

Abstract
The activation of group I metabotropic glutamate receptors (mGluRs) produces a variety of actions that lead to alterations in excitability and synaptic transmission in the CA1 region of the hippocampus. The group I mGluRs, mGluR1 and mGluR5, are activated selectively by (S)-3,5-dihydroxyphenylglycine (DHPG). To identify which of these mGluR subtypes are responsible for the various actions of DHPG in area CA1, we took advantage of two novel subtype-selective antagonists. (S)-(+)-α-amino-a-methylbenzeneacetic acid (LY367385) is a potent competitive antagonist that is selective for mGluR1, whereas 2-methyl-6-(phenylethynyl)-pyridine (MPEP) is a potent noncompetitive antagonist that is selective for mGluR5. The use of these compounds in experiments with whole-cell patch-clamp recording and Ca2+-imaging techniques revealed that each group I mGluR subtype plays distinct roles in regulating the function of CA1 pyramidal neurons. The block of mGluR1 by LY367385 suppressed the DHPG-induced increase in intracellular Ca2+concentration ([Ca2+]i) and the direct depolarization of CA1 hippocampal neurons. In addition, the increase in the frequency of spontaneous IPSCs (sIPSCs) caused by the DHPG-induced depolarization of inhibitory interneurons also was blocked by LY367385, as was the DHPG-induced inhibition of transmission at the Schaffer collateral→CA1 synapse. On the other hand, the block of mGluR5 by MPEP antagonized the DHPG-induced suppression of the Ca2+-activated potassium current (IAHP) and potentiation of the NMDA receptor. Finally, antagonism of the DHPG-induced suppression of evoked IPSCs required the blockade of both mGluR1 and mGluR5. These data suggest that mGluR1 and mGluR5 play distinct roles in the regulation of the excitability of hippocampal CA1 pyramidal neurons.
Keywords: mGluR; mGluR1; mGluR5; (S)-3,5-dihydroxyphenylglycine (DHPG); (S)-(+)-α-amino-a-methylbenzeneaceticacid (LY367385); 2-methyl-6-(phenylethynyl)-pyridine (MPEP); IAHP; IPSC; EPSC; hippocampus
Metabotropic glutamate receptors (mGluRs) have been implicated in a number of physiological and pathological responses to glutamate in CA1 hippocampal region. These include the modulation of neuronal excitability and synaptic transmission (for review, see Anwyl, 1999) as well as the induction of long-term potentiation (Bashir et al., 1993), generation of epileptiform activity (Aronica et al., 1997; Merlin, 1999; Galoyan and Merlin, 2000), and postischemic injury (Bond et al., 1999, 2000;Pellegrini-Giampietro et al., 1999). The family of mGluRs is composed of three distinct groups that are based on sequence homology, pharmacology, and coupling to second messenger systems (for review, seeNakanishi, 1992; Schoepp and Conn, 1993; Pin and Duvoisin, 1995). Although all three groups of mGluRs play roles in regulating hippocampal function (Anwyl, 1999), group I mGluRs are especially important for the regulation of pyramidal cell excitability. The group I mGluRs include mGluR1 and mGluR5, both of which are activated selectively by (S)-3,5-dihydroxyphenylglycine (DHPG), and couple to Gq and to the activation of phosphoinositide hydrolysis.
The activation of group I mGluRs has a number of direct excitatory effects on CA1 pyramidal cells, including depolarization and increased cell firing (Charpak et al., 1990; Desai and Conn, 1991; Pedarzani and Storm, 1993; Davies et al., 1995; Gereau and Conn, 1995b; Mannaioni et al., 1999). These effects have been ascribed to the activation of Ca2+-activated and Ca2+-independent cationic conductances (Crepel et al., 1994; Guerineau et al., 1995) and the inhibition of at least four different K+ currents. These include the AHP current (Charpak et al., 1990; Desai and Conn, 1991), a leak current (Guerineau et al., 1994), the M current (Charpak et al., 1990), and a voltage-dependent slow-inactivating current (Luthi et al., 1996). In addition, the activation of group I mGluRs increases CA1 pyramidal cell excitability by decreasing GABA-mediated inhibition (Desai and Conn, 1991; Gereau and Conn, 1995a; Fitzsimonds and Dichter, 1996).
Immunocytochemistry studies reveal that mGluR5 is localized in CA1 pyramidal cells, whereas mGluR1a is not detectable (Baude et al., 1993; Romano et al., 1995). Therefore, mGluR5 is thought to play a predominant role in regulating CA1 pyramidal cells. However, although mGluR5 is the most abundant group I mGluR in CA1 pyramidal cells, these cells also express mGluR1 mRNA (Shigemoto et al., 1992; Berthele et al., 1998), and a recent immunohistochemical study with antibodies that react with all splice forms of mGluR1 revealed low levels of mGluR1 immunoreactivity in this region (Ferraguti et al., 1998). Therefore, it is possible that both mGluR1 and mGluR5 participate in regulating CA1 pyramidal cell function.
We have used new pharmacological tools to determine the physiological roles of mGluR1 and mGluR5 in CA1 pyramidal cells. We report that both mGluR1 and mGluR5 participate in regulating synaptic transmission and pyramidal cell excitability in hippocampal area CA1. However, each of these group I mGluR subtypes has distinct physiological roles and shows little overlap. Thus, although mGluR1 and mGluR5 are highly homologous and display similar effector coupling, these receptors are functionally distinct when present in a single neuronal population.
MATERIALS AND METHODS
Materials.(S)-3,5-dihydroxyphenylglycine (DHPG), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX),d(−)-2-amino-5-phosphonopentanoicacid (d-AP5), (S)-(+)-α-amino-a-methylbenzeneacetic acid (LY367385), and 2-methyl-6-(phenylethynyl)-pyridine (MPEP) were obtained from Tocris (Balwin, MO). QX 314 was purchased from Alomone Laboratories (Jerusalem, Israel). The cell-impermeant fluo-3 pentapotassium salt was obtained from Molecular Probes (Eugene, OR). All other materials were obtained from Sigma (St. Louis, MO).
Electrophysiological recording from rat hippocampal slices.Preparation of hippocampal slices was performed as described previously (Marino et al., 1998). Young rats [Sprague Dawley, age postnatal day (P) P15–P20] were anesthetized deeply with isoflurane and decapitated. The brain was removed rapidly and submerged in an ice-cold artificial CSF (aCSF) of the following composition (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 3 MgCl, and 10 glucose saturated with 95% O2/5% CO2, pH 7.4. The hemisected brain was glued onto the stage of a vibrating microtome (Vibratome series 1000, Ted Pella, Redding, CA), and sections of 300 μm thickness were cut and stored in an incubation chamber at room temperature for ∼1 hr before use. The Emory University Institutional Animal Care and Use Committee approved all procedures.
Conventional blind and visually guided whole-cell patch recordings were obtained from CA1 pyramidal neurons both in voltage-clamp and in current-clamp configuration with an Axopatch 200A (Axon Instruments, Foster City, CA) and a pipette resistance of 5–7 MΩ. The solution used to fill the electrodes and the holding current in the voltage-clamp configuration varied with the different experimental approaches that were used as described below. The standard recording solution was composed of (in mm) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1.5 CaCl2, 1.5 MgCl and 10 glucose saturated with 95% O2/5% CO2, pH 7.4. All neurons included in this study had a resting membrane potential below −55 mV (−58 ± 1.1; n = 125) and an access resistance in the range of 10–20 MΩ that showed only minimal variations during the recordings that were included in this study. Records were filtered at 5 kHz and digitized at 20 kHz with a Digidata 1200 analog-to-digital board. All data were acquired, stored, and analyzed on a PC with the pClamp and Origin software (Axon Instruments and Microcal Software, Northampton, MA, respectively) and the Mini Analysis Program (Synaptosoft, Leonia, NJ). In all of the experiments the drugs were administered by addition to the superfusing medium and were applied for a sufficient period to allow for their full equilibration. All of the data were collected at room temperature (23–26°C).
