

Abstract
To investigate the role of the Egr family of transcription regulatory factors in neuronal apoptosis, we examined the effect of a dominant negative Egr inhibitor construct in a well characterizedin vitro paradigm, cerebellar granule cell death induced by withdrawal of depolarizing concentrations of extracellular potassium. We found that this apoptotic stimulus increases the activity of a reporter gene driven by the Egr response element and that a dominant negative inhibitor of Egr-mediated transcription blocks granule cell apoptosis. In contrast, apoptosis of immature granule cells induced by cytosine arabinoside is not inhibited by the Egr inhibitor construct. Because activation of c-Jun is an essential step in granule cell death induced by potassium deprivation, but not cytosine arabinoside, we asked whether the Egr inhibitor acts by influencing c-Jun activation or its ability to induce apoptosis. We found that the Egr inhibitor does not block the ability of a constitutively active c-Jun construct to induce apoptosis in these cells but does suppress activation of c-Jun-mediated transcription induced by lowering extracellular potassium concentration. Furthermore, the Egr inhibitor blocks the ability of MEKK1 [mitogen-activated protein kinase (MAPK) kinase kinase 1], an upstream kinase capable of stimulating the JNK (c-Jun N-terminal protein kinase)–c-Jun pathway, to induce apoptosis and activate c-Jun. Together, these studies indicate that the Egr family of transcription factors plays a critical role in neuronal apoptosis and identify c-Jun activation as an important downstream target of the Egr family in this process.
Keywords: Egr1, zif268, c-Jun, MEKK1, JNK, cytosine arabinoside
The central role of apoptosis in both normal development of the nervous system and the pathophysiology of neuropsychiatric disorders has focused attention on deciphering the complex array of intracellular signaling pathways that control neuronal survival and death (Yuan and Yankner, 2000). Recent studies have provided evidence that Egr1, a transcription regulatory factor that is robustly induced by a broad range of extracellular stimuli (Gashler and Sukhatme, 1995), may be involved in triggering apoptosis under certain circumstances. For example, Egr1 appears to be required for thapsigargin-induced apoptosis in a melanoma cell line (Muthukkumar et al., 1995). In addition, Egr1 enhances sensitivity of fibroblasts to apoptosis induced by ionizing radiation (Das et al., 2001). However, in other studies, Egr1 appears to confer resistance to apoptotic stimuli (Hallahan et al., 1995; Huang et al., 1998).
Although these studies implicate Egr1 in apoptotic signaling in peripheral cell types, little attention has been paid to investigating the involvement of Egr1 or other Egr family members in neuronal apoptosis. Recently, Egr1 has been implicated in neuronal apoptosis induced by zinc toxicity (Park and Koh, 1999); however its role in other, widely studied neuronal apoptosis paradigms has not been clarified. The paucity of information on Egr family members in neuronal apoptosis may stem, in part, from findings linking this transcription factor family to neuronal plasticity (O'Donovan et al., 1999), which may have diverted attention from considering its role in neuronal apoptosis. However, there are clear precedents for transcription factors, such as p53 or c-Jun (Behrens et al., 1999; Vogelstein et al., 2000), being involved in regulating multiple cellular processes. In addition, there is now compelling evidence that cAMP response element-binding protein plays pivotal roles in both neuronal survival and plasticity (Dash et al., 1990; Guzowski and McGaugh, 1997;Lamprecht et al., 1997; Ahn et al., 1999; Riccio et al., 1999). In light of these considerations, we have conducted studies aimed at assessing the role of the Egr family in a well established neuronal apoptosis paradigm, cerebellar granule cell death induced by withdrawal of depolarizing concentrations of potassium (D'Mello et al., 1993;Miller and Johnson, 1996; Tanabe et al., 1998; Watson et al., 1998). This neuronal apoptotic paradigm appeared to be well suited to addressing this issue because recent studies have indicated that Egr1 expression is induced in these cells by this apoptotic stimulus (Catania et al., 1999).
MATERIALS AND METHODS
Chemical reagents and plasmids. Cytosine α-d-arabinofuranoside (AraC) (Sigma, St. Louis, MO) and Hoechst 33258 (Molecular Probes, Eugene, OR) were obtained commercially. Preparation of ZnEgr3, an expression construct encoding the zinc finger DNA binding domain of Egr3, has been described previously (O'Donovan et al., 2000; Levkovitz et al., 2001). Expression plasmids encoding the zinc finger domains of Egr1 and Sp1, ZnEgr1 and ZnSp1, respectively (Chapman and Perkins, 2000), were provided by N. D. Perkins (Department of Biochemistry, University of Dundee, Dundee, UK). An expression plasmid for a constitutively active form of c-Jun, c-Jun(Asp) (Leppa et al., 1998), was provided by D. Bohmann (European Molecular Biology Laboratory, Heidelberg, Germany). An expression plasmid encoding a truncated, constitutively active form of MEKK1 [mitogen-activated protein kinase (MAPK) kinase kinase 1] was purchased from Stratagene (La Jolla, CA). The full-length rat Egr1 expression plasmid was provided by J. Milbrandt (Washington University, St. Louis, MO).
To monitor transcription mediated by the Egr response element (ERE), we used an ERE reporter plasmid provided by S. J. Kim (National Institutes of Health, Bethesda, MD) in which luciferase is under the control of an ERE sequence located in the TGFβ1 promoter (phTG-5), as described previously (Levkovitz et al. 2001). To monitor transcription mediated by c-Jun, we used both the PathDetect c-Jun Trans Reporting System, which uses both the Pfa2-c-Jun plasmid and Pfr-Luc, and the PathDetect AP-1 cis Reporting System (Stratagene, La Jolla, CA). To assess activation of ATF2, we used the PathDetect ATF2 Trans Reporting System. The green fluorescent protein (GFP) expression plasmid was obtained from Clontech (Palo Alto, CA).
Preparation of cultures. Cerebellar granule cell cultures were prepared as described by Thangnipon et al. (1983). Briefly, cerebella from 7- to 9-d-old Sprague Dawley rat pups were dissected from their meninges and minced. The tissue was then trypsinized for 20 min, triturated, and filtered. Cells were seeded onto dishes coated with poly-d-lysine (50 μg/ml) and laminin (5 μg/ml) at a density of 3–5 × 106cells per 60 mm well and cultured in Eagle's basal medium supplemented with 10% heat-inactivated fetal serum, 25 mmKCl, 0.5%(v/v) penicillin–streptomycin, and 2 mm glutamine. To prevent growth of glial cells, AraC (5 μm) was added to the cultures 24 hr after seeding.
