

Abstract
Previous studies have shown that transgenic (Tg) mice overexpressing human tau protein develop filamentous tau aggregates in the CNS. The most abundant tau aggregates are found in spinal cord and brainstem in which they colocalize with neurofilaments (NFs) as spheroids in axons. To elucidate the role of NF subunit proteins in tau aggregate formation and to test the hypothesis that NFs are pathological chaperones in the formation of intraneuronal tau inclusions, we crossbred previously described tau (T44) Tg mice overexpressing the smallest human tau isoform with knock-out mice devoid of NFL (NFL−/−) or NFH (NFH−/−). Depletion of NF subunit proteins from the T44 mice (i.e., T44;NFL−/− and T44;NFH−/−), in particular NFL, resulted in a dramatic decrease in the total number of tau-positive spheroids in spinal cord and brainstem. Concomitant with the reduction in spheroid number, the bigenic mice showed delayed accumulation of insoluble tau protein in the CNS, increased viability, reduced weight loss, and improved behavioral phenotype when compared with the single T44 Tg mice. These results imply that NFs are pathological chaperones in the development of tau spheroids and suggest a role for NFs in the pathogenesis of neurofibrillary tau lesions in neurodegenerative disorders that contain both NFs and tau proteins.
Keywords: tau, neurofilaments, neurodegeneration, neurofibrillary pathology, animal models, cytoskeleton
Filamentous tau inclusions, accompanied by extensive neuron loss and gliosis, are the neuropathological hallmarks of an expanding family of neurodegenerative diseases that are now collectively known as tauopathies (Lee et al., 2001). Prototypical tauopathies are exemplified by frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), amyotrophic lateral sclerosis/parkinsonism–dementia complex (ALS/PDC), Alzheimer's disease (AD), and progressive supranuclear palsy (PSP). Like other tauopathies, FTDP-17, PSP, AD, and ALS/PDC are characterized by numerous inclusions formed by aggregated paired helical filaments (PHFs) and/or straight filaments composed of aberrantly phosphorylated tau proteins (PHF-tau) in widespread regions of the CNS (Hong et al., 2000). The discovery of tau gene mutations in FTDP-17 kindreds provides unique opportunities to elucidate the disease mechanisms that underlie FTDP-17 as well as related brain disorders characterized by abundant insoluble intracellular filamentous tau inclusions, including AD (Clark et al., 1998; Hong et al., 1998; Hutton et al., 1998; Poorkaj et al., 1998;Spillantini et al., 1998). The FTDP-17 mutations occur in exons and introns of the tau gene, and they may cause FTDP-17 by altering the functions or levels of specific tau isoforms in the CNS (Hong et al., 1998; Hutton et al., 1998; D'Souza et al., 1999).
To begin elucidating the role of tau inclusions in the pathogenesis of tauopathies, we generated transgenic (Tg) mice that overexpressed the shortest human brain tau isoform (T44) in CNS (Ishihara et al., 1999,2001). As reported previously, the T44 Tg mice acquired age-dependent CNS pathology similar to the filamentous tau inclusions found in FTDP-17 and ALS/PDC, including insoluble, hyperphosphorylated intraneuronal aggregates formed by tau-immunoreactive filaments (Ishihara et al., 1999). These inclusions, mostly in the form of spheroids, were abundant in spinal cord neurons, in which they were associated with axon degeneration and reduced axonal transport in ventral roots, as well as spinal cord gliosis and motor weakness. Thus, these Tg mice recapitulate some of, but not all, key aspects of prototypical tauopathies. For example, tau-positive spheroids are strongly positive for neurofilaments (NFs) in the spinal cord and brainstem of T44 Tg mice. Significantly, NF subunit proteins have long been associated with pathogenic protein aggregates in a number of neurodegeneration diseases. For example, spheroids in the T44 Tg mice and in the spinal cord of ALS/PDC patients contain both tau and NF-immunoreactive filaments, and all three NF subunits (NFL, NFM, and NFH) are found in neurofibrillary tangles (NFTs) at late stages of the disease in AD patients (Schmidt et al., 1989; Ishihara et al., 1999). NFs are also detected in Lewy bodies in which they coexist with α-synuclein (Tu et al., 1998). Based on these and other observations, we hypothesize that NFs may play a role as “pathological chaperones” (Wisniewski and Frangione, 1992) in facilitating the aggregation of tau in our Tg mice and in the human neurodegenerative diseases discussed above.
To test this hypothesis, we generated bigenic mice overexpressing tau protein but lacking NFL (Zhu et al., 1997) or NFH (Elder et al., 1998) by crossbreeding T44 Tg mice with NFL knock-out (NFL−/−) or NFH knock-out (NFH−/−) mice, respectively. Our studies showed that depletion of NF subunit proteins, especially NFL, in T44 Tg mice reduces or delays the pathological phenotype of T44 Tg mice as demonstrated histologically, biochemically, and clinically. These results show that NFs could play an important role as pathological chaperones in the pathogenesis of tau aggregates with both tau and NF proteins in a number of neurodegenerative tauopathies.
MATERIALS AND METHODS
Generation of mice. Tau Tg mice overexpressing the shortest human brain tau isoform (T44) were generated as described previously (Ishihara et al., 1999). We have produced stable transgenic lines (line 7, line 27, and line 43) of T44 mice. Although the heterozygous line 27 mice had the highest level of Tg tau, they are not viable beyond 3 months, and homozygous mice generated from each of the three lines died in utero or within 3 months postnatal. Therefore, all of the T44 Tg mice used in this study were heterozygotes from line 7 unless specified.
T44 Tg mice were crossbred with either NFL−/− or NFH−/− mice to generate bigenic T44;NFL−/− and T44;NFH−/− mice. The characterization of the T44 Tg mice, as well as the NFL−/− and NFH−/− knock-out mice, has been well documented previously (Elder et al., 1998; Ishihara et al., 1999, 2001; Julien, 1999). As a consequence of crossbreeding between T44 Tg and NFL or NFH knock-out mice, lines of mice with eight different genotypes were generated, including wild-type (WT) littermates of T44 Tg mice (Fig.1). The genotype of these mice was identified by Southern blot analysis of tail DNA.
Fig. 1.