Imaging of fluo-3 fluorescence. In the calcium-imaging experiments the electrodes were filled with (in mm) 140 K-gluconate, 10 HEPES, 7 NaCl, 4 Mg-ATP, 0.3 Na3-GTP, and 100 μm fluo-3. To eliminate synaptic activity, we added 1 μmtetrodotoxin (TTX) to the aCSF. Throughout the experiments 15 msec, −5 mV voltage steps were applied at 30 sec intervals from a holding potential of −70 mV to monitor the holding current, series resistance, and membrane input resistance continuously. After entering whole-cell mode, the cells were maintained for ∼20 min to allow for filling with fluo-3 before image acquisition. After the baseline period, DHPG was applied, and images were acquired every 5 sec after a 25 msec exposure to 450–490 nm light. Fluorescence was recorded through a bandpass filter (500–550 nm) with a Princeton Micromax camera (Trenton, NJ). Fluorescence intensity was measured in cell bodies by using the Axon Imaging Workbench program (v2.2.1; Axon Instruments) and expressed asF/Fo, whereFo is the fluorescence intensity before drug treatment. Increases in fluorescence >1.2-fold were considered to be real changes. Baseline fluorescence values possessed a peak F/Fo ratio of 1.01 ± 0.01 over a typical experiment (see Fig. 1C,filled bar). Peaks ofF/Fo ratio and of ΔI holding during the DHPG application were used as a measure of the actions of DHPG to remove the variability associated with the time required for DHPG to reach cells at different depths in the slices (see Fig. 1C,D).
Fig. 1.

mGluR1 mediates the DHPG-induced increase in [Ca2+]i and inward currents simultaneously recorded from CA1 hippocampal neurons. A, B, Time course of the effect of 30 μm DHPG alone (left) and in the presence of 10 μm MPEP (middle) or 100 μm LY367385 (right) on fluo-3 fluorescence and on holding current obtained in four different CA1 pyramidal neurons. Eachsymbol represents a different cell. Antagonists and 1 μm TTX were applied for 10 min before the recordings were started. The horizontal bars for drug application inA also apply to B. C, A typical example of mGluR-mediated increase in somatic fluo-3 fluorescence measured in three different CA1 pyramidal neurons. Shown are DHPG alone (left) or in the presence of 10 μm MPEP (middle) or 100 μmLY367385 (right). D–F, Bar graphs expressing the highest values ofF/Fo ratio of fluo-3 fluorescence (D), peak inward current (E), and peak depolarization (F) induced by DHPG application alone or in the presence of 10 μm MPEP or 100–300 μm LY367385. Values are the means ± SEM; n = 4 in Dand E and is indicated in parentheses inF. *p < 0.05; **p < 0.01; Student's t test.
Current-clamp experiments. Electrodes were filled with (in mm) 140 K-gluconate, 10 HEPES, 7 NaCl, 4 Mg-ATP, and 0.3 Na3-GTP. Standard aCSF was used with the addition of 1 μm TTX.
I–V relationship. Electrodes were filled with (in mm) 140 K-gluconate, 10 HEPES, 7 NaCl, 4 Mg-ATP, and 0.3 Na3-GTP. Standard aCSF was used with the addition of (in μm) 10 MPEP, 1 TTX, 10 bicuculline, 25 CNQX, and 50 AP-5. Depolarizing pulses (−10 mV amplitude, 750 msec long) were applied periodically to monitor membrane conductance, and a chart recorder was used to monitor the holding current. The I–Vrelationship was assessed by ramping the membrane potential from +10 to −130 mV (20 mV/sec) before drug application and at the time of maximal DHPG-induced inward current. Voltage-dependent calcium currents were inactivated by holding the membrane potential at +10 mV for 1 sec before initiating the ramp.
Ca2+-activated potassium current (IAHP) measurements. For measurement of IAHP the aCSF contained 0.5 μm TTX, 1 mmtetraethylammonium (TEA), 10 μm bicuculline, and 2.5 mm CaCl2. Electrodes were filled with (in mm) 140 K-methylsulfate, 10 HEPES, 2 Na2-ATP, 3 MgCl2, and 0.4 Na3-GTP.IAHP was elicited once every 60 sec by applying 100–200 msec depolarizing steps to +60 and to +70 mV from a holding potential of −50 mV. This depolarizing step elicits a robust, unclamped Ca2+ action current (Pedarzani and Storm, 1993; Stocker et al., 1999), andIAHP was measured as the outward tail current that immediately followed the depolarizing step. The access resistance and the amplitude and time course of the Ca2+ current during the step were monitored continuously and showed only minimal variations during the recordings that were included in this study.
NMDA-evoked currents. Measurement of NMDA-evoked currents was performed as described previously (Marino et al., 1998; Alagarsamy et al., 1999). NMDA (100 μm) was applied directly above the recording site by a modified U-tube application system. NMDA-evoked currents were recorded from a holding potential of −60 mV in slices bathed in aCSF containing 0.5 μm TTX. The percentage of potentiation was defined by using the ratio of maximum current amplitude during DHPG application (Imax) to the average current amplitude of three trials immediately preceding the drug application (Ibase) in the equation: % potentiation = [((Imax/Ibase) − 1) · 100]. Patch solution was identical to the K-gluconate solution described above except that the K-gluconate was replaced with Cs-methanesulfonate.
Measurement of IPSCs. For recordings of monosynaptic IPSCs and spontaneous IPSCs, electrodes were filled with (in mm) 120 CsCl, 10 HEPES, 4 Mg-ATP, 0.3 Na3-GTP, and 5 QX 314. IPSCs were evoked from a holding potential of −70 mV by stimulation at a frequency of 0.1 Hz through a bipolar stimulating electrode (240 μm spacing; FHC, Bowdoinham, ME). Stimulating electrodes were placed within 100 μm of the patched cell. The ionotropic glutamate receptor blockers AP-5 (50 μm) and CNQX (25 μm) were included in all of the IPSC studies. Because of the high intracellular chloride current, IPSCs were inward currents at −70 mV.
Spontaneous IPSCs were detected automatically. Both the frequency and the peak amplitude of detected events were analyzed. The GABAA receptor blocker bicuculline (10 μm) was added routinely at the end of experiments to verify that both the evoked IPSCs and the spontaneous IPSCs were abolished completely, confirming that they were GABAA receptor-mediated.
Measurement of EPSCs and paired pulse facilitation recording.For the recording of evoked EPSCs and paired pulse facilitation, electrodes were filled with (in mm) 140 K-gluconate, 10 HEPES, 7 NaCl, 4 Mg-ATP, and 0.3 Na3-GTP. Pairs of EPSCs were evoked from a holding potential of −60 mV by stimulation at a frequency of 0.06 Hz with an interpulse interval of 100 msec. Stimuli were delivered through a bipolar stimulating electrode (240 μm spacing; FHC) placed in the stratum radiatum within 100 μm of the patched cell. Standard aCSF was used with the addition of bicuculline (10 μm); a preventive cut between areas CA3 and CA1 was made to avoid recurrent excitation. The AMPA receptor blocker CNQX (25 μm) was added routinely at the end of the experiments to verify that the evoked EPSCs were abolished completely, confirming that they were AMPA receptor-mediated.