Human embryonic kidney 293 (HEK293) cells were used to express recombinant rat Egr1 as described previously (O'Donovan and Baraban, 1999).
Transfection. Cerebellar granule cell cultures were transfected using Lipofectamine 2000 (Life Technologies, Gaithersburg, MD). For studies with the zinc finger or c-Jun(Asp) expression constructs, each culture well received ∼5 μg of each plasmid. Briefly, DNA plasmids containing expression or reporter constructs were mixed with the GFP expression vector, in a ratio of 3–4:1 by weight and diluted in Eagle's basal medium. The Lipofectamine 2000 reagent was diluted in Eagle's basal medium and then incubated for 5 min at room temperature. Then, the diluted lipofectamine reagent was mixed with the plasmid DNA at least 20 min before adding this mixture to the cells. Before adding the lipofectamine–DNA mixture to the cultures, half of the culture medium from a well was removed and saved. The lipofectamine–DNA mixture was added dropwise to the culture well, and then the saved medium was placed back in the culture well after 4 hr.
Induction and assessment of apoptosis. The procedure used byDudek et al. (1997) was used to induce apoptosis of cerebellar granule cells by potassium deprivation. For these studies, cultures were maintained for 5–6 d before transfection. One day after transfection, cerebellar granule cells were placed in fresh media containing either 5 mm KCl and no serum (potassium deprivation) or 25 mm KCl and 10% fetal bovine serum (control). In some studies, granule cell apoptosis was triggered by addition of AraC. In these experiments, granule cells were cultured for only 1 d before initiating apoptosis by replacing the medium with fresh medium supplemented with 500 μm AraC.
One or 2 d after initiating either of these apoptosis paradigms, Hoechst 33258 was added at 2 μg/ml, and GFP-positive cells were inspected by an observer who was unaware of the treatment condition being evaluated. The “blind” observer scored each cell as apoptotic or not based on standard morphological criteria, including nuclear condensation and margination (visualized by Hoechst 33258 fluorescence) as well as cytoplasmatic blebbing and fragmentation of neuronal processes (visualized by GFP fluorescence) (Dudek et al., 1997). We estimate that ∼0.1 to 1.0% of granule cells were GFP-positive.
Reporter assays. Sixty millimeter wells were transfected with one of the reporter plasmids, expression plasmids or the corresponding empty vector (2–4 μg/well), and a GFP expression plasmid (1 μg/well). For the GAL4/c-Jun or GAL4/ATF2 reporter assays, cells were transfected with both Pfr-Luc (5 μg), a luciferase reporter plasmid driven by the GAL4 response element, and either Pfa2-c-Jun (1 μg) or Pfa2-ATF2 (1 μg), expression plasmids encoding fusion proteins containing the N-terminal activation domain of c-Jun or ATF2 and the GAL4 DNA binding domain. For the AP-1 reporter assay, cells were transfected with the AP-1 reporter plasmid, in which luciferase expression is under the control of the AP-1 response element (5 μg). In experiments examining the effect of apoptotic stimuli on reporter activity, cells were harvested 6–12 hr after these treatments were initiated. Cells were rinsed twice with warm PBS, harvested in reporter lysis buffer (Promega, Madison, WI), and placed in 1.5 ml tubes on ice. Extracts were vortexed and centrifuged. Supernatants were collected and used for the luciferase assays according to the protocol of the manufacturer. To help control for variability in transfection efficiency, the number of GFP-positive cells was used to normalize the luciferase values obtained. In each reporter experiment, four 60 mm wells were transfected with the constructs being assayed. Each construct was assayed in at least two independent experiments.
Immunoblotting. To monitor the levels of Egr1 protein, cells extracts were harvested in a lysis buffer containing 20 mm HEPES, pH 7.9, 0.4 mNaCl, 20% glycerol, 5 mmMgCl2, 0.5 mm EDTA, 0.1 mm EGTA, and 1% Nonidet P-40, supplemented with freshly added 5 mm dithiothreitol, 10 μg/ml leupeptin, 0.1 mm benzamidine, and 0.5 mm phenylmethylsulfonyl fluoride. The cell extracts were clarified by centrifugation, and ∼40 μg of supernatant protein from each sample were processed for immunoblotting with antibodies to the C terminal of Egr1 (Santa Cruz Biotechnology, Santa Cruz, CA) and α-tubulin (sc-5546, 1:500; Santa Cruz Biotechnology).
RESULTS
Role of the Egr family in apoptosis induced by potassium deprivation
Previous studies have provided immunocytochemical evidence that Egr1 protein levels are increased in granule cells 6 hr after reducing the potassium concentration in the medium (Catania et al., 1999). To confirm those results, we conducted immunoblotting studies on cerebellar granule cell culture extracts harvested before or after potassium deprivation. Consistent with the immunocytochemical findings, we found that Egr1 protein levels are increased at 6 hr after potassium deprivation (Fig. 1A). However, in contrast to previous studies that reported that Egr1 expressed in these cultures has an aberrantly low molecular weight (Catania et al., 1999), we found that Egr1 migrated at the expected molecular weight, ∼80 kDa, matching that displayed by recombinant rat Egr1 expressed in HEK293 cells. As reported previously, Egr1 migrates as a broad band, a feature that presumably reflects posttranslational modification of the protein (Cao et al., 1990; Grimes and Jope, 1999).
Fig. 1.
