

Abstract
In the mammalian retina, rods feed into the cone pathway through electrotonic coupling, and recent histological data suggest the involvement of connexin36 (Cx36) in this pathway. We therefore generated Cx36 null mice and monitored the functional consequences of this deficiency on early visual transmission. The homozygous mutant mice had a normally developed retina and showed no changes in the cellular organization of the rod pathway. In contrast, the functional coupling between AII amacrine cells and bipolar cells was impaired. Recordings of electroretinograms revealed a significant decrease of the scotopic b-wave in mutant animals and an increased cone threshold that is compatible with a distorted, gap junctional transmission between AII amacrine cells and cone bipolar cells. Recordings of visual evoked potentials showed extended latency in mutant mice but unaffected ON and OFF components. Our results demonstrate that Cx36-containing gap junctions are essential for normal synaptic transmission within the rod pathway.
Keywords: Cx36, connexin36, gap junctions, knock-out, visual transmission, electroretinogram, ERG, visual evoked potential, VEP, AII amacrine cell, cone ON bipolar cell, retina, mouse
Connexins are channel-forming proteins building up hexameric hemichannels in membranes of apposed cells that form the intercellular gap junctional channels. Gap junctions provide electrotonic and metabolic communication between neurons and contribute to neuronal development and processing. The lateral pathways established by gap junctions are crucial for pattern formation of the developing neuronal networks and neuron differentiation (Bennett et al., 1991; Peinado et al., 1993; Bruzzone et al., 1996; Rozental et al., 1998; Roerig and Feller, 2000). A growing body of morphological and physiological evidence indicates that gap junctional communication persists in certain areas of the adult brain. Gap junctions mediate temporal coordination of neuronal activity (Strata et al., 1997; Mann-Metzer and Yarom, 1999), form inhibitory cortical networks (Galarreta and Hestrin, 1999; Gibson et al., 1999;Tamas et al., 2000; Venance et al., 2000), and generate high-frequency oscillations (Traub et al., 1999). The multigene family of vertebrate connexins comprises at least 16 different connexins. Only some of these have been shown to be expressed in the CNS so far (Bruzzone et al., 1996; Feigenspan et al., 2001; Teubner et al., 2001). Among these, murine connexin36 (Cx36) and its homologs Cx35 and Cx34.7 in fish have emerged recently as prime candidates for neuronal connexins (O'Brien et al., 1996; Condorelli et al., 1998; Söhl et al., 1998). Cx36 expression is prominent in various regions of the brain and particularly in the retina (Al-Ubaidi et al., 2000; Belluardo et al., 2000; Güldenagel et al., 2000; Rash et al., 2000; Teubner et al., 2000; Feigenspan et al., 2001). Its expression appears to be modulated during development showing different temporal and spatial peaks (Söhl et al., 1998; Belluardo et al., 2000; Gulisano et al., 2000). The level of Cx36 transcripts in the retina has been shown to depend on lighting conditions during rearing of the mice (Al-Ubaidi et al., 2000).
The functional role of Cx36 in neurons of the adult CNS has so far not been clarified because of the lack of specific blockers and distinct cellular localization. Recently, Cx36 transcripts have been detected in interneurons that were functionally coupled in the visual somatosensory and hippocampal cortex (Venance et al., 2000).
In mouse retina, Cx36 is predominantly expressed in a subclass of amacrine cells, the AII or rod amacrine cell (Feigenspan et al., 2001). This cell type represents the major output of rod bipolar cells and feeds the rod signals into the cone pathway by chemical synapses with OFF cone bipolar cells and electrical synapses with ON cone bipolar cells, respectively (Kolb and Famiglietti, 1974; Strettoi et al., 1992). Besides being electrically coupled with ON cone bipolar cells, AII cells also form coupled networks through direct coupling with neighboring AII cells. Cx36 protein has been located on contact membranes toward both types of coupled neighboring cells of AII cells (Feigenspan et al., 2001), which, however, are differently modulated (Hampson et al., 1992; Mills and Massey, 1995). Cx36-containing gap junctions, therefore, could mediate the very early stage of visual processing, in particular the interaction between rod and cone systems. To test this hypothesis, we have generated transgenic mice deficient in Cx36 and have analyzed the functional consequences on the visual pathway.
MATERIALS AND METHODS
Construction of the targeting vector. The targeting vector was constructed from the vector pBCx36 that contained 9.8 kb of the mouse Cx36 locus. The targeting construct (pCx36KO) consisted of two arms of homology of mouse Cx36 genomic DNA: a 5′ 1.7 kbNheI/BamHI and a 3′ 5.5 kb BbrPI/XhoI fragment separated by a hypoxanthine phosphoribosyltransferase (HPRT) minigene that was flanked by lox P sites and driven by the mouse phosphoglycerate kinase promoter (Magin et al., 1992). The HPRT minigene replaced the complete coding region of Cx36 exon2 and 170 bp of the flanking 3′ untranslated region (UTR) (Fig.1A). The final targeting vector was restriction mapped, and both Cx36/HPRT transition regions were sequenced. Two hundred micrograms of the targeting vector were linearized by NotI digestion and transfected by electroporation (800 V, 3 μF) into HPRT-deficient and feeder-independent HM-1 embryonic stem (ES) cells (Magin et al., 1992;Magin, 1998). Cell culture and selection of targeted colonies using hypoxanthine–aminopterine–thymidine (HAT) medium was performed according to Selfridge et al. (1992) and Theis et al. (2000).
Fig. 1.

Generation of connexin36-deficient mice.A, Targeting scheme. The coding region of the wild-type Cx36 gene consists of two exons (boxes) that are separated by a 1100 bp intron. Exon1 contains a 530 bp noncoding region (hatched) and a 70 bp coding region (black). Exon 2 consists of a 900 bp coding region (black) and a 1400 bp noncoding region (hatched). After electrotransfecting the linearized targeting vector into HM-1 ES cells, homologous recombination occurred in 3% of all clones analyzed. In the mutated Cx36 allele, the complete coding region of exon2 and 170 bp of the flanking noncoding region were replaced by an HPRT minigene that was flanked by loxP sites (arrowheads) and transcribed in opposite direction (arrow) to Cx36. B, Diagnostic PCR of tail-cut DNA was performed to screen pedigree mice for the targeting effort. PCR was established using a three primer approach with a Cx36 intron-specific upstream primer (P1) and either a Cx36 exon2-specific downstream primer (P2), resulting in a 510 bp wild-type amplicon, or an HPRT-specific downstream primer (P3), giving a 230 bp amplicon for the mutated allele.C, Southern blot analysis was used to confirm PCR screening. KpnI-digested (K) tail-cut DNA was probed with an 860 bp externalXhoI/HindIII fragment. The smaller fragment (8.5 kb) resulted from the wild-type allele, the larger band (10.3 kb) derived from the mutated allele. D, RT-PCR. cDNA preparations of wild-type and Cx36 −/− tissues were used to check for the presence of Cx36 transcripts. PCR using an intron-spanning primer pair yielded a 520 bp amplicon specific for Cx36 cDNA in wild-type retina and brain but not in the corresponding Cx36 −/− cDNA preparations. The use of equal amounts of cDNA was verified by a separate β-actin PCR that yielded a 243 bp amplicon.
Screening of ES cell clones. Homologous recombination in HAT-resistant ES cell clones was screened using PCR and subsequently confirmed by Southern blot hybridization.