Statistical analysis. All numerical data are expressed as means ± SEM. Data were analyzed statistically by paired or unpaired Student's t test or by the Kolmogorov–Smirnov test. A value of p ≤ 0.05 was considered statistically significant.
RESULTS
The highly selective, noncompetitive mGluR5 antagonist MPEP has been shown previously to inhibit recombinant mGluR5 with an IC50 value of 39 nm with no detectable effect on mGluR1-mediated responses at much higher concentrations (Gasparini et al., 1999). The highly selective, competitive mGluR1 antagonist LY367385 has been found to block responses selectively to recombinant mGluR1a with an IC50 value of 8.8 μm and with no detectable effect on mGluR5-mediated responses (Clark et al., 1997). Therefore, these compounds were used to determine which group I mGluR underlies the calcium responses and direct excitatory effects of DHPG onto CA1 hippocampal pyramidal neurons, as well as DHPG-induced modulation of the inhibitory transmission in CA1 hippocampal area.
mGluR1 mediates the DHPG-induced increase in [Ca2+]i and depolarization in CA1 hippocampal neurons
A common response to the activation of group I mGluRs that has been observed in both recombinant and native systems is the release of calcium from internal stores. Consistent with this and with previous studies in hippocampal pyramidal cells (Abe et al., 1992; Pozzo-Miller et al., 1996; Bianchi et al., 1999), DHPG induced a robust increase in fluorescence in CA1 pyramidal cells that were loaded with the calcium-sensitive dye fluo-3. DHPG (30 μm) elicited a robust increase in fluo-3 fluorescence in both the soma (Fig.1A,C, left panels, D) and the dendrites (data not shown) of all of the cells that were examined, suggesting that this treatment increases intracellular [Ca2+]. Also consistent with previous studies (Guerineau et al., 1995), simultaneous voltage-clamp recordings in the same neurons revealed a DHPG-induced inward current in CA1 pyramidal cells (Fig. 1B,left panel, E).
Surprisingly, neither of these effects of DHPG was blocked by maximal concentrations of the mGluR5-selective antagonist MPEP (10 μm) (Fig. 1A–C, middle panels, D,E), suggesting that mGluR5 plays little or no role in these group I-mediated responses. In contrast, the mGluR1-selective antagonist LY367385 (100 μm) completely blocked the DHPG-induced increase in the fluo-3 fluorescence (Fig. 1A,C, right panels, D) and significantly reduced the DHPG-induced inward current (Fig.1B, right panel, E).
The DHPG-induced inward current underlies the previously described 1-aminocyclopentane-1,3-dicarboxylic acid (ACPD) and DHPG-induced depolarization of CA1 pyramidal neurons recorded in current-clamp mode (Desai and Conn, 1991; Gereau and Conn, 1995b; Mannaioni et al., 1999). Figure 1F summarizes the depolarizing effect of 30 μm DHPG application (ΔVm of 4.2 ± 0.3 mV;n = 18) on CA1 pyramidal cells. Consistent with the voltage-clamp studies, DHPG-induced depolarization was not blocked by 10 μm MPEP (ΔVm of 4.6 ± 1.1 mV;n = 10) (Fig. 1F, open bar). Because the slight DHPG-induced inward current that remained in the presence of 100 μm LY367385 could be sufficient for a substantial depolarization, we slightly increased the concentration of this competitive mGluR1 antagonist used in the current-clamp experiments. Consistent with the voltage-clamp studies, the DHPG-induced depolarization was blocked completely by 300 μm LY367385 (ΔVm of 1.20 ± 0.1 mV;n = 5) (Fig. 1F).
In other brain regions, group I mGluR activation depolarizes neurons by modulating a variety of conductances. Notably, in hippocampal area CA3 DHPG depolarizes neurons by the inhibition of a leak potassium conductance (Guerineau et al., 1994) or by an increase in a nonselective cationic conductance (Guerineau et al., 1995). The DHPG-induced inward current was associated with an increase in input resistance (ΔRin 9.3 ± 1.8 MΩ; n = 4) (Fig.2A). This suggests that an inhibition of a leak potassium conductance is the most likely mechanism underlying this effect. We therefore examined the current–voltage relationship of the mGluR1-mediated inward current. Application of DHPG (30 μm) in the presence of the mGluR5 blocker MPEP (10 μm) induced a change in the slope of the whole-cell current–voltage relationship (Fig. 2B). Subtracting the predrug I–Vtrace from the trace in the presence of DHPG reveals a near-linearI–V relationship for the DHPG-induced current that reverses near the calculated potassium equilibrium potential in three of the four cells that were tested (−104.5 ± 8 mV; n = 3) (Fig. 2B, inset), suggesting that the activation of mGluR1 depolarizes CA1 pyramidal cells by an inhibition of a leak potassium conductance.
Fig. 2.

DHPG-induced inward current and depolarization in CA1 hippocampal neurons most likely are mediated by decreasing a leak potassium conductance. A, The mGluR1-mediated inward current observed in CA1 pyramidal neurons is associated with a decrease in membrane conductance (top vs bottomtrace). B, This decrease in membrane conductance is evident in the whole-cell current–voltage relation. Theinset shows the subtraction of the currents that reveals a linear I–V relationship, which is inward at normal resting potentials and reverses near the predicted potassium equilibrium potential. Axis titles in B apply in theinset. The figure is representative of the results that were observed in three of four cells.
Taken together, these data suggest that the DHPG-induced increase in intracellular calcium concentration and the DHPG-induced inward current and depolarization in CA1 hippocampal neurons are mediated by mGluR1 and not by mGluR5.
mGluR5 mediates the DHPG-induced suppression ofIAHP and potentiation of NMDA receptor currents in CA1 pyramidal neurons
The finding that DHPG-induced calcium transients and depolarization of CA1 pyramidal cells is mediated by mGluR1 alone was surprising in light of previous studies that revealed that mGluR5 is expressed abundantly in these neurons. However, DHPG induces a number of other physiological responses in these cells, some of which could be mediated by mGluR5. Two of the most prominent effects of DHPG in these cells are the inhibition of IAHP and the potentiation of NMDA receptor currents. Thus, we determined the effects of the mGluR1 and mGluR5-selective antagonists on each of these responses to DHPG. As reported previously (Gereau and Conn, 1995b), DHPG (30 μm) suppressedIAHP in a reversible manner (73.2 ± 3% of control) (Fig. 3A,C). Application of the selective mGluR5 antagonist MPEP (10 μm) completely blocked the DHPG-induced suppression ofIAHP (Fig. 3B), whereas the selective mGluR1 antagonist LY367385 (100 μm) had no effect (Fig. 3C). Neither of the antagonists had an effect on the amplitude of IAHP when applied alone.
Fig. 3.