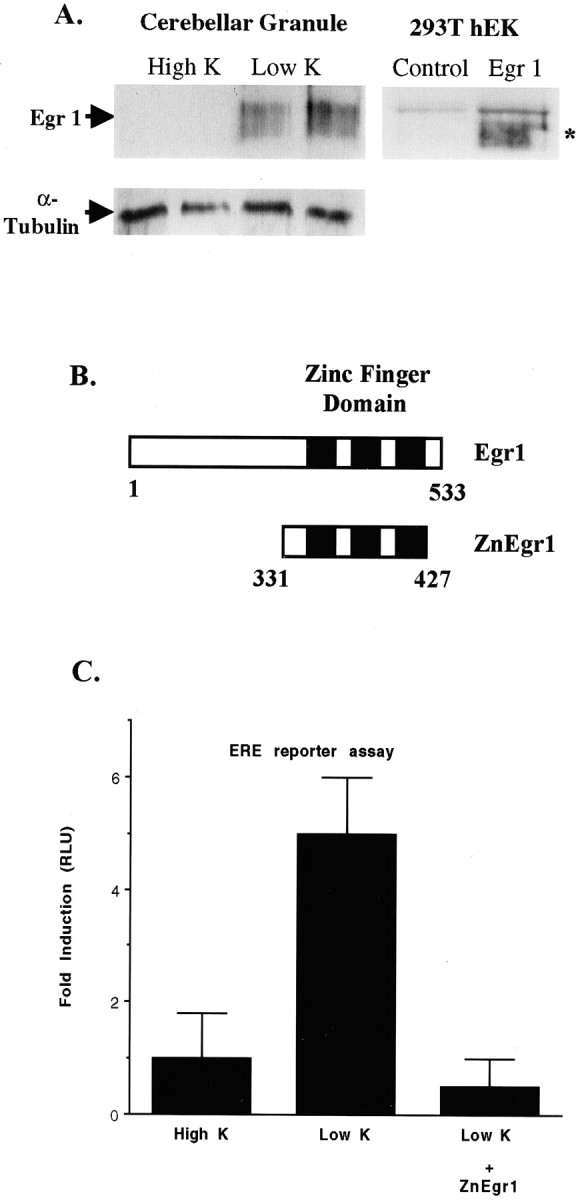
Potassium deprivation increases Egr1 protein levels and ERE reporter activity. A, Immunoblot analysis of Egr1 levels. The left panel shows an immunoblot of extracts prepared from either control (High K) or potassium-deprived (Low K) granule cell cultures harvested 6 hr after switching to potassium deprivation. The broad protein band corresponding to Egr1, which migrates with a molecular weight of ∼80 kDa, is indicated by the arrow. Samples in each lane were obtained from separate culture dishes. Immunoblot analysis of rat Egr1 expressed in HEK293T cells, shown in the right panel, demonstrates that recombinant Egr1 also yields a broad protein band (indicated by asterisk), which comigrates with the band identified as Egr1 in cerebellar granule cell extracts. The Egr1 band is not detected in extracts from HEK293T cells that were transfected with an empty vector (Control). B, Schematic diagram of Egr1 and ZnEgr1. The diagram presented in this panel shows the portion of Egr1 contained in the zinc finger DNA binding domain construct, ZnEgr1, being used as an inhibitor of ERE-mediated transcription.C, Stimulation of an ERE reporter by potassium deprivation: blockade by ZnEgr1. Reducing the concentration of potassium in the medium from 25 mm (High K) to 5 mm (Low K) triggers a fivefold increase in ERE reporter activity (p < 0.01). This increase is abolished by cotransfection of the ZnEgr1 construct (Low K vs Low K + ZnEgr1,p < 0.005). Reporter activity is presented as the ratio of luciferase activities, or relative luciferase units (RLU), detected in extracts from treated and control cultures. Cells were transfected with the ERE reporter and either ZnEgr1 or an empty vector. On the second day after transfection, cell extracts were harvested for luciferase assay, 6 hr after switching the potassium concentration. Error bars shown in this and subsequent figures depict SEM values.
Because these results indicated that Egr1 is induced in this apoptotic paradigm, we wanted to examine whether it plays a critical role in this process. To accomplish this, we used a dominant negative approach. The defining feature of the Egr family is a highly conserved zinc finger DNA binding domain shared by all four Egr family members (Gashler and Sukhatme, 1995; O'Donovan et al., 1999). This domain mediates their interaction with a consensus DNA binding site referred to as the ERE (Christy and Nathans, 1989; Swirnoff and Milbrandt, 1995). In previous studies conducted in PC12 cells, we demonstrated that this zinc finger domain is an effective and selective inhibitor of Egr-mediated transcription (Levkovitz et al., 2001). Therefore, we reasoned that a dominant negative Egr construct of this type would be useful in determining whether Egr1 or other Egr family members play a critical role in mediating apoptosis in this paradigm.
Before investigating the effect of the Egr inhibitor construct on apoptosis in this paradigm, we first wanted to check whether it effectively blocks ERE-mediated transcription in these cells. To this end, we assessed the effect of the zinc finger domain of Egr1, ZnEgr1 (Fig. 1B), on the activity of a luciferase reporter placed under the control of an ERE. Consistent with our immunoblotting studies, we found that potassium deprivation stimulated the activity of the ERE reporter. Furthermore, as expected, we found that ZnEgr1 abolishes this increase (Fig. 1C).
To monitor the effect of ZnEgr1 on apoptosis in this preparation, we cotransfected a GFP expression plasmid with either a plasmid containing the ZnEgr1 insert or the corresponding vector without an insert. We then determined the percentage of GFP-positive cells that displayed pyknotic nuclei, as revealed by Hoechst staining. We found that the ZnEgr1 plasmid exerted a high degree of protection from apoptosis induced by switching to low potassium (Fig.2). The protective effect of ZnEgr1 appears to be attributable to its blockade of endogenous Egr family members, because the corresponding zinc finger domain from Egr3, ZnEgr3, mimics this effect, whereas a structurally similar zinc finger DNA binding domain from Sp1, which does not bind to the ERE with high affinity, does not (Nardelli et al., 1991).
Fig. 2.

ZnEgr constructs block granule cell death induced by potassium deprivation. A, Quantitative analysis. In control cultures, lowering extracellular potassium concentration to 5 mm (Low K) from 25 mm(High K) greatly increases the percentage of apoptotic cells (p < 0.003). Transfection with either ZnEgr1 or ZnEgr3 blocks the increase in apoptosis elicited by potassium deprivation (Low K/control vs Low K/ZnEgr1,p < 0.008; Low K/control vs Low K/ZnEgr3,p < 0.01), whereas ZnSp1 does not. At least 200 cells were counted in each group. Scoring of GFP-positive cells was performed 2 d after initiating potassium deprivation.B, Hoechst staining of GFP-positive cells. Left column shows examples of GFP-positive granule cells. Note the presence of extensive neuronal processes studded with strongly fluorescent varicosities, typical of granule cells in culture.Right column shows the Hoechst staining of the GFP-positive cell shown in the same row along with a few surrounding GFP-negative cells. Note that potassium deprivation produces an increase in the percentage of cells with pyknotic nuclei that display stronger fluorescence, characteristic of apoptotic cells. However, the GFP-positive neuron shown from cultures transfected with ZnEgr1 and switched to 5 mm potassium (Low K+ ZnEgr1) displays dim, diffuse Hoechst staining characteristic of normal cells. Cultures shown in thefirst and third rows were transfected with an empty vector instead of the ZnEgr1 expression plasmid.C, Effects of Egr1 and ZnEgr1 at 1 d after potassium deprivation. Cells were transfected with ZnEgr1, Egr1, or empty vector (Control) and then either maintained in 25 mm KCl (High K) or switched to 5 mm potassium (Low K). One day later, GFP-positive cells were scored as apoptotic or normal. One hundred twenty cells were scored for each group. Transfection with Egr1 potentiated the apoptotic response (control/Low K vs Egr1/Low K,p < 0.03), whereas ZnEgr1 conferred protection (control/Low K vs ZnEgr1/Low K, p < 0.02).
