

Abstract
Lipodystrophies are the result of a range of inherited and acquired causes, but all are characterized by perturbations in white adipose tissue function and, in many instances, its mass or distribution. Though patients are often nonobese, they typically manifest a severe form of the metabolic syndrome, highlighting the importance of white fat in the “safe” storage of surplus energy. Understanding the molecular pathophysiology of congenital lipodystrophies has yielded useful insights into the biology of adipocytes and informed therapeutic strategies. More recently, genome-wide association studies focused on insulin resistance have linked common variants to genes implicated in adipose biology and suggested that subtle forms of lipodystrophy contribute to cardiometabolic disease risk at a population level. These observations underpin the use of aligned treatment strategies in insulin-resistant obese and lipodystrophic patients, the major goal being to alleviate the energetic burden on adipose tissue.
Introduction
Lipodystrophies are a heterogeneous group of conditions characterized by a lack of, and/or dysfunctional, white adipose tissue. They may be genetic (inherited) or acquired in origin and localized, partial, or generalized in distribution; however, despite the etiological heterogeneity, other than localized forms and most cases of Barraquer-Simons acquired partial lipodystrophy, they almost all cause insulin resistance, nonalcoholic fatty liver disease (NAFLD), and dyslipidemia (characterized by high triglyceride and low HDL cholesterol concentrations) (1). Therefore, even though patients are often (though not always) nonobese or lean, lipodystrophies mirror the metabolic syndrome associated with obesity.
Although lipodystrophies are relatively rare, studying these patients has advanced understanding of adipose biology and the pathophysiology of the metabolic syndrome. They act as very informative models of inadequate adipose storage capacity in the face of excess energy intake, resulting in ectopic fat accumulation and insulin resistance. Accumulating evidence suggests that similar mechanisms of adipose overload are responsible for insulin resistance in patients with obesity (2, 3), and therefore treatment is conceptually similar for lipodystrophies and obesity; for example, limited data in patients with familial partial lipodystrophy suggest that bariatric surgery is highly effective in alleviating the metabolic consequences of lipodystrophy (4–6), as has been widely reported in obesity (7).
White adipose tissue is the primary site for physiological energy storage in humans (ref. 8 and Figure 1). Surplus energy can only really be stored as glycogen (carbohydrate) or triglyceride (neutral lipid), and the latter represents a more concentrated/energy-dense (9 vs. 4 kcal/g) and “lighter” reserve, so it is not surprising that a lean adult human stores approximately 100-fold more energy as triglyceride compared with glycogen (9). In healthy adult humans almost all triglyceride is stored within white adipose tissue, which regulates the uptake of substrates (e.g., glucose and nonesterified fatty acids), the synthesis of neutral lipid for storage, and the release of stored triglycerides through lipolysis. These processes are under nervous, hormonal, and nutritional regulation to facilitate homeostatic energy balance but can be perturbed by chronic overnutrition (resulting in obesity) or inherited disorders of white adipose tissue (lipodystrophies). Adipocytes signal the status of their energy reserves by secreting leptin, which in turn acts centrally to influence energy balance and reproductive capacity. They also secrete a host of other proteins (collectively known as adipokines) with a range of purported functions (10, 11). Clearly, in addition to perturbing energy storage, lipodystrophies may well alter adipokine secretion and/or interactions with stromovascular cells present in adipose tissue. These changes could also contribute to the physiological changes associated with lipodystrophy but have, for the most part, not been extensively studied and so are not considered in detail here.
Figure 1. White adipose tissue contains the body’s major store of energy.

Even lean adults store 600–800 mJ of energy as triglyceride in adipose tissue compared with 6 to 8 mJ as glycogen in liver and muscle. The physiological regulation of triglyceride stores varies in different adipose tissue depots. Gluteofemoral subcutaneous white adipose is relatively insulin sensitive, and its expansion is not associated with cardiometabolic disease, whereas visceral and abdominal (upper body) subcutaneous adipose tissue has a higher rate of lipolysis and is more closely linked with insulin resistance. WAT, white adipose tissue.
White fat is distributed in several characteristic sites (or “depots”; Figure 1), which differ substantially between humans and mice. For example, humans do not have a large gonadal fat pad, which is a commonly isolated and studied fat depot in mice. Importantly, adipogenesis, lipogenesis, and lipolysis vary in their physiological regulation in these depots (12–14). For example, human studies have suggested that lipolysis is higher in visceral (intra-abdominal) than in subcutaneous white adipose tissue (15). In this Review, we highlight examples of inherited lipodystrophies where selective loss of gluteofemoral fat can recapitulate features of the metabolic syndrome, even if upper body fat depots are unaffected. In other cases, where fat loss is restricted to the face, upper trunk, and arms, patients do not typically manifest metabolic disease.