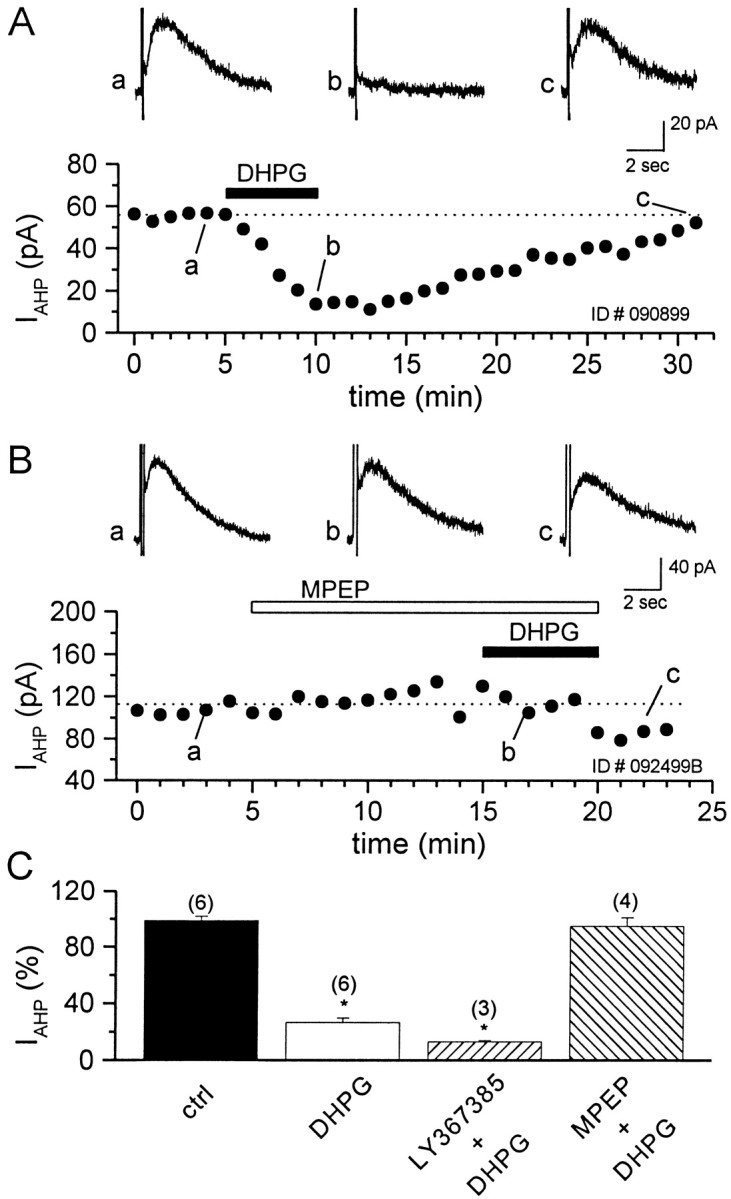
The DHPG-induced suppression ofIAHP in CA1 pyramidal neurons is mediated by mGluR5. A, Time course from a single experiment showing the suppression of IAHP induced by the application of 30 μm DHPG. Lettersindicate the time of the corresponding traces that are shown at thetop. B, The 10 min preapplication of 10 μm MPEP antagonizes the DHPG-induced suppression ofIAHP. C, Bar graph summarizing mean data showing that the DHPG-induced suppression ofIAHP is blocked selectively by MPEP but is not altered by preincubation with the mGluR1-selective antagonist LY367385 (100 μm). The number of cells that have been tested is in parentheses; values are the means ± SEM of the data, expressed as a percentage of controlIAHP peak values. *p < 0.05 versus control; Student's t test.
We also investigated which group I mGluR subtype mediates the DHPG-induced potentiation of NMDA-induced currents. Consistent with previous studies (Aniksztejn et al., 1991, 1992; Fitzjohn et al., 1996;Pisani et al., 1997), the bath application of DHPG (30 μm) increased the peak amplitude of the current induced by fast local application of NMDA (100 μm) (Fig.4A, top panels). The potentiation of NMDA receptor currents was inhibited by preincubation with MPEP (Fig. 4A–C), but not by LY367385 (Fig. 4C).
Fig. 4.

MPEP antagonizes the DHPG-induced potentiation of NMDA currents in CA1 pyramidal cells. A, Representative traces demonstrate the DHPG-induced potentiation of NMDA currents (top traces) and its block by preincubation with 10 μm MPEP (bottom traces). B, Average time course showing the effect of DHPG (open circles) and its antagonism by 10 μm MPEP (filled circles). C, Bar graph showing the mean effect of DHPG on NMDA current alone and in the presence of MPEP or LY367385. The number of cells that have been tested is in parentheses; values are the means ± SEM of the data, expressed as peak potentiation of control values. *p < 0.05; Student'st test.
The DHPG-induced increase in the frequency of spontaneous IPSCs is mediated by mGluR1
In addition to its direct actions on CA1 pyramidal cells, the activation of group I mGluRs impacts CA1 pyramidal cell excitability indirectly by regulating GABA-mediated inhibitory transmission. Two distinct effects of group I mGluR activation on inhibitory transmission can be seen in recordings from CA1 pyramidal cells. First, group I mGluR agonists induce depolarization and increase firing of inhibitory interneurons (McBain et al., 1994; van Hooft et al., 2000), leading to a robust increase in spontaneous IPSCs. Second, group I mGluR agonists reduce the amplitude of evoked monosynaptic IPSCs (Desai et al., 1994;Gereau and Conn, 1995a). Activation of group I mGluRs does not alter the amplitude of miniature IPSCs, suggesting that the reduction of evoked IPSCs is mediated by a presynaptic action. However, analysis of the effect of mGluR activation on monosynaptic IPSCs is complicated by the high frequency of DHPG-induced spontaneous IPSCs. Thus, we took advantage of the mGluR1 and mGluR5-selective antagonists to determine whether we could differentiate between these two actions of DHPG.
Figure 5A, left, shows examples of sIPSCs recorded under control conditions and during the bath application of 10 μm DHPG. Similar to results reported previously in area CA3 (Miles and Poncer, 1993) and in rat frontal cortex (Chu and Hablitz, 1998), the application of DHPG (10 μm) increased the frequency of sIPSCs in all of the cells that were tested (n = 4). These effects were reversible on drug washout from the recording chamber (data not shown). Preincubation with the mGluR5-selective antagonist MPEP (10 μm) did not block the DHPG-induced increase in sIPSC frequency (Fig. 5A, middle), whereas preincubation with the selective mGluR1 antagonist LY367385 (100 μm) completely blocked this effect (Fig.5A, right). DHPG produced a leftward shift of the cumulative probability distributions of sIPSC inter-event intervals (Fig. 5B, left), indicating a DHPG-induced increase in the frequency of sIPSCs. This effect was blocked by LY367385, but not by MPEP (Fig. 5B, right andmiddle, respectively). The average frequency was calculated for each cell, and the means of these values are shown in Figure5D. Application of 10 μm DHPG induced a significant increase in mean frequency (Fig. 5D,left). Preincubation with either MPEP or LY367385 alone did not change the mean frequency of sIPSCs significantly; however, LY367385 blocked the DHPG-induced increase in sIPSC frequency, whereas MPEP was without effect (Fig. 5D, right andmiddle, respectively). In contrast, the application of 10 μm DHPG alone or in combination with the subtype-selective antagonists produced no significant effect on sIPSC amplitude (Fig. 5C, left, middle,right). This is consistent with the previously reported lack of effect of DHPG on mIPSC amplitude (Gereau and Conn, 1995a) and suggests that DHPG does not alter postsynaptic GABAA receptor currents.
Fig. 5.

mGluR1 mediates the DHPG-induced increase in the frequency of pharmacologically isolated spontaneous IPSCs (sIPSCs) in CA1 pyramidal neurons. A, Four consecutive traces under the control condition (top left) and during the bath application of 10 μm DHPG (bottom left) demonstrate the DHPG-induced increase in sIPSCs. This effect was not altered by DHPG preincubation with MPEP (top middle,bottom middle) but was decreased significantly by preincubation with LY367385 (top right, bottom right). Traces are representative of results obtained in four independent experiments for each condition. B, Cumulative probability plots demonstrate the effect of DHPG on sIPSC inter-event interval. DHPG applied alone (left) or after preincubation with MPEP (middle) caused a significant shift in the inter-event interval distributions toward a shorter interval, indicating a significant increase in the frequency of sIPSCs [Kolmogorov–Smirnov (K–S) statistic;p < 0.01]. The mGluR1-selective antagonist LY367385 completely antagonized this DHPG-mediated effect (right; K–S statistic; p > 0.05).C, Cumulative probability plots demonstrate the lack of effect of DHPG on sIPSC amplitude in all of the conditions that were tested (K–S statistic; p > 0.05).D, Bar graph showing the increase in the frequency of sIPSCs, induced by DHPG alone and in the presence of MPEP or LY367385. Note that antagonist alone has no effect on the mean frequency (filled bars). The number of cells that have been tested is in parentheses; values are the means ± SEM. *p < 0.05 versus control; Student's t test.