Genomic DNA of HAT-resistant ES cell clones was prepared according toTheis et al. (2000). PCR screening was performed using a 5′ upstream primer external to the targeting vector (Cx36tUSP; 5′CTACTTAAGGGCGGATTAGAGGCG) specific for Cx36 5′ UTR and a 3′ HPRT-specific downstream primer (P3; 5′CAGTAAATCGTTGTCAACAGTTCC) with the following parameters: 95°C for 1 min, 64°C for 1.30 min, and 72°C for 2 min, for 40 cycles. PCR was performed using a Peltier PTC200 thermocycler (Biozym, Oldendorf, Germany) and Taq DNA polymerase purchased from Promega (Madison, WI). The resulting diagnostic amplicon had an estimated size of 1.8 kb (data not shown).
For genomic Southern blot hybridization, DNA of PCR-positive clones was digested by KpnI and electrophoresed in a 0.4% agarose gel. After blotting the digested DNA onto Hybond N+ membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK), it was fixed by UV cross-linking. As hybridization probe, we used an external, 860 bpXhoI/HindIII fragment probe corresponding to part of the 3′ homologous region and an internal, 330 bp HPRT-specificHindIII/XhoI fragment probe. Hybridization was performed under stringent conditions (two washes at 55°C for 10 min in 2× SSC and 0.1% SDS) using Quick Hyb hybridization solution (Stratagene, La Jolla, CA) according to instructions provided by the manufacturer. The diagnostic wild-type DNA fragment had an estimated size of 8.5 kb. Southern blot hybridization using either the external 860 bp XhoI/HindIII probe or the internal 330 bpHindIII/XhoI probe resulted in a DNA fragment of ∼10.3 kb containing the targeted Cx36 allele.
Generation of Cx36 knock-out mice. Successfully targeted ES cell clones were prepared for injection into C57BL/6 blastocysts as described previously (Theis et al., 2000). Blastocyst injection resulted in the birth of coat-color chimeric mice that were then crossed with C57BL/6 partners. Offspring mice were checked for germ line transmission by PCR of tail-cut DNA using a Cx36 intron-specific upstream primer (P1; 5′CTGTTCAAGGACTGGTAAGCGCTG) which was combined with Cx36 exon2-specific (P2; 5′GTCTCCTTACTGGTGGTCTCTGTG) and HPRT-specific (P3) downstream primers in a three primer experiment that was run under conditions mentioned above. The PCR reaction yielded a 510 bp amplicon specific for the wild-type and a 230 bp fragment specific for the mutated allele (Fig. 1B). Additionally, Southern blot hybridization described above was used to confirm the targeting effort (Fig. 1C).
Mice homozygous for the Cx36 null allele were crossed with C57BL/6 partners to obtain heterozygous offspring mice with a 75% C57BL/6 genetic background. Appropriate progenies were then intercrossed to obtain Cx36 −/−, +/−, and +/+ littermates with a 75% C57BL/6 background that were used throughout the studies.
Reverse transcription-PCR analysis. Total RNA was isolated using the RNeasy kit (Qiagen, Hilden, Germany) according to instructions provided by the manufacturer. To remove any residual genomic DNA, RNA preparations were incubated with DNase I (Roche, Mannheim, Germany) according to the manufacturer. First-strand cDNA was synthesized as described previously (Söhl et al., 1998).
To selectively amplify Cx36 cDNA, PCR was performed using an intron-spanning primer pair (exon1-specific 5′ primer, 5′CGGAATTCCGCCATGGGGGAATGGACCATC; exon2-specific 3′ primer, 5′GTCTCCTTACTGGTGGTCTCTGTG) under the following conditions: 95°C for 1 min, 63°C for 1.30 min, 72°C for 1.30 min, for 40 cycles. The resulting Cx36 amplicon had an estimated size of 520 bp. As a control, an additional β-actin-specific PCR (De Sousa et al., 1993) was run in parallel to check for equal cDNA amounts applied for the PCR reactions. The resulting β-actin amplicon had an estimated size of 243 bp.
Histological analysis. Nissl and immunohistochemical stainings were performed with frozen sections (15–20 μm thickness) from paraformaldehyde-fixed (4%, 40 min) retinas according to standard protocols (Feigenspan et al., 2001).
The antibodies used were as follows: a mouse monoclonal antibody to PKC (Amersham Pharmacia Biotech), and rabbit polyclonal antibodies to a 15-mer peptide corresponding to part of the cytoplasmic loop of mouse Cx36 (Teubner et al., 2000). To visualize immunoreactivity, the following secondary antibodies were used: goat anti-mouse Alexa 488 and goat anti-rabbit Alexa 568 (diluted 1:200; Molecular Probes, Eugene, OR). Glycine was detected in vertical sections of Cx36 +/+ and Cx36 −/− retinas that were incubated overnight with rat polyclonal antibodies directed to glycine (1:1000 in 0.1 m phosphate buffer containing 3% NGS and 0.5% Triton X-100) and visualized with anti-rat fluorescein isothiocyanate (FITC). The antibodies were a generous gift of Dr. David Pow (Department of Physiology and Pharmacology, University of Queensland, Brisbane, Australia).
For tissue slices, the retina was removed from the sclera and cut into quarters that were cut with a tissue chopper (McIlwain) into slices of 100–200 μm thickness. The slices were briefly (5 min) fixed in 4% paraformaldehyde, rinsed in PBS, and mounted in the microscope. AII amacrine cells were injected with 0.5% Lucifer yellow (Sigma, Deisenhofen, Germany) and 3% Neurobiotin (Vector Laboratories, Burlingame, CA) in 0.1 m Tris buffer, pH 7.6, using sharp electrodes. Cells were randomly chosen according to their size and localization using infrared video microscopy. After penetrating, a negatively charged current (−1 nA) was applied for 1 min to inject Lucifer yellow into the cell. When the evaluated dendritic morphology confirmed an AII cell identity, Neurobiotin was injected with positive current (5 nA, 15 min). After the injection, the slices were fixed once more in 4% paraformaldehyde (15 min) and rinsed in PBS before overnight incubation with rabbit anti-Cx36 antibodies, followed by incubation with a mixture of goat anti-rabbit Alexa 568 and FITC–streptavidin conjugate for 2 hr.
Images were taken on a confocal laser scanning microscope (Leica TCS SP; Leica, Nussloch, Germany). For double-labeling experiments, optical sections of 200 nm thickness were cut for each channel (488 and 568 nm lines of a krypton–argon laser) and processed in Adobe Photoshop (Adobe Systems, San Jose, CA).
Experiments were always performed in parallel with homozygous mutant mice and their wild-type littermates.
Electrophysiological measurements. Before an experimental session, animals were dark adapted for at least 12 hr. Mice (8–12 weeks old) were anesthetized by intraperitoneal injections of xylazine (50 mg/kg) and ketamine (20 mg/kg), and the pupils were dilated with 1% atropine sulfate. Surgery and subsequent handling were done under dim red dark-room light. For electroretinogram (ERG) recordings, a continuously moistened Ag/AgCl cotton-wick electrode was placed on the corneal surface, and a Ag/AgCl reference electrode was placed in the mouth. A needle grounding electrode was inserted subcutaneously into the tail. The mouse was laid on its side with the head fixed with surgical tape. For recordings of visual evoked potentials (VEP), the head of the animal was fixed in a stereotactic Plexiglas holder, and a needle scalp electrode was placed underneath the skin. Electrode position on the skull was chosen according to the high-resolution mouse brain atlas (www.hms.harvard.edu/research/brain) and was located at the posterior end of the underlying optic tectum. As for ERG recordings, the reference and ground electrodes were placed in the mouth and tail, respectively. Electrical potentials were recorded, bandpass filtered (1–500 Hz), and averaged online with the MacLab system, equipped with the ML-bioamplifier (AD Instruments, Hastings, UK).