Because these studies implicated Egr1 or other Egr family members in mediating apoptosis in this paradigm, we wanted to check whether increased expression of Egr family members might be sufficient to activate the apoptotic process. However, transfection with a full-length Egr1 expression construct did not induce apoptosis (control, 10 ± 7%; Egr1, 16 ± 10%), indicating that elevated expression of Egr family members acts in concert with other factors to induce apoptosis in these cells. As a positive control to check that the Egr1 expression construct is effective in this preparation, we confirmed that it stimulated the ERE reporter by ∼10-fold over control levels (data not shown).
To assess whether Egr1 might potentiate the apoptotic response to potassium deprivation, we examined its effect 24 hr after initiating this apoptotic stimulus. At this time point, Egr1 augmented the apoptotic response induced by potassium deprivation. In addition, we found that ZnEgr1, which confers protection after 2 d (Fig.2A), also does so at the 1 d time point (Fig.2C).
To characterize further the role of the Egr family in apoptotic signaling, we examined whether it is also involved in apoptosis induced in these cells by another stimulus. Previous studies have demonstrated that immature granule cells undergo apoptosis when exposed to high concentrations of AraC (Dessi et al., 1995; Courtney and Coffey, 1999). In contrast to the results obtained in the potassium deprivation paradigm, AraC did not trigger an increase in the activity of the ERE reporter and ZnEgr1 did not inhibit cell death induced by AraC (Fig.3A,B).
Fig. 3.
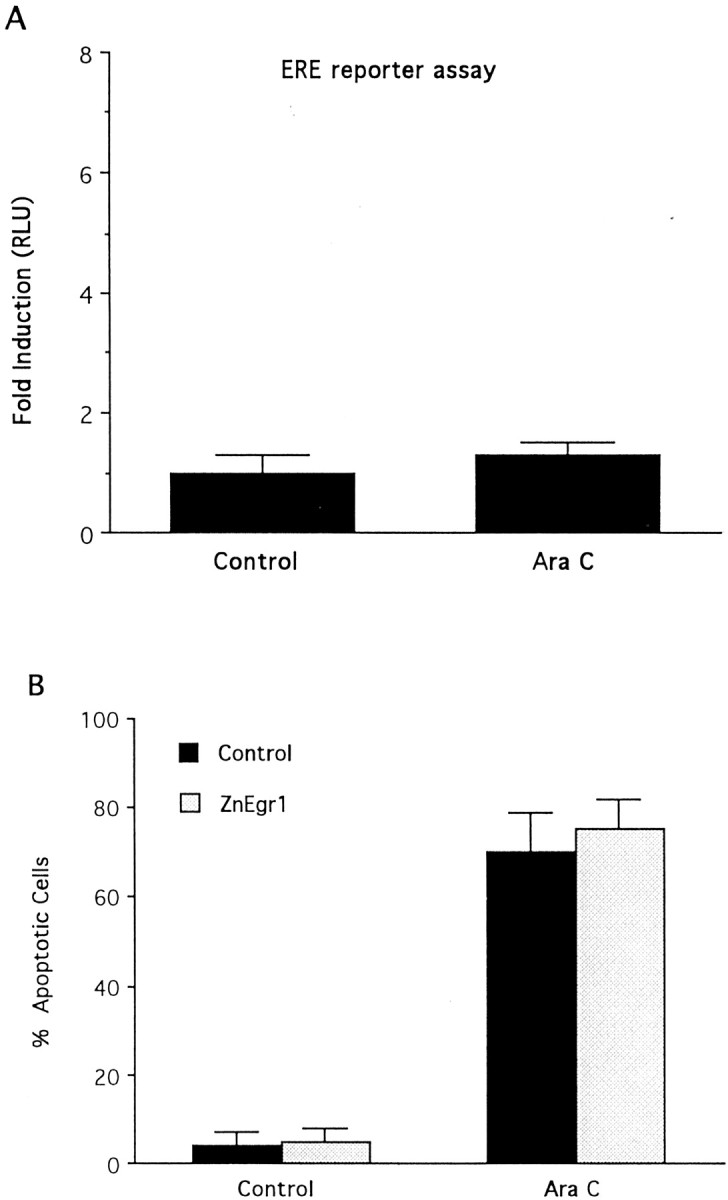
Egr proteins do not mediate apoptosis induced in immature granule cells by cytosine arabinoside. A, AraC (500 μm) does not stimulate ERE reporter activity. Cells were transfected with the ERE reporter plasmid and then treated with either AraC (500 μm) or control medium. Eight hours later, cell extracts were harvested for luciferase assays.B, ZnEgr1 does not protect immature granule cells from apoptosis induced by AraC (500 μm). Cells were transfected with GFP and either empty vector or ZnEgr1. Two days after AraC treatment, GFP-positive cells were scored as apoptotic or normal based on Hoechst staining. Two hundred cells were scored in each group.
ZnEgr1 protects by acting upstream of c-Jun activation
Because ZnEgr1 confers protection against apoptosis induced by potassium deprivation but not AraC, these studies suggest that it acts on signaling pathways involved selectively in mediating apoptosis triggered by potassium deprivation. Comparison of the signaling pathways implicated in these apoptotic paradigms revealed that one salient difference is the involvement of the c-Jun N-terminal protein kinase (JNK)–c-Jun pathway in apoptosis induced by potassium withdrawal but not by AraC (Miller and Johnson, 1996; Tanabe et al., 1998; Watson et al., 1998; Courtney et al., 1999; Le-Niculescu et al., 1999). Accordingly, we started to analyze the site of action of ZnEgr1 by assessing whether it blocks apoptosis mediated by a constitutively active c-Jun construct, c-Jun(Asp). In contrast to its protective effect in the potassium deprivation paradigm, ZnEgr1 did not inhibit cell death induced by c-Jun(Asp) (Fig.4).
