

Abstract
Neointima formation is a major contributor to arteriovenous fistula (AVF) failure. We have previously shown that activation of the Notch signaling pathway contributes to neointima formation by promoting the migration of vascular smooth muscle cells (VSMCs) into the venous anastomosis. In the current study we investigated the mechanisms underlying the dedifferentiation and migration of VSMCs, and in particular the role of bone marrow-derived fibroblast specific protein 1 (FSP-1)+ cells, another cell type found in models of vascular injury. Using VSMC-specific reporter mice, we found that most of the VSMCs participating in AVF neointima formation originated from dedifferentiated VSMCs. We also observed infiltration of bone marrow-derived FSP-1+ cells into the arterial anastomosis where they could interact with VSMCs. In vitro, conditioned media from FSP-1+ cells stimulated VSMC proliferation and phenotype switching. Activated Notch signaling transformed FSP-1+ cells into type I macrophages and stimulated secretion of cytokines and growth factors. Pretreatment with a Notch inhibitor or knockout of the canonical downstream factor RBP-Jκ in bone marrow-derived FSP1+ cells decreased FSP1+ cell infiltration into murine AVFs, attenuating VSMC dedifferentiation and neointima formation. Our results suggest that targeting Notch signaling could provide a new therapeutic strategy to improve AVF patency.
Keywords: arteriovenous fistula, dedifferentiation, vascular smooth muscle cell, fibroblast specific protein 1, Notch signaling
Graphical abstract
Introduction
Arteriovenous fistulas (AVFs), the Achilles’ heel of hemodialysis patients, lose function primarily due to the formation of neointima that can cause venous stenosis and promote thrombosis.1 Although the sociological and economic costs of a failed AVF are well known,2 the pathophysiology of AVF occlusion has not been established. Advances in understanding the molecular and cellular mechanisms underlying neointima formation are required to alleviate the problem.
Our results and those of others indicate that chronic kidney disease (CKD) accelerates the neointima formation of AVFs, resulting in AVF failure. The principal cells involved in this growth of the neointima are vascular smooth muscle cells (VSMCs).3, 4 Some neointima VSMCs in the neointima arises from the arterial anastomosis.5 We have found that the stimulus for VSMCs to participate in the growth of the neointima is activation of Notch which promotes migration of VSMC into the venous anastomosis to stimulate neointima growth and ultimately, occlusion of the AVF.5
Several strategies including anti-thrombotic strategies have been evaluated to determine if the failure of AVFs can be suppressed or blocked. Unfortunately, clinical trials of these strategies have been unsuccessful.6-10 As noted in the NIH/NIDDK symposium, there is a need for ongoing research directed at improving the duration of functional AVFs (NIH/NIDDK Hemodialysis Vascular Access: Taking the Next Steps (http://niddk.nih.gov/news/events-calendar/Pages/hemodialysis-faxcular-access-2015).
Positive results from this research may not be limited to AVF because in other models of vascular disease including hypertension, atherosclerosis and post-angioplasty stenosis (e.g., balloon or wire injury of arteries), VSMCs have been implicated as key mediators of vascular occlusion.11-14 For example, Rotllan and colleagues reported that a VSMC phenotype switch participates in the neointima formation after femoral artery injury.15 They concluded that VSMC proliferation is the principal source of these cells in injured arteries. This conclusion is consistent with our finding that VSMCs from the arterial anastomosis comprise as many as 50% and 95% of VSMCs forming the neointima in AVFs5 and in arteriovenous grafts,14 respectively.
The biologic factors mediating changes in VSMC are incompletely identified: in the anastomosis of artery and vein in an AVF, there are cellular and molecular events that could initiate neointima formation. For example, Notch signaling is activated,16 there is accumulation of VEGF and monocyte chemotactic protein-1 (MCP-1)17-19 and other factors that could promote neointima formation in AVFs.20, 21 In the present report, we have concentrated on identifying mechanisms that cause VSMC phenotype switching and their migration to the venous anastomosis of the mouse AVF resulting in the development of a neointima and loss of AVF function. One type of the cells that is of particular interest is the bone marrow-derived fibroblast-specific protein-1 (FSP-1) positive cells. These cells are found in organs that are developing inflammation and fibrosis, and FSP-1+ cells are also present in models of vascular injury including AVFs.22 The role of FSP-1 cells in the development of the neointima and whether Notch signaling promote its activation have not been fully identified. To overcome this deficit, we have examined inter-connections among Notch signaling, FSP-1 cell activation and the phenotype switching of VSMCs that occurs in the AVF anastomosis.
Results
Dedifferentiated VSMCs contribute to neointima formation in AVFs.
Phenotypic switching in VSMCs can occur in injured vessels. However, there is no direct evidence that this phenotype alteration causes neointima formation in AVFs. To analyze this shortcoming, we examined longitudinal sections of AVFs obtained at two weeks after creation of the AVF. VSMCs present in the wall of the artery near the AVF anastomosis were found to change distinctive markers: VSMCs became SMA-α negative, while SMA-α-positive VSMCs were present in the media of the distal artery as well as in the anastomosed vein of AVFs. These results suggest that SMA-α negative cells of the venous anastomosis derive from either dedifferentiated VSMCs or cells infiltrating the AVF (Figure 1A). Double immunofluorescent staining confirmed that VSMC markers (SMMHC and SMA-α) were absent in cells from the arterial anastomoses of the AVFs (Figure 1B). We recognize that VSMC marker-negative cells (see arrow in Figure 1A & B) could be derived from dedifferentiated VSMCs. To evaluate this possibility, we use RFPf/f-GFP/SMMHC-ERCre+ mice (VSMCGFP mice) to label and track VSMCs.14 This strategy was based on SMMHC-ERCre, which will be activated by tamoxifen to induce GFP expression in VSMCs and their progeny in VSMCGFP mice. The strategy successfully and permanently labelled SMMHC-expressing VSMCs in common carotid artery, recognized as GFP positive cells (Figure 1C & D).