Test and background lights were generated with two 150 W halogen light sources and focused onto the cornea. Intensities were adjusted with neutral-density filters, and test flash duration was controlled with an electromagnetic shutter. Corneal illuminance for white light (in lux) was measured with a calibrated luxmeter (Palux, Gossen GmbH, Nürnberg, Germany) at the position of the cornea.
Two different experimental paradigms were chosen for ERG measurements. Scotopic intensity–response functions were measured with 10 msec, white light flashes and a stimulus interval of 20.2 sec. At each intensity, 16 responses were averaged. For the increment threshold functions, steady white background lights with increasing corneal illuminance were applied by the second beam. Each background was given for 5 min before white light test flashes (10 msec duration, 0.9 sec interval, 32 responses averaged) of increasing illuminance were superimposed. The threshold illuminance was determined by fitting a Michaelis–Menten function through the response amplitude data of the a-wave and b-wave, respectively, for each background intensity. A 20 μV criterion response was chosen as threshold for the b-wave, and a 5 μV criterion was chosen for the a-wave. The illuminance required to produce the threshold was interpolated from the fitting curves (Green et al., 1991). A total of 10 mutant mice and their corresponding wild-type littermates were included in the ERG analysis.
For the VEP, stimulus duration was 10 or 800 msec, with a 2 sec stimulus interval. Sixty-four responses were averaged. Each experiment was performed with five wild-type and five mutant mice, respectively.
RESULTS
Generation of Cx36KO mice
We disrupted the mouse Cx36 gene by homologous recombination (Fig.1A) in HPRT-deficient and feeder-independent HM-1 embryonic stem cells (Magin et al., 1992, 1998). Of 337 HAT-resistant clones, screened for homologous recombination by PCR, 10 clones were correctly targeted as verified by Southern blot analysis. Three of these targeted clones were microinjected into C57BL/6 blastocysts (10–15 mutant ES cells per blastocyst); one of these clones yielded coat-color chimeric mice. One male mouse of high percentage coat-color chimerism was crossed to C57BL/6 females, which led to germ line transmission of the altered Cx36 allele that was analyzed by PCR (Fig.1B) and Southern blotting (Fig. 1C). Mice with a heterozygous deletion of Cx36 (Cx36 +/−) and a C57BL/6 genetic background of 75% were intercrossed to obtain Cx36 −/−, +/−, and +/+ littermates. Of 469 animals analyzed, Cx36 mutant mice were born at the expected Mendelian frequencies (24% −/−, 26% +/+, and 50% +/−), as well as distribution of gender (male, 49%; female, 51%), and showed no obvious behavioral abnormalities. Reverse transcription (RT)-PCR analyses (Fig. 1D) revealed the absence of Cx36 transcripts in both retina and brain cDNA preparations of Cx36 −/− mice.
Cellular organization of the retina
The retina is a highly organized neuronal network with distinctly ordered layers (Dowling, 1987). Among the neuronal subsets responsible for specific visual task processing, the functional architecture of the rod pathway is particularly well described (Vaney et al., 1991;Wässle et al., 1995). This pathway could be associated with Cx36 (Feigenspan et al., 2001), and we therefore investigated histologically and immunocytochemically whether the absence of Cx36 affects the cellular organization of this pathway. Nissl staining revealed that the retinas from homozygous mutant mice were normally organized in layered structures of similar dimensions (Fig.2A,B). Cx36 deficiency did not affect number, size, and density of the nuclei in the three nuclear layers, and mutant mice did not show morphological alterations. Because rods form ∼97% of the photoreceptor cells in mice (Jeon et al., 1998), the persistent high number of nuclei in the outer nuclear layer suggested that rods were present.
Fig. 2.
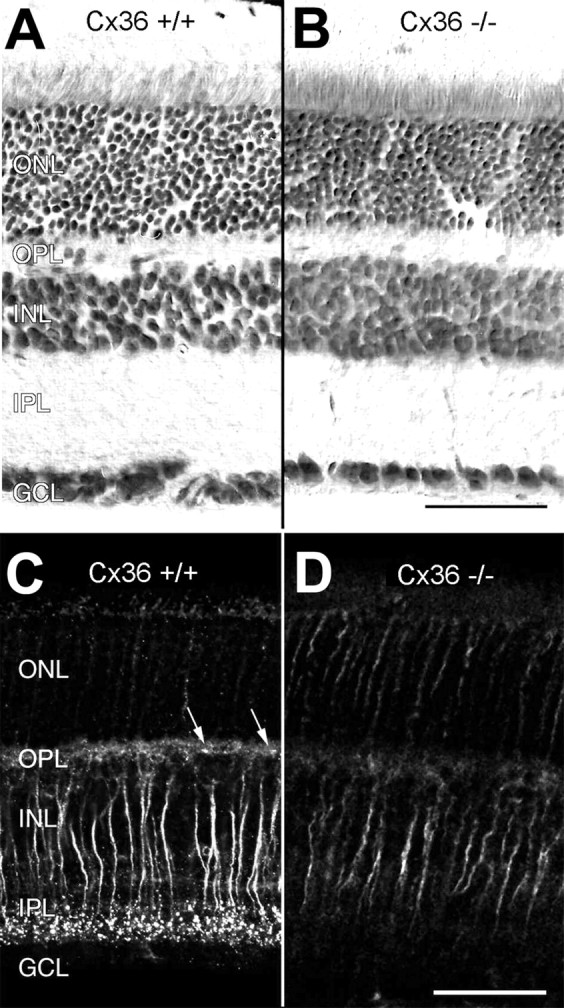
Retinal structure and immunoreactivity for connexin36 in wild-type (Cx36 +/+) and Cx36-deficient (Cx36 −/−) retinas. Vertical sections of Cx36 +/+ (A) and Cx36 −/− (B) retina counterstained with toluidine blue show no obvious differences in their overall structure. When immunolabeled with antibodies to Cx36, paraformaldehyde-fixed, frozen vertical sections of wild-type retina display the typical strong, punctate staining pattern in the inner plexiform layer and a weaker, punctate staining pattern (arrows) in the outer plexiform layer (C) that cannot be detected in the retinas of Cx36 −/− animals (D). Nonspecific staining of transretinal fibers is present in both Cx36 −/− and Cx36 +/+ mice and is attributable to paraformaldehyde fixation. ONL, Outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer;IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars, 50 μm.
Punctate immunostaining for Cx36 within the outer and inner plexiform layers of the wild-type retina (Fig. 2C) was absent in the Cx36 −/− retina (Fig. 2D). Some transretinal fibers, most likely bipolar cell axons, were unspecifically labeled because of paraformaldehyde fixation, which was necessary for processing Neurobiotin-injected slices for Cx36 immunofluorescence analysis (Güldenagel et al., 2000; Feigenspan et al., 2001).
In the mammalian retina, rods synapse predominantly on rod bipolar cells, which can be specifically labeled with a monoclonal antibody directed to protein kinase C (PKC) (Greferath et al., 1990). PKC immunostaining revealed a population of neurons in mutant mice that was identified as rod bipolar cells based on their morphology, in particular their dendritic and axonal arborization. This immunostaining pattern, including cell shapes and axonal arborization in the inner third of the inner plexiform layer, was similar to that observed in wild-type animals (Fig. 3). No significant reduction in the number of rod bipolar cells was observed throughout the retina.
Fig. 3.