Fig. 4.

Effect of ZnEgr1 on apoptosis induced by c-Jun(Asp). Transfection of granule cells with ZnEgr1, which protects these cells from apoptosis induced by potassium deprivation, does not reduce the percentage of cells scored as apoptotic after transfection with a constitutively active c-Jun construct, c-Jun(Asp) [control vs Jun(Asp), p < 0.01]. Cells were scored 2.5 d after transfection with an empty vector (Control), c-Jun(Asp), or both c-Jun(Asp) and ZnEgr1 [Jun(Asp) + ZnEgr1]. Over 200 cells were scored in each group.
Because ZnEgr1 blocks cell death induced by potassium deprivation, which is mediated by c-Jun, but does not block apoptosis induced by c-Jun(Asp), we reasoned that its protective effects may be mediated by acting upstream of c-Jun to block its activation. To test this inference, we examined the effect of ZnEgr1 on two reporter assays that are useful for monitoring activation of c-Jun. In one, we used a conventional AP-1 reporter that would be sensitive to changes in either the levels of c-Jun or its phosphorylation. In the other, we used a c-Jun/GAL4 reporter system that monitors stimulation of c-Jun-mediated transcription mediated by phosphorylation of its N-terminal activation domain. In this assay, cells are transfected with both a GAL4 reporter plasmid and a plasmid encoding a fusion protein composed of the GAL4 DNA binding domain and the N-terminal activation domain of c-Jun. Consistent with previous studies demonstrating that c-Jun activation plays a key role in this apoptosis paradigm, we found that potassium deprivation increases the activity of both of these reporters (Fig.5A,B). Furthermore, we found that ZnEgr1 abolished these increases, suggesting that it confers protection by blocking c-Jun activation. To control for the possibility that ZnEgr1 might exert a nonspecific inhibitory effect on these reporter assays, we confirmed that it does not suppress activation of the AP-1 reporter by c-Jun(Asp) (Fig. 5C), a finding that is also consistent with its inability to block apoptosis induced by this construct (Fig. 4).
Fig. 5.

Effect of Egr1 and ZnEgr1 on c-Jun-mediated transcription. A, ZnEgr1 blocks stimulation of AP-1 reporter activity induced by potassium deprivation. All groups were transfected with the AP-1 reporter plasmid and either ZnEgr1 or empty vector. After transfection, cells were either maintained in 25 mm KCl (High K) or switched to 5 mm KCl (Low K). Cell extracts were harvested for luciferase assays 6 hr later. Control versus Low K,p < 0.02; Low K versus Low K + ZnEgr1,p < 0.09. B, ZnEgr1 blocks activation of c-Jun induced by potassium deprivation. All groups of cells were transfected with the GAL4/c-Jun reporter system plasmids and either ZnEgr1 or empty vector. Potassium deprivation stimulates GAL4/c-Jun reporter activity (control vs Low K, p< 0.02), and this effect is blocked by ZnEgr1 (Low K vs Low K + ZnEgr1, p < 0.04). C, ZnEgr1 does not inhibit the ability of c-Jun(Asp) to stimulate AP-1 reporter activity. Cells were transfected with the AP-1 reporter plasmid and empty vector (control), c-Jun(Asp), or both c-Jun(Asp) with ZnEgr1. Two days after transfection, cell extracts were harvested for luciferase assays. c-Jun(Asp) stimulates AP-1 reporter activity [control vs c-Jun(Asp), p < 0.02]; however, this response is not blocked by cotransfection with ZnEgr1. D, Effect of Egr1 on c-Jun activation. Cells were transfected with the Gal4/c-Jun reporter system plasmids and either Egr1 or empty vector. Two days after transfection, some cells were switched to Low K, as indicated. Cell extracts were harvested for luciferase assays 6 hr later. As shown in B, potassium deprivation stimulates GAL4/c-Jun reporter activity. Egr1 overexpression augments the response to Low K; however, the difference between these two groups does not meet the conventional criterion for statistical significance (Low K vs Low K + Egr1, p < 0.065).
Together, these findings indicate that endogenous Egr family members induced in this paradigm play a critical role in activating c-Jun. To assess this inference directly, we examined the effect of Egr1 on the GAL4/c-Jun reporter. Consistent with our observation that Egr1 by itself does not trigger apoptosis in these cells, it is also unable to activate this reporter in cells maintained in elevated potassium. Because we had found that Egr1 enhances the apoptotic effect of potassium deprivation (Fig. 2C), we also assessed whether it might potentiate the increase in this reporter assay elicited by switching to low-potassium medium (Fig. 5D). Consistent with this possibility, we found that Egr1 elevates the level of GAL4/c-Jun reporter activity above that triggered by potassium deprivation. However, this result did not reach the conventional threshold used to assess statistical significance (p < 0.065).
ZnEgr1 acts downstream of MEKK1
Although the signaling pathways linking potassium deprivation to c-Jun activation have not been defined in great detail, it is widely assumed that they involve activation of one or more the kinase cascades capable of activating c-Jun. One candidate upstream kinase implicated in this process is MEKK1, because it triggers c-Jun activation and induces apoptosis in neurons (Eilers et al., 1998). Accordingly, to help define which aspect of the signaling pathway upstream of c-Jun is blocked by ZnEgr1, we examined the effect of this Egr inhibitor construct on these effects of MEKK1. We found that ZnEgr1 is highly effective in protecting cerebellar granule cells from apoptosis induced by MEKK1 and that it also inhibits the ability of MEKK1 to stimulate the GAL4/c-Jun reporter (Fig.6A,B). These inhibitory effects of ZnEgr1 do not appear to be attributable to an inadvertent suppression of MEKK1 expression, because ZnEgr1 does not diminish MEKK1 activation of an ATF2 reporter (Fig. 6C). Thus, these findings indicate that ZnEgr1 selectively blocks c-Jun activation by interfering with the signaling pathway linking upstream kinases, such as MEKK1, to c-Jun activation.
Fig. 6.