Figure 1. Dedifferentiated VSMCs contribute to neointima formation in AVF.

A. VSMC marker, SMA-α, was absent in arterial media in the anastomosis of AVF The anastomosis regions in the longitudinal section of AVFs were showed in frames, the black arrows point to the SMA-α negative cells (A, arterial side; V, venous side; L, lumen, scale bar = 500 μm). B. Double immunofluorescent staining of the SMMHC/SMA-α in anastomosed area of AVFs (2 weeks). The arrows point to the VSMC marker negative cells in the media of the arterial side of the anastomosis. C. Generating VSMC reporter transgenic mice. After tamoxifen induction, only VSMCs and their lineages will be labeled with GFP. D. VSMCs (SMMHC+) were labeled with GFP in the common carotid artery in RFPflox/flox-GFP/SMMHC-ERCre+ mice (VSMCGFP mice) after tamoxifen treatment. E. AVFs were created in VSMCGFP mice after 14 days of last dose of tamoxifen treatment. The expression of SMMHC and GFP were determined in anastomosis (E, left panel) of the 2 week AVFs (yellow arrow points SMMHC+/GFP+ cells; white arrows point SMMHC−/GFP+ cells); the percentage of the two types of cells were counted (E, right panel). F. mRNA ratio of SMMHC and GFP were determined from common carotid artery and anastomosis of AVFs that were created in VSMCGFP mice (representative data was shown from 3 repeated experiments, *, p < 0.05). G. AVFs were created in VSMCGFP mice after 14 days of last dose of tamoxifen treatment. The expression of SMA-α and GFP were determined in neointima area in the venous side of the 4 week AVFs. The total GFP+ cells and GFP+/SMA-α− cells were counted (right panel. n = 6). Scale bar = 50 μm in panel B, D, E, and G.
Two weeks after the last dose of the tamoxifen, AVFs were created in VSMCGFP mice, so that all GFP+ cells found in AVFs would be derived from pre-labeled VSMCs. We found that about 50% of GFP+ cells lost expression of SMMHC in the AVF anastomosis (Figure 1E, white arrow) indicating that these VSMCs in the AVF had dedifferentiated. To further confirm the VSMC dedifferentiation in anastomosis of AVFs, the RNAs from common carotid artery and anastomosis of AVFs in VSMCGFP mice were collected and the mRNA levels of SMMHC and GFP were determined. There are much decreased mRNA ratio of SMMHC/GFP in anastomosis of AVFs vs. that in control arteries (Figure 1F), indicating loss of VSMC markers in GFP labeled cells (VSMC lineages).
We also found that GFP+/SMA-α+ double positive cells were present in ~80% of neointima cells detected in venous arm of the 1 month AVFs (Figure 1G), indicating dedifferentiated VSMCs regain VSMC markers at later stage. These results demonstrate that VSMCs are the major source that form the neointima in AVFs.
Bone marrow-derived FSP-1 positive cells are associated with dedifferentiated VSMCs in AVFs.
Infiltration of CD45-positive inflammatory cells occurred in the arterial media of AVF anastomoses (Figure 2A). Double immunofluorescent staining results showed that a large fraction of FSP-1 positive cells were accumulated in the arterial anastomosis. Notably, these FSP-1+ cells were mainly positive for CD45 and macrophage marker, Mac2 (Figure 2B & C). About 40 −60% of cells in the anastomosis area of the AVFs were FSP-1+ inflammatory cells (Figure 2D). Since bone marrow is the major source for inflammatory cells in AVFs, we next determined if bone marrow-derived FSP-1+ cells are linked to VSMC activation. Wild type mice were transplanted with bone marrow of FSP-1-GFP transgenic mice to get WTFSP-1-GFP BM mice. In AVFs created in WTFSP-1-GFP BM mice, 30 – 40% of GFP-positive cells costaining with CD45 were located both in the anastomosis and in the neointima of the 2 week AVFs (Figure 2E & F). These results indicate that bone marrow-derived FSP-1+ cells infiltrated into the medium and could interact with VSMCs and leads to VSMC activation.
Figure 2. Bone marrow-derived FSP-1 positive cells infiltrate in the arterial anastomosis of the AVF.

A, CD45 positive cells were detected by immunostaining, and the red arrows pointed the CD45 positive cells in the media of arterial side of AVF anastomosis. B-D. FSP-1+ cell infiltration in AVF were revealed by immunofluorescent staining with CD45 (B) or macrophage marker, Mac-2 (C). The double positive cells in B & C were counted and summarized (D). E & F. FSP-1+ inflammatory cells derived from bone marrow of FSP-1-GFP transgenic mice. AVFs were created in wild type mice with bone marrow from FSP-1-GFP mice. The Bone marrow derived-FSP-1+ cells in the media of anastomosed artery (left panel) or in the neointima area (right panel) of the 2 week AVFs were detected and co-immunostained with CD45. The GFP+ cells and the GFP+/CD45+ double positive cells in the areas were counted and calculated (F) (n = 6). G. Double immunostaining of GFP or CD45 in the anastomosis of AVFs created from VSMCGFP mice (arrows point CD45+/GFP− cells), the positive cells were counted (n = 6 mice). Scale bar = 50 μm in all panels.