Immunostaining for protein kinase C in wild-type (Cx36 +/+) and homozygous knock-out (Cx36 −/−) retina. Immunolabeling for protein kinase C displays no differences for Cx36 +/+ (A) and Cx36 −/− (B) retinas. In both types of animals, rod bipolar cells, few amacrine cells, and some outer segments of photoreceptor cells are labeled. Corresponding Nomarski images are shown on the left. Scale bars, 50 μm.
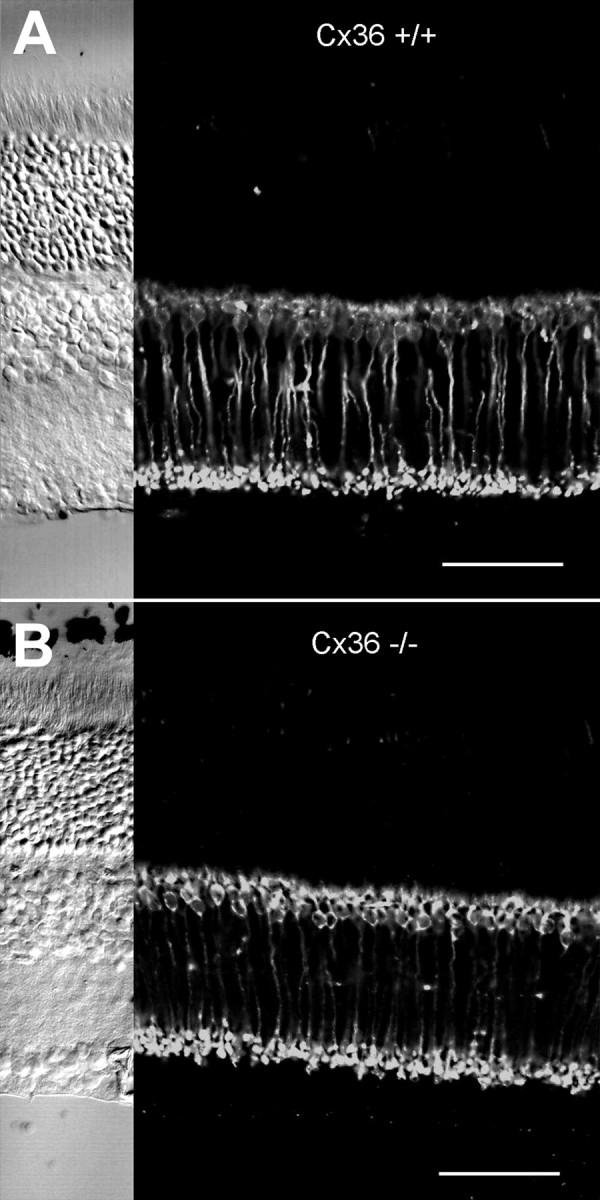
AII amacrine cells are the most numerous amacrine cell type in the rodent retina (Jeon et al., 1998). They exhibit a typical narrow-field morphology with distinctive lobular dendrites in the outer one-third of the inner plexiform layer and extensive arboreal dendrites in the inner two-thirds (Vaney, 1985; Mills and Massey, 1991). The presence of AII amacrine cells was demonstrated by intracellular dye injection in retinal slices with the aid of infrared video microscopy. Cells were randomly chosen according to their location at the border between the inner nuclear and inner plexiform layers, microinjected with Neurobiotin, and subsequently detected with FITC-conjugated streptavidin. A total of 10 AII amacrine cells were filled intracellularly and immunocytochemically processed. AII amacrine cells were present in both mutant and wild-type mice with no obvious difference in their characteristic morphology (Fig. 3). Slices with successfully injected AII amacrine cells were further processed and incubated with antibodies directed to Cx36 (Teubner et al., 2000). Whereas in wild-type retina the known localization of Cx36 immunosignals on arboreal dendrites of AII cells was revealed as expected (Feigenspan et al., 2001), no such colocalization was found in mutant mice (Fig. 4). Although the punctate labeling was strongly decreased in the Cx36-deficient mice, there was still some residual labeling, but there was no colocalization of this label with arboreal processes of AII cells in the mutant mice. This confirmed that the colocalized label in the wild-type retina resulted from Cx36, i.e., the connexin involved in homologous and heterologous electrotonic coupling of AII amacrine cells.
Fig. 4.

Confocal analysis of AII amacrine cells counterstained with antibodies to Cx36. AII amacrine cells injected with Neurobiotin and visualized with FITC show their typical morphology in vertical slices of both Cx36 −/− and Cx36 +/+ animals.A and C represent 200 nm optical sections of Cx36 −/− and Cx36 +/+ mice, respectively. When double-labeled with antibodies to Cx36, only wild-type mice display significant overlap in the dendritic region of AII cells (C,arrows). In B and D, a stack of superimposed optical sections is displayed to show the overall morphology of AII amacrine cells and the extent of double-labeling in the ON sublamina of the inner plexiform layer. Scale bars, 50 μm.
Does the lack of Cx36 expression by AII amacrine cells also result in a lack of functional coupling between these neurons and ON cone bipolar cells? It has been shown that this coupling forms the basis of neurotransmitter coupling (Vaney et al., 1998). Cone bipolar cells show an elevated level of glycine that is not derived by high-affinity uptake or de novo synthesis but is obtained by neurotransmitter coupling through gap junctions with glycinergic amacrine cells, in particular the glycinergic AII amacrine cells. If the functional coupling between these neurons and the bipolar cells is impaired in the Cx36-deficient mice, one would expect a strongly reduced level of glycine in ON cone bipolar cells. We used antibodies against glycine to compare neuronal glycine levels in wild-type and mutant mice (Fig. 5). The number of heavily labeled amacrine cell somata was approximately the same in both populations; the number of labeled cone bipolar cells, however, was strongly reduced in the mutant animals. Previous studies have shown that AII amacrine cells are the source of the glycine in all ON cone bipolar cells, which form the major group of glycine-labeled bipolar cells (Cohen and Sterling, 1986; Marc, 1986; Vaney et al., 1998). The strong reduction of labeled cone bipolar cells in the Cx36-deficient mice therefore indicates that functional coupling between AII amacrine cells and ON cone bipolar cells is impaired in these animals.
Fig. 5.
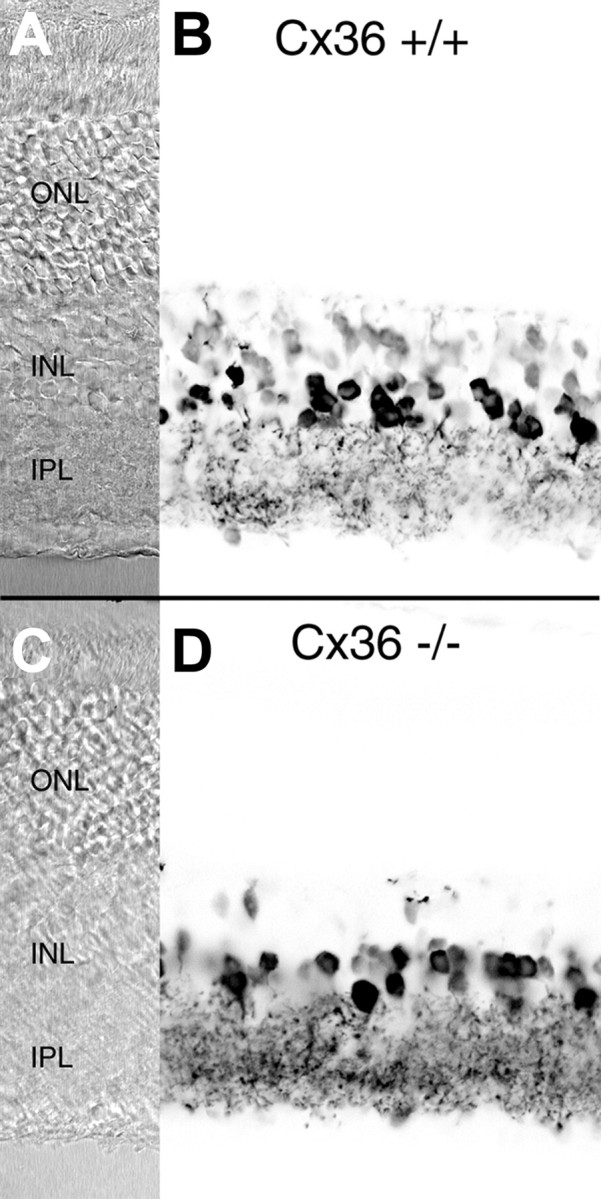
Immunostaining of wild-type (Cx36 +/+) and Cx36-deficient (Cx36 −/−) mouse retina with polyclonal antibodies against glycine. A, C, Normarski images.B, D, Negative confocal micrographs of glycine immunofluorescence. In the wild-type retina (B), glycinergic amacrine cells display strong immunoreactivity. Cone bipolar cells show a weaker but still significant labeling. Staining of glycinergic amacrine cells in the Cx36 −/− retina (D) is comparable with that of the wild-type, whereas only a few bipolar cells appear glycine positive. Scale bars: 25 μm. ONL, Outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer.