Analysis of protein expression in cortices. Western blot analysis of tau, NFs, and α-tubulin isolated from cortices of the different lines of mice. Six-month-old mice from each group were used. 17026, an anti-recombinant tau antibody that recognizes both human and mouse tau, shows the expression level of tau in each line of mice. Endogenous mouse tau protein levels are comparable in WT, NFL−/−, and NFH−/− mice, and overexpressed human tau protein levels in T44 Tg, T44 Tg;NFL+/−, T44;NFL−/−, T44;NFH+/−, and T44;NFH−/− mice were ∼10-fold higher than endogenous mouse tau. NFH and NFM protein levels were decreased ∼85 and 50%, respectively, in the cortices of NFL−/− and T44 Tg;NFL−/− mice, whereas the level of α-tubulin was dramatically increased compared with WT mice. NFL protein levels were decreased ∼30% in NFH−/− and T44 Tg;NFH−/− mice without a change in NFM protein levels. Equal amounts (10 μg for tau; 20 μg for NFs and α-tubulin) of mouse cortical samples were loaded on each gel lane.
Immunohistochemical analyses. Tg mice aged 1–15 months were lethally anesthetized and perfused transcardially using fixative including 70% ethanol in isotonic saline and 4% paraformaldehyde in PBS as described previously (Ishihara et al., 1999) in accordance with protocols approved by University of Pennsylvania University Laboratory Animal Research. Immunohistochemistry were performed on representative 6 μm paraffin sections from brain, spinal cord, and spinal roots. Three animals from each mouse line and at each specific age were used for the immunohistochemical studies. Anti-NF antibodies were used to detect the presence or absence of NF subunit proteins in the CNS of Tg mice, and sections were also stained with anti-tau antibodies to localize the tau aggregates (Table 1). In addition, mouse monoclonal antibodies (mAbs) to α- and β-tubulin and antibodies to α-internexin and to peripherin were used to assess whether or not these molecules also accumulate in tau aggregates (Table 1).
Table 1.
Summary of the antibodies used in this study
| Antibody | Antigen | References or company |
|---|---|---|
| T14, mouse monoclonal | Human tau | Kosik et al., 1988 |
| 17026, rabbit polyclonal | Largest human recombinant tau | Ishihara et al., 1999 |
| T1, mouse monoclonal | Nonphosphorylated tau | Binder et al., 1985; Szendrei et al., 1993 |
| T46, mouse monoclonal | Tau | Mawal-Dewan et al., 1994 |
| T49, mouse monoclonal | Rodent tau | Mawal-Dewan et al., 1994 |
| PHF1, mouse monoclonal | Phosphorylated tau | Greenberg and Davies, 1990; Greenberg et al., 1992; Lang et al., 1992; Otvos et al., 1994 |
| PHF6, mouse monoclonal | Phosphorylated tau | Hoffmann et al., 1997 |
| AT8, mouse monoclonal | Phosphorylated tau | Goedert et al., 1994; Matsuo et al., 1994 |
| AT270, mouse monoclonal | Phosphorylated tau | Goedert et al., 1994; Matsuo et al., 1994 |
| 12E8, mouse monoclonal | Phosphorylated tau | Seubert et al., 1995 |
| T3P, rabbit polyclonal | Phosphorylated tau | Lee et al., 1991 |
| DP1, rat monoclonal | Nonphosphorylated NFH | Lee et al., 1986 |
| RMdO9, mouse monoclonal | Nonphosphorylated NFH | Carden et al., 1987 |
| RMO24, mouse monoclonal | Phosphorylated NFH | Carden et al., 1987 |
| RMO55, mouse monoclonal | Phosphorylated NFM | Carden et al., 1987 |
| RMO189, mouse monoclonal | Nonphosphorylated NFM | Carden et al., 1987 |
| NFL, rabbit polyclonal | NFL | Tu et al., 1995 |
| α-Tubulin, mouse monoclonal | α-Tubulin | Santa Cruz Biotechnology (Santa Cruz, CA) |
| β-Tubulin, mouse monoclonal | β-Tubulin | Sigma (St. Louis, MO) |
| α-Internexin, mouse monoclonal | α-Internexin | Chemicon (Temecula, CA) |
| Peripherin, mouse monoclonal | Peripherin | Chemicon |
Axonal tau pathology in Tg mice was quantified by counting the number of tau-positive spheroids with a diameter of >5 μm. This quantitative analysis was performed for 20 lumber spinal cord sections from each Tg mouse, and the average number of spheroids per section was used as a representative value. The mean and SD values of the number of spheroids in three Tg mice at the same age were estimated in each line, and the difference in the number of spheroids among all of the lines was examined statistically by one-way ANOVA.
To see colocalization of the cytoskeletal components in the spheroids, selected sections were also double- and triple-labeled by immunofluorescence using anti-tau, anti-NF subunit, and anti-tubulin antibodies (Table 1).
Western blot analysis of Tau, NFs, peripherin, α-internexin, and tubulin expressed in the CNS of Tg mice.Tissues were carefully dissected after mice were lethally anesthetized. The tissues were homogenized in 2 ml/gm ice-cold high salt reassembly buffer (RAB) [0.1 m MES, 1 mm EGTA, 0.5 mmMgSO4, 0.75 m NaCl, 0.02m NaF, 1 mm PMSF, and 0.1% protease inhibitor cocktail (100 μg/ml each of pepstatin A, leupeptin, N-tosyl-l-phenylalanyl cloromethyl ketone, N-tosyl-lisine cloromethyl ketone, soybean trypsin inhibitor, and 100 mm EDTA), pH 7.0] and centrifuged at 50,000 × g for 40 min at 4°C in the Beckman Instruments (Fullerton, CA) TL-100 ultracentrifuge. The pellets were saved for NF–peripherin–internexin analyses. Half of the supernatants were saved for tubulin analyses, and the rest were boiled for 5 min, chilled on ice for 5 min, and recentrifuged at 10,000 × g for 20 min at 4°C, and the supernatant were saved for tau Western blotting. Protein concentration was then determined for the samples using the BCA assay kit (Pierce, Rockford, IL). Equal amounts of samples were subsequently resolved on 7.5% SDS-PAGE gels and transferred onto nitrocellulose membranes. Quantitative Western blot analyses (three animals from each line) were performed by using either [125I]-labeled goat anti-mouse IgG or [125I]-labeled Protein A (NEN, Boston, MA) as secondary antibodies as described previously (Ishihara et al., 1999).