Role of Egr proteins in apoptosis and c-Jun activation induced by MEKK1. A, ZnEgr1 suppresses apoptosis induced by transfection of granule cells with a constitutively active MEKK1 construct. Cells were transfected with a GFP expression vector, along with one or more of the following plasmids, as indicated below each bar: a constitutively active MEKK1 construct (MEKK), ZnEgr1, or empty vector (Control). Two days after transfection, GFP-positive cells (120 per group) were scored as normal or apoptotic. MEKK mimics the ability of potassium deprivation to induce apoptosis (control vs MEKK, p < 0.002). This effect is blocked by cotransfection with ZnEgr1 (MEKK vs MEKK + ZnEgr1,p < 0.003). ZnEgr1 does not induce apoptosis.B, ZnEgr1 inhibits the ability of MEKK1 to activate c-Jun. Cells were transfected with the GAL4/c-Jun reporter system plasmids and the plasmids indicated below each bar. Eight hours after transfection, cell extracts were harvested for luciferase assays. MEKK1 stimulates GAL4/c-Jun reporter activity (control vs MEKK, p < 0.02). This effect is blocked by ZnEgr1 (MEKK vs MEKK + ZnEgr1, p < 0.02). Coexpression of Egr1 with MEKK does not further potentiate reporter activity. C, ZnEgr1 does not inhibit MEKK1-induced activation of ATF2. Cells were transfected with the ATF2 reporter system plasmids and the plasmids indicated below eachbar. Eight hours after transfection, cells were harvested for luciferase assays. MEKK stimulates ATF2 reporter activity (control vs MEKK, p < 0.009). However, this effect is not inhibited by cotransfection with ZnEgr1. D, MEKK1 strongly stimulates ERE reporter activity. Cells were transfected with the ERE reporter plasmid and either empty vector or a constitutively active MEKK1 plasmid (MEKK). Eight hours after transfection, cells were harvested for luciferase assays. Control versus MEKK, p < 0.006.
In these studies, we have used ZnEgr1 as a tool to obtain information about the role of endogenous Egr family members in apoptosis. The ability of ZnEgr1 to block activation of the c-Jun/GAL4 reporter induced by MEKK1, as found for potassium deprivation, suggests that this kinase is able to induce expression of endogenous Egr family members that play a critical role in activating c-Jun. We obtained direct support for this inference by demonstrating that MEKK1 strongly increases the activity of the ERE reporter (Fig. 6D). However, cotransfection of Egr1 with MEKK1 does not significantly potentiate the ability of MEKK1 to stimulate the c-Jun/GAL4 reporter (Fig. 6B), presumably because the effect of heterologously expressed Egr1 is occluded by the strong induction of MEKK1 of endogenous Egr family members.
DISCUSSION
The two major findings of this study are as follows: (1) that Egr inhibitor constructs block granule cell apoptosis induced by potassium deprivation, and 2) that they act upstream of c-Jun activation, an essential step in engaging the apoptotic process in this paradigm. Because the signaling pathways linking survival factor withdrawal to c-Jun activation are poorly understood, these findings provide an important new clue to deciphering these critical, early steps in the apoptotic process.
Several lines of evidence support our conclusion that endogenous Egr family members play a critical role in activating the cell death program in this paradigm. First, Egr1 protein levels are increased in this paradigm, as is the activity of an ERE reporter. Second, ZnEgr1, which confers protection, blocks the increase in ERE reporter activity. Third, this protective effect is mimicked by ZnEgr3, which also binds to the ERE consensus sequence with a high degree of specificity, but not ZnSp1, which recognizes a distinct response element. Fourth, Egr1 overexpression potentiates the apoptotic response induced by potassium deprivation.
Although we have demonstrated that Egr1 is induced in this paradigm, our results do not rule out the possibility that other Egr family members may be involved in this apoptotic process as well. Because previous studies have demonstrated that neuronal stimulation often induces expression of multiple Egr family members (O'Donovan et al., 1999), we have conducted gel-shift studies to check for the presence of other Egr family members in this paradigm. These pilot studies indicate that both Egr1 and Egr3 are induced by potassium deprivation (Y. Levkovitz, unpublished observations). Because the Egr inhibitor constructs used in this study would be expected to block transcription mediated by any of the four Egr family members, our studies leave open the possibility that multiple Egr family members are involved in this neuronal apoptosis paradigm.
In addition to providing compelling evidence that the Egr family plays a critical role in mediating apoptosis in the potassium deprivation paradigm, we have also made substantial inroads into understanding where it interacts with apoptotic signaling pathways. In particular, our results localize its site of action upstream of c-Jun. This conclusion is based on multiple lines of evidence. First, ZnEgr1 does not block apoptosis induced by c-Jun(Asp), a constitutively active version of c-Jun. Second, ZnEgr1 blocks the potassium deprivation-induced increase in two reporter assays used to monitor activation of c-Jun. Third, ZnEgr1 inhibits the ability of MEKK1 to induce apoptosis and activate the c-Jun/GAL4 reporter. Thus, together, these results indicate that the Egr family plays an essential role in enabling upstream signaling events, such as potassium deprivation or MEKK1 activation, to activate c-Jun-mediated transcription (Fig.7).
Fig. 7.