Insights from inherited lipodystrophy syndromes with well-defined mechanisms
Clearly defined monogenic diseases can provide unique insights into the biological function of specific genes and the proteins they encode, somewhat akin to observations made in knockout or hypomorphic mouse models. In several cases (for example, PPARG, encoding peroxisome proliferator–activated receptor γ), the link between specific genes implicated in causing lipodystrophy and adipose dysfunction is clear, whereas in others (such as LMNA, encoding lamin A/C), the mechanism is yet to be fully understood. Below, we briefly review selected situations in which the link between particular genes and adipose dysfunction is relatively clear; these include examples involving genes implicated in the transcriptional regulation of adipogenesis, triglyceride synthesis, lipid droplet morphology, and lipolysis (Figure 2).
Figure 2. Some of the genes/proteins in which mutations cause lipodystrophy have well-characterized roles in the function of adipocytes.

PPARγ (mutated in FPLD3) is the “master regulator” of adipogenesis. It heterodimerizes with retinoid X receptor and coordinates the transcription of multiple proteins central to adipocyte function (e.g., perilipin, CD36, and lipoprotein lipase). BSCL2, or seipin (mutated in CGL2), is an ER protein required for early lipid droplet (LD) biogenesis. AGPAT2 (mutated in CGL1) is necessary for the conversion of glycerophosphates (G-3-P) into triacylglycerols (TAGs) using fatty acids linked to coenzyme A (FA-CoA). CAV1 (mutated in CGL3) and PTRF (mutated in CGL4) are required for the formation of caveolae, which may be sites for nonesterified fatty acid (NEFA) uptake. PLIN1 (mutated in FPLD4) regulates lipolysis from lipid droplets, and HSL (mutated in FPLD6) is one of the lipases involved in this process. Finally, CIDEC (mutated in FPLD5) is required for the formation of unilocular lipid droplets, though how this is achieved is unclear.
PPARG.
Peroxisome proliferator–activated receptors (PPARs) α, δ, and γ were identified nearly 30 years ago (16). They all have a DNA-binding domain (DBD) as well as a ligand-binding domain (LBD) that is believed to bind, somewhat promiscuously, a range of putative fatty acid ligands (17–20). Seminal in vitro studies undertaken in the 1990s established that PPARγ is essential for adipocyte differentiation (21–24), and it is now generally considered to be the “master regulator” of both adipogenesis, the process through which fibroblast-like precursors are converted into mature adipocytes, and mature adipocyte function (21, 23). It is therefore not surprising that PPARG mutations cause lipodystrophy, but why these mutations are usually associated with a stereotypical pattern of partial lipodystrophy remains an unresolved puzzle.
Heterozygous, dominant-negative mutations in PPARG are associated with autosomal dominant familial partial lipodystrophy type 3 (FPLD3). Several case studies have reported patients with severe insulin resistance, dyslipidemia in which hypertriglyceridemia appears to be exquisitely sensitive to high fat intake (25), type 2 diabetes mellitus, reduced subcutaneous gluteofemoral and leg fat, and hypertension (26–35). Affected women frequently manifest features of polycystic ovary syndrome (26, 36, 37) and premature-onset cardiovascular disease (26), and cirrhosis has been reported (28, 38). The majority of patients with FPLD3 are diagnosed with lipodystrophy in early adulthood, though affected children have been identified through family screening studies (35) and men typically present later than women.
Heterozygous autosomal dominant mutations in either the DBD or LBD of PPARγ have been associated with lipodystrophy (27, 39), though debate continues about whether or not these mutations truly manifest dominant-negative properties (40). LBD variants can bind to DNA response elements but manifest impaired transcriptional responses to agonists or coactivators (35). DBD mutants impair DNA binding, but they can also inhibit wild-type function, possibly via sequestration of coactivators (36). Heterozygous PPARγ missense variants are present in as many as 1 in 500 people, though prospective functional classification of all possible missense variants in PPARγ suggests that many of these are benign and that gene-environment interactions are important (17, 41).
Some human evidence supports the notion that the degree of loss of PPARγ function correlates with the severity of lipodystrophy. A child harboring biallelic PPARG mutants, a frameshift mutation and a DBD mutation that were predicted to result in near-complete loss of PPARγ function, presented with a congenital generalized lipodystrophy phenotype (42). In contrast, the most common PPARγ variant, p.Pro12Ala (rs1801282, minor allele frequency 0.11), which is thought to only mildly modify PPARγ function, reduces the risk of developing type 2 diabetes without obviously causing a lipodystrophic phenotype (43).