Tau and NF protein solubility in the CNS of Tg mice. To study the solubility of tau and NF proteins in the mouse CNS, cerebral cortical and spinal cord tissues of 1-, 3-, 6-, 9-, and 12-month-old mice in each group (n = 3) were extracted with RAB to generate the RAB-soluble fractions as described above. The pellets were rehomogenized with 1 m sucrose–RAB (0.1m MES, 1 mm EGTA, and 0.5 mm MgSO4, pH 7.0) and centrifuged at 50,000 × g for 20 min at 4°C to eliminate myelin and related lipids. The resulting pellets were extracted with 1 ml/gm radioimmunoprecipitation assay buffer (RIPA) buffer (50 mm Tris, 150 mm NaCl, 1% NP-40, 5 mmEDTA, 0.5% sodium deoxycholate, and 0.1% SDS, pH 8.0) and centrifuged to generate RIPA-soluble samples. Finally, the RIPA-insoluble pellets were reextracted with 70% formic acid (FA) to recover the most insoluble cytoskeletal aggregates. Quantitative Western blot analyses were used to determine tau and NF levels in each fraction.
Transmission electron microscopy. To determine the ultrastructural features of the inclusions, transmission EM (TEM) was performed in T44, T44;NFL−/− mice, as well as WT littermates, at 6 and 12 months of age (n = 6). These mice were deeply anesthetized and killed by intracardiac perfusion with 10 ml of 0.05% glutaraldehyde and 0.5% paraformaldehyde in 0.1m cacodylate buffer, pH 7.4, followed by 50 ml fixative of 2% glutaraldehyde and 2% paraformaldehyde in 0.1m cacodylate buffer. The L5 segments of the spinal cord and ventral root were removed and post-fixed in 2% osmium tetroxide for 60 min at 4°C. After dehydration with graded alcohol and propylene oxide, the blocks were embedded in Epon-812 and polymerized at 60°C for 72 hr. Sixty-five nanometer thin sections were cut and mounted on 200 mesh copper grids, stained with 1% uranyl acetate in 50% ethanol and bismuth subnitrate, and examined with a JEM1010 electron microscope (Jeol, Peabody, MA) at 80 kV.
Preembedding immunoelectron microscopy. To determine components of proteins in the inclusions, preembedding immunoelectron microscopy (immuno-EM) was performed in the T44, T44;NFL−/−, and age-matched WT mice at 6 and 12 months of age (n = 4). These mice weredeeply anesthetized and killed by intracardiac perfusion with 10 ml of 0.05% glutaraldehyde and 0.5% paraformaldehyde in 0.1m cacodylate buffer, pH 7.4, followed by 50 ml fixative of 0.2% glutaraldehyde and 2% paraformaldehyde in 0.1m cacodylate buffer. The L5 segments of the spinal cord and ventral root were removed and post-fixed in fixative of 4% paraformaldehyde, 0.2% glutaraldehyde, and 0.2 picric acid in 0.1m cacodylate buffer overnight. The spinal cords were cut into 50-μm-thick sections with a vibratome, followed by quenching in 0.1% sodium borohydride in Tris-buffered saline for 10 min and treatment for another 10 min with 20% ethanol. The sections were blocked in 5% donor horse serum in PBS with 0.1% cold-water fish skin gelatin and 1% chicken egg albumin for 60 min and then incubated with 17026, the recombinant anti-tau antiserum (dilution of 1:500), in 0.1% bovine serum albumin and PBS overnight at 4°C. By using diaminobenzidine (DAB) plus the silver–gold enhancement immuno-EM method, biotinylated goat anti-rabbit IgG (dilution of 1:100; Vector Laboratories, Houston, TX) secondary antibody was applied for 2 hr at room temperature for each set of sections. After visualizing the DAB-positive staining labeled by routine immuno-EM methods, silver–gold intensification was performed by incubating the sections in silver methenamine developer containing 3% methenamine, 5% silver nitrate, and 1% sodium tetraborate at 60°C for 10 min as described previously (Teclemariam-Mesban et al., 1997). The reaction was stopped with 2% sodium acetate and then stabilized in 3% sodium thiosulphate for 5 min. Gold toning was obtained by incubating the sections in 0.1% gold chloride for 5 min, followed by stabilizing with 3% sodium thiosulfate for 5 min. Sections were fixed within 2% glutaraldehyde in PBS buffer overnight and processed and examined as described above for TEM.
Tail suspension test. The tail suspension test was performed as described by Yamamoto et al. (2000). Briefly, mice from each experimental group (n = 4) were suspended by their tails for 15 sec and videotaped at 5 and 11 months of age. Animals were assessed for clasping score. The test period was divided into 2 sec slots. An animal would receive a score of 1 point if they displayed any abnormal movements during the given time slot. An abnormal movement was defined as dystonic movements of the hindlimbs or a combination of hindlimbs and forelimbs and trunk, during which the limbs were pulled into the body in a manner not observed in WT mice.
Antibodies. The antibodies used in this study are summarized in Table 1.
RESULTS
Generation and characterization of bigenic mice
To generate the mice used in these experiments, T44 Tg mice were crossbred with either NFL−/− or NFH−/− mice. As a consequence of crossbreeding, mice with eight different genotypes i.e., WT, T44, NFL−/−, NFH−/−, T44;NFL+/−, T44;NFL−/−, T44;NFH+/−, and T44;NFH−/−, were generated (Fig. 1). Western blot analyses were conducted to determine whether or not the expression of transgenic human fetal tau or the elimination of NFL and NFH in mice alter the level of expression of the relevant endogenous cytoskeletal proteins in each of the lines. For example, previous studies have shown that, in the absence of NFL, there is a downregulation of NFH and NFM levels and an upregulation of tubulin subunits (Zhu et al., 1997), and our data (Fig. 1) confirmed reduction in both NFH and NFM (85 and 50%, respectively) and an increase in α-tubulin levels in the NFL−/− and T44;NFL−/− mice. Similar to previous studies on NFH−/− mice (Elder et al., 1998), in the absence of NFH, NFL protein levels were decreased by ∼30% in the CNS. However, the endogenous mouse tau protein levels remained unchanged in cortices of WT, NFL−/−, and NFH−/− mice (Fig. 1). The expression of human tau in the bigenic T44;NFL−/− and T44;NFH−/− mice also has no effect on the levels of any of the relevant endogenous cytoskeletal proteins, including peripherin and α-internexin (Fig. 1 and data not shown). The same pattern of protein expression was also observed in the spinal cord of these mice (data not shown). Thus, the expression of the human fetal tau transgenic protein was not altered by crossbreeding the T44 mice with NFL−/− and NFH−/− mice, nor did the expression of the tau transgene affect the pattern of relevant endogenous gene expression in the CNS.