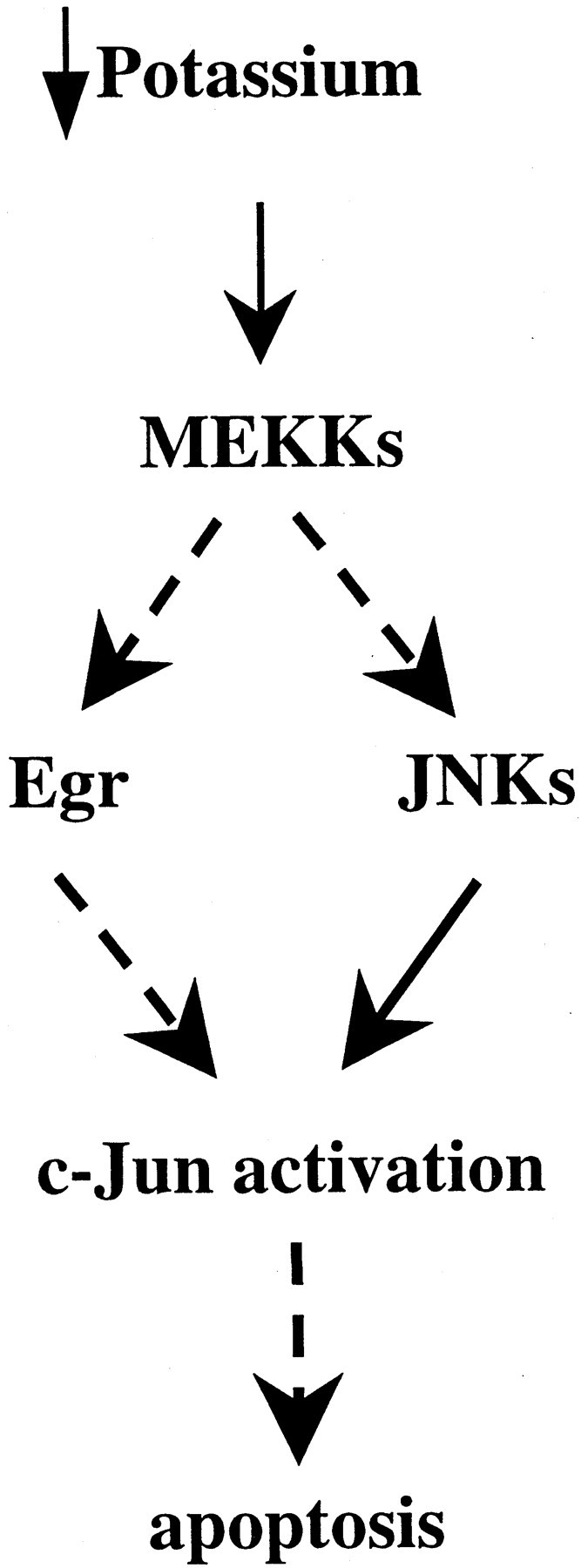
Model of apoptotic signaling pathway linking potassium deprivation to c-Jun activation: site of the protective action of ZnEgr1. As shown in this schematic diagram, our findings indicate that ZnEgr protects granule cells from apoptosis induced by potassium deprivation by suppressing c-Jun activation. Because ZnEgr1 does not protect against the apoptotic effects of c-Jun(Asp), we infer that it acts upstream of c-Jun activation. This inference is corroborated by studies demonstrating that ZnEgr1 blocks c-Jun activation triggered by potassium deprivation. Furthermore, we have placed ZnEgr1 downstream of MEKKs because it blocks the ability of MEKK1 to stimulate c-Jun activation and induce apoptosis. Because these findings indicate that Egr1 or other Egr family members are necessary for mediating c-Jun activation by MEKK1, but Egr1 is not sufficient to stimulate c-Jun reporter activity on its own, we hypothesize that it acts in concert with JNK kinases downstream of MEKKs to activate c-Jun. Thus, according to this model, MEKK1 is able to stimulate c-Jun by triggering both induction of Egr proteins and activation of JNKs.
Although it is unclear whether endogenous MEKK1 is involved in apoptotic signaling in this paradigm, the ability of ZnEgr1 to block the actions of transfected MEKK1 suggest that it acts on a portion of the signaling pathway between MEKK1 and c-Jun activation. Because recent studies have implicated p38 MAPK in playing a critical role in c-Jun phosphorylation in this apoptotic paradigm (Yamagishi et al., 2001), it will be interesting in future experiments to determine whether Egr target genes are involved in activation of p38 elicited by potassium deprivation. Conceivably, these could act by blocking dephosphorylation of p38, thereby enhancing or prolonging its activity. Precedent for the proposal that inducible genes may regulate the MAPK signaling pathway is provided by MKP, an inducible MAPK phosphatase (Hutter et al., 2000; Sanchez-Perez et al., 2000). Another plausible scenario is that Egr family members control expression of scaffolding proteins, such as JIPs (JNK interacting proteins), which plays a central role in allowing MEKK1 or related kinases to activate c-Jun efficiently (Yasuda et al., 1999; Harding et al., 2001).
Although we favor the view that ZnEgr1 acts by blocking ERE-mediated changes in transcription, it is conceivable that ZnEgr1 blocks these actions of MEKK1 by acting as a substrate of MEKK1 or one of its downstream kinases and in this way interferes with c-Jun activation. However, ZnEgr1 does not block MEKK1 activation of ATF2, which argues against this possibility. Furthermore, if this were the case, then one would expect that full-length Egr1, which contains the ZnEgr1 domain, would exert a similar effect. Because this is not the case, this explanation seems unlikely. Accordingly, we propose that ZnEgr1 confers protection by opposing the action of endogenous Egr family members on one or more ERE-regulated target genes that play an essential role in allowing potassium deprivation to increase c-Jun-mediated transcription.
Because it is presumably advantageous to target early steps in apoptotic signaling pathways as a means of maximizing protection of neurons from cellular damage or death, these studies indicate that Egr family members or their target genes may be attractive candidates for developing agents that combat neuronal apoptosis. Because recent studies emphasize that apoptotic mechanisms exhibit cell-specific variations, it will also be important in future studies to determine whether Egr family members play a similar role in other neuronal apoptosis paradigms that are critically dependent on c-Jun activation (Estus et al., 1994; Ham et al., 1995; Yang et al., 1997; Behrens et al., 1999).
Footnotes
This work was supported by grants from the National Institute on Drug Abuse and the National Alliance for Research on Schizophrenia and Depression. We thank N. Perkins and D. Bohmann for providing plasmids and Weizhu Tang for expert technical assistance.
Correspondence should be addressed to Dr. J. M. Baraban, Department of Neuroscience, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, Baltimore, MD 21205. E-mail:jbaraban@jhmi.edu.