No specific immunocytochemical marker for cone bipolar cells was available. Antibodies to recoverin, which had been reported to infrequently label ON cone bipolar cells (Haverkamp et al., 2000), did not label bipolar cells in wild-type mice, although they strongly labeled photoreceptor cells (data not shown). However, because the number of ganglion cells was not reduced in Cx36 null mice (Fig.2A) and because ganglion cells in mammals receive massive input from cone bipolar cells, their existence suggests the presence of these neurons also in the Cx36-deficient mice. Thus, there were no gross changes in the overall retinal cellular arrangement and in particular in the rod cellular pathway of Cx36 −/− mice.
With the aid of an anterograde neuronal tract tracing technique, we also examined the central termination of ganglion cell axons in wild-type and homozygous mutant mice. Wheat germ agglutinin-conjugated horseradish peroxidase was injected into the vitreous body of the right eye of mutant and wild-type mice. In both types of animals, the optic fibers projected into the pretectal regions (data not shown).
Together, these results demonstrate that the gross morphology of the visual pathway from eye to brain was not affected by the absence of Cx36.
Impairment of ERG responses in Cx36-deficient mice
To examine the function of Cx36 in the early visual pathway, we first compared ERG recordings between the wild-type and homozygous mutant mice. The ERG response to a light flash can basically be divided into three major components, the a-, b-, and c-waves (Dowling, 1987;Steinberg et al., 1991). Each wave represents a mass response generated by complex interactions in the retina. Although the exact origin and generation are still partially elusive, it has become clear that the a-wave mainly originates in photoreceptor cells and the b-wave principally arises from depolarizing bipolar cells, whereas the c-wave reflects activity of Müller cells and the pigment epithelium, respectively (Steinberg et al., 1991; Masu et al., 1995).
Scotopic ERG recordings from dark-adapted wild-type mice after a 10 msec white light flash showed all three wave components (Fig.6A, top traces). In mutant mice, all three wave components were also present; however, the b-wave and the c-wave amplitudes were considerably smaller than in wild-type animals. Because the b-wave has a lower threshold than the a-wave, both components were recorded as a function of illumination intensities. In wild-type mice, the b-wave appeared ∼2 log units earlier than the a-wave (Fig.6B), increased with light intensity, and was overridden by typical oscillations at higher intensities. Although the threshold of the b-wave in mutant animals was similar, the amplitude was decreased and showed no increase with higher intensities. Oscillations could be discerned but were weakly developed. The threshold of the a-wave was similar in both types of animals and gradually increased with higher intensities. The time-to-peak intensity functions for both waves were identical in both animals (Fig.6C). In all experiments, the c-wave of the mutant mice was only visible at very strong intensities and could hardly be quantified. Because the c-wave reflects the activity of non-neuronal elements, it was not further analyzed.
Fig. 6.
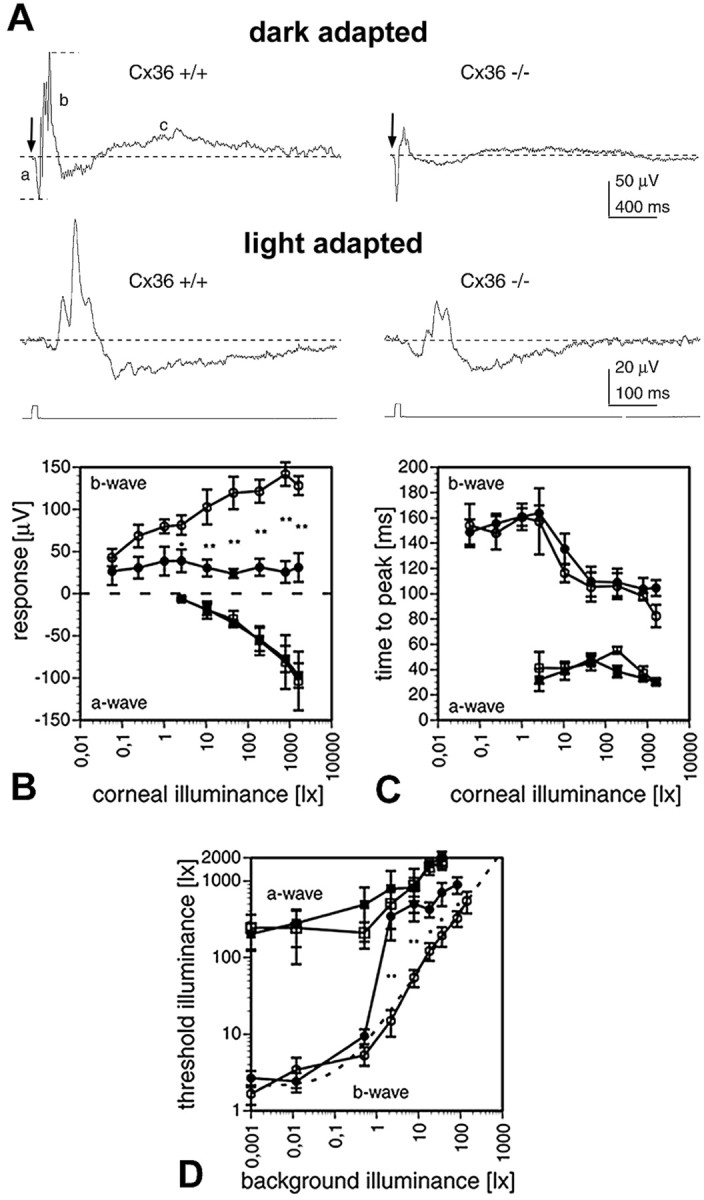
ERG recordings. A, Examples for dark- and light-adapted ERGs of Cx36 wild-type (+/+) and deficient (−/−) mice in response to 10 msec, white light flashes. Top traces, Responses to 520 lux light flashes (arrows) under dark-adapted conditions. Short dashed lines indicate how response amplitudes of a-wave (a) and b-wave (b) were measured with respect to the base level (long dashed lines). In mutant mice, the b- and c-wave amplitudes were reduced compared with wild type. The c-wave (c) was not studied in further detail. Bottom traces, Responses to 784 lux light flashes superimposed to 7.8 lux white background light. B, Intensity–response curves for the a-waves (squares) and b-waves (circles) of wild-type (open symbols) and mutant (filled symbols) mice under dark-adapted conditions. The data points plot the mean ± SEM (n = 5) in B–D. Wild-type mice showed significantly larger b-wave amplitudes for corneal illuminances in excess of 1 lux (Mann–Whitney U test; *p < 0.05; **p < 0.01).C, Time-to-peak versus intensity curves of a- and b-waves. Time-to-peak was identical for wild-type and mutant mice, for both a- and b-waves. Symbols as in B.D, Increment threshold curves for mutant (filled symbols) and wild-type (open symbols) a-waves (squares) and b-waves (circles). Each data point plots the mean ± SEM threshold illuminance for evoking a threshold response on a steady background. Mutant mice showed significantly increased b-wave thresholds for background illuminances in excess of 1 lux (Mann–Whitney U test; *p < 0.05; **p < 0.01), whereas the a-wave thresholds were not affected. The dotted line indicates a Weber–Fechner fit of the wild-type b-wave data points (ΔL/ΔLo =k(L −Lo)n;R2 = 0.99; ΔLo = 1.037 lux;Lo = 0.127; k = 10.372; n = 0.789; ΔL, incremental threshold illuminance; ΔLo, absolute threshold; L, background illuminance;Lo, “dark” light or Fechner's “Eigengrau;” k and n are constants).