A reduction in the number of spheroids in the CNS of T44 mice lacking NFs
Because the major pathology found in the T44 mice are spheroids containing both human tau and NF subunit proteins, we sought to determine whether or not the removal of NFL or NFH would have any effect on the number and the composition of the spheroids. As reported previously (Ishihara et al., 1999) and as shown in Figure2, the number of tau-positive spheroids in the spinal cord of the T44 mice increased with age until ∼6 months, and they decreased thereafter. The number of spheroids in the T44;NFH+/− and T44;NFH−/− bigenic mice also peak at 6 months of age, but there is a significant reduction in the total number of spheroids in the T44;NFH+/− (∼80% reduction at the age of 6 months) and T44;NFH(−/−) mice (∼90% reduction at the age of 6 months) compared with the single T44 Tg mice (Fig. 2). The T44;NFL−/− mice showed an even greater reduction in the number of spheroids than T44;NFH−/− mice. Compared with the T44 Tg mice, there is an ∼85% reduction in the number of spheroids in T44;NFL+/− mice at the age of 6 months. Notably, almost no spheroids were found in T44;NFL−/− mice until ∼12 months of age, although dense tau staining was observed in axons in spinal cord white matter (Fig. 3). However, the number of spheroids in the T44;NFL−/− mice increased with age, demonstrating that tau aggregates occur over a more prolonged time period in the absence of NFL. Figure 3 illustrates the progressive reduction of the total number of spheroids in the spinal cords of T44 (A), T44;NFL+/− (B), and T44;NFL−/− (C) mice at 6 months.
Fig. 2.

Quantification of spheroids in the mouse spinal cord at different ages. Quantification of spheroids is described in Materials and Methods. The mean value among three mice is shown for the number of spheroids summarized here. The error bars represent SEM. Statistical analysis was performed for the mice at the same age by ANOVA. Significant differences between T44 and other Tg mice are indicated by asterisks (*p < 0.001; **p < 0.0001).
Fig. 3.

Comparison of the number of spheroids in T44, T44;NFL+/−, and T44;NFL−/− Tg mice. The sections were stained with 17026, a polyclonal antibody to recombinant tau protein. Note the reduction in the number of tau-positive spheroids in the bigenic mice relative to the T44 mice at 6 months of age. All photomicrographs are at the same magnification. Scale bar, 10 μm.
Because we showed previously that all three NF subunit proteins coexisted with tau protein in the spheroids of the T44 mice, we performed triple-labeled indirect immunofluorescence to evaluate the consequences of elimination one of the three NF subunits on the composition of the spheroids. In the T44;NFL−/− mice, although NFH was found to colocalize with tau protein in the spheroids, the intensity of NFH staining was dramatically reduced compared with the T44 Tg mice (Fig. 4, compareA–D with E–H), indicating that tau protein is the major component of spheroids in this mouse. A slight reduction in the intensity of NFL stain was also observed in spheroids of T44;NFH−/− mice (Fig. 4I–L). The intensity of NFM immunoreactivity in the spheroids also was reduced dramatically in T44;NFL−/− mice and slightly diminished in the T44;NFH−/− mice (data not shown). Previous studies also identified α- and β-tubulin in the tau-immunoreactive spheroids in the T44 mice, and the increase in tubulin subunits observed in the T44;NFL−/− bigenic lines (Fig. 1) also resulted in an increase of α- and β-tubulin staining in the spheroids compared with the T44 and T44;NFH−/− mice (data not shown). In contrast, although spheroids also contained α-internexin, there was no difference in the intensity of the staining for this neuronal intermediate filament protein among all lines of mice, and none of the spheroids were stained with the anti-peripherin antibody (data not shown). Thus, the reduction in the intensity of staining described here reflects the overall change in the neuronal cytoskeleton of the NFL−/− and NFH−/− mice (Zhu et al., 1997; Elder et al., 1998).
Fig. 4.
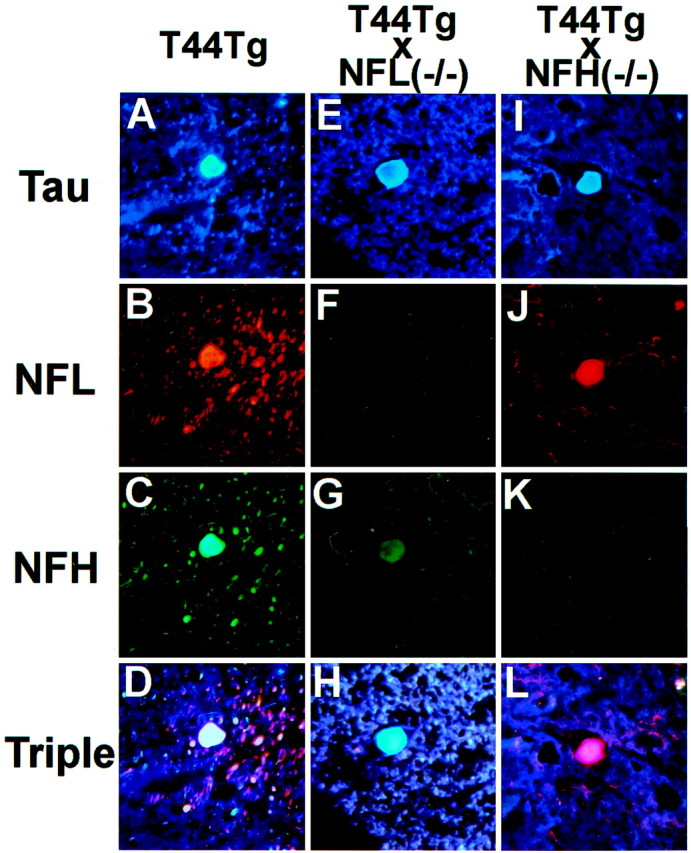
Triple immunofluorescence staining of spheroids in the spinal cord of 6-month-old T44, 15-month-old T44;NFL−/−, and 6-month-old T44 Tg;NFH−/− mice. The sections were stained with the T14 mouse anti-human tau mAb (A, E,I), the rabbit anti-NFL antibody (B, F, J), and the DP1.6 rat anti-NFH mAb (C, G,K). A triple fluorescent filter cube was used to visualize colocalization of tau, NFL, and NFH (D,H, L). All photomicrographs are at the same magnification. Scale bar, 10 μm.