Activation of both mGluR1 and mGluR5 reduces evoked monosynaptic IPSCs
The activation of group I mGluRs also has been demonstrated to induce a reduction in evoked monosynaptic IPSCs (Desai et al., 1994;Gereau and Conn, 1995a). Consistent with this, the bath application of DHPG (10 μm) produced a reversible depression of IPSCs by 80 ± 4% (means ± SEM) of baseline amplitude (Fig.6A,E) that was antagonized only partially by preincubation with LY367385 (100 μm; 48 ± 9% of baseline amplitude; means ± SEM) (Fig. 6B) or MPEP (10 μm; 54 ± 10% of baseline amplitude; means ± SEM) (Fig. 6C). However, preincubation with a combination of LY367685 (100 μm) and MPEP (10 μm) fully blocked the DHPG-induced suppression of evoked IPSCs (7 ± 2% of baseline amplitude) (Fig.6D,E). These data suggest that the group I mGluR-induced suppression of monosynaptic IPSCs is mediated by both mGluR1 and mGluR5. Therefore, DHPG induces an increase in sIPSC frequency by the activation of mGluR1 and an inhibition of evoked IPSCs by activation of both mGluR1 and mGluR5.
Fig. 6.

Both mGluR1 and mGluR5 play a role in the DHPG-induced decrease in monosynaptic evoked IPSCs.A–C, Time course of the DHPG-induced decrease in IPSC amplitude (means ± SEM) alone (A) and in the presence of LY367385 (100 μm; B) or MPEP (10 μm; C). IPSCs are normalized to predrug values. Sample traces (at top) were obtained before (left), during (middle), and after the washout of DHPG (right) and represent averages of 10 traces. D, Time course of the effect of DHPG on IPSC amplitude (means ± SEM) in the presence of the combined group I antagonists demonstrates that blockade of both mGluR1 and mGluR5 is necessary to inhibit fully the DHPG-induced suppression of inhibitory transmission. E, Bar graphs representing mean peak IPSC depression during DHPG application alone and in the presence of MPEP, LY367385, and the combination. Mean data represent the means of the lowest values during DHPG application expressed as a percentage of predrug values. The number of cells is indicated inparentheses. Values are the means ± SEM. *p < 0.05 versus control; Student'st test.
Activation of mGluR1 reduces evoked EPSCs via a presynaptic mechanism
Previous studies have demonstrated that multiple mGluRs modulate excitatory synaptic transmission. In particular, the group I-selective agonist DHPG has been shown to induce a presynaptically mediated depression of excitatory transmission at the Schaffer collateral→CA1 synapse (Gereau and Conn, 1995a; Rodriguez-Moreno et al., 1998). Consistent with these previous reports, 30 μm DHPG induced a 54 ± 5% (means ± SEM) suppression in EPSCs evoked by electrical stimulation of the stratum radiatum (Fig.7A). We used LY367385 and MPEP to determine which of the group I mGluRs mediate this response. Interestingly, we found that the DHPG-induced suppression of transmission at the Schaffer collateral→CA1 synapse was not affected by preincubation with MPEP (10 μm) but was antagonized completely by the preapplication of LY367385 (300 μm) (Fig. 7B,C, respectively). Moreover, in experiments in which paired pulse facilitation was measured, DHPG induced a 46.3% increase in the paired pulse ratio (PPR), which was significant, and reversed with the washout of the drug (PPR predrug, 2.08 ± 0.16; DHPG, 3.03 ± 0.37;p < 0.05, paired t test; n= 9) (Fig. 8). This effect on PPR was antagonized by preincubation with 300 μmLY367385 (DHPG alone, 46.3 ± 15%; n = 9; DHPG plus LY367385, 6.3 ± 2.1%; n = 7;p < 0.05; t test) but was not affected by 10 μm MPEP (DHPG alone, 46.3 ± 15%;n = 9; DHPG plus MPEP, 32.4 ± 10.4%;n = 9; p > 0.05; t test) (Fig. 8). Because an increase in PPR is generally indicative of a presynaptic mechanism, these results suggest that the mGluR1 activation suppresses excitatory transmission at the Schaffer collateral→CA1 synapse via a presynaptic mechanism.
Fig. 7.
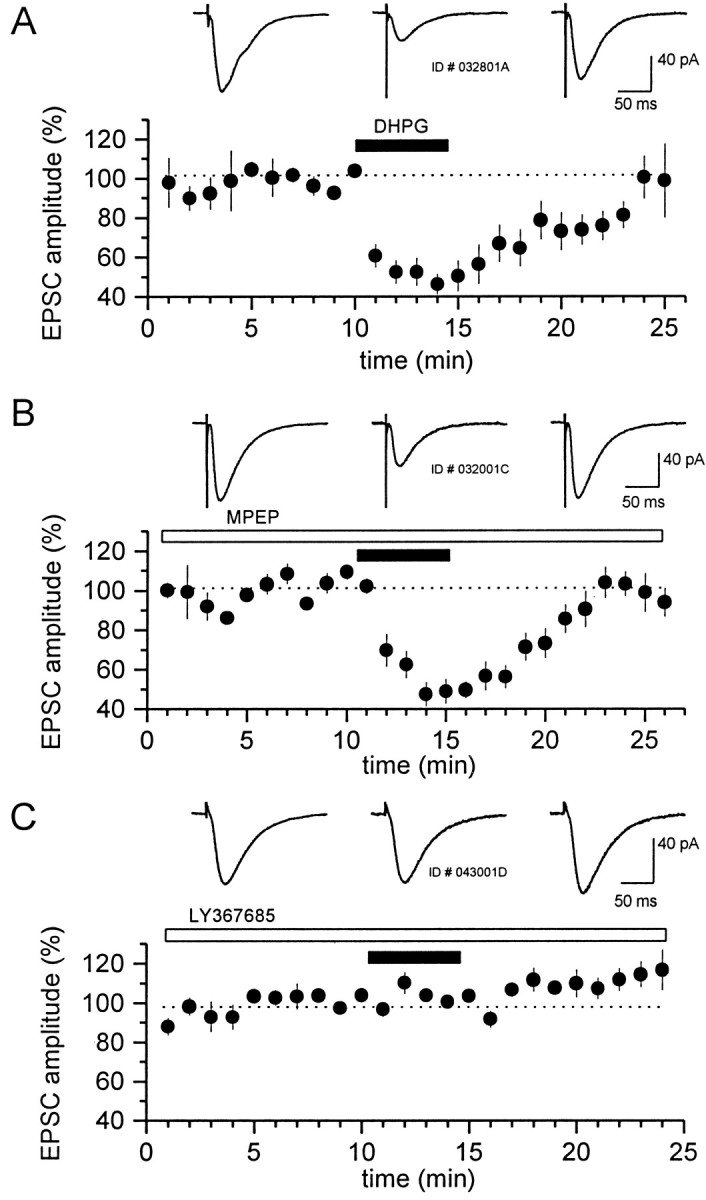
The DHPG-induced inhibition of transmission at the Schaffer collateral→CA1 synapse is mediated by mGluR1.A–C, Time courses of the DHPG-induced decrease in evoked EPSC amplitude alone (A) or in the presence of 10 μm MPEP (B) or 100 μm LY367385 (C). Consistent with mediation by mGluR1, the effect of DHPG is blocked fully by the preapplication of LY367385. Each point represents the average amplitude of four evoked responses acquired during 1 min by stimulation once every 15 sec. Data are presented as the means ± SEM of n = 11 cells (A),n = 9 cells (B), andn = 7 cells (C). EPSCs were normalized to predrug values. Sample traces (at top in each panel) were obtained before (left), during (middle), and after (right) DHPG application.
