. 8A). The muscimol-induced facilitation was significantly reduced from 280.3 ± 30.3% facilitation observed in response to the first application to 181.1 ± 13.7 and 140.3 ± 17.3% for the second and third applications, respectively (p< 0.05; n = 6) (Fig. 8Ab). It should be noted, however, that the first application of muscimol in the presence of bumetanide induced nearly the same facilitatory effect as that observed in the absence of bumetanide (Fig. 8B). It is unlikely that the lack of effect is caused by a short bumetanide preincubation time, because even the first application of muscimol after longer (>10 min) preincubation with bumetanide induced nearly the same facilitatory effect (n = 4; data not shown). On the other hand, the muscimol facilitation slowly recovered after washing out of bumetanide (151.9 ± 16.9 and 213.6 ± 19.2% for 8 and 18 min, respectively) (Fig. 8A). However, the effects of bumetanide were not complicated by direct GABAA receptor blockade, because 10 μm bumetanide did not influence the GABAergic sIPSC amplitude (Fig. 8B).
Fig. 8.
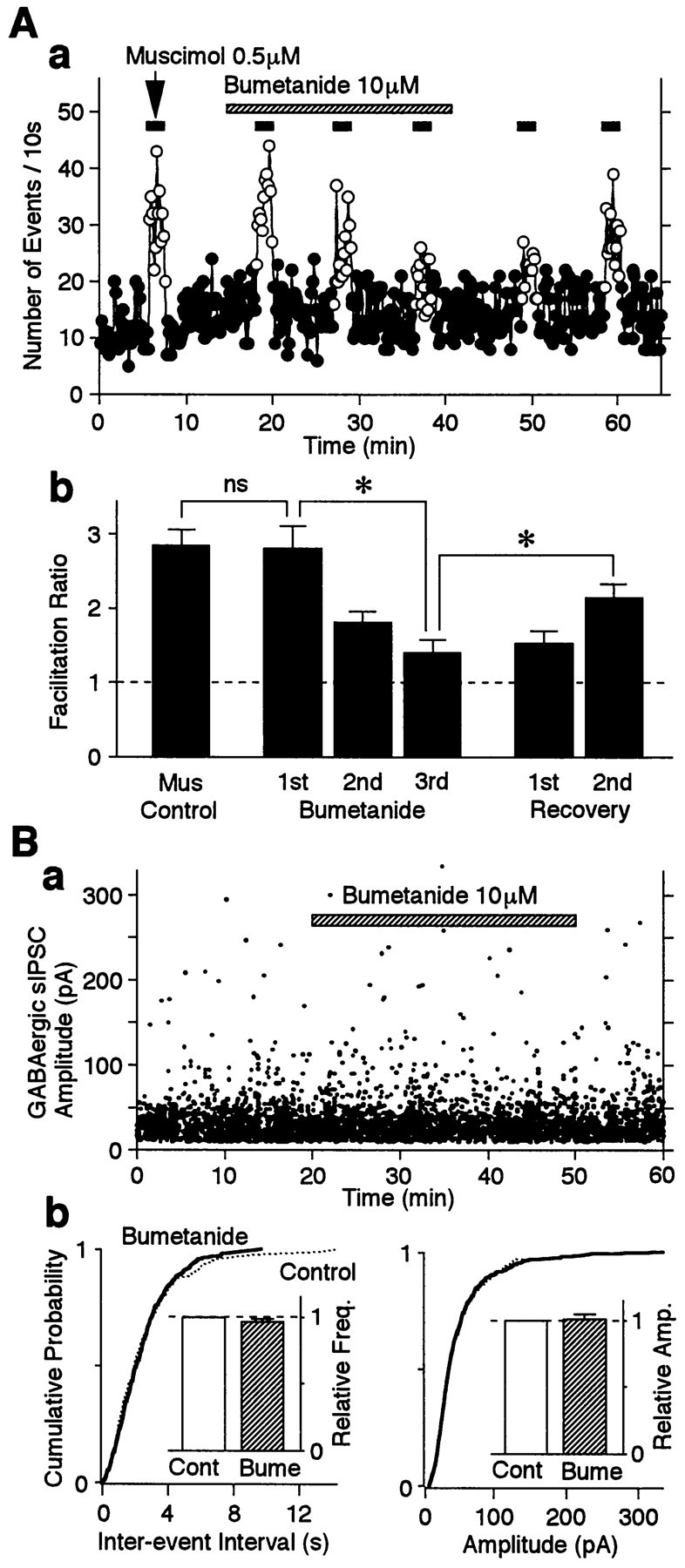
Bumetanide attenuates muscimol-induced sEPSC frequency facilitation without affecting the GABAAresponse. A, a, Typical time course of sEPSC frequency before, during, and after repeated applications of 0.5 μmmuscimol in the absence and presence of 10 μm bumetanide as indicated. The number of events in every 10 sec period was summed and plotted. Note that the muscimol-induced facilitation of sEPSC frequency was gradually reduced in the presence of bumetanide.b, Muscimol-induced facilitation ratios in the absence and presence of bumetanide. All columns are the mean of six neurons and normalized to the control (dotted line). B, The effect