REFERENCES
- 1.Ahn S, Ginty DD, Linden DJ. A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron. 1999;23:559–568. doi: 10.1016/s0896-6273(00)80808-9. [DOI] [PubMed] [Google Scholar]
- 2.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 3.Cao X, Koski RA, Gashler A, McKiernan M, Morris CF, Gaffney R, Hay RV, Sukhatme VP. Identification and characterization of the Egr-1 gene product, a DNA-binding zinc finger protein induced by differentiation and growth signals. Mol Cell Biol. 1990;10:1931–1939. doi: 10.1128/mcb.10.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Catania MV, Copani A, Calogero A, Ragonese GI, Condorelli DF, Nicoletti F. An enhanced expression of the immediate early gene, Egr-1, is associated with neuronal apoptosis in culture. Neuroscience. 1999;91:1529–1538. doi: 10.1016/s0306-4522(98)00544-2. [DOI] [PubMed] [Google Scholar]
- 5.Chapman NR, Perkins ND. Inhibition of the RelA(p65) NF-kappaB subunit by Egr-1. J Biol Chem. 2000;275:4719–4724. doi: 10.1074/jbc.275.7.4719. [DOI] [PubMed] [Google Scholar]
- 6.Christy B, Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proc Natl Acad Sci USA. 1989;86:8737–8741. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtney MJ, Coffey ET. The mechanism of Ara-C-induced apoptosis of differentiating cerebellar granule neurons. Eur J Neurosci. 1999;11:1073–1084. doi: 10.1046/j.1460-9568.1999.00520.x. [DOI] [PubMed] [Google Scholar]
- 8.Das A, Chendil D, Dey S, Mohiuddin M, Mohiuddin M, Milbrandt J, Rangnekar VM, Ahmed MM. Ionizing radiation down-regulates p53 protein in primary Egr-1 −/− mouse embryonic fibroblast cells causing enhanced resistance to apoptosis. J Biol Chem. 2001;276:3279–3286. doi: 10.1074/jbc.M008454200. [DOI] [PubMed] [Google Scholar]
- 9.Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- 10.Dessi F, Pollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C. Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem. 1995;64:1980–1987. doi: 10.1046/j.1471-4159.1995.64051980.x. [DOI] [PubMed] [Google Scholar]
- 11.D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 13.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death: identification of c-Jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 16.Grimes CA, Jope RS. Cholinergic stimulation of early growth response-1 DNA binding activity requires protein kinase C and mitogen-activated protein kinase kinase activation and is inhibited by sodium valproate in SH-SY5Y cells. J Neurochem. 1999;73:1384–1392. doi: 10.1046/j.1471-4159.1999.0731384.x. [DOI] [PubMed] [Google Scholar]
- 17.Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci USA. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hallahan DE, Dunphy E, Virudachalam S, Sukhatme VP, Kufe DW, Weichselbaum RR. c-Jun and Egr-1 participate in DNA synthesis and cell survival in response to ionizing radiation exposure. J Biol Chem. 1995;270:30303–30309. doi: 10.1074/jbc.270.51.30303. [DOI] [PubMed] [Google Scholar]
- 19.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 20.Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM, Uney JB. Inhibition of JNK by overexpression of the JNK binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem. 2001;276:4531–4534. doi: 10.1074/jbc.C000815200. [DOI] [PubMed] [Google Scholar]
- 21.Huang R-P, Fan Y, deBelle I, Ni Z, Matheny W, Adamson ED. Egr-1 inhibits apoptosis during the UV response: correlation of cell survival with Egr-1 phosphorylation. Cell Death Differ. 1998;5:96–106. doi: 10.1038/sj.cdd.4400322. [DOI] [PubMed] [Google Scholar]
- 22.Hutter D, Chen P, Barnes J, Liu Y. Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: critical role of the p38 C-terminal domain in its negative regulation. Biochem J. 2000;352:155–163. [PMC free article] [PubMed] [Google Scholar]
- 23.Lamprecht R, Hazvi S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long- but not short-term conditioned taste aversion memory. J Neurosci. 1997;17:8443–8450. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret F-X, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leppa S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levkovitz Y, O'Donovan KJ, Baraban JM. Blockade of NGF-induced neurite outgrowth by a dominant-negative inhibitor of the Egr family of transcription regulatory factors. J Neurosci. 2001;21:45–52. doi: 10.1523/JNEUROSCI.21-01-00045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller TM, Johnson EM., Jr Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muthukkumar S, Nair P, Sells SF, Maddiwar NG, Jacob RJ, Rangnekar VM. Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma cell line A375–C6. Mol Cell Biol. 1995;15:6262–6272. doi: 10.1128/mcb.15.11.6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nardelli J, Gibson TJ, Vesque C, Charnay P. Base sequence discrimination by zinc finger DNA binding domains. Nature. 1991;349:175–178. doi: 10.1038/349175a0. [DOI] [PubMed] [Google Scholar]
- 30.O'Donovan KJ, Baraban JM. Major Egr3 isoforms are generated via alternate translation start sites and differ in their abilities to activate transcription. Mol Cell Biol. 1999;19:4711–4718. doi: 10.1128/mcb.19.7.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Donovan KJ, Tourtellotte WG, Milbrandt J, Baraban JM. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–173. doi: 10.1016/s0166-2236(98)01343-5. [DOI] [PubMed] [Google Scholar]
- 32.O'Donovan KJ, Levkovitz Y, Ahn D, Baraban JM. Functional comparison of Egr3 transcription factor isoforms: identification of an activation domain in the N-terminal segment absent from Egr3β, a major isoform expressed in brain. J Neurochem. 2000;75:1352–1357. doi: 10.1046/j.1471-4159.2000.0751352.x. [DOI] [PubMed] [Google Scholar]
- 33.Park JA, Koh JY. Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- 34.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez-Perez I, Martinez-Gomariz M, Williams D, Keyse SM, Perona R. CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene. 2000;19:5142–5152. doi: 10.1038/sj.onc.1203887. [DOI] [PubMed] [Google Scholar]
- 36.Swirnoff AH, Milbrandt J. DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol Cell Biol. 1995;15:2275–2287. doi: 10.1128/mcb.15.4.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanabe H, Eguchi Y, Shimizu S, Martinou J-C, Tsujimoto Y. Death-signalling cascade in mouse cerebellar granule neurons. Eur J Neurosci. 1998;10:1403–1411. doi: 10.1046/j.1460-9568.1998.00148.x. [DOI] [PubMed] [Google Scholar]
- 38.Thangnipon W, Kingsbury A, Webb M, Balazs R. Observations on rat cerebellar cells in vitro: influence of substratum, potassium concentration and relationship between neurones and astrocytes. Brain Res. 1983;313:177–189. doi: 10.1016/0165-3806(83)90215-8. [DOI] [PubMed] [Google Scholar]
- 39.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 40.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamagishi S, Yamada M, Ishikawa Y, Matsumoto T, Ikeuchi T, Hatanaka H. p38 mitogen-activated protein kinase regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J Biol Chem. 2001;276:5129–5133. doi: 10.1074/jbc.M007258200. [DOI] [PubMed] [Google Scholar]
- 42.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 43.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]