Electron microscopy detect tau filaments in the absence of NFs filaments
To determine whether or not tau aggregates contain filaments in the absence of NFL, which is the backbone of NFs, TEM and immuno-EM were performed in the T44;NFL−/− mice. In control WT mice, NFs were evenly distributed in the normal axon of the spinal cord (Fig.5A), but less NFs and more microtubules were found in the NFL−/− and T44;NFL−/− mice (Fig.5B,C). TEM studies of the tau inclusions revealed masses of loosely (Fig. 5D–F) or tightly packed aggregates (Fig. 5G–I) of randomly arranged 10–20 nm straight filaments. These aggregates were found in myelinated (Fig. 5G–L) and unmyelinated axons in peripheral white matter of the spinal cord (Fig. 5D–F) of 12-month-old T44;NFL−/− mice (Fig. 5D–L) but not in age-matched control WT mice (Fig. 5A). Preembedding immuno-EM studies showed that the aggregates were immunolabeled by antibodies to tau (Fig. 5J–L). Different packing densities of tau filamentous aggregates could represent different stages of aggregate formation, from loose to tightly packed filamentous aggregates, or from small to big aggregates. We also observed a shift in the location of tau aggregates: from the junction of the gray and white matter in the T44 mice to the peripheral white matter of the T44;NFL−/− mice. Because our previous studies in the T44 mice suggest that axonal transport was compromised in the presence of tau axonal aggregates, the detection of tau aggregates in peripheral white matter of the T44;NFL−/− mice may indicate a possible endogenous rescue of axonal transport in the bigenic mice. Together, these results indicate that filamentous tau aggregates develop without NFL, but the delay in the formation of aggregates suggests that NFs play an important role as a pathological chaperone in tau aggregate formation.
Fig. 5.

Tau-rich aggregates in the axons of the peripheral white matter of spinal cord contain straight tau filaments.A, NFs are evenly distributed in a spinal cord myelinated axon of a WT mouse. B, C, Few NFs (arrowheads) and more microtubules (short arrows) are seen in the NFL−/− (B) and T44;NFL−/− (C) mice. D–F, A mass of loosely packed disorganized filaments (large arrow) in a spinal cord unmyelinated axon of a 12-month-old T44;NFL−/− mouse. D–F show the same aggregate at different magnification. G–I, A mass of tightly packed disorganized filaments in a myelinated axon of spinal cord in a 12-month-old T44;NFL−/− mouse. G–I show the same aggregate at increased magnification. J–L, Preembedding immuno-EM labeled the aggregates with antibody 17026 in a 12-month-old T44;NFL−/− mouse. J–L show the same aggregate at different magnifications. Note that silver enhancement was performed for 17026 staining. Scale bars: A–C, 500 nm;D, G, J, 10 μm;E, H, K, 500 nm;F, I, L, 100 nm.
The absence of NFL delayed accumulation of insoluble tau protein in the CNS of T44;NFL−/− bigenic mice
As reported previously, tau protein becomes progressively more insoluble and hyperphosphorylated with age in the T44 Tg mice as in human tauopathies (Ishihara et al., 1999, 2001). To determine the effect of crossbreeding of T44 with NFL−/− and NFH−/− mice on the accumulation of insoluble tau, we analyzed the solubility of tau protein in the different lines by extracting brain and spinal cord samples using buffers with increasing extraction strengths. The spinal cord and brain samples from 6- and 12-month-old mice from each group of single and bigenic mice were sequentially extracted with RAB, RIPA buffer, and 70% FA. The three fractions were then analyzed by quantitative Western blotting with antibody 17026. As shown in Figure6, over 90% of endogenous mouse tau from both the brain and the spinal cord of the WT mice was primarily RAB soluble, and no tau immunoreactivity was detected in the FA-soluble fraction. At 6 months of age, ∼76 and ∼74% of total tau protein were RAB soluble, and ∼1.2 and ∼1.7% were in the FA-soluble fraction in the brain and spinal cord of the T44 Tg mice, respectively. Although the RAB-soluble tau remained relatively constant (at ∼74–80% in each group) in all lines of mice overexpressing human tau protein, the amount of FA-soluble fraction as a percentage of total tau protein in T44;NFL−/− mice was significantly decreased compared with that of T44 Tg mice at 6 months of age. However, this formic acid extractable pool of tau in the T44;NFL−/− mice increased by 90% (brain) and 78% (spinal cord) between 6 and 12 months of age, whereas a similar increase in the other mouse lines was more modest at ∼18–43% (Fig. 6C,D). These results are consistent with the immunohistochemical findings showing that there were no detectable tau aggregates in the spinal cord of T44;NFL−/− mice at 6 months of age but that they appeared at ∼12 months of age (Fig. 2).
Fig. 6.

Accumulation of insoluble tau protein in the CNS of mice. A, B, Neocortical (A) and spinal cord (B) tissues of 6-month-old mice from each group were sequentially extracted with RAB, RIPA buffer, and 70% FA, and the tau levels were determined by quantitative Western blot analysis with antibody 17026.C, D, Alteration in the FA-soluble tau as a percentage of total tau in the neocortex (C) and spinal cord (D) of T44 tau-overexpressing mice. The percentage was calculated from quantitative Western blot analysis at 6 and 12 months of age (n = 3). The amount of FA-soluble fraction as a percentage of total tau protein in T44;NFL−/− mice was significantly decreased (*p< 0.01) compared with that of T44 mice at 6 month of age, and it increased by 90.2% (neocortex) and 77.6% (spinal cord) at 12 months of age, whereas the increase in the other group of mice was 17.5–42.9%.
To determine whether or not tau proteins in these bigenic mice are also hyperphosphorylated at the same sites as in PHF-tau in the AD and ALC/PDC brain, we performed Western blot analysis of RAB- and FA-soluble tau extracted from the cerebral cortex of Tg mice. Both RAB- and FA-soluble Tg tau protein in these mice were recognized by the phosphorylation-dependent antibodies PHF-1 (phosphoserine 396 and 404), T3P (phosphoserine 396), and AT270 (phosphothreonine 181), indicating that the phosphorylation state of Tg tau recapitulates that of PHF-tau found in human tauopathies (including AD and ALS/PDC) and human fetal tau but is different from that of normal adult human brain tau (Fig.7). There was no obvious difference in the extent of phosphorylation between T44 Tg mice and the other T44 overexpressed mice, including T44;NFL+/−, T44;NFL−/−, T44;NFH+/−, and T44;NFH−/− mice (Fig. 7).
Fig. 7.

Phosphorylation state of tau in the CNS of single Tg and bigenic mice. Immunoblots were performed using PHF tau from AD brain, autopsy-derived normal human adult tau and autopsy-derived human fetal tau samples, as well as soluble and insoluble fractions of tau from the neocortex of 12-month-old T44 Tg, T44 Tg;NFL−/−, and T44;NFH−/− mice. Antibodies 17026 and T14 recognized total tau proteins regardless of the phosphorylation state. Antibody T1 was specific to nonphosphorylated tau and did not react with PHF-tau. Phosphorylation-dependent antibodies PHF1, T3P, and AT270 did not recognize normal adult tau but reacted with PHF-tau and fetal tau, as well as both soluble and insoluble fraction of tau from the single Tg and bigenic mice.