of bumetanide on GABAergic sIPSCs in the absence and presence of 10 μmbumetanide as indicated. The pipette solution contained 4 mm Mg-ATP. a, A typical scatter plot for all sIPSCs recorded at a VH of 0 mV. b, Cumulative distributions of sEPSC interevent intervals (p = 0.392) and current amplitudes (p = 0.784) recorded from the same neuron shown in B, a (1251 events for the control and 1861 events for bumetanide). Insets, The mean sEPSC frequency (left) and amplitude (right) from four neurons. All columns were normalized to the control (dotted lines). Bume, Bumetanide.
The effects of a higher concentration of muscimol on sEPSC frequency were also tested in the presence of bumetanide. The facilitatory effects of 10 μm muscimol on sEPSC facilitation were also sensitive to bumetanide and seemed to be more rapidly attenuated (210.3 ± 19.0, 121.4 ± 15.3, and 104.6 ± 7.6% facilitation for the first, second, and third applications, respectively) than was observed with the lower muscimol concentration. The facilitatory action of the higher muscimol concentration also recovered more slowly after washout of bumetanide (118.7 ± 9.8 and 155.4 ± 10.7% facilitation for 8 and 18 min, respectively;n = 5) (Fig.9A). In addition, 300 μm furosemide, a less potent NKCC blocker, mimicked the action of bumetanide on muscimol-induced facilitation of sEPSC frequency (n = 4) (Fig. 9B). The results clearly indicate that functional NKCC exists on these glutamatergic nerve terminals and plays an important role in the maintenance of high [Cl−]i.
Fig. 9.
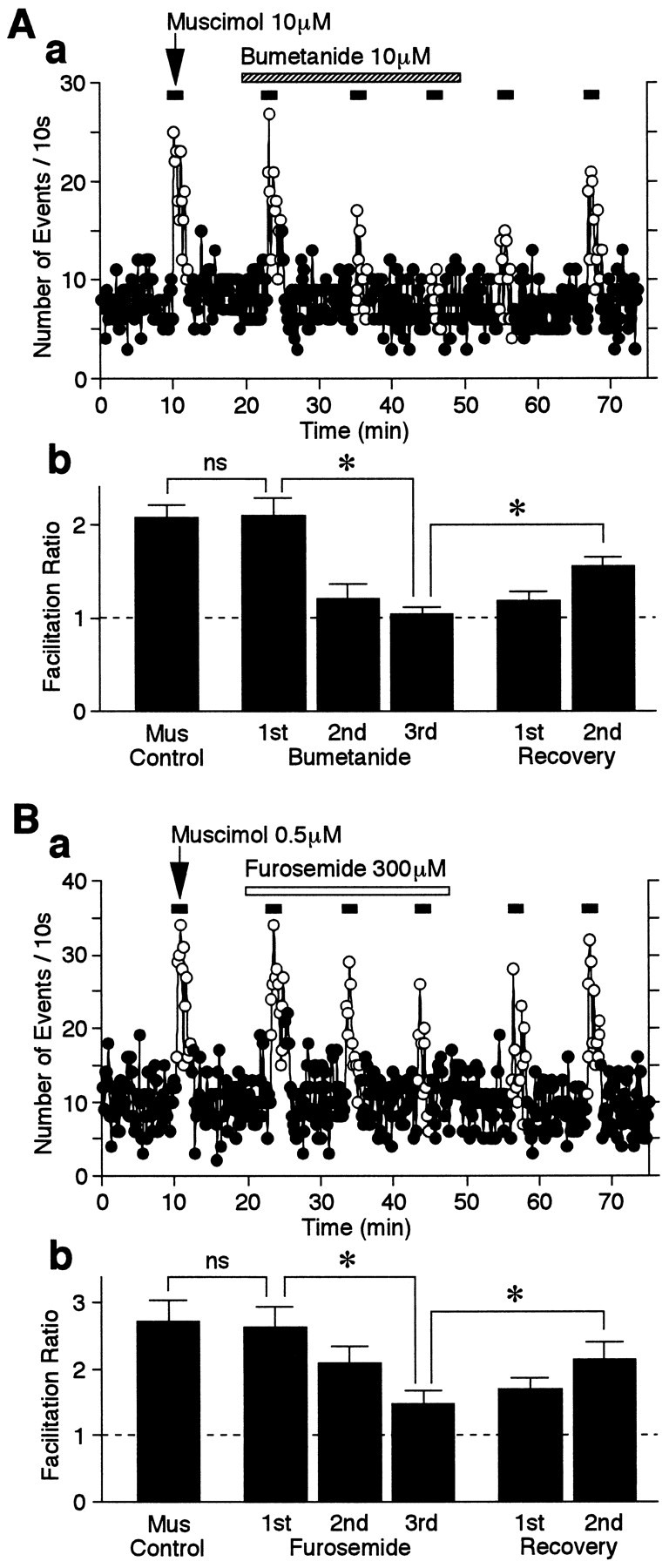