To further confirm that the dedifferentiated VSMCs are different population from the inflammatory cells, the VSMCs in AVFs created in VSMCGFP mice were characterized. There were ~45% of GFP positive cells found in anastomosis area in 2 week AVFs, only few (1-2%) of these cells were positive for markers of inflammatory cells (CD45) or endothelial cells (CD31) (Figure 2G). This result indicates that inflammatory marker-positive cells in the AVF anastomoses did not originate from VSMCs.
CKD promotes VSMC differentiation and stimulates the secretion of cytokines of M1 macrophages in FSP-1+ cells.
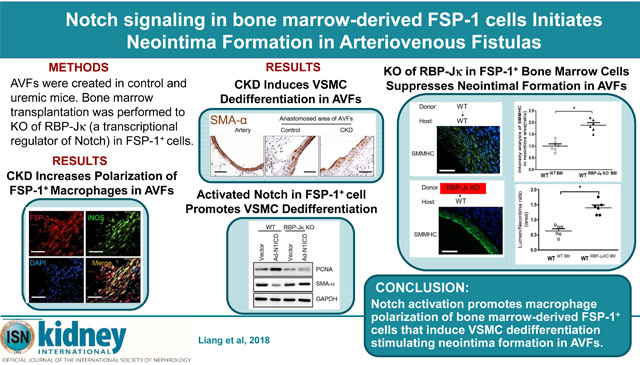
CKD enhances VSMC accumulation and neointima formation in AVFs.3 VSMC dedifferentiation stimulates VSMC proliferation.15 We created CKD mice and determined the renal function. The values of BUN and serum creatinine were significantly increased in CKD mice vs. that in sham control mice (Figure 3A & B). We found that more VSMCs lost SMA-α expression plus an increase in the accumulation of CD45 positive cells in the anastomosis area of the AVF created in uremic mice vs. results from sham control mice (Figure 3C). Consistently, in AVFs created in VSMCGFP mice, the mRNA levels of SMMHC in GFP+ cells were decreased vs. that in control AVFs (Figure 3D). This result indicates that CKD accelerates the loss of VSMC terminal differentiation marker, SMMHC in the anastomosis.
Figure 3. FSP-1+ cells express cytokines of type I macrophages in AVFs with CKD.

A & B. Serum BUN and creatinine were carried out in control and CKD mice. (n = 6, *, p <0.05, vs control). C & D. CKD increases VSMC dedifferentiation. The expression of VSMC markers were determined by immunostaining (C) and RT-PCR (D) in the 2 week AVFs that were created in VSMCGFP mice after sham and CKD. Representative images and RT-PCR results were shown (n = 6 mice; mean ± SD,* p < 0.05 compared with artery; #, p < 0.05, compared with AVF control). E. AVF were created in control and CKD mice and collected after 2 weeks, co-immunostaining of FSP-1 and type I macrophages markers, iNOS, were performed (left panel). Statistical analysis of FSP-1+/iNOS+ cell numbers is shown in right panel. F. Arrays of cytokines in the AVFs from control or CKD mice were detected by real-time RT-RCR, indicating that cytokines from macrophage type 1 were induced by CKD. (n = 6; mean ± SD; * p < 0.05 compared with AVF control). Scale bar = 50 μm in all panels.
FSP-1+ inflammatory cells that infiltrated in AVFs co-expressed type I macrophage markers including nitric oxide synthase (iNOS) (Figure 3E). At 2 weeks after placing the AVF in mice with CKD, there were more FSP-1+ M1 pro-inflammatory macrophages in AVFs vs. results from control mice (Figure 3E). Next, we measured cytokines in the AVFs of control and CKD mice: there were increased levels of monocyte chemoattractant protein (MCP-1), interleukin-6 (IL-6), interleukin-1β (IL-1β) and iNOS in AVFs created in CKD mice (Figure 3F). These findings suggest that CKD induces the infiltration of FSP-1+ cells and this is associated with decreased expression of VSMC markers.
FSP-1+ cell-secreted cytokines and growth factors induce VSMC dedifferentiation.
Mechanism of Notch signaling pathway underlying the regulation of macrophage polarization is well established. The ligand, Dll4, binds to the Notch1 receptor, promotes M1-like polarization.23 To examine the function of FSP-1+ inflammatory cells, the FSP-1+ cells were isolated from bone marrows of FSP-1-GFP transgenic mice and transfected with Ad-N1ICD. Overexpression of N1ICD significantly induced the production of M1 cytokines including MCP-1, IL-1β, IL-6, IL-12β and iNOS mRNA levels in cultured FSP-1+ cells (Figure 4A). The expression of PDGF-BB and bFGF-2 were also up-regulated (Figure 4B). When we incubated VSMCs with individual cytokines (e.g., MCP-1, IL-1β or growth factors, PDGF-BB or FGF-2), we found that IL-1β, PDGF-BB or bFGF-2 stimulated dedifferentiation of VSMCs. This was accompanied by a reduction in the expression of SMA-α (Figure 4C). Next, we incubated VSMCs with conditioned media from FSP-1+ cells. Conditioned media from Ad-NICD-transfected FSP-1+ cells was found to induce VSMC proliferation and dedifferentiation that was associated with up-regulating PCNA and down-regulating SMA-α expression (Figure 4D & E). Taken together, these results suggest that bone marrow-derived FSP-1+ cells synthesize specific cytokines and growth factors which induce VSMC proliferation and dedifferentiation.
Figure 4. FSP-1+ type I macrophages induce VSMC dedifferentiation.

A-B. FSP-1-GFP+ bone marrow cells were infected with Ad-N1ICD for 48 hrs, the expression of cytokines of type 1 macrophages (A) and growth factors (B) were determined by real time RT-PCR. C. The cytokines of type 1 macrophages or growth factors induce VSMCs dedifferentiation. VSMCs were treated with indicated factors, VSMC markers were determined by Western blot. D. Conditioned medium (CM) from vector or Ad-N1ICD-infected FSP-1/GFP+ bone marrow cells were used to treat VSMCs. The VSMC dedifferentiation and proliferation were determined by western blotting. E. Density analysis of Western blot in Panel D. Data are representative of 3 independent experiments. *, p < 0.05.