Bigenic mice lacking NF subunits showed increased viability, reduced weight loss, and improved behavioral phenotype
We showed previously a correlation between reduced viability and increased expression of transgenic tau protein in our three stable tau Tg lines (Ishihara et al., 1999). For example, the heterozygous mice in the highest tau-expressing line were not viable beyond 3 months, and homozygous mice generated from each of the lower expressing lines diedin utero or within 3 months postnatal. Although ∼80% of the heterozygous mice in the lowest expressing T44 line used in this study survived >3 months, only ∼60% lived >1 year (Fig.8). As a consequence of crossbreeding T44 Tg mice with NF knock-out mice, the impaired viability of these bigenic mice was reduced. For example, although only 67% of T44 Tg mice lived for 9 months, the 9 month survival of the T44;NFL−/− and T44;NFH−/− mice was ∼90 or 78%, respectively, whereas the viability of NFL−/− and NFH −/− mice was comparable with that of WT mice at 9 months (data not shown).
Fig. 8.

Longevity of mice. Approximately 67% of T44 Tg mice survived until 9 months of age, but after crossbreeding with NFL−/− or NFH−/− mice, the survival went up to 90 or 78%, respectively. Longevity of mice was evaluated using cohorts of pups weaned at 3–4 weeks old because some mice died before 4 weeks from poor nursing. WT, n = 30; T44;NFL−/−,n = 20; T44;NFH−/−, n = 18; T44+/−, n = 24; T44+/+, n = 8.
As reported previously, T44 Tg mice develop progressive motor weakness as demonstrated by an impaired ability to stand on a slanted surface and by the clasping of their hindlimbs when lifted by the tail. These impairments may explain why T44 Tg mice weighed ∼30–40% less than age-matched WT littermates. Crossbreeding T44 Tg mice with NF knock-out mice reduced the weight loss and abnormal clasping behavior that were seen in T44 Tg mice. As shown in Figure9, T44 Tg mice weighed 35% less than WT littermates at 12 months of age, although no weight loss was observed after crossbreeding the T44 mice with the NFL−/− mice, but significant albeit moderate (∼8%) weight loss was observed when T44 Tg mice were crossbred with NFH−/− mice. Crossbreeding T44 Tg mice with NFL−/− mice, but not with NFH−/− mice, significantly reduced the abnormal clasping behavior at 5 and 11 months of age (Fig.10). For example, the T44 mice had a clasping score of ∼4 at 11 months of age, but the T44;NFL−/− mice had a clasping score of only 2 by the same age, suggesting a dramatic reduction in hindlimb weaknesses. Together with the change in viability, our data suggest that the reduction of tau aggregates by crossbreeding T44 Tg mice with NF knock-out mice partially rescues the pathological symptoms caused by overexpression of human tau protein.
Fig. 9.
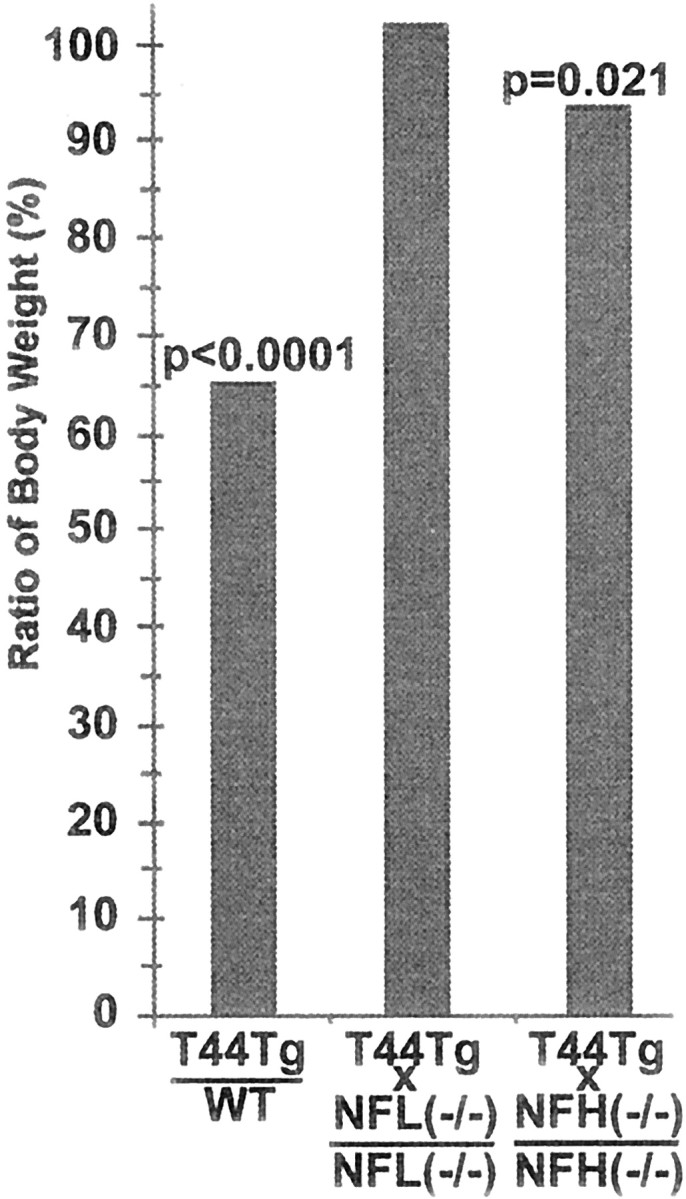
Weight change of mice. Bar graphs illustrate average body weight of T44+/−: WT, T44;NFL−/−: NFL−/−, and T44;NFH−/−: NFH−/−, respectively, at 12 months of age. T44 Tg mice weighed 35% less than WT littermates, although similar weight loss was no longer observed after crossbreeding T44 mice with NFL−/− mice. More modest (8%) but significant weight loss was observed in T44;NFH−/− mice (n = 8–12).
Fig. 10.

Clasping phenotype of mice. Mice were videotaped during a 15 sec tail suspension test at 5 and 11 months of age and analyzed for clasping score as described in Materials and Methods. T44;NFL−/− mice showed a significantly lower score than T44 Tg mice at 5 and 11 months. Bar graphs show mean ± SE of five trials.p values are shown in the graph.