In addition to the in vitro data referred to above, mouse studies strongly support observations that varying levels of PPARγ function produce a spectrum of phenotypes. Complete knockout of Pparg is lethal for mice because of its requirement in placental and cardiac development (44). However, mice with embryonal-only Pparg knockout (45), heterozygous Pparg knockout, Pparg hypomorphism (46), or adipose-specific Pparg knockout all have a lipodystrophic phenotype (including insulin resistance and elevated triglycerides) (47). Several of these models also manifested abnormalities in blood pressure: hypotension in embryonal-only knockout and hypertension in P465L knockin mice (consistent with the human condition), though this model did not demonstrate insulin resistance or hypertriglyceridemia (48).
Therefore, impairment in the transcriptional control of adipogenesis results in a reproducible white adipose tissue–mediated partial lipodystrophy associated with all the characteristic features of the metabolic syndrome, the severity of which correlates, at least broadly, with residual PPARγ activity.
AGPAT2.
The most severe form of lipodystrophy is congenital generalized lipodystrophy (CGL), which is inherited in an autosomal recessive manner (49). Mutations in AGPAT2 (encoding 1-acylglycerol-3-phosphate O-acyltransferase 2) are responsible for a substantial proportion of these patients (50). AGPAT2 is a lysophosphatidic acid acyltransferase that plays a key role in the synthesis of triglycerides from glycerol-3-phosphate (51).
Affected patients present from infancy with an almost complete lack of adipose tissue, extreme insulin resistance, hypertriglyceridemia (potentially leading to pancreatitis), and severe hepatic steatosis (52). Many have acanthosis nigricans, a condition characterized by dark discoloration and velvety thickening of flexural skin, and children may have a progeroid-like appearance due to the lack of facial adipose tissue. Patients are essentially aleptinemic, and leptin replacement therapy is now a mainstay of therapy, as discussed below.
Biallelic AGPAT2 mutations associated with CGL are predicted to profoundly disrupt the protein product. Many are splice site mutations that result in a frameshift with premature stop codons or exon skipping (53). Only a few missense variants have been identified in patients with AGPAT2-associated CGL, including p.Glu172Lys, which is thought to prevent the binding of substrate and catalytic activity (54). Functional studies have consistently demonstrated that mutants linked to CGL result in almost complete loss of AGPAT2 enzymatic activity (55).
AGPAT2 demonstrates tissue-selective expression in both visceral and subcutaneous adipose tissue, unlike AGPAT1, which is widely expressed (56). Homozygous loss-of-function variants appear to cause a failure of early adipogenesis due to several proposed interlinked mechanisms: perturbation of phospholipids, inhibition of PPARγ, and adipocyte apoptosis (57–59). There is a profound alteration of the lipidome, including reduced phosphatidylinositol and elevated lysophosphatidyl choline, in AGPAT2-knockdown 3T3-L1 adipocytes (60), the result of which is reduced activity in the PI3K/AKT pathway and inhibition of PPARγ activity. The lack of AKT signaling results in adipocyte apoptosis, and though PPARγ overexpression may partially restore the adipogenic potential in AGPAT2-knockout cells, they still undergo apoptosis (61). These conclusions are supported by a lipodystrophic phenotype in Agpat2-null mice (62).
These findings highlight the devastating metabolic consequences of near-total fat loss due to a major impairment in the ability of adipocytes to synthesize triglyceride. This defect in neutral lipid synthesis is compounded by sustained hyperphagia due to severe leptin deficiency in CGL.
PLIN1.
The perilipins are a group of proteins that were originally identified as highly abundant proteins covering the surface of lipid droplets (63–65). Since then they have been found to be critical for the regulation of lipolysis from lipid droplets (66–69); specifically, PLIN1 regulates the first two steps in triglyceride hydrolysis, namely those catalyzed by adipose tissue triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL; encoded by LIPE), the major diglyceride lipase.