Activated Notch signaling in FSP-1+ type I macrophages in AVFs promotes VSMC dedifferentiation.
To investigate how Notch signaling influences infiltration of macrophages in AVFs, we investigated the expression of Notch target genes, Hes1, Hey1 and Hes5 in AVFs at 2 weeks after their placement. These genes were significantly induced in mice with CKD vs. results in control mice (Figure 5A). These responses suggest that Notch/RBP-Jκ signaling is activated by CKD. We also found that the activated Notch signal, N1ICD, was co-expressed in FSP-1+ cells and Mac2+ macrophages (Figure 5 B& C), and that there was a higher expression of RBP-Jκ in Mac2+ cells vs. the results in neointima cells (Figure 5D). These results suggest that Notch/RBP-Jκ signaling might involve in M1 polarization of FSP-1+ cells.
Figure 5. Activated Notch signaling in FSP-1+ type I macrophages in AVFs promotes VSMC dedifferentiation.

A. mRNA levels in AVFs from control and CKD mice. The expression levels of Notch downstream targets were determined by RT-PCR. B & C. Double immunofluorescence analyses of N1ICD with FSP-1 (B) or Mac2 (C) in 2 week AVFs of CKD mice. White arrows refer to double staining in the macrophages. D. Immunostaining showed the colcalization of RBP-Jκ and Mac2 in 2 week AVFs. E. Forced N1ICD expression induces the Notch signaling in FSP-1/GFP+ bone marrow cells. The GFP+ cells were isolated from the bone marrow of FSP-1-GFP transgenic mice through flow cytometry. The cells were infected with Ad-N1ICD for 48 hrs. Notch downstream targets were determined (E) and density analysis of the western blots is shown (F). G. Immunofluorescence staining for iNOS in FSP-1-GFP+ bone marrow cells with or without RBP-Jκ after these cells were infected with Ad-N1ICD or Ad-Vector for 48 hours. Fluorescence intensity analysis of iNOS shown in red were quantified. H. VSMCs were cultured with the conditioned medium from FSP-1-GFP+ bone marrow cells with or without RBP-Jκ after these cells were infected with Ad-N1ICD or Ad-Vector for 48 hours. Western blot analysis of SMA-α and PCNA in VSMCs were performed (H) and quantified (I). Data are representative from 3 independent experiments. *p < 0.05 versus control groups. Scale bar = 50 μm in all panels.
In bone marrow-derived FSP-1+ cells, Forced expression of N1ICD stimulated activation of Notch downstream signals, Hes5 and Hes1 (Figure 5E & F). In addition, Over-expression of N1ICD also induced M1 polarization of FSP-1+ cells as well as the expression of iNOS. These responses were suppressed in FSP-1+ cells lack of RBP-Jκ (Figure 5G). Furthermore, conditioned media from FSP-1+ cells that were specifically knocked out of RBP-Jκ blocked dedifferentiation and proliferation of VSMCs that were treated with CM from Ad-N1ICD-infected FSP-1+ cells (Figure 5H & I). These results implicate Notch signaling is a regulator of M1 activation of FSP-1+ cells. Activated Notch signaling in FSP-1+ cells stimulates the secretory factors directly driving VSMC dedifferentiation.
RBP-Jκ KO in Bone Marrow-derived FSP-1+ Cells Suppresses Neointimal Formation of AVFs.
We next determined whether KO of RBP-Jκ in bone marrow-derived FSP-1+-cells would rescue CKD-induced AVF failure. To examine this possibility, we created FSP-1+ cell-specific RBP-Jκ KO mice. This was accomplished by breeding RBP-jκf/f mice with FSP-1-Cre transgenic mice (Figure 6A). The expression of RBP-Jκ in bone marrow FSP-1+ cells was significantly decreased in RBP-Jκf/f/FSP-1-Cre+ mice vs. results in control mice (Figure 6B). To assess the contribution of bone marrow–derived FSP1+ cells to the formation of the neointima, we performed a bone marrow transplantation of irradiated wild-type mice with bone marrow from RBP-Jκf/f/FSP-1-Cre+ mice or WT mice, to generate WTRBP-Jκ BM-KO mice or control WTWT BM) mice, respectively. CKD and AVFs were created in these mice. We found that growth factors were significantly lower in AVFs created in WTRBP-Jκ BM-KO mice vs. results in control mice (Figure 6C). In AVFs of WTWT BM mice, immunostaining demonstrated that almost all cells were RBP-Jκ positive (Figure 6D). In AVFs created in in WTRBP-Jκ BM-KO mice, no expression of RBP-Jκ was detected in the FSP1+-inflammatory cells (Figure 6E), while the RBP-Jκ positive signals were only found in endothelium and neointima VSMCs (Figure 6E). FSP-1 positive cells were significantly decreased in AVFs from in WTRBP-Jκ BM-KO mice (Figure 6D & E). SMMHC immunostaining of AVFs showed that KO RBP-Jκ in bone marrow-derived FSP-1+ cells increased the SMMHC expression and VSMC differentiation, and attenuates accumulation of VSMCs in AVFs (Figure 6F & G). Notably, the increased ratio of the lumen to neointima areas was found in AVFs created in WTRBP-Jκ BM-KO mice compared to results obtained in control mice (Figure 6H). Taken together, our results indicate that RBP-Jκ deficiency in FSP-1+ bone marrow cells reduces the neointima formation by inhibiting dedifferentiation and activation of VSMCs.