Next, we analyzed the cone pathway by recording light-adapted ERGs (Fig. 6A, bottom traces). Light flashes were presented on a background illumination of increasing intensities, and significant differences between wild-type and mutant mice were found in the increment threshold functions for the b-wave (Fig.6D). Whereas the threshold function of the b-wave in wild-type retina closely followed a Weber–Fechner relation, the function for mutant mice b-wave significantly deviated from this relation at background illuminances larger than 1 lux. At these higher background illuminances, mutant mice had significantly higher increment thresholds. The increment threshold functions for the a-wave, on the other hand, were similar in both populations, indicating that the photoreceptors were most likely not affected by the depletion of Cx36.
Central projection
The histological examination of the central projection did not reveal a difference in the mutant mice at the light microscopic level. The ERG data, on the other hand, revealed a significant decrease of the b-wave and therefore most likely of the ON pathway, because this pathway constitutes the major part of the b-wave. We next examined the effects of Cx36 deficiency on visual transmission from the retina to the brain and recorded light-evoked field potentials (VEP) from the optic tectum, the prime target of retinal ganglion cells.
In wild-type animals, short flashes of strong white light given to one eye produced a large negative potential, followed by oscillations on the contralateral side (Fig.7A). Similar responses were recorded from the Cx36-deficient animals. The amplitude of the initial negativity in both wild-type and mutant mice did not show a clear intensity dependence over the range tested (Fig. 7B). The time-to-peak intensity function of the initial negativity, however, was different in mutant mice. There was a significant increase at lower intensities in which the latency was doubled (Fig. 7C). ON and OFF components of VEP become more obvious with longer light stimuli (Masu et al., 1995). When the duration of illumination was increased to 800 msec, two negative components at the beginning and end of the light stimulus could be separated in wild-type mice (Fig. 7D). Similar negativities at ON and OFF set were observed in the mutant mice.
Fig. 7.

VEP recordings. A, Example for a VEP of a wild-type mouse in response to a 10 msec white light flash.B, Intensity–response curves were identical for wild-type (open symbols) and mutant (filled symbols) mice. Data points plot mean ± SEM (n = 5). C, Time-to-peak versus intensity curves of the VEP. Time-to-peak was significantly longer for mutant mice at the two lowest intensities used (Mann–Whitney U test; *p < 0.05; **p < 0.01; n = 5 for each data point). D, VEP examples of Cx36 +/+ and Cx36 −/− mice in response to 800-msec-long light flashes both showed ON and OFF components. Corneal illuminance was 784 lux in A andD.
DISCUSSION
In this study, targeted disruption of the Cx36 gene in transgenic mice was used to examine the role of Cx36 in visual transmission. Cx36 is the first connexin that could be localized to identified retinal neurons and that showed highest expression in murine retina. As expected, a deletion of the Cx36 coding region resulted in complete loss of Cx36 transcript. The homozygous mutant mice developed normally and showed no apparent abnormalities. Their retinas displayed all nuclear and plexiform layers, and the histology of retinal projection to the optic tectum was not distorted. ERG analysis revealed a significant decrease in the b-wave amplitude, and VEP analysis yielded an extended latency of the initial negativity. Thus, our results provide clear evidence that Cx36 is essential for electrical synaptic interactions in early visual transmission, whereas it does not appear to cause obvious developmental abnormalities when deleted by targeted disruption of its gene.
In the retina, Cx36 protein is expressed in AII amacrine cells (Feigenspan et al., 2001). Within the inner plexiform layer, it forms homotypic gap junctional channels that connect AII cells with their neighboring AII cells and forms heterotypic gap junctional channels with cone ON bipolar cells. This electrotonic pathway is supposed to transmit the rod signal to the cone ON pathway (Vaney, 1997).
Our characterization of Cx36-deficient mice shows that the b-wave is primarily affected, suggesting that Cx36 is involved in the pathways generating the b-wave. Because the increment threshold function of the a-wave under light-adapted conditions was not affected, the functional deficit resulting from Cx36 deficiency is expected to be located downstream of the photoreceptors. Previously, it was reported that the b-wave was completely lacking in mice deficient for metobotropic glutamate receptor 6 (mGluR6), the major glutamate receptor of ON-type bipolar cells (Masu et al., 1995), and the b-wave was therefore linked to activity in the ON pathway. Both rod bipolar cells and ON-type cone bipolar cells are part of the ON pathway, and their relative contribution to the b-wave is not known. In light-adapted retinas in which the rods and rod bipolar cells are saturated and unresponsive to light stimuli, light flashes still produce a b-wave that is larger than in the dark-adapted retina (Peachey et al., 1993), indicating that cone ON bipolar cells could be the major source of the b-wave. The decrease of the b-wave in Cx36 mutant mice then likely resulted from the elimination of activity in the ON-type cone bipolar cells because of the missing gap junctional communication between AII amacrine cells and ON-type cone bipolar cells. That this communication is impaired in the mutant mice was demonstrated by decreased neurotransmitter coupling between AII amacrine cells and ON cone bipolar cells. Because the number of PKC-labeled rod bipolar cells in Cx36 mutant mice did not differ from that of wild-type mice, it is likely that the residual b-wave present in Cx36-deficient mice reflects the activity of rod bipolar cells. An alternative explanation would be that some rod activity reaches the cone pathway via an alternative route. It has been proposed by others (Smith et al., 1986; Soucy et al., 1998; Hack et al., 1999; Sharpe and Stockman, 1999) that such alternative pathways may exist. Among these, gap junctions found between rods and cones could indeed activate ON-type cone bipolar cells. The connexin involved in coupling of these photoreceptor cells has not yet been identified, and it is not known whether or not depletion of Cx36 affects this pathway.