Gandotra et al. identified the first three families affected with heterozygous loss-of-function PLIN1 mutations, all of which were inherited in an autosomal dominant fashion (70). They presented with partial lipodystrophy, particularly manifesting a lack of subcutaneous lower limb and gluteofemoral fat. Biochemically, they had hypoadiponectinemia, hyperinsulinemia, NAFLD, and profound hypertriglyceridemia. Two different frameshift mutations were reported, both of which were shown to be expressed and to be targeted to the surface of lipid droplets (70). The mutant PLIN1 reported in these cohorts increased basal lipolysis in an in vitro model (67) by failing to effectively bind ABHD5, a key activator of ATGL (71–73). PLIN1 is almost exclusively expressed in white and brown adipocytes, where it is very specifically involved in stabilizing lipid droplets and in regulating lipolysis; thus the phenotype described in patients with PLIN1 mutations highlights the fact that a highly specific defect affecting energy storage in adipocytes is sufficient to produce almost all the features of the metabolic syndrome.
CIDEC.
Cell death–inducing DFFA-like effector C (CIDEC; also known as the murine form, Fsp27) is another lipid droplet–associated protein that is expressed in white adipose tissue (74, 75), where it is required for the formation of large unilocular lipid droplets (76). Mice lacking Fsp27 uniformly manifest reduced fat mass with multilocular lipid droplets in all white adipocytes (77, 78), and overexpression of Fsp27/CIDEC in a range of cell types consistently increases lipid droplet size (75, 79–81). Many human cell types can contain lipid droplets, including myocytes, hepatocytes, and pancreatic islet cells. However, these lipid droplets always adopt a multilocular form, whereas the ability to form a single massive lipid droplet is a unique property of the white adipocyte (82). This high volume-to-surface-area ratio facilitates very precise regulation of lipolysis, which is clearly important in a cell type responsible for bulk storage (and release) of surplus energy as neutral lipid for the whole organism.
In 2009, a single patient was identified with a partial lipodystrophy phenotype caused by a homozygous nonsense mutation in CIDEC (83). Clinically, the patient had absent lower limb and gluteofemoral adipose tissue with reduced total body fat mass. The proband had poorly controlled diabetes mellitus with a propensity for ketoacidosis despite elevated C-peptide and negative anti-islet and anti–glutamic acid decarboxylase antibodies. She also had severe hypertriglyceridemia (resulting in acute pancreatitis), acanthosis nigricans, hepatic steatosis, and hypertension. Fat biopsy demonstrated a mixed population of white adipocytes with many, though not all, containing multilocular lipid droplets. This unusual phenotype was recapitulated in an adipocyte-specific Fsp27-knockout mouse (84), which similarly demonstrated multilocular white adipocytes, in addition to insulin resistance. When fed a chow diet, Fsp27-null mice are lean and insulin sensitive; however, when challenged over many weeks with a high-fat diet, or when crossed with obesity-prone ob/ob mice or mice lacking brown adipose tissue, these mice do manifest NAFLD and severe insulin resistance (78).
Though apparently a very rare condition in humans, these data further demonstrate that disruption of the ability of white adipocytes to form unilocular lipid droplets is sufficient to cause partial lipodystrophy and features of the metabolic syndrome.
Other genetic lipodystrophies
Several other human genetic lipodystrophies have been recognized, though exactly how these cause lipodystrophy is less clear. Biallelic loss-of-function mutations in BSCL2 (encoding seipin) on chromosome 11 are a major cause of CGL (52). Affected patients have almost complete absence of body fat from birth, severe insulin resistance, and hypertriglyceridemia (85). The syndrome is also associated with cardiomyopathy and intellectual disability. BSCL2 is clearly critical for the development and function of cultured white adipocytes (86, 87), and both whole-body and adipose-specific Bscl2-knockout mice manifest a lipodystrophic phenotype (88). Recent cryogenic electron microscopy–derived structural models suggest that both the Drosophila ortholog (89) and human seipin (90) oligomerize to form a ring-like structure in the ER membrane. These data are consistent with seipin’s proposed involvement in the early formation of lipid droplets from the ER, a model supported by a series of yeast (91) and other cell-based experiments (92, 93).
Heterozygous mutations in lamin A/C are probably the most common cause of monogenic partial lipodystrophy (94–96). Lamin A/C is a well-established component of the nuclear lamina network expressed in almost all cells. Different LMNA mutations have also been linked to cardiomyopathy and muscular dystrophy, and some patients with LMNA-associated partial lipodystrophy do also manifest variable degrees of cardiac and skeletal muscle impairment. As intermediate filaments, lamins clearly impact nuclear structure and transcriptional regulation of gene expression, but exactly why specific mutations are more strongly associated with particular phenotypes and why nuclear perturbation leads to partial lipodystrophy remain unclear (97, 98).