Figure 6. KO RBP-Jκ in bone marrow-derived FSP-1+ cells increases AVF patency.

A. Generation of RBP-jκflox/flox/FSP-1-Cre+ mice. B. KO of RBP-Jκ in bone marrow FSP-1+ cells was verified by Western blot. C. Decreases in cytokine production in AVFs (4 weeks) from specific RBP-Jκ deficiency mice were detected by real-time RT-PCR. D & E. At 4 weeks after creating the AVF in CKD mice, specific KO of RBP-Jκ in FSP-1 cells in AVFs were determined by double immunostaining of FSP-1 and RBP-Jκ. White arrows point to RBP-Jκ−/FSP-1+ inflammatory cells. F & G. Representative image and densitometry analysis of SMMHC in AVFs created in WTRBP-Jκ BM-KO mice or control WTWT BM mice. H. Summary of morphometrical analysis shown as area ratio of lumen and neointima (n = 6, *, p < 0.05 vs WT). Scale bar = 50 μm in all panels.
Discussion:
We have uncovered two linked cellular events that contribute to neointima formation resulting in reduced function of a mouse AVF. The events include macrophage polarization of bone marrow-derived FSP-1+ cells and phenotypic switch of VSMCs. We identified that infiltration of bone marrow-derived FSP-1+ cells trigger VSMC dedifferentiation and proliferation (i.e., the phenotype switch of VSMCs). We demonstrated that activation of Notch/RBP-Jκ in FSP-1+ cells arises from a pathway that is stimulated by CKD. The FSP-1+ cells infiltrate the arterial anastomosis of AVF and they produce cytokines and growth factors which induce VSMC dedifferentiation and neointima formation. Finally, we documented that this pathway is important by showing that blocking Notch/RBP-Jκ signaling in bone marrow-derived FSP-1+ cells suppresses the neointima formation in AVFs. We believe these results are the first evidence of a pathway composed of cellular and molecular events results in AVF failure.
It has been reported that myofibroblasts, fibroblasts, macrophages and VSMCs all play a role in the pathogenesis of failed dialysis accesses,24-26 but most of the conclusions in these reports are based on immunostaining results which have led to controversial conclusions.24-26 To avoid these problems, we used GFP-Stopf/f/SMMHC-ERCre+ mice to label VSMCs with GFP and identify cells involved in neointima formation in the AVF. Following placement of the AVF, GFP+ cells in the anastomosis as well as in the neointima of AVFs were detected, demonstrating that VSMCs are the major contributor to the forming neointima. Fortunately, our earlier publication led to a similar conclusion when we used Wnt1-Cre reporter mice27 to label and track VSMC lineages in the outflow track of the developing heart. Results in that publication indicated that as many as 50% of the VSMCs of the neointima arise from the anastomosed artery.5 Other report also indicated that local vein VSMCs could be the neointima cells in an end-to-side configuration in rat AVFs.28 These evidences demonstrate that VSMC activation and dedifferentiation are important cellular events leading to neointima formation in AVFs.
FSP-1 has been implicated in the control of the motility of bone marrow-derived monocytes (inflammatory cells). These processes occur early in dysfunction of a vein graft.29 In mice with CKD, we found that an excess of FSP-1+cells could be detected in AVF samples, suggesting that FSP-1 cells adversely influence formation of the neointima in AVFs. How the activated FSP-1+ monocytes induce the VSMC phenotype switch is not known. We determined the function of bone marrow-derived, FSP-1+cells and found that conditioned media obtained from cultures of these FSP-1+ cells contain significant levels of proinflammatory cytokines and growth factors (MCP-1, IL-1β, IL-6, IL-12β and PDGF-BB). These potential mediators promote phenotype switching of VSMC and are consistent with the report of Nath et al.: they showed that production of proinflammatory cytokines and growth factors promote VSMC activation and neointima formation in models of vascular injury.17 In short, our results demonstrate that bone marrow-derived FSP-1+ cells accumulate in the AVFs and participate in the neointima formation.
Based on the present results and our recent publications, we conclude that there are three major cellular events affecting neointima formation in AVFs. 1) VSMC migration. We have used Wnt1-Cre/reporter mice to label and track VSMCs in the outflow tract (e.g., common carotid artery) and explored the contribution of VSMCs migrating from the arterial anastomosis to neointima formation. Our results indicate that VSMCs from anastomosed artery contribute to as much as 50% of SMCs in the neointima.5 Furthermore, on evaluating the mouse vein grafts, we find that about 60% SMA-α-positive cells in the neointima were migrated from anastomosing arteries.14 2) VSMC de-differentiation. In the current study, we found that GFP-specifically labeled VSMCs lose VSMC markers in AVFs. Bone marrow-derived FSP-1+ inflammatory cells promote VSMC de-differentiation. Our results indicate that local VSMCs de-differentiate and migrate into the AVF venous arm, forming the neointima. 3) VSMC maturation. In these experiments, we found that GFP-labeled VSMCs regain VSMC markers in the neointima in AVFs (Figure 1G). These findings suggest that de-differentiated VSMCs migrate from the arterial media into the venous arm of the AVFs, where the de-differentiated VSMCs regain the expressions of contractile markers of VSMCs.