Effect of NKCC blockers on muscimol-induced facilitation of sEPSC frequency. A, a, Typical time course of sEPSC frequency before, during, and after repeated applications of 10 μm muscimol in the absence and presence of 10 μm bumetanide as indicated. The number of events in every 10 sec period was summed and plotted. Note that 10 μm muscimol-induced facilitation of sEPSC frequency was more quickly attenuated than was that of 0.5 μm muscimol (compare Fig. 8A). b, Muscimol-induced facilitation ratios in the absence and presence of bumetanide. All columns are the mean of five neurons and normalized to the control (dotted line). B, a, Typical time course of sEPSC frequency before, during, and after repeated applications of 0.5 μm muscimol in the absence and presence of 300 μm furosemide as indicated. The number of events in every 10 sec period was summed and plotted.b, Muscimol-induced facilitation ratios in the absence and presence of furosemide. All columns are the mean of four neurons and normalized to the control (dotted line).
The effect of muscimol on eEPSCs
We also examined whether muscimol affects electrically evoked and spontaneous release from the excitatory amygdala neurons that synapse onto VMH neurons in the slice preparation This amygdala–VMH connection has been well documented both morphologically (McBride and Sutin, 1977) and electrophysiologically (Renaud, 1976). Muscimol (0.5 μm) slightly decreased eEPSC amplitude to 84.3 ± 6.7% of the control (p < 0.05;n = 4) (Fig.10A), whereas it facilitated sEPSC frequency (Fig. 10Ba). The decrease in eEPSC amplitude does not seem to be caused by postsynaptic effects, because the distribution of sEPSC amplitudes did not change during the application of muscimol (Fig. 10Bb). At a higher concentration (5 μm), muscimol further decreased eEPSC amplitude, but it also slightly decreased sEPSC amplitude (n = 4; data not shown).
Fig. 10.