Fig. 8.

Preapplication of the mGluR1 antagonist LY367385 blocks the DHPG-induced increase in paired pulse ratio (PPR).A, Consistent with previous studies, the application of DHPG induces a significant and reversible enhancement of the PPR. This is evident both in the superimposed scaled traces (toppanel) and in the scatter plot illustrating the effect of DHPG on the average PPR in each experiment (bottom panel). The DHPG-induced increase in PPR is not affected by preincubation with 10 μm MPEP (B) but is antagonized by the preapplication of 100 μm LY367385 (C). The traces represent averages of 40 EPSCs before DHPG, superimposed on an average of four traces obtained during the maximum DHPG effect. The traces are scaled to the peak value of the first EPSC to illustrate the DHPG-induced change in facilitation.
DISCUSSION
Previous studies have revealed that the activation of group I mGluRs has a number of actions on hippocampal CA1 pyramidal cells that increase excitability and synaptic excitation of these neurons. These include direct depolarization (Charpak et al., 1990; Desai and Conn, 1991; Davies et al., 1995; Mannaioni et al., 1999), reduction in the slow afterhyperpolarization (Pedarzani and Storm, 1993; Abdul-Ghani et al., 1996), and potentiation of NMDA receptor currents (Aniksztejn et al., 1991, 1992; Fitzjohn et al., 1996). In addition, the activation of group I mGluRs decreases evoked synaptic inhibition in area CA1 (Gereau and Conn, 1995a). This combination of effects can act to increase the net excitatory drive through this portion of the hippocampal circuit. Although the specific group I mGluR subtypes that mediate each of these effects have not been determined, mGluR5 has been considered to be the most likely candidate for the receptor responsible for the actions of group I mGluR agonists on CA1 pyramidal cells because of its high abundance in these neurons (Luján et al., 1996). In addition, although some splice variants of mGluR1 are present at lower levels in CA1 pyramidal cells, it has been assumed that these two receptors serve similar roles because both mGluR1 and mGluR5 couple to Gq and to the activation of phosphoinositide hydrolysis.
The most important finding of the present study is that mGluR1 and mGluR5 do not serve similar or redundant roles in CA1 pyramidal cells but, rather, that the roles of these receptors are highly segregated. The finding that DHPG-induced somatic calcium transients are mediated exclusively by mGluR1 was especially surprising, because both mGluR1 and mGluR5 couple to the activation of phosphoinositide hydrolysis and inositol trisphosphate-mediated calcium release in other systems. It should be noted that measurements of calcium transients in soma and proximal dendrites in these studies do not rule out the possibility that mGluR5 activation induces calcium release in small subcellular compartments (such as distal dendrites or spines) that were not detected in these studies. However, regardless of the reason for the lack of a calcium response to mGluR5 activation, these findings, along with the other data that have been reported, reveal that mGluR1 and mGluR5 display a high specificity of function when present in the same neuronal population.
Until recently, the selective tools required for determining the functions of mGluR1 and mGluR5 in native preparations have not been available. However, a growing body of literature suggests distinct roles for mGluR1 and mGluR5 when these receptors are present in the same neurons. For instance, neurons in the subthalamic nucleus (STN) contain both mGluR1 and mGluR5, and DHPG induces membrane depolarization and oscillations in these cells (Awad et al., 2000). However, this response to DHPG is mediated exclusively by mGluR5, with no defined role for mGluR1. Conversely, GABAergic neurons in the substantia nigra pars reticulata (SNr) express both mGluR1 and mGluR5, but depolarization of these neurons is mediated exclusively by mGluR1 (Marino et al., 1999). At present, the roles of mGluR1 and mGluR5 in STN and SNr neurons, respectively, are not known. However, the studies that have been reported make it clear that these receptors are not playing redundant roles in these neuronal populations.
At present, the molecular mechanisms involved in the segregation of function of mGluR1 and mGluR5 are not clear. However, because these two receptor subtypes are generally capable of coupling to the same GTP effector proteins (i.e., Gq), it is possible that the receptors have differential access to signaling partners. For instance, mGluR1 might be localized in close proximity to leak potassium channels and inositol trisphosphate receptors, whereas mGluR5 is coupled functionally to NMDA receptors and potassium channels that are responsible for the AHP current. Recent evidence suggests that mGluR5 and NMDA receptors may interact physically via interactions with a series of scaffolding proteins that include Homer, Shank, PSD-95, and others (Ehlers, 1999; Naisbitt et al., 1999; Tu et al., 1999). A key component of this interaction is the binding of mGluR5 to Homer. Homer is known to interact with the group I mGluRs via a specific sequence found in the C-terminal portion of the receptor (Tu et al., 1998). Although both splice forms of mGluR5 can bind to Homer, mGluR1a is the only mGluR1 splice form that contains the Homer binding domain (Houamed et al., 1991; Masu et al., 1991; Pin et al., 1992;Tanabe et al., 1992). Because the mGluR1a splice variant is not present in CA1 pyramidal cells (Shigemoto et al., 1992; Hampson et al., 1994;Berthele et al., 1998), the splice forms that are present in these neurons may not be capable of this interaction. It is conceivable that other signaling complexes exist that include mGluR1 and associated signaling partners. It is also possible that some of the differences between mGluR1 and mGluR5 actions are attributable to differential coupling to specific G-protein subunits that are present in CA1 pyramidal cells. Indeed, early studies in recombinant systems revealed that, whereas group I mGluRs couple primarily to Gq, these receptors can couple to Gs in several cell types (Aramori and Nakanishi, 1992; Joly et al., 1995). Interestingly, some cell types are permissive for mGluR1 coupling to Gs but restrict mGluR5 coupling to Gq (Abe et al., 1992). This suggests that the G-protein coupling of these two receptors is not identical and raises the possibility that the different actions of these receptor subtypes are defined in part by the G-protein subunits.
It is important to point out that our finding that the depolarization of CA1 pyramidal cells in rats is mediated by mGluR1 differs from the conclusion of Lu and colleagues (Lu et al., 1997), who found that the depolarization induced by 1S,3R-ACPD is absent in CA1 pyramidal cells from mGluR5 knock-out mice. Although we do not have an explanation for this difference, it is possible that this represents a species difference between rats and mice. Also, it is possible that there is a developmental compensation in the knock-out animals such that the role of mGluR1 is altered in the absence of mGluR5 expression. Finally, there also might be a developmental regulation of the receptors that mediates this response. In the present studies we restricted our analysis to young animals (P15–P20). Group I mGluRs are known to be regulated tightly during postnatal development, and it is possible that the receptor that mediates this response could be different in the older animals used by Lu and colleagues (Lu et al., 1997). In future studies it will be useful to use these new pharmacological tools in wild-type mice as well as in mGluR5 and mGluR1 knock-out animals across developmental stages to differentiate among these possibilities.