Which signaling pathway promotes FSP-1+ cell activation in AVFs? One possibility is the Notch cell signaling pathway because it is an important regulator of stem or progenitor cell proliferation and differentiation.30, 31 In response to Notch activation, the intracellular domain of Notch, NICD, translocates to the nucleus and binds the transcriptional repressor, RBP-Jκ. This converts RBP-Jκ into a transcriptional activator which induces the expression of downstream target genes.30, 32 Xu et al. have suggested that Notch-RBP-Jκ signaling controls the expression of the transcription factor, interferon regulatory factor 8 (IRF8), inducing M1 macrophage-associated genes.33 Fung et al reported that cross-talk between the NF-κB and Notch pathways regulates M1 polarization23 and we find that Notch signaling is activated in FSP-1+ monocytes in vitro and in AVFs from mice. KO of RBP-Jκ in FSP-1+ monocytes decreases the expression of M1 cytokines and the production of growth factors. Bone marrow-derived FSP-1+ cells initially promote an inflammatory response and to modulate the phenotype of VSMC by a mechanism mediated by Notch. This result suggests that the formation of neointimas in AVFs could be therapeutically regulated by inhibiting Notch signaling in FSP-1+.
Other investigators report that VSMCs that become activated to express the inflammatory markers during injury stimuli.34, 35 We explored whether cells expressing inflammatory markers could be either monocytes or dedifferentiated VSMCs.36 Specifically, we examined AVFs created in GFP-Stopf/f/SMMHC-Cre+ transgenic mice because VSMC lineages were GFP+. Our results indicate that GFP+ VSMC-derived cells do not express CD45 or Mac2, instead, the CD45 and Mac2 positive cells are bone marrow-derived inflammatory cells and most are FSP-1 positive.
Our mouse AVF model mimics the development of stenosis near the anastomosis, which is similar to events in ESRD patients, we recognize that the mouse AVF can be used to test how AVF maturation failure proceeds with neointima formation in the anastomosis. Our results were obtained with end-to-end rather than end-to-side anastomosis. Moreover, the AVF in mice is not suitable to repeated needle puncture.
In summary, we find that Notch/RBP-Jκ signaling activates bone marrow-derived FSP-1+ monocytes. The monocytes infiltrate the media of the artery in an AVF resulting in polarization and maturation of the FSP-1+ cells and become macrophage type 1 cells. These cells, in turn, secret cytokines and growth factors which initiate dedifferentiation of the VSCM, resulting in a phenotype switch. These events increase the accumulation of activated VSMC that will form a neointima and ultimately, cause failure of the AVF. This new pathway to AVF failure is enhanced by the presence of CKD. Notably, KO of RBP-Jκ in bone marrow-derived FSP-1+ cells blocks the polarization and secretion of cytokines, preventing VSMC dedifferentiation and neointima formation in AVFs.
Concise Methods
Animals.
All studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine, Houston and performed in accordance with National Institutes of Health (NIH) guidelines. Mice were housed in an animal facility with a 12-h light/dark cycle. FSP-1-Cre mice, FSP-1-GFP transgenic mice were purchased from Jackson Laboratories (Bar Harbor, ME). SMMHC-ERCre mice were used as described.5 RBP-Jκf/f mice were generously provided by Dr. Susztaka from Albert Einstein College of Medicine and permission from T. Honjo (Kyoto University Faculty of Medicine). To generate mice with KO of RBP-Jκ knockout in FSP-1+ cells, mice with a floxed RBP-Jκ allele37 were bred with FSP1-Cre mice. After a backcross, RBP-Jκf/f/FSP1-Cre+ (RBP-Jκ KO) mice were obtained. Male mice from RBP-Jκ KO and littermate controls (FSP-1-Cre negative) mice were studied.
Generation of Reagents and Viruses.
Penicillin, streptomycin, DMEM, Dynabeads of sheep anti-rat IgG and FBS were obtained from Invitrogen (Life Technologies, Carlsbad, CA). The protein assay kit was from Bio-Rad (Hercules, CA). Monoclonal α-smooth muscle actin (α-SMA)-FITC antibodies were from Sigma-Aldrich (St. Louis, MO) and Hes1, Hey1 and Hes5 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against Notch1, against the active cleaved Notch1 (N1ICD), RBP-Jκ, and iNOS were from Cell Signaling Biotechnology (Cell Signaling, Danvers, MA). The fluorescent-700/800 secondary antibodies were obtained from Invitrogen (Carlsbad, CA), while an antibody against CD45 was from Millipore (Billerica, MA) and antibodies against F4/80, PCNA, GFP, rabbit anti-α-SMA were from Abcam (Cambridge, MA). The GFP antibody was purchased from Rockland (Limerick, PA) and recombinant human TGF-β1, LPS, IL-1β, FGF-2 were purchased from R&D Systems (Minneapolis, MN). Monocyte chemotactic protein-1 (Cedarlane, Burlington, NC). The adenovirus vector N1ICD were generously provided by Dr. Liaw (Maine Medical Center Research Institute, Scarborough, ME).38 Cell lysates were prepared 48–72 hours after adenovirus infection for western blot.
Chronic kidney disease model:
CKD was induced by a two-step, subtotal nephrectomy as described.3, 16 The mice were kept warm after surgery, and the analgesic (slow release buprenorphine) was given before surgery. After removing about 2/3 of the left kidney, mice were fed 6% Protein Rodent Diet Chow (Harlan Teklad, Madison, WI) ad libitum to reduce mortality and limit kidney hypertrophy. One week later, the right kidney was removed and after recovery for another week, mice were fed 40% protein chow. Control mice were sham-operated and pair fed. Blood urea nitrogen (BUN) and serum creatinine were measured after 2 - 3 weeks, AVFs were created in control and CKD mice; some control and CKD mice underwent sham surgery.
Assessment of Renal Function:
Blood samples were collected for creatinine measurement in 2–3 weeks after sham surgery or CKD. Serum creatinine was measured by using the Quanti Chrom Creatinine Assay Kit (Jaffe method) as the manufacturer’s instructions (Sigma-Aldrich). The BUN was measured by the Comparative Pathology Laboratory Center at Baylor College of Medicine.