GABAA receptor-mediated presynaptic depolarization induces presynaptic inhibition of evoked glutamate release. All recordings were obtained using a slice preparation.A, The effect of muscimol on eEPSCs. a, A time course of eEPSC amplitude before, during (closed circles), and after the application of 0.5 μmmuscimol (n = 3). b, The absolute changes in eEPSC amplitude in response to muscimol.Open circles represent the individual results from four experiments, whereas the closed circles and error bars indicate the mean ± SEM. Insets, Typicaltraces of eEPSCs in the control condition and in the presence of muscimol. B, Cumulative probability plots of sEPSC interevent intervals (a) and amplitudes (b). Note that the cumulative distribution of sEPSC amplitudes was not changed by adding 0.5 μmmuscimol, indicating that no change in the postsynaptic membrane properties occurred.
DISCUSSION
GABA receptors on the excitatory presynaptic nerve terminals
GABA activates three classes of ligand-operated receptors, GABAA, GABAB, and GABAC receptors. In the present study, GABA strongly facilitated sEPSC frequency in the presence of the GABAB receptor antagonist. Furthermore, this facilitation was completely and reversibly blocked by bicuculline, a selective GABAA receptor antagonist, and mimicked by muscimol, a GABAAreceptor agonist. GABAC receptor participation appeared to be negligible. In contrast, GABABreceptor activation by baclofen reduced sEPSC frequency. The results clearly indicate that functional GABAA and GABAB receptors modulate the probability of spontaneous glutamate release onto VMH neurons.
As the concentration of muscimol was increased from 0.5 to 50 μm, there was no further facilitation of sEPSC frequency. One possibility is that muscimol at high concentrations might actually change the relative Cl− concentrations, e.g., by reducing intraterminal Cl−, so that ECl is no longer so depolarizing during the prolonged agonist application (see also below). Alternatively, muscimol at lower concentrations induces a mild presynaptic depolarization, which elicits repeated Na+ spikes during the agonist application. At a high concentration, muscimol induces a greater depolarization, which only transiently increases Na+ spike frequency and is followed by inactivation of Na+ channels, thereby attenuating the muscimol-induced sEPSC facilitation over the period of agonist application.
GABA-induced presynaptic depolarization activates both Na+ and Ca2+ channels
In immature rat hypothalamic nerve cell bodies, GABAA-mediated depolarization, which arises fromEGABA being positive to the resting membrane potential, plays an excitatory role by directly evoking APs (Chen et al., 1996). In immortalized hypothalamic (GT1–7) neurons, GABA evokes APs after the activation of Ca2+ channels, which increases [Ca2+]i (Hales et al., 1994). In rat embryonic and neonatal cortical slices, GABAA receptor-mediated spontaneous increases of [Ca2+]i are abolished by TTX (Owens et al., 1996). Additionally, GABAA receptor-mediated depolarization directly increases [Ca2+]ieven after blockade of TTX-sensitive Na+channels in some developing neuronal systems (Yuste and Katz, 1991;Horvath et al., 1993; Reichling et al., 1994). Thus, GABAA receptor-mediated depolarization is thought to exceed the threshold for the activation of both Na+ channels and VDCCs.
In the present study, muscimol-induced sEPSC facilitation was completely blocked by TTX in most neurons tested, suggesting that muscimol-induced depolarization might directly contribute to the activation of voltage-dependent Na+channels in the presynaptic nerve terminals. Muscimol-induced sEPSC facilitation was also abolished in the Ca2+-free external solution and in the presence of a solution containing either Cd2+ or a mixture of high-threshold VDCC antagonists. Together, these results strongly suggest that the activation of GABAA receptors facilitates spontaneous release of glutamate by activating TTX-sensitive Na+ channels, followed by the opening of VDCCs and the subsequent extracellular Ca2+ influx. Occasionally, however, muscimol-induced sEPSC facilitation occurred even in the presence of TTX. In these cases, muscimol-induced depolarization may be sufficiently large enough to activate VDCCs directly without the involvement of Na+ channels.