Finally, one specific variant (R707W) in mitofusin 2 (MFN2), the gene classically mutated in Charcot-Marie-Tooth type 2A, has been linked to a striking adipose overgrowth–lipodystrophy phenotype known as multiple symmetric lipomatosis (99). Patients with biallelic R707W mutations develop fat hyperplasia on their back and neck with lower limb lipodystrophy, NAFLD, insulin resistance, and peripheral neuropathy (100). A particularly remarkable aspect of this phenotype is very low leptin levels despite the fact that patients retain excess upper body fat (100, 101). Mitochondrial fusion-fission dynamics are essential for all metabolically active tissues, but how this single variant confers an adipose phenotype is yet to be established (102).
As summarized in Tables 1 and 2, several additional genetic variants have been linked to specific forms of lipodystrophy. Further details related to all these disorders can be found in the references cited in Tables 1 and 2.
Table 1. The main congenital generalized and familial partial lipodystrophies with their involved genes and proposed pathogenic mechanisms.

Table 2. Other genetic and acquired lipodystrophies with their involved genes and proposed pathogenic mechanisms.

Lipodystrophy and insulin resistance
The central hallmark of the metabolic syndrome is insulin resistance, and, other than obesity itself, compelling arguments exist linking almost all the features of the metabolic syndrome to underlying insulin resistance (144). Insulin resistance is a consistent feature of lipodystrophy and reveals a number of specific insights:
(a) Despite the range of different genetic and acquired causes of lipodystrophy, almost all “appreciable” (by which we mean at least partial in terms of extent) lipodystrophies are associated with insulin resistance. Exceptions that we are aware of include patients with FBN1 mutations, though in this setting, the “apparent lipodystrophy” is more likely to be secondary to reduced food intake. In all other instances, lipodystrophies are associated with very low or at least relatively low leptin levels, and thus a tendency to hyperphagia.
(b) The severity of insulin resistance is broadly proportional to the extent of fat loss or dysfunction. In other words, generalized lipodystrophy is typically associated with more severe metabolic manifestations than partial lipodystrophy. Furthermore, upper body fat loss is less prone to be associated with metabolic disease than gluteofemoral fat loss. This is best exemplified by cases of Barraquer-Simons acquired partial lipodystrophy, in which the fat loss proceeds in a cephalocaudal pattern: this entity is often associated with the presence of C3 nephritic factor and sometimes with renal glomerular disease, and insulin resistance is usually not a feature unless fat loss extends down to the gluteofemoral adipose depot and/or patients are otherwise obese (140).
Another intriguing form of partial lipodystrophy is that associated with dermatomyositis; in these cases, lipodystrophy most prominently affects subcutaneous depots, whereas visceral fat is often preserved (145). The patients frequently manifest advanced NAFLD and insulin resistance.
Finally, in this context, metabolic disease is also typically more severe in affected girls/women than in boys/men, presumably because under normal circumstances fat mass in a lean woman is roughly twice that in a lean man (146). Collectively, the two points above indicate that lack and/or dysfunction of white fat is consistently associated with insulin resistance and the metabolic syndrome, particularly when the defect in white fat is compounded by hyperphagia due to relative leptin deficiency (147, 148).
(c) NAFLD is a very consistent feature in insulin-resistant lipodystrophies, and is typically associated with metabolic dyslipidemia (high triglycerides and low HDL cholesterol). While there are limited data on liver histology in patients with lipodystrophy (149, 150), one study found that 40% of biopsied patients had bridging fibrosis or cirrhosis and 62% had definite steatohepatitis at a mean age of 29 years (38).
(d) Patients with lipodystrophy are prone to early-onset cardiovascular disease. In some ways, this appears to be as severe as the cardiovascular disease associated with heterozygous familial hypercholesterolemia, as we have had several women present with ischemic events well before the age of 50 years (151, 152).
(e) Circulating markers of cellular or mitochondrial stress (e.g., FGF21 and GDF15) are elevated in both obesity (153, 154) and lipodystrophy (155). Patel et al. recently showed that chronic energetic overload results in an increase in serum GDF15 and FGF21 in mice (156), and at least in the case of GDF15, this change may help to alleviate ongoing surplus energy intake.
However, this potentially “corrective” GDF15 signal is offset in lipodystrophic patients by relative or near-complete leptin deficiency, a key signal for the persistent hyperphagia observed in many patients with lipodystrophy. Unfortunately, hyperphagia compounds the relative deficiency of adipocyte energy storage capacity and is considered a major factor in the pathogenesis of metabolic disease in this disorder. Obese patients have elevated leptin levels, in keeping with their increased total fat mass (157), though it does not seem to suppress hunger, and therefore the term “leptin resistance” has been coined. In both lipodystrophy and obesity (158), the relative lack of leptin action contributes to ongoing surplus energy intake and insulin resistance, although it should be noted that leptin signaling at its receptor is actually increased in obesity and that a lack of response to additional exogenous leptin is complex and incompletely understood (159).