Mouse AVF Model:
Mouse AVFs were created as described.3 Briefly, mice of 12 weeks of age were anesthetized, and the right internal jugular vein was isolated using a dissecting microscope (Leica MZ6; Leica, Germany). Its distal end was clamped and ligated, the common carotid artery was ligated below its bifurcation, and the proximal end was clamped. An end-to-end anastomosis was created using 12–0 nylon suture with an interrupted stitch. After unclamping, patency was confirmed visually. The mice were kept warm after surgery, and the analgesic (slow release buprenorphine) was given before surgery. At 2 and 4 weeks after surgery, mice with AVFs or sham-operated, pair-fed controls were anesthetized by intraperitoneal injection and euthanized by perfusing the left ventricle with PBS and 10% formalin for 10 minutes (to maintain the endothelium and morphology of the AVF). AVFs were collected and slides from 0.5 to 1 mm of the venous anastomosis were prepared for hematoxylin/eosin staining. Mice dying before AVF collection were excluded from analysis. Bone marrow transplantation and CKD each caused ~20% of mortality. 10-12 mice in each group were used to keep AVFs for statistical analysis.
Conditioned media preparation:
Bone marrow-derived FSP-1+ cells were cultured in DMEM media. Cells were collected, centrifuged for 5 min at 1400 rpm and the supernatant was added to VSMCs.
Cell culture:
To evaluate how specific cytokines affected cultured VSMCs, MCP-1 (5 ng/ml), interleukin-6 (IL-6, 1 ng/ml), IL-1β (2 ng/ml), FGF-2 (5 ng/ml), TGF-β1 (2 ng/ml) or PDGF-BB (10 ng/ml) were added individually to VSMCs and cultured for 72h.
Histology and Immunohistochemistry.
For histological analyses, mice were perfused through the left ventricle and slides of AVFs were prepared as described.5 Immunohistochemical staining and H & E-stained sections from AVFs in each group were examined by a pathologist who was masked as to treatment. For double immunofluorescence staining, primary antibodies were added followed by fluorescent secondary antibodies; DAPI was used to stain nuclear DNA. To capture images, the Nikon Eclipse 80i fluorescence microscope (Melville, NY) was used. The negative controls were either addition of isotype-matched IgG or PBST. The areas of positive signal were measured using the NIS-Elements BR 3.0 program. Images (× 400) from each section were analyzed in a blinded manner and quantified using Image-Pro Plus software (Media Cybernetics, Silver Spring, Md., USA).
Measurements of mRNA Expression.
Total RNA was isolated by the RNeasy kit (Qiagen, Valencia, CA). Real-time RT-PCR was performed with the primers.
Western blot analysis.
The protein content of cell extracts prepared in RIPA buffer, was determined using the Bradford protein assay kit (BioRad, Hercules, CA). About 30 μg of proteins were separated by SDS-PAGE and after transferring to nitrocellulose membranes, immunoblots were probed separately with various primary antibodies. Subsequently, the immunoblots were blocked with 5% skimmed milk in TBS. Fluorescently labeled secondary antibodies were detected by the Odyssey Infrared Imaging System (LI-COR, Inc, Lincoln, Nebraska, USA).
Isolation of bone marrow-derived FSP-1+ and bone marrow transplantation.
Bone marrow cells were obtained from mouse tibias and femurs of FSP-1–GFP transgenic mice. GFP+ bone marrow cells were isolated by cell sorting (Becton Dickinson LSRII flow cytometer, BD Biosciences). Approximately, 2×105 of the purified cells were cultured in DMEM (Invitrogen) plus 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT); 20% L929 supernatant containing macrophage-stimulating factor for 6 days were tested. Bone marrow transplantation was performed by injecting 5×106 BM cells into the lateral tail vein of lethally irradiated (1100 rads) mice recipients.39
Statistical Analysis.
All data are presented as the mean ± standard deviation (SD). Comparisons between groups were analyzed using one-way ANOVA; p< 0.05 was considered to be statistically significant.
Acknowledgements and Sources of Funding
This work was supported by American Heart Association grants 15GRNT25700209 and R01-DK095867 (to J.C.), National Institutes of Health grants R37 and DK37175, and a grant from Dr. and Mrs. Harold Selzman. National Natural Science Foundation of China (NSFC81570689 and 81770677 [M.L.]), Science & Technology Planning Project of Guangzhou (201707010290 [M.L.]), Natural Science Foundation of Guangdong Province (2017A030313566 [M.L.]).