GABA-induced presynaptic depolarization and NKCC
Our results suggest that, in hypothalamic neurons, the [Cl−]i of glutamatergic nerve terminals is maintained higher than predicted for a passive distribution. This would require the presence of an inwardly directed Cl− transport system, such as the NKCC, the Cl−– HCO exchanger, and the Na+-dependent Cl−– HCO exchanger (Alvarez-Leefmans, 1990; Kaila, 1994; Russell, 2000). However, because HEPES-buffered external solution (rather than a bicarbonate buffer) was used in the present study, the influences of both the Cl−– HCO exchanger and the Na+-dependent Cl−– HCO exchanger should be negligible. Of the two known types of NKCC, NKCC-1 is detected at significant levels in the brain and expressed maximally by postnatal days 7–14 (Plotkin et al., 1997; Clayton et al., 1998). Thus, NKCC-1 may be responsible for generating this intracellular accumulation of [Cl−]i, thereby creating an outwardly directed Cl−driving force and the GABA-induced presynaptic depolarization.
In the present study, this possibility was tested using a pharmacological approach. Bumetanide (10 μm) gradually attenuated muscimol-induced sEPSC facilitation. It was unlikely that a direct blockade of GABAA receptors contributed to this, because 10 μm bumetanide did not change either amplitude or frequency of spontaneous GABAergic events. Furthermore, at a higher concentration of muscimol, muscimol-induced sEPSC facilitation was more quickly attenuated in the presence of bumetanide. These results suggest that muscimol reduces the magnitude of the presynaptic terminal [Cl−] gradient and that the restoration of this gradient is dependent on the activity of NKCC. Higher concentrations of muscimol may more potently dissipate the Cl− gradient, and hence attenuation of the muscimol-induced sEPSC facilitation by bumetanide was more rapid and potent. Furosemide also mimicked the bumetanide action on muscimol-induced sEPSC facilitation. In conclusion, our pharmacological results suggest that presynaptic NKCCs generate an outwardly directed Cl− driving force for GABAA receptor-mediated presynaptic depolarization. It should also be noted that muscimol induced a facilitatory effect in response to the first muscimol application in the presence of bumetanide that was similar to the control. Such results are consistent with the previous finding (Xu et al., 1994) that a decrease of [Cl−]i is an important stimulus for the activation of bumetanide-sensitive NKCCs. Our data are clearly consistent with the presence of functioning NKCCs on the presynaptic nerve terminals, although more direct evidence demonstrating the localization of NKCCs on the presynaptic nerve terminals using an immunohistochemical study with an NKCC-specific antibody would be useful.
The K–Cl cotransporter is a major Cl− extrusion mechanism the increased expression levels of which during the second week of postnatal development have been shown to contribute to the conversion of GABA-induced postsynaptic responses from depolarization to hyperpolarization (Kakazu et al., 1999; Rivera et al., 1999; Vu et al., 2000). It would also be interesting to determine whether this outwardly directed Cl− transporter can also regulate presynaptic nerve terminal [Cl−]i during neuronal development.
Physiological implications
Most axoaxonic GABAergic synapses, which represent the morphological substrate of presynaptic inhibition, have been found on the terminal arbor between the axon and the output synapses as well as close to the presynaptic release sites (Atwood et al., 1984;Lamotte d'Incamps et al., 1998; Cattaert and El Manira, 1999). In the crayfish locomotor system, in which PADs do not directly act on the transmitter release machinery, the activation of presynaptic GABAA receptors reduces transmitter release at the presynaptic nerve terminals by decreasing the amplitude of action potentials (Cattaert and El Manira, 1999). These studies suggest that presynaptic inhibition because of the activation of presynaptic GABAA receptors acts as one of the inhibitory feedback systems involved in the regulation of neuronal excitability. Despite these previous findings, the present study clearly shows that GABAA receptor-mediated presynaptic depolarization can facilitate spontaneous glutamate release. In the slice preparation, muscimol again increased spontaneous release but also slightly decreased eEPSC amplitude. However, it is still unclear whether at higher concentrations muscimol-induced presynaptic depolarization induces presynaptic inhibition for evoked release. Further studies need to address the relationship between the presynaptic GABAA receptor activation and presynaptic inhibition at these synapses.