Adiponectin is an adipokine that paradoxically falls in obesity and insulin resistance, though the mechanisms underlying control of its release are unclear. Adiponectin levels are also low in lipodystrophy, and this is generally in proportion to the loss of adipose tissue (i.e., lower in CGL than in FPLD) and the severity of insulin resistance (160).
Insulin receptor signaling defects
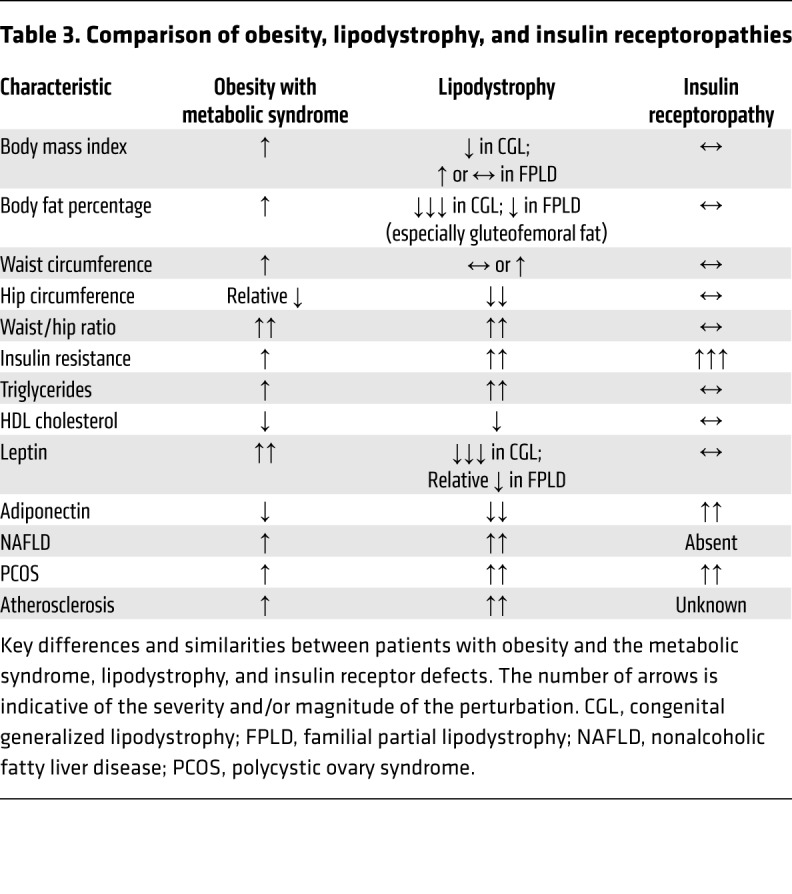
“Insulin receptoropathies,” a term used to describe insulin-resistant states caused by a mutation or acquired defect in one of the proximal insulin signaling components (most commonly the insulin receptor [INSR] itself), represent another cluster of monogenic disorders associated with severe insulin resistance (161). Examination of the differences between patients with obesity-related metabolic syndrome, lipodystrophy, and insulin receptoropathies (Table 3) indicates that whereas lipodystrophy-associated and obesity-associated metabolic syndrome are strikingly concordant, there are several marked differences between the latter and insulin receptoropathies. In particular, NAFLD and dyslipidemia are typically not seen in insulin receptoropathies (162), and adiponectin levels tend to be high rather than low in these conditions (163).
Table 3. Comparison of obesity, lipodystrophy, and insulin receptoropathies.
Adipose inflammation
Histological assessment of adipose tissue from patients with the metabolic syndrome consistently demonstrates features of inflammation (164–166), the extent of which correlates with the severity of insulin resistance and NAFLD in humans (167). Inflammation appears to be more prominent in adipose tissue with higher lipolytic capacity (particularly visceral adipose); however, opinion remains divided on the extent to which macrophage infiltration is pathogenic in adipose dysfunction and/or insulin resistance (168). Obesity-associated insulin resistance is a state of chronic, systemic inflammation, as evidenced by elevated IL-6, high-sensitivity C-reactive protein, and GDF15, among other markers, but these derive from more than just adipose tissue. Nevertheless, adipose tissue macrophages certainly act as one source of inflammatory cytokines that contribute to the systemic inflammatory state, and their contribution appears to be ameliorated by weight loss (169).