DISCUSSION
In the present study, we have provided compelling evidence that NF subunit proteins act as pathological chaperones that facilitate the formation of intraneuronal filamentous tau aggregates that cause neurodegeneration in the T44 Tg mice. To accomplish this, we eliminated either NFL or NFH by crossbreeding the T44 Tg mice with NFL−/− or NFH−/− mice, and our analyses of the T44;NFL−/− and T44;NFH−/− bigenic mice provide unequivocal evidence supporting a pathogenic role for NFs in the formation of these filamentous tau lesions. For example, we showed a dramatic reduction in the number of tau-positive aggregates in the spinal cord of T44 mice lacking either NFH or NFL, suggesting that both of these NF subunit proteins play an important role in facilitating filamentous tau aggregate formation. Furthermore, this reduction in aggregate number appears to correlate with the overall improvement in the health of the bigenic mice because increased survival and reduced weight loss were observed in these mice compared with the T44 single Tg mice. Finally, the reduction in tau spheroids in the T44;NFL−/− mice also correlated with attenuation of the motor weaknesses observed in the T44 mice, suggesting a pathologic role of the aggregates in causing motor neuron degeneration.
As discussed above, although NFs have long been associated with a number of neurodegenerative diseases because NF immunoreactivities have been detected in well known neuropathologic lesions, including NFTs in AD (Schmidt et al., 1989, 1990), Lewy bodies in Parkinson's disease and diffuse Lewy body disease (Nakazato et al., 1984; Trojanowski and Lee, 1998), as well as in the axonal spheroids in classic ALS (Nakazato et al., 1984) and in ALS/PDC (Shankar et al., 1989; Ishihara et al., 1999), the precise role NFs play in the pathogenesis of these neurodegenerative diseases remains unclear. Studies reported in the early 1980s implicated NFs as the building blocks of NFTs because NFTs are immunopositive for NFs and because 10-nm-diameter NFs are the most abundant cytoskeletal structures in mature neurons (Dahl et al., 1982;Perry et al., 1985). However, subsequent biochemical and genetic studies demonstrated unequivocally that tau proteins rather than NFs are the building blocks of NFTs (Goedert et al., 1989; Lee et al., 1991; Hutton et al., 1998; Poorkaj et al., 1998). Other studies support a secondary role for NFs in the pathogenesis of NFTs in AD because NF immunoreactivity is only detected in NFTs at late stages of the disease, but NF proteins are not detected in pretangles or in the majority of the NFTs (Schmidt et al., 1989). In this regard, it is tempting to speculate that the involvement of NFs at late stages in the neurodegenerative disease mentioned above may be a harbinger of the final collapse of NFs, the most abundant cytoskeletal structure in neurons, followed by the trapping of NFs within NFTs and other disease-specific intraneuronal inclusions.
Although NFTs containing both NFs and PHFs are found in neuronal perikarya, the formation of NF-positive spheroids in the axon hillock of spinal cord motor neurons of ALS, ALS/PDC, and FTDP-17 patients is likely to be mediated by different pathogenic mechanism(s). Significantly, NF-containing spheroids have been detected in the spinal cord and ventral roots of a number of Tg mouse models overexpressing the following: (1) different NF subunits (Xu et al., 1993; Wong et al., 1995); (2) mutant superoxide dismutase 1 (Zhang et al., 1997); and (3) tau proteins (Ishihara et al., 1999; Spittaels et al., 1999; Probst et al., 2000). These Tg mice all showed selective axonal degeneration and motor weaknesses most likely attributable to the interruption of fast and slow axonal transport by the NF-rich spheroids (Collard et al., 1995; Zhang et al., 1997; Ishihara et al., 1999). Although the exact mechanisms for the development of the spheroids in these Tg mice is unknown, it is possible that selective aggregation of NFs occur because of the overexpression of specific transgenes that perturb the transport of a large number of slow-moving NFs in motor neurons. Regardless of the exact mechanism of pathogenesis, the results of previous studies in Tg mice point to a role of NFs in spheroid formation and motor neuron degeneration. The production of bigenic mice overexpressing tau but lacking either NFL or NFH allow the dissection of the role of each of these NF subunits in the development of tau pathology in our model of tauopathies.
Our analyses on the T44;NFL−/− and T44/NFH−/− mice suggest that the elimination of NFL rather than NFH is more efficacious in reducing the number of spheroids and ameliorating the phenotypes of the T44 mice. This observation is reasonable because NFL forms the backbone of intact NFs and NFL−/− mice contain no intact NFs, but they contain reduced levels of NFH and NFM (Zhu et al., 1997). In contrast, abundant NFs comprised of NFL and NFM are present in the NFH−/− mice (Elder et al., 1998; Rao et al., 1998; Zhu et al., 1998). Thus, our data suggest that intact NFs are involved in the coaggregation with tau filaments in the spheroids. However, it is surprising that the T44;NFH−/− mice also showed a dramatic reduction (fourfold reduction) in the number of tau-positive spheroids compared with the single T44 mice because previous studies have shown only a 20% reduction in the total number of NFs in the NFH−/− mice compared with WT mice (Elder et al., 1998). This observation suggests that, in addition to the filament backbone, NFH, the sidearms of NFs also participate in coaggregation with tau filaments. Indeed, the presence of NFH in tau-positive spheroids of T44;NFL−/− mice support an independent role of NFH in tau aggregation formation.
In our previous studies on the T44 Tg mice, we were able to define two different types of neuronal tau protein aggregates based on different criteria, including the age of onset, the location within the CNS, and the protein composition (Ishihara et al., 1999, 2001). The first type of tau inclusions consists of mostly spheroids found predominantly in the spinal cord and brainstem. The spheroids contain NFs and other cytoskeletal proteins appear at ∼1 month of age and peak at ∼6 months of age. However, a second type of tau aggregates developed in the hippocampus and associated areas of the T44 mice as they advance in age to over 18 months. These tau inclusions are not immunopositive for NFs and appear to comprise only of tau proteins, although ubiquitin immunoreactivities also are detected (Ishihara et al., 2001). In this regard, the second type of tau aggregates are more similar to authentic AD NFTs because the second but not the first are stained by histochemical dyes such as Congo red, Thioflavin S, and Gallyas Silver. These hippocampal tau tangles are relative few in number, and we hypothesize that the development of these tangles is an age-related phenomenon much like normal aging in humans. Interestingly, although the absence of NFL in the T44 mice essentially abrogates the first type of tau inclusions, it has no effect on the development of congophilc tau tangles in the hippocampus of aged T44;NFL−/− mice (data not shown). Thus, our analyses of the bigenic mice define an NF-dependent and an NF-independent mechanism for the pathogenesis of tau inclusions. Although much still needs to be done to elucidate how different types of tau aggregates develop in different tauopathies, the analyses of tau Tg mouse models provide important insights into the pathogenesis of diverse neurodegenerative diseases, including AD, ALS/PDC, and related tauopathies.