The finding that the DHPG-induced increase in the frequency of spontaneous IPSCs is mediated by mGluR1 is consistent with previous findings that mGluR1a is expressed heavily in the stratum oriens/alveus interneurons (Baude et al., 1993; Luján et al., 1996; Shigemoto et al., 1997). The activation of group I mGluRs induces a robust depolarization of these neurons (Miles and Poncer, 1993; McBain et al., 1994; van Hooft et al., 2000) that could contribute to the DHPG-induced increase in sIPSCs. These findings must be kept in mind when interpreting the role that mGluR1 plays in the modulation of evoked IPSCs as demonstrated in the present study. It is possible that an mGluR1-mediated direct depolarization of interneurons effectively could reduce the ability of an electrical stimulus to evoke an IPSC without affecting transmitter release at the level of the terminal.
It has been known for some time now that the activation of group I mGluRs inhibits excitatory transmission at the Schaffer collateral→CA1 synapse and that this inhibition of transmission occurs via a presynaptic mechanism (Gereau and Conn, 1995a;Rodriguez-Moreno et al., 1998). However, it has been difficult to predict which group I mGluR might mediate this response, because immunocytochemical studies have not been able to detect mGluR1 or mGluR5 presynaptically localized in area CA1 (Baude et al., 1993;Luján et al., 1996; Shigemoto et al., 1997). Interestingly, we have found that this decrease in excitatory drive through the hippocampal circuit is mediated by mGluR1. Presently, it is unclear whether the activation of presynaptic mGluR1 by glutamate release also would activate postsynaptic mGluR1 to mediate a localized increase in input resistance, depolarization, and Ca2+release. Understanding the temporal–spatial nature of synaptic mGluR1 signaling in CA1 pyramidal cells will be necessary before the role of this receptor can be appreciated fully.
Our finding that mGluR1 and mGluR5 play different and nonoverlapping roles in CA1 pyramidal cells has a number of physiological and therapeutic implications. For instance, selective antagonists of mGluR5 might be useful as novel anticonvulsant agents by selectively reducing mGluR-mediated disinhibition and inhibition of the AHP current. Such compounds would leave the mGluR1-mediated excitation of inhibitory interneurons intact, which could provide a therapeutic advantage. Consistent with this, it was reported recently that selective mGluR5 antagonists have robust anticonvulsant actions in rats (Chapman et al., 2000). Conversely, the finding that mGluR5 is responsible for the potentiation of NMDA receptor currents raises the possibility that agonists of this receptor could be useful as cognitive-enhancing agents or novel antipsychotic agents that act by selectively enhancing NMDA receptor function. The combined use of mGluR1- and mGluR5-selective reagents for studies at the behavioral, systems, and cellular levels eventually should allow for a detailed understanding of the physiological roles and therapeutic potential of each of these receptors in the hippocampus as well as other CNS circuits.
Footnotes
This work was supported by the National Institutes of Health and by the National Alliance for Research on Schizophrenia and Depression. We thank Dr. Gianmaria Maccaferri for critical reading and comments on this manuscript.
Correspondence should be addressed to Dr. Jeffrey Conn, Senior Director, Neuroscience Division, Department of Pharmacology, Merck Research Laboratories, Merck & Company, Incorporated, 770 Sumneytown Pike, P.O. Box 4, WP 46-300, West Point, PA 19486-0004. E-mail:jeff_conn@merck.com.
REFERENCES
- 1.Abdul-Ghani MA, Valiante TA, Carlen PL, Pennefather PS. Metabotropic glutamate receptors coupled to IP3 production mediate inhibition of IAHP in rat dentate granule neurons. J Neurophysiol. 1996;76:2691–2700. doi: 10.1152/jn.1996.76.4.2691. [DOI] [PubMed] [Google Scholar]
- 2.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- 3.Alagarsamy S, Marino MJ, Rouse ST, Gereau RW, 4th, Heinemann SF, Conn PJ. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat Neurosci. 1999;2:234–240. doi: 10.1038/6338. [DOI] [PubMed] [Google Scholar]
- 4.Aniksztejn L, Bregestovski P, Ben-Ari Y. Selective activation of quisqualate metabotropic receptor potentiates NMDA but not AMPA responses. Eur J Pharmacol. 1991;205:327–328. doi: 10.1016/0014-2999(91)90921-c. [DOI] [PubMed] [Google Scholar]
- 5.Aniksztejn L, Otani S, Ben-Ari Y. Quisqualate metabotropic receptors modulate NMDA currents and facilitate the induction of long-term potentiation through protein kinase C. Eur J Neurosci. 1992;4:500–505. doi: 10.1111/j.1460-9568.1992.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 6.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 7.Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8:757–765. doi: 10.1016/0896-6273(92)90096-v. [DOI] [PubMed] [Google Scholar]
- 8.Aronica EM, Gorter JA, Paupard MC, Grooms SY, Bennett MVL, Zukin RS. Status epilepticus-induced alterations in metabotropic glutamate receptor expression in young and adult rats. J Neurosci. 1997;17:8588–8595. doi: 10.1523/JNEUROSCI.17-21-08588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 11.Baude A, Nusser Z, Roberts JDB, Mulvihill E, McIlhinney RAJ, Somogyi P. The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- 12.Berthele A, Laurie DJ, Platzer S, Zieglgansberger W, Tolle TR, Sommer B. Differential expression of rat and human type I metabotropic glutamate receptor splice variant messenger RNAs. Neuroscience. 1998;3:733–749. doi: 10.1016/s0306-4522(97)00670-2. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi R, Young SR, Wong RK. Group I mGluR activation causes voltage-dependent and -independent Ca2+ rises in hippocampal pyramidal cells. J Neurophysiol. 1999;81:2903–2913. doi: 10.1152/jn.1999.81.6.2903. [DOI] [PubMed] [Google Scholar]
- 14.Bond A, Ragumoorthy N, Monn JA, Hicks CA, Ward MA, Lodge D, O'Neill MJ. LY379268, a potent and selective group II metabotropic glutamate receptor agonist, is neuroprotective in gerbil global, but not focal, cerebral ischaemia. Neurosci Lett. 1999;273:191–194. doi: 10.1016/s0304-3940(99)00663-1. [DOI] [PubMed] [Google Scholar]
- 15.Bond A, Jones NM, Hicks CA, Whiffin GM, Ward MA, O'Neill MF, Kingstom AE, Monn JA, Ornstein PL, Schoepp DD, Lodge D, O'Neill MJ. Neuroprotective effects of LY379268, a selective mGlu2/3 receptor agonist: investigations into possible mechanism of action in vivo. J Pharmacol Exp Ther. 2000;294:800–809. [PubMed] [Google Scholar]
- 16.Chapman AG, Nanan K, Williams M, Meldrum B. Anticonvulsant activity of two metabotropic glutamate group I antagonists selective for the mGlu5 receptor: 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and (E)-6-methyl-2-styryl-pyridine (SIB 1893). Neuropharmacology. 2000;39:1567–1574. doi: 10.1016/s0028-3908(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 17.Charpak S, Gahwiler BH, Do KQ, Knoepfel T. Potassium conductances in hippocampal neurons blocked by excitatory amino acid transmitters. Nature. 1990;347:765–767. doi: 10.1038/347765a0. [DOI] [PubMed] [Google Scholar]
- 18.Chu Z, Hablitz JJ. Activation of group I mGluRs increases spontaneous IPSC frequency in rat frontal cortex. J Neurophysiol. 1998;80:621–627. doi: 10.1152/jn.1998.80.2.621. [DOI] [PubMed] [Google Scholar]
- 19.Clark BP, Baker SR, Goldsworthy J, Harris JR, Kingston AE. (+)-2-Methyl-4-carboxyphenylglycine (LY367385) selectively antagonizes metabotropic glutamate mGluR1 receptors. Bioorg Med Chem Lett. 1997;7:2777–2780. [Google Scholar]