The mouse retina is rod dominated, and cones constitute only ∼3% of the photoreceptor cells (Jeon et al., 1998). Nevertheless, cone contribution to the ERG can be physiologically isolated if rods are saturated with a background light of appropriate intensity. Under these conditions, the b-wave represents merely the activity of the ON-type cone bipolar cells, which is cone generated, and Cx36 null mice showed a reduced sensitivity. Because ON-type cone bipolar cells are directly driven by the cones and not indirectly by the gap junctional input from AII cells under these circumstances, the lack of this electrotonic pathway in Cx36-deficient mice cannot explain this result. ON-type cone bipolar cells, on the other hand, do not express Cx36 and appear to make use of a yet unknown connexin for heterologous coupling with AII amacrine cells. It may well be that they continue to express this connexin and even insert hemichannels into their membrane, but, because of a lack of Cx36 hemichannels in AII amacrine cells, they do not form gap junctional channels. This continuous but temporary insertion of hemichannels would lower the input resistance of the cell and as a consequence reduce any voltage signal across the membrane. That hemichannels can be present on retinal neurons and do not compromise viability has indeed been shown (DeVries and Schwartz, 1992; Malchow et al., 1993). Such a decrease of input resistance could also explain the flat profile of the residual b-wave in mutant mice. An alternative explanation for the observed reduction in sensitivity could arise from the observation that Cx36 is also present in the outer plexiform layer; the low density of the immunosignals and the regular mosaic suggest that it is linked to the cone system (Feigenspan et al., 2001). Thus, Cx36 might be involved in the cone system at a very early stage.
Despite the decreased b-wave in mutant mice, the VEP of these animals exhibits both ON and OFF components. This contrasts to findings of Masu et al. (1995), in which the lack of a b-wave in mGluR6-deficient mice corresponded to the lack of an ON component in VEPs recorded from the colliculus. Because in Cx36-deficient mice the glutamatergic transmission was not interrupted but only the transmission between rods and ON-type cone bipolar cells was, the cone pathway must be functioning. The light intensities needed to evoke VEPs from the skull surface and to demonstrate the ON and OFF components are compatible with a major contribution of the cone pathway. A major contribution of the cone system would also partially explain the extended latency observed in the VEP of mutant mice because, in these animals, the cone threshold was increased.
Our data show that Cx36 is an essential component of the mouse visual pathway, without which perception of light is severely impaired. Because Cx36 is also expressed in other cerebral and cerebellar neurons (Parenti et al., 2000; Teubner et al., 2000), as well as in excitable β-cells of the pancreas (Serre-Beinier et al., 2000), Cx36-deficient mice can also be used to study other possible functions of Cx36-containing gap junctions in the corresponding tissues. Moreover, disruption of the Cx36 gene in different types of neurons, for example by using the cre/lox system (Theis et al., 2000), will allow to dissect additional functions of this connexin in electrical synapses between central neurons.
Footnotes
M.G. received a stipend of the Graduierten Kolleg “Pathogenesis of central nervous diseases.” Work in the Bonn laboratory was supported by German Research Association Grant Wi270/22-2 and Funds of the Chemical Industry (to K.W.). Work in the Oldenburg laboratory was supported by German Research Association Grant SFB 517/A2 (to R.W.). We thank Thomas Hennek for excellent technical assistance.
Correspondence should be addressed to Dr. K. Willecke, Institute of Genetics, Division of Molecular Genetics, University of Bonn, Römerstraße 164, 53117 Bonn, Germany. E-mail:genetik@uni-bonn.de.
REFERENCES
- 1.Al-Ubaidi MR, White TW, Ripps H, Poras I, Avner P, Gomes D, Bruzzone R. Functional properties, developmental regulation, and chromosomal localization of murine connexin36, a gap-junctional protein expressed preferentially in retina and brain. J Neurosci Res. 2000;59:813–826. doi: 10.1002/(SICI)1097-4547(20000315)59:6<813::AID-JNR14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- 3.Bennett MV, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: new tools, new answers, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- 4.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 5.Cohen E, Sterling P. Accumulation of (3H)glycine by cone bipolar neurons in the cat retina. J Comp Neurol. 1986;250:1–7. doi: 10.1002/cne.902500102. [DOI] [PubMed] [Google Scholar]
- 6.Condorelli DF, Parenti R, Spinella F, Trovato Salinaro A, Belluardo N, Cardile V, Cicirata F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci. 1998;10:1202–1208. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- 7.De Sousa PA, Valdimarsson G, Nicholson BJ, Kidder GM. Connexin trafficking and the control of gap junction assembly in mouse preimplantation embryos. Development. 1993;117:1355–1367. doi: 10.1242/dev.117.4.1355. [DOI] [PubMed] [Google Scholar]
- 8.DeVries SH, Schwartz EA. Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J Physiol (Lond) 1992;445:201–230. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dowling JE. The retina. Belknap; Cambridge, MA: 1987. [Google Scholar]
- 10.Feigenspan A, Teubner B, Willecke K, Weiler R. Expression of neuronal connexin36 in AII amacrine cells of the mammalian retina. J Neurosci. 2001;21:230–239. doi: 10.1523/JNEUROSCI.21-01-00230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature. 1999;402:72–75. doi: 10.1038/47029. [DOI] [PubMed] [Google Scholar]
- 12.Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- 13.Green DG, Herreros de Tejada P, Glover MJ. Are albino rats night blind? Invest Ophthalmol Vis Sci. 1991;32:2366–2371. [PubMed] [Google Scholar]
- 14.Greferath U, Grünert U, Wässle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- 15.Güldenagel M, Söhl G, Plum A, Traub O, Teubner B, Weiler R, Willecke K. Expression patterns of connexin genes in mouse retina. J Comp Neurol. 2000;425:193–201. [PubMed] [Google Scholar]
- 16.Gulisano M, Parenti R, Spinella F, Cicirata F. Cx36 is dynamically expressed during early development of mouse brain and nervous system. NeuroReport. 2000;11:3823–3828. doi: 10.1097/00001756-200011270-00045. [DOI] [PubMed] [Google Scholar]
- 17.Hack I, Peichl L, Brandstatter JH. An alternative pathway for rod signals in the rodent retina: rod photoreceptors, cone bipolar cells, and the localization of glutamate receptors. Proc Natl Acad Sci USA. 1999;96:14130–14135. doi: 10.1073/pnas.96.24.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. J Neurosci. 1992;12:4911–4922. doi: 10.1523/JNEUROSCI.12-12-04911.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haverkamp S, Grünert U, Wässle H. The cone pedicle, a complex synapse in the retina. Neuron. 2000;27:85–95. doi: 10.1016/s0896-6273(00)00011-8. [DOI] [PubMed] [Google Scholar]
- 20.Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolb H, Famiglietti EV. Rod and cone pathways in the inner plexiform layer of cat retina. Science. 1974;186:47–49. doi: 10.1126/science.186.4158.47. [DOI] [PubMed] [Google Scholar]
- 22.Magin TM. Lessons from keratin transgenic and knockout mice. Subcell Biochem. 1998;31:141–172. [PubMed] [Google Scholar]