There have been only a few studies examining the histology of adipose tissue in patients with inherited lipodystrophy. Patients with FPLD4 secondary to PLIN1 mutations had similar features of inflammation and fibrosis to those reported in obese people, whereas patients with CIDEC-associated lipodystrophy (83), MFN2-associated lipodystrophy (100), and LMNA mutations (170) did not demonstrate gross inflammation. Collectively, evidence from genetic lipodystrophies that almost always cause severe insulin resistance tends to suggest that adipose inflammation is a second “hit” rather than the primary driver of adipose dysfunction and so, at least in our view, raises questions about the widespread interest in anti-inflammatory strategies for obesity-associated metabolic disease.
NAFLD is a very consistent feature of lipodystrophies that are severe enough to cause insulin resistance and other features of the metabolic syndrome. In this scenario, nonalcoholic steatohepatitis is very common and could conceivably be involved in causing hepatic insulin resistance. However, here too the liver pathology is very likely to occur secondary to adipose tissue dysfunction.
Evidence for subtle lipodystrophy in the general population
The striking overlap between lipodystrophy and more prevalent forms of the metabolic syndrome (Table 3) has long suggested that subtle forms of lipodystrophy may be relevant to the pathogenesis of metabolic syndrome and type 2 diabetes in particular (171–173). However, direct supportive evidence has been hard to come by, so it has remained a largely hypothetical premise.
In 2014, Scott et al. undertook a GWAS that used fasting insulin as a proxy for insulin resistance (174). Intriguingly, just over half the loci associated with a higher fasting insulin were also associated with higher triglycerides, lower HDL cholesterol, and a lower BMI and/or a reduction in gluteofemoral fat mass as measured by dual x-ray absorptiometry (DXA) scanning. Lotta et al. later performed a much larger GWAS focused on loci associated with a combination of BMI-adjusted insulin, higher triglycerides, and lower HDL cholesterol (175). This analysis identified 53 loci, which were replicated in a second cohort and shown to be significantly associated with gold-standard hyperinsulinemic-euglycemic clamp–based measures of insulin resistance. In a large human cohort in whom fat mass and distribution were documented with DXA scans, there was a significant association between higher insulin-resistant SNP scores and lower levels of gynoid and leg fat mass (175). Interestingly, in this study, reduced expansion of gluteofemoral fat depot was also documented in response to weight gain (175). Importantly, this 53 SNP score was enriched in patients with FPLD1, which is in many ways an extreme form of apple-shaped fat distribution, implying that these common alleles contribute to both common insulin resistance and a specific form of partial lipodystrophy known as FPLD1 (Figure 3). This is also consistent with observations that there are a large number of patients with clinical features of FPLD1 (or FPLD2) with no known genetic diagnosis.
Figure 3. The severity of lipodystrophy and the degree of adipose dysfunction correlate broadly with the severity of insulin resistance.

This principle extends from the most extreme form of lipodystrophy, congenital generalized lipodystrophy (CGL), through familial partial lipodystrophies (FPLDs) to the general population. Individuals in the highest quintile (Q5) for a polygenic risk score for insulin resistance (see Lotta et al.; ref. 175) have less gluteofemoral fat, resulting in an “apple-shaped” fat distribution, whereas those in the lowest quintile (Q1) manifest a protective “pear-shaped” fat distribution and are less insulin resistant. FPLD type 1 (FPLD1) represents an intermediate state between other monogenic forms of FPLD and the highest-risk individuals from the general population. The degree of genetic disruption of adipose tissue also correlates with these phenotypes, as exemplified by the impact of a range of PPARG mutations: complete loss of PPARγ function can cause CGL; dominant-negative PPARG mutations cause FPLD3; and common PPARG variants affect insulin resistance at a population level. Exemplars of PPARG mutations in each of these categories have been included. Each black dot represents a distinct monogenic disease (see Table 1 for classifications), and red diamonds schematically represent common genetic variants that influence adipogenesis and insulin resistance.
Waist/hip ratio (WHR) is a widely used noninvasive measure of adipose tissue distribution, and its relationship to diabetes and cardiovascular risk is well established (176, 177). Given that WHR is a ratio, it can be modified by changes in either the numerator or the denominator. However, visceral fat accumulation (i.e., an increase in the numerator) is usually assumed to be the driving factor behind the link between this index of fat distribution and insulin resistance. This notion is also supported by considerable scientific evidence (178–182). However, when Lotta et al. generated SNP risk scores for loci shown to be associated with a higher WHR, through a specific association with either a reduction in hip circumference (22 of a total of 202 loci) or an increase in waist circumference (36 of 202 loci), both scores were associated with cardiometabolic risk factors as well as an increase in the odds ratio of developing type 2 diabetes mellitus and coronary disease (183). Intriguingly, the odds ratio for the risk of type 2 diabetes was statistically significantly greater for the hip-specific risk score than the waist-specific score, whereas the odds ratios for cardiovascular disease were similar. Collectively these data are at least consistent with the notion that subtle partial lipodystrophy is a major factor in the pathogenesis of the metabolic syndrome in the general population (Figure 3). Put another way, people with an impaired capacity to increase hip fat mass in response to weight gain are more likely to develop insulin resistance and type 2 diabetes if they do increase their weight.