GABA is closely related to VMH function. Experimental manipulations of GABAergic systems influence many functions that are, in part, regulated via the VMH, including reproductive functions and behavior (McCarthy, 1995), autonomic functions (Takenaka et al., 1995), and feeding behaviors (Dube et al., 1995). In addition, a recent study has suggested that intrinsic GABA within the VMH directly influences the embryonic development and organization of the VMH (Tobet et al., 1999). Thus, GABA plays a pivotal role in both the development and the regulation of VMH functions. Consequently, the present results suggest that GABAergic modulation of spontaneous glutamatergic transmission may contribute to the tropic and trophic roles of GABA in VMH function.
Footnotes
This study was supported by Grants-in-Aid for Scientific Research, The Ministry of Education, Science, and Culture, Japan (Grant 13307003), The Japan Health Sciences Foundation (Grant 21279, Research on Brain Science), and Kyushu University Interdisciplinary Programs in Education and Projects in Research Development (N.A.). We thank Dr. K. Kaila for his valuable comments and Dr. A. Moorhouse for critically reading this manuscript and correcting the English.
Correspondence should be addressed to Dr. Norio Akaike, Cellular and System Physiology, Graduate School of Medical Sciences, Kyushu University, Maidashi 3-1-1, Fukuoka 812-8582, Japan. E-mail:akaike@physiol2.med.kyushu-u.ac.jp.
REFERENCES
- 1.Akaike N. Time-dependent rundown of GABA response in mammalian CNS neuron during experimental anoxia. Obes Res. 1995;5:769S–777S. doi: 10.1002/j.1550-8528.1995.tb00498.x. [DOI] [PubMed] [Google Scholar]
- 2.Akaike N, Harata N. Nystatin perforated patch recording and its application to analysis of intracellular mechanism. Jpn J Physiol. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez-Leefmans FJ. Intracellular Cl− regulation and synaptic inhibition in vertebrate and invertebrate neurons. In: Alvarez-Leefmans FJ, Russell JM, editors. Chloride channels and carriers in nerve, muscle, and glial cells. Plenum; New York: 1990. pp. 109–158. [Google Scholar]
- 4.Atwood HL, Stevens JK, Marin L. Axoaxonal synapse location and consequences for presynaptic inhibition in crustacean motor axon terminals. J Comp Neurol. 1984;225:64–74. doi: 10.1002/cne.902250108. [DOI] [PubMed] [Google Scholar]
- 5.Bading H, Ginty D, Greenberg M. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 6.Cattaert D, El Manira A. Shunting versus inactivation: analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish. J Neurosci. 1999;19:6079–6089. doi: 10.1523/JNEUROSCI.19-14-06079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cattaert D, El Manira A, Clarac F. Direct evidence for presynaptic inhibitory mechanisms in crayfish sensory afferents. J Neurophysiol. 1992;67:610–624. doi: 10.1152/jn.1992.67.3.610. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Trombley PQ, van den Pol AN. Excitatory actions of GABA in developing rat hypothalamic neurons. J Physiol (Lond) 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Res Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- 10.Decavel C, van den Pol AN. GABA: a dominant transmitter in the hypothalamus. J Comp Neurol. 1990a;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- 11.Decavel C, van den Pol AN. Converging GABA- and glutamate-immunoreactive axons make synaptic contact with identified hypothalamic neurosecretory neurons. J Comp Neurol. 1990b;316:104–116. doi: 10.1002/cne.903160109. [DOI] [PubMed] [Google Scholar]
- 12.Dube MG, Kalra PS, Crowley WR, Kalra SP. Evidence of a physiological role for neuropeptide Y in ventromedial hypothalamic lesion-induced hyperphagia. Brain Res. 1995;690:275–278. doi: 10.1016/0006-8993(95)00644-6. [DOI] [PubMed] [Google Scholar]