- 23.Magin TM, McWhir J, Melton DW. A new mouse embryonic stem cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 1992;20:3795–3796. doi: 10.1093/nar/20.14.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magin TM, Schröder R, Leitgeb S, Wanninger F, Zatloukal K, Grund C, Melton DW. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J Cell Biol. 1998;140:1441–1451. doi: 10.1083/jcb.140.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malchow RP, Qian H, Ripps H. Evidence for hemi-gap junctional channels in isolated horizontal cells of the skate retina. J Neurosci Res. 1993;35:237–245. doi: 10.1002/jnr.490350303. [DOI] [PubMed] [Google Scholar]
- 26.Mann-Metzer P, Yarom Y. Electrotonic coupling interacts with intrinsic properties to generate synchronized activity in cerebellar networks of inhibitory interneurons. J Neurosci. 1999;19:3298–3306. doi: 10.1523/JNEUROSCI.19-09-03298.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marc RE. Neurochemical stratification in the inner plexiform layer of the vertebrate retina. Vision Res. 1986;26:223–238. doi: 10.1016/0042-6989(86)90017-9. [DOI] [PubMed] [Google Scholar]
- 28.Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- 29.Mills SL, Massey SC. Labeling and distribution of AII amacrine cells in the rabbit retina. J Comp Neurol. 1991;304:491–501. doi: 10.1002/cne.903040312. [DOI] [PubMed] [Google Scholar]
- 30.Mills SL, Massey SC. Differential properties of two gap junctional pathways made by AII amacrine cells. Nature. 1995;377:734–737. doi: 10.1038/377734a0. [DOI] [PubMed] [Google Scholar]
- 31.O'Brien J, Al-Ubaidi MR, Ripps H. Connexin 35: a gap-junctional protein expressed preferentially in the skate retina. Mol Biol Cell. 1996;7:233–243. doi: 10.1091/mbc.7.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parenti R, Gulisano M, Zappala A, Cicirata F. Expression of connexin36 mRNA in adult rodent brain. NeuroReport. 2000;11:1497–1502. [PubMed] [Google Scholar]
- 33.Peachey NS, Goto Y, Al-Ubaidi MR, Naash MI. Properties of the mouse cone-mediated electroretinogram during light adaptation. Neurosci Lett. 1993;162:9–11. doi: 10.1016/0304-3940(93)90547-x. [DOI] [PubMed] [Google Scholar]
- 34.Peinado A, Yuste R, Katz LC. Gap junctional communication and the development of local circuits in neocortex. Cereb Cortex. 1993;3:488–498. doi: 10.1093/cercor/3.5.488. [DOI] [PubMed] [Google Scholar]
- 35.Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci USA. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roerig B, Feller MB. Neurotransmitters and gap junctions in developing neural circuits. Brain Res Brain Res Rev. 2000;32:86–114. doi: 10.1016/s0165-0173(99)00069-7. [DOI] [PubMed] [Google Scholar]
- 37.Rozental R, Morales M, Mehler MF, Urban M, Kremer M, Dermietzel R, Kessler JA, Spray DC. Changes in the properties of gap junctions during neuronal differentiation of hippocampal progenitor cells. J Neurosci. 1998;18:1753–1762. doi: 10.1523/JNEUROSCI.18-05-01753.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selfridge J, Pow AM, McWhir J, Magin TM, Melton DW. Gene targeting using a mouse HPRT minigene/HPRT-deficient embryonic stem cell system: inactivation of the mouse ERCC-1 gene. Somat Cell Mol Genet. 1992;18:325–336. doi: 10.1007/BF01235756. [DOI] [PubMed] [Google Scholar]
- 39.Serre-Beinier V, Le Gurun S, Belluardo N, Trovato-Salinaro A, Charollais A, Haefliger JA, Condorelli DF, Meda P. Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes. 2000;49:727–734. doi: 10.2337/diabetes.49.5.727. [DOI] [PubMed] [Google Scholar]
- 40.Sharpe LT, Stockman A. Rod pathways: the importance of seeing nothing. Trends Neurosci. 1999;22:497–504. doi: 10.1016/s0166-2236(99)01458-7. [DOI] [PubMed] [Google Scholar]
- 41.Smith RG, Freed MA, Sterling P. Microcircuitry of the dark-adapted cat retina: functional architecture of the rod-cone network. J Neurosci. 1986;6:3505–3517. doi: 10.1523/JNEUROSCI.06-12-03505.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Söhl G, Degen J, Teubner B, Willecke K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998;428:27–31. doi: 10.1016/s0014-5793(98)00479-7. [DOI] [PubMed] [Google Scholar]
- 43.Soucy E, Wang Y, Nirenberg S, Nathans J, Meister M. A novel signaling pathway from rod photoreceptors to ganglion cells in mammalian retina. Neuron. 1998;21:481–493. doi: 10.1016/s0896-6273(00)80560-7. [DOI] [PubMed] [Google Scholar]
- 44.Steinberg RH, Frishman LJ, Sieving PA. Progress in retinal research. Pergamon; Oxford: 1991. [Google Scholar]
- 45.Strata F, Atzori M, Molnar M, Ugolini G, Tempia F, Cherubini E. A pacemaker current in dye-coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. J Neurosci. 1997;17:1435–1446. doi: 10.1523/JNEUROSCI.17-04-01435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strettoi E, Raviola E, Dacheux RF. Synaptic connections of the narrow-field, bistratified rod amacrine cell (AII) in the rabbit retina. J Comp Neurol. 1992;325:152–168. doi: 10.1002/cne.903250203. [DOI] [PubMed] [Google Scholar]
- 47.Tamas G, Buhl EH, Lorincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat Neurosci. 2000;3:366–371. doi: 10.1038/73936. [DOI] [PubMed] [Google Scholar]
- 48.Teubner B, Degen J, Söhl G, Güldenagel M, Bukauskas FF, Trexler EB, Verselis VK, De Zeeuw CI, Lee CG, Kozak CA, Petrasch-Parwez E, Dermietzel R, Willecke K. Functional expression of the murine connexin 36 gene coding for a neuron-specific gap junctional protein. J Membr Biol. 2000;176:249–262. doi: 10.1007/s00232001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teubner B, Odermatt B, Güldenagel M, Söhl G, Degen J, Bukauskas FF, Kronengold J, Verselis VK, Jung YT, Kozak CA, Schilling K, Willecke K. Functional expression of the new gap junction gene connexin47 transcribed in mouse brain and spinal cord neurons. J Neurosci. 2001;21:1117–1126. doi: 10.1523/JNEUROSCI.21-04-01117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Theis M, Magin TM, Plum A, Willecke K. General or cell type-specific deletion and replacement of connexin-coding DNA in the mouse. Methods. 2000;20:205–218. doi: 10.1006/meth.1999.0938. [DOI] [PubMed] [Google Scholar]
- 51.Traub RD, Schmitz D, Jefferys JG, Draguhn A. High-frequency population oscillations are predicted to occur in hippocampal pyramidal neuronal networks interconnected by axoaxonal gap junctions. Neuroscience. 1999;92:407–426. doi: 10.1016/s0306-4522(98)00755-6. [DOI] [PubMed] [Google Scholar]
- 52.Vaney DI. The morphology and topographic distribution of AII amacrine cells in the cat retina. Proc R Soc Lond B Biol Sci. 1985;224:475–488. doi: 10.1098/rspb.1985.0045. [DOI] [PubMed] [Google Scholar]
- 53.Vaney DI. Neuronal coupling in rod-signal pathways of the retina. Invest Ophthalmol Vis Sci. 1997;38:267–273. [PubMed] [Google Scholar]
- 54.Vaney DI, Young HM, Gynther IC. The rod circuit in the rabbit retina. Vis Neurosci. 1991;7:141–154. doi: 10.1017/s0952523800011019. [DOI] [PubMed] [Google Scholar]
- 55.Vaney DI, Nelson JC, Pow DV. Neurotransmitter coupling through gap junctions in the retina. J Neurosci. 1998;18:10594–10602. doi: 10.1523/JNEUROSCI.18-24-10594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venance L, Rozov A, Blatow M, Burnashev N, Feldmeyer D, Monyer H. Connexin expression in electrically coupled postnatal rat brain neurons. Proc Natl Acad Sci USA. 2000;97:10260–10265. doi: 10.1073/pnas.160037097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wässle H, Grünert U, Chun MH, Boycott BB. The rod pathway of the macaque monkey retina: identification of AII-amacrine cells with antibodies against calretinin. J Comp Neurol. 1995;361:537–551. doi: 10.1002/cne.903610315. [DOI] [PubMed] [Google Scholar]