Footnotes
This work was supported by grants from the National Institute on Aging. We thank Drs. Jean-Pierre Julien and Robert Lazzarini for providing the NFL−/− and NFH−/− mice, respectively, and the Biomedical Imaging Core Facility of the University of Pennsylvania for assistance in the electron microscopic studies.
Correspondence should be addressed to Dr. Virginia M.-Y. Lee, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, Maloney 3, HUP, 3600 Spruce Street, Philadelphia, PA 19104-4283. E-mail: vmylee@mail.med.upenn.edu.
REFERENCES
- 1.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM-Y. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci. 1987;7:3489–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM-Y, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 5.Dahl D, Selkoe DJ, Pero RT, Bignami A. Immunostaining of neurofibrillary tangles in Alzheimer's senile dementia with a neurofilament antiserum. J Neurosci. 1982;2:113–119. doi: 10.1523/JNEUROSCI.02-01-00113.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM-Y, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elder GA, Friedrich VL, Kang C, Bosco P, Gourov A, Tu PH, Zhang B, Lee VM-Y, Lazzarini RA. Requirement of heavy neurofilament subunit in the development of axons with large calibers. J Cell Biol. 1998;143:195–205. doi: 10.1083/jcb.143.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 9.Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5931. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 12.Hoffmann R, Lee VM-Y, Leight S, Varga I, Otvos L. Unique Alzheimer's disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry. 1997;36:8114–8124. doi: 10.1021/bi970380+. [DOI] [PubMed] [Google Scholar]
- 13.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM-Y. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 14.Hong M, Trojanowski JQ, Lee VM-Y. Tau-based neurofibrillary lesions. In: Clark CM, Trojanowski JQ, editors. Neurodegenerative dementias: clinical features and pathological mechanisms. McGraw-Hill; New York: 2000. pp. 161–176. [Google Scholar]
- 15.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 16.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 17.Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VM-Y. Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol. 2001;158:555–562. doi: 10.1016/S0002-9440(10)63997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Julien JP. Neurofilament functions in health and disease. Curr Opin Neurobiol. 1999;9:554–560. doi: 10.1016/S0959-4388(99)00004-5. [DOI] [PubMed] [Google Scholar]
- 19.Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–825. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 20.Lang E, Szendrei GI, Lee VM-Y, Otvos L. Immunological and conformation characterization of a phosphorylated immunodominant epitope on the paired helical filaments found in Alzheimer's disease. Biochem Biophys Res Commun. 1992;187:783–790. doi: 10.1016/0006-291x(92)91264-q. [DOI] [PubMed] [Google Scholar]
- 21.Lee VM-Y, Carden MJ, Trojanowski JQ. Novel monoclonal antibodies provide evidence for the in situ existence of a nonphosphorylated form of the largest neurofilament subunit. J Neurosci. 1986;6:850–858. doi: 10.1523/JNEUROSCI.06-03-00850.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee VM-Y, Balin BJ, Otvos L, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 23.Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 24.Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM-Y. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 25.Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM-Y. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–30987. [PubMed] [Google Scholar]
- 26.Nakazato Y, Sasaki A, Hirato J, Ishida Y. Immunohistochemical localization of neurofilament protein in neuronal degenerations. Acta Neuropathol (Berl) 1984;64:30–36. doi: 10.1007/BF00695603. [DOI] [PubMed] [Google Scholar]
- 27.Otvos L, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM-Y. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 28.Perry G, Rizzuto N, Autilio-Gambetti L, Gambetti P. Paired helical filaments from Alzheimer disease patients contain cytoskeletal components. Proc Natl Acad Sci USA. 1985;82:3916–3920. doi: 10.1073/pnas.82.11.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 30.Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM-Y, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol (Berl) 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 31.Rao MV, Houseweart MK, Williamson TL, Crawford TO, Folmer J, Cleveland DW. Neurofilament-dependent radial growth of motor axons and axonal organization of neurofilaments does not require the neurofilament heavy subunit (NF-H) or its phosphorylation. J Cell Biol. 1998;143:171–181. doi: 10.1083/jcb.143.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt ML, Lee VM-Y, Trojanowski JQ. Analysis of epitopes shared by Hirano bodies and neurofilament proteins in normal and Alzheimer's disease hippocampus. Lab Invest. 1989;60:513–522. [PubMed] [Google Scholar]
- 33.Schmidt ML, Lee VM-Y, Trojanowski JQ. Relative abundance of tau and neurofilament epitopes in hippocampal neurofibrillary tangles. Am J Pathol. 1990;136:1069–1075. [PMC free article] [PubMed] [Google Scholar]
- 34.Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VM-Y. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- 35.Shankar SK, Yanagihara R, Garruto RM, Grundke-Iqbal I, Kosik KS, Gajdusek DC. Immunocytochemical characterization of neurofibrillary tangles in amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Ann Neurol. 1989;25:146–151. doi: 10.1002/ana.410250207. [DOI] [PubMed] [Google Scholar]
- 36.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szendrei GI, Lee VM-Y, Otvos L. Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J Neurosci Res. 1993;34:243–249. doi: 10.1002/jnr.490340212. [DOI] [PubMed] [Google Scholar]
- 39.Teclemariam-Mesbah R, Wortel J, Romijin HJ, Buijs RM. A simple silver-gold intensification procedure for double DAB labeling studies in electron microscopy. J Histochem Cytochem. 1997;45:619–621. doi: 10.1177/002215549704500414. [DOI] [PubMed] [Google Scholar]
- 40.Trojanowski JQ, Lee VM-Y. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- 41.Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM-Y. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol. 1995;129:1629–1640. doi: 10.1083/jcb.129.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- 43.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 44.Wong PC, Marszalek J, Crawford TO, Xu Z, Hsieh ST, Griffin JW, Cleveland DW. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol. 1995;30:1413–1422. doi: 10.1083/jcb.130.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Z, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 47.Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM-Y. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Q, Couillard-Despres S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Q, Lindenbaum M, Levavasseur F, Jacomy H, Julien JP. Disruption of the NF-H gene increases axonal microtubule content and velocity of neurofilament transport: relief of axonopathy resulting from the toxin beta,beta′-iminodipropionitrile. J Cell Biol. 1998;143:183–193. doi: 10.1083/jcb.143.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]