Abbreviation:
- AVF
arteriovenous fistula
- CKD
chronic kidney disease
- VSMC
vascular smooth muscle cell
- FSP-1
fibroblast specific protein
Footnotes
Disclosure
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Roy-Chaudhury P, Sukhatme VP and Cheung AK. Hemodialysis vascular access dysfunction: a cellular and molecular viewpoint. J Am Soc Nephrol. 2006;17:1112–27. [DOI] [PubMed] [Google Scholar]
- 2.Allon M and Robbin ML. Increasing arteriovenous fistulas in hemodialysis patients: problems and solutions. Kidney Int. 2002;62:1109–24. [DOI] [PubMed] [Google Scholar]
- 3.Liang A, Wang Y, Han G, et al. Chronic kidney disease accelerates endothelial barrier dysfunction in a mouse model of an arteriovenous fistula. Am J Physiol Renal Physiol. 2013;304:F1413–F1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kokubo T, Ishikawa N, Uchida H, et al. CKD Accelerates Development of Neointimal Hyperplasia in Arteriovenous Fistulas. J Am Soc Nephrol. 2009;20:1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang M, Wang Y, Liang A, et al. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires Notch activation to form neointima. Kidney Int. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crowther MA, Clase CM, Margetts PJ, et al. Low-intensity warfarin is ineffective for the prevention of PTFE graft failure in patients on hemodialysis: a randomized controlled trial. J Am Soc Nephrol. 2002;13:2331–7. [DOI] [PubMed] [Google Scholar]
- 7.Dember LM, Beck GJ, Allon M, et al. Effect of Clopidogrel on Early Failure of Arteriovenous Fistulas for Hemodialysis. JAMA. 2008;299:2164–2171.18477783 [Google Scholar]
- 8.Kaufman JS, O'Connor TZ, Zhang JH, et al. Randomized controlled trial of clopidogrel plus aspirin to prevent hemodialysis access graft thrombosis. J Am Soc Nephrol. 2003;14:2313–21. [DOI] [PubMed] [Google Scholar]
- 9.Dixon BS, Beck GJ, Dember LM, et al. Use of Aspirin Associates with Longer Primary Patency of Hemodialysis Grafts. J Am Soc Nephrol. 2011;22:773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon BS, Beck GJ, Vazquez MA, et al. Effect of dipyridamole plus aspirin on hemodialysis graft patency. N Engl J Med. 2009;360:2191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabas I, Garcia-Cardena G and Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemenoff RA, Horita H, Ostriker AC, et al. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:1300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang M, Liang A, Wang Y, et al. Smooth muscle cells from the anastomosed artery are the major precursors for neointima formation in both artery and vein grafts. Basic research in cardiology. 2014;109:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rotllan N, Wanschel AC, Fernandez-Hernando A, et al. Genetic Evidence Supports a Major Role for Akt1 in VSMCs During Atherogenesis. Circ Res. 2015;116:1744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y Liang A, Luo J, et al. Blocking Notch in Endothelial Cells Prevents Arteriovenous Fistula Failure Despite CKD. J Am Soc Nephrol. 2014;25:773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juncos JP, Grande JP, Kang L, et al. MCP-1 Contributes to Arteriovenous Fistula Failure. J Am Soc Nephrol 2011;22:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang B, Janardhanan R, Vohra P, et al. Adventitial transduction of lentivirus-shRNA-VEGF-A in arteriovenous fistula reduces venous stenosis formation. Kidney Int. 2014;85:289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang B, Brahmbhatt A, Nieves Torres E, et al. Tracking and Therapeutic Value of Human Adipose Tissue-derived Mesenchymal Stem Cell Transplantation in Reducing Venous Neointimal Hyperplasia Associated with Arteriovenous Fistula. Radiology. 2016;279:513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nath KA, Grande JP, Kang L, et al. beta-Catenin is markedly induced in a murine model of an arteriovenous fistula: the effect of metalloproteinase inhibition. Am J Physiol Renal Physiol. 2010;299:F1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misra S, Doherty MG, Woodrum D, et al. Adventitial remodeling with increased matrix metalloproteinase-2 activity in a porcine arteriovenous polytetrafluoroethylene grafts. Kidney Int. 2005;68:2890–900. [DOI] [PubMed] [Google Scholar]
- 22.Rossini M, Cheunsuchon B, Donnert E, et al. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005;68:2621–8. [DOI] [PubMed] [Google Scholar]
- 23.Fung E, Tang SM, Canner JP, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007;115:2948–56. [DOI] [PubMed] [Google Scholar]
- 24.Roy-Chaudhury P, Arend L, Zhang J, et al. Neointimal Hyperplasia in Early Arteriovenous Fistula Failure. Am J Kidney Dis. 2007;50:782–790. [DOI] [PubMed] [Google Scholar]
- 25.Roy-Chaudhury P, Wang Y Krishnamoorthy M, et al. Cellular phenotypes in human stenotic lesions from haemodialysis vascular access. Nephrol Dial Transplant. 2009;24:2786–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duque JC and Vazquez-Padron RI. Myofibroblasts: the ideal target to prevent arteriovenous fistula failure? Kidney Int. 2014;85:234–6. [DOI] [PubMed] [Google Scholar]
- 27.Lewis AE, Vasudevan HN, O'Neill AK, et al. The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev Biol. 2013;379:229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skartsis N, Manning E, Wei Y, et al. Origin of neointimal cells in arteriovenous fistulae: bone marrow, artery, or the vein itself? Semin Dial. 2011;24:242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng J, Wang Y, Liang A, et al. FSP-1 silencing in bone marrow cells suppresses neointima formation in Vein Graft. Circ Res. 2012;110:230–240. [DOI] [PubMed] [Google Scholar]
- 30.Kopan R and Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mead TJ and Yutzey KE. Notch pathway regulation of neural crest cell development in vivo. Developmental dynamics : an official publication of the American Association of Anatomists. 2012;241:376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mumm JS and Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–65. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Zhu J, Smith S, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol. 2012;13:642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fouillade C, Monet-LeprÃatre M, Baron-Menguy Cl and Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res. 2012. [DOI] [PubMed] [Google Scholar]
- 35.He C, Medley SC, Hu T, et al. PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat Commun. 2015;6:7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreeva ER, Pugach IM and Orekhov AN. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis. 1997;135:19–27. [DOI] [PubMed] [Google Scholar]
- 37.Cheng J and Du J. Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arterioscler Thromb Vasc Biol. 2007;27:1744–51. [DOI] [PubMed] [Google Scholar]
- 38.Tang Y Urs S and Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang M, Wang Y, Liang A, et al. Impaired integrin beta3 delays endothelial cell regeneration and contributes to arteriovenous graft failure in mice. Arterioscler Thromb Vasc Biol. 2015;35:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]