- 13.Dyball REJ, Shaw FD. Inhibition by GABA of hormone release from the neurohypophysis in the cat. J Physiol (Lond) 1978;283:78–79. [PubMed] [Google Scholar]
- 14.Fisher JL, Macdonald RL. The role of an α subtype M2–M3 His in regulating inhibition of GABAA receptor current by zinc and other divalent cations. J Neurosci. 1998;18:2944–2953. doi: 10.1523/JNEUROSCI.18-08-02944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham B, Redman SA. Simulation of action potentials in synaptic boutons during presynaptic inhibition. J Neurophysiol. 1994;71:538–549. doi: 10.1152/jn.1994.71.2.538. [DOI] [PubMed] [Google Scholar]
- 16.Haas M. Properties and diversity of (Na-K-Cl) cotransporters. Annu Rev Physiol. 1989;51:443–457. doi: 10.1146/annurev.ph.51.030189.002303. [DOI] [PubMed] [Google Scholar]
- 17.Hales TG, Sanderson MJ, Charles AC. GABA has excitatory actions on GnRH-secreting immortalized hypothalamic (GT1–7) neurons. Neuroendocrinology. 1994;59:297–308. doi: 10.1159/000126671. [DOI] [PubMed] [Google Scholar]
- 18.Harata N, Wu J, Ishibashi H, Ono K, Akaike N. Run-down of the GABAA response under experimental ischaemia in acutely dissociated CA1 pyramidal neurones of the rat. J Physiol (Lond) 1997;500:673–688. doi: 10.1113/jphysiol.1997.sp022052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvath G, Acs Z, Mergl Z, Nagy I, Makara GB. Gamma-aminobutyric acid-induced elevation of intracellular calcium concentration in pituitary cells of neonatal rats. Neuroendocrinology. 1993;57:1028–1034. doi: 10.1159/000126467. [DOI] [PubMed] [Google Scholar]
- 20.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 21.Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumamoto E, Murata Y. Characterization of GABA current in rat septal cholinergic neurons in culture and its modulation by metal cations. J Neurophysiol. 1995;74:2012–2027. doi: 10.1152/jn.1995.74.5.2012. [DOI] [PubMed] [Google Scholar]
- 23.Lamotte d'Incamps B, Destombes J, Thiesson D, Hellio R, Lasserre X, Kouchtir-Devanne N, Jami L, Zytnicki D. Indications for GABA-immunoreactive axo-axonic contacts on the intraspinal arborization of a Ib fiber in cat: a confocal microscope study. J Neurosci. 1998;18:10030–10036. doi: 10.1523/JNEUROSCI.18-23-10030.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat hippocampal slices. J Physiol (Lond) 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy RA. The role of GABA in primary afferent depolarization. Prog Neurobiol. 1977;9:211–267. doi: 10.1016/0301-0082(77)90002-8. [DOI] [PubMed] [Google Scholar]
- 26.Mattson MP, Kater SB. Calcium regulation of neurite elongation and growth cone motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McBride RL, Sutin J. Amygdaloid and pontine projections to the ventromedial nucleus of the hypothalamus. J Comp Neurol. 1977;174:377–396. doi: 10.1002/cne.901740302. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy MM. Functional significance of steroid modulation of GABAergic neuro-transmission: analysis at the behavioral, cellular, and molecular levels. Horm Behav. 1995;29:131–140. doi: 10.1006/hbeh.1995.1010. [DOI] [PubMed] [Google Scholar]
- 29.Nicoll RA, Alger BE. Presynaptic inhibition: transmitter and ionic mechanism. Int Rev Neurobiol. 1979;21:217–258. doi: 10.1016/s0074-7742(08)60639-x. [DOI] [PubMed] [Google Scholar]
- 30.Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obrietan K, van den Pol AN. Growth cone calcium elevation by GABA. J Comp Neurol. 1996;372:167–175. doi: 10.1002/(SICI)1096-9861(19960819)372:2<167::AID-CNE1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Owens DF, Boyce LH, Davis BE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickles HG. Presynaptic γ-aminobutyric acid responses in the olfactory cortex. Br J Pharmacol. 1979;65:223–228. doi: 10.1111/j.1476-5381.1979.tb07822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brain: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Reichling DB, Kyrozis A, Wang J, MacDermott AB. Mechanisms of GABA and glycine depolarization-induced calcium transients in rat dorsal horn neurons. J Physiol (Lond) 1994;476:411–421. doi: 10.1113/jphysiol.1994.sp020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renaud LP. An electrophysiological study of amygdalohypothalamic projections to the ventromedial nucleus of the rat. Brain Res. 1976;105:45–58. doi: 10.1016/0006-8993(76)90921-5. [DOI] [PubMed] [Google Scholar]