- 20.Crepel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 21.Davies CH, Clarke VR, Jane DE, Collingridge GL. Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurones. Br J Pharmacol. 1995;116:1859–1869. doi: 10.1111/j.1476-5381.1995.tb16674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desai MA, Conn PJ. Excitatory effects of ACPD receptor activation in the hippocampus are mediated by direct effects on pyramidal cells and blockade of synaptic inhibition. J Neurophysiol. 1991;66:40–52. doi: 10.1152/jn.1991.66.1.40. [DOI] [PubMed] [Google Scholar]
- 23.Desai MA, McBain CJ, Kauer JA, Conn PJ. Metabotropic glutamate receptor-induced disinhibition is mediated by reduced transmission at excitatory synapses onto interneurons and inhibitory synapses onto pyramidal cells. Neurosci Lett. 1994;181:78–82. doi: 10.1016/0304-3940(94)90564-9. [DOI] [PubMed] [Google Scholar]
- 24.Ehlers MD. Synapse structure: glutamate receptors connected by the Shanks. Curr Biol. 1999;9:R848–R850. doi: 10.1016/s0960-9822(00)80043-3. [DOI] [PubMed] [Google Scholar]
- 25.Ferraguti F, Conquet F, Corti C, Grandes P, Kuhn R, Knoepfel T. Immunohistochemical localization of the mGluR1β metabotropic glutamate receptor in the adult rodent forebrain: evidence for a different distribution of mGluR1 splice variants. J Comp Neurol. 1998;400:391–407. [PubMed] [Google Scholar]
- 26.Fitzjohn SM, Irving AJ, Palmer MJ, Harvey J, Lodge D, Collingridge GL. Activation of group I mGluRs potentiates NMDA responses in rat hippocampal slices. Neurosci Lett. 1996;203:211–213. doi: 10.1016/0304-3940(96)12301-6. [DOI] [PubMed] [Google Scholar]
- 27.Fitzsimonds RM, Dichter MA. Heterologous modulation of inhibitory synaptic transmission by metabotropic glutamate receptors in cultured hippocampal neurons. J Neurophysiol. 1996;75:885–893. doi: 10.1152/jn.1996.75.2.885. [DOI] [PubMed] [Google Scholar]
- 28.Galoyan SM, Merlin LR. Long-lasting potentiation of epileptiform bursts by group I mGluRs is NMDA receptor independent. J Neurophysiol. 2000;83:2463–2467. doi: 10.1152/jn.2000.83.4.2463. [DOI] [PubMed] [Google Scholar]
- 29.Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliçelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective, and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 30.Gereau RW, 4th, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995a;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gereau RW, 4th, Conn PJ. Roles of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995b;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 32.Guerineau NC, Gahwiler BH, Gerber U. Reduction of resting K+ current by metabotropic glutamate and muscarinic receptors in rat CA3 cells: mediation by G-proteins. J Physiol (Lond) 1994;474:27–33. doi: 10.1113/jphysiol.1994.sp019999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerineau NC, Bossu JL, Gahwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hampson DR, Theriault E, Huang XP, Kristensen P, Pickering DS, Franck JE, Mulvihill ER. Characterization of two alternatively spliced forms of a metabotropic glutamate receptor in the central nervous system of the rat. Neuroscience. 1994;60:325–336. doi: 10.1016/0306-4522(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 35.Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O'Hara PJ, Mulvihill ER, Almers W, Hagen FS. Cloning, expression, and gene structure of a G-protein-coupled glutamate receptor from rat brain. Science. 1991;252:1318–1321. doi: 10.1126/science.1656524. [DOI] [PubMed] [Google Scholar]
- 36.Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin J-P. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder J. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luján R, Nusser Z, Roberts JC, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 39.Luthi A, Gahwiler BH, Gerber U. A slowly inactivating potassium current in CA3 pyramidal cells of rat hippocampus in vitro. J Neurosci. 1996;16:586–594. doi: 10.1523/JNEUROSCI.16-02-00586.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mannaioni G, Attucci S, Missanelli A, Pellicciari R, Corradetti R, Moroni F. Biochemical and electrophysiological studies on (S)-(+)-2-(3′-carboxybicyclo[1.1.1]pentyl)-glycine (CBPG), a novel mGlu5 receptor agonist endowed with mGlu1 receptor antagonist activity. Neuropharmacology. 1999;38:917–926. doi: 10.1016/s0028-3908(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 41.Marino MJ, Rouse ST, Levey A, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-d-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci USA. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marino MJ, Risso Bradley S, Wittmann M, Conn PJ. Direct excitation of GABAergic projection neurons of the rat substantia nigra pars reticulata by activation of the mGluR1 metabotropic glutamate receptor. Soc Neurosci Abstr. 1999;25:446. doi: 10.1523/JNEUROSCI.21-18-07001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- 44.McBain CJ, DiChiara TJ, Kauer JA. Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. J Neurosci. 1994;14:4433–4445. doi: 10.1523/JNEUROSCI.14-07-04433.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merlin LR. Group I mGluR-mediated silent induction of long-lasting epileptiform discharges. J Neurophysiol. 1999;82:1078–1081. doi: 10.1152/jn.1999.82.2.1078. [DOI] [PubMed] [Google Scholar]
- 46.Miles R, Poncer JC. Metabotropic glutamate receptors mediate a post-tetanic excitation of guinea-pig hippocampal inhibitory neurones. J Physiol (Lond) 1993;463:461–473. doi: 10.1113/jphysiol.1993.sp019605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 48.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 49.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 50.Pellegrini-Giampietro DE, Cozzi A, Peruginelli F, Leonardi P, Meli E, Pellicciari R, Moroni F. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)-2-(3′-carboxybicyclo[1.1.1]pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischemia. Eur J Neurosci. 1999;11:3637–3647. doi: 10.1046/j.1460-9568.1999.00786.x. [DOI] [PubMed] [Google Scholar]
- 51.Pin J-P, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 52.Pin J-P, Waeber C, Prezeau L, Bockaert J, Heinemann SF. Alternative splicing generates metabotropic glutamate receptors inducing different patterns of calcium release in Xenopus oocytes. Proc Natl Acad Sci USA. 1992;89:10331–10335. doi: 10.1073/pnas.89.21.10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pisani A, Calabresi P, Centonze D, Bernardi G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pozzo-Miller LD, Petrozzino JJ, Golarai G, Connor JA. Ca2+ release from intracellular stores induced by afferent stimulation of CA3 pyramidal neurons in hippocampal slices. J Neurophysiol. 1996;76:554–562. doi: 10.1152/jn.1996.76.1.554. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez-Moreno A, Sistiaga A, Lerma J, Sánchez-Prieto J. Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron. 1998;21:1477–1486. doi: 10.1016/s0896-6273(00)80665-0. [DOI] [PubMed] [Google Scholar]
- 56.Romano C, Sesma MA, McDonald CT, O'Malley K, van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 57.Schoepp DD, Conn PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13–20. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- 58.Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol. 1992;322:121–135. doi: 10.1002/cne.903220110. [DOI] [PubMed] [Google Scholar]
- 59.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stocker M, Krause M, Pedarzani P. An apamin-insensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 62.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group I metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 63.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 64.van Hooft JA, Giuffrida R, Blatow M, Monyer H. Differential expression of group I metabotropic glutamate receptors in functionally distinct hippocampal interneurons. J Neurosci. 2000;20:3544–3551. doi: 10.1523/JNEUROSCI.20-10-03544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]