- 37.Rhee JS, Ishibashi H, Akaike N. Calcium channels in the GABAergic presynaptic nerve terminals projecting to Meynert neurons of the rat. J Neurochem. 1999;72:800–807. doi: 10.1046/j.1471-4159.1999.0720800.x. [DOI] [PubMed] [Google Scholar]
- 38.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 39.Rudomin P. Presynaptic inhibition of muscle spindle and tendon organ afferents in the mammalian spinal cord. Trends Neurosci. 1990;13:499–505. doi: 10.1016/0166-2236(90)90084-n. [DOI] [PubMed] [Google Scholar]
- 40.Rudomin P, Schmidt RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res. 1999;129:1–37. doi: 10.1007/s002210050933. [DOI] [PubMed] [Google Scholar]
- 41.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 42.Saridaki E, Carter DA, Lightman SL. γ-Aminobutyric acid regulation of neurohypophysial hormone secretion in male and female rats. J Endocrinol. 1989;121:343–349. doi: 10.1677/joe.0.1210343. [DOI] [PubMed] [Google Scholar]
- 43.Segev I. Computer study of presynaptic inhibition controlling the spread of action potentials into axon terminals. J Neurophysiol. 1990;63:987–998. doi: 10.1152/jn.1990.63.5.987. [DOI] [PubMed] [Google Scholar]
- 44.Shirasaki T, Aibara K, Akaike N. Direct modulation of GABAA receptor by ATP in dissociated nucleus tractus solitarii neurones of rat. J Physiol (Lond) 1992;449:551–572. doi: 10.1113/jphysiol.1992.sp019101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tachibana M, Kaneko A. γ-Aminobutyric acid exerts a local inhibitory action on the axon terminal of bipolar cells: evidence for negative feedback from amacrine cells. Proc Natl Acad Sci USA. 1987;84:3501–3505. doi: 10.1073/pnas.84.10.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takenaka K, Sasaki S, Nakamura K, Uchida A, Fujita H, Itoh H, Nakata T, Takeda K, Nakagawa M. Hypothalamic and medullary GABAA and GABAB-ergic systems differently regulate sympathetic and cardiovascular systems. Clin Exp Pharmacol Physiol. 1995;22:S48–S50. doi: 10.1111/j.1440-1681.1995.tb02966.x. [DOI] [PubMed] [Google Scholar]
- 47.Tobet SA, Henderson RG, Whiting PJ, Sieghart W. Special relationship of γ-aminobutyric acid to the ventromedial nucleus of the hypothalamus during embryonic development. J Comp Neurol. 1999;405:88–98. doi: 10.1002/(sici)1096-9861(19990301)405:1<88::aid-cne7>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 48.Vaccarino F, Haywark M, Nestler E, Duman R, Tallman J. Dif- ferential induction of immediate early genes by excitatory amino acid receptor types in primary cultures of cortical and striatal neurons. Mol Brain Res. 1992;12:233–241. doi: 10.1016/0169-328x(92)90089-t. [DOI] [PubMed] [Google Scholar]
- 49.Vu TQ, Payne JA, Copenhagen DR. Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. J Neurosci. 2000;20:1414–1423. doi: 10.1523/JNEUROSCI.20-04-01414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J-C, Lytle C, Zhu TT, Payne JA, Benz E., Jr Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci USA. 1994;91:2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuste R, Katz LC. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:333–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]
- 52.Zhang SJ, Jackson MB. GABAA receptor activation and the excitability of nerve terminals in the rat posterior pituitary. J Physiol (Lond) 1995;483:583–595. doi: 10.1113/jphysiol.1995.sp020608. [DOI] [PMC free article] [PubMed] [Google Scholar]