Shared treatment strategies for lipodystrophy and obesity
The implications of these observations are that therapeutic strategies in both lipodystrophy and obesity-related metabolic syndrome should aim to minimize adipocyte overload. This could be achieved by increasing adipocyte number/function or by reducing the energetic load on adipose tissue.
Increasing fat mass is a highly effective method for treating the metabolic syndrome in mice. Evidence from animal models has demonstrated the profound benefits associated with adipose transplant in severely lipodystrophic mice (184, 185). But, aside from being cosmetically unappealing, fat transplantation would also be technically challenging in humans. However, thiazolidinediones, which selectively activate PPARγ, very effectively improve insulin sensitivity in the clinical setting, primarily by increasing subcutaneous fat mass (186).
Thus, reducing caloric intake is the mainstay of treatment for the metabolic syndrome, and it is highly effective. Data suggest that while there are some mild benefits of diets of various composition (the Mediterranean diet, for example), the most effective therapy is considerable calorie restriction. Limiting intake to less than 650 kcal per day can result in complete remission of diabetes (187) in patients with a relatively short duration of type 2 diabetes. A similar approach can be achieved through bariatric surgery (188); though the metabolic impact varies depending on the technique used, the greater the reduction in net energy intake, the greater the reduction in weight and improvement in insulin sensitivity (7). Bariatric surgery has also been demonstrated to be very effective in a small number of patients with FPLD, despite these patients having a BMI lower than would be typically used as an eligibility criterion for surgery (4).
Lastly, as mentioned previously, leptin replacement therapy is highly effective in lipodystrophy (38, 189), but patients with obesity (who have high circulating leptin) do not appear to show a response to additional exogenous leptin unless they are already in a weight-reduced state (190). However, there is ongoing interest in targeting leptin therapy to obese subjects with relatively low leptin levels.
Conclusion
Inherited lipodystrophies are a complex group of conditions, all of which ultimately impair adipose tissue function and particularly its capacity to efficiently store surplus energy. The metabolic sequelae of lipodystrophy are remarkably similar to those associated with obesity, and compelling human genetic evidence now suggests that this similarity reflects adipocyte overload in both settings. Thus the goal of treatment in both states is to alleviate the energetic burden on adipocytes by inducing negative energy balance and/or weight loss.
Acknowledgments
JPM is supported by a Wellcome Trust Fellowship (216329/Z/19/Z). DBS is supported by the Wellcome Trust (WT 107064), the Medical Research Council (MRC) Metabolic Diseases Unit (MRC_MC_UU_12012/2), and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre and NIHR Rare Disease Translational Research Collaboration.
Version 1. 08/05/2019
Electronic publication
Version 2. 10/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4009–4021.https://doi.org/10.1172/JCI129190.
Contributor Information
Jake P. Mann, Email: jake.mann@mrc-epid.cam.ac.uk.
David B. Savage, Email: Dbs23@medschl.cam.ac.uk.
References
- 1.Melvin A, O’Rahilly S, Savage DB. Genetic syndromes of severe insulin resistance. Curr Opin Genet Dev. 2018;50:60–67. doi: 10.1016/j.gde.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome — an allostatic perspective. Biochim Biophys Acta. 2010;1801(3):338–349. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Cuthbertson DJ, et al. What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int J Obes (Lond) 2017;41(6):853–865. doi: 10.1038/ijo.2017.4. [DOI] [PubMed] [Google Scholar]
- 4.Melvin A, et al. Roux-en-Y gastric bypass surgery in the management of familial partial lipodystrophy type 1. J Clin Endocrinol Metab. 2017;102(10):3616–3620. doi: 10.1210/jc.2017-01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundfest-Broniatowski S, Yan J, Kroh M, Kilim H, Stephenson A. Successful treatment of an unusual case of FPLD2: the role of Roux-en-Y gastric bypass-case report and literature review. J Gastrointest Surg. 2017;21(4):739–743. doi: 10.1007/s11605-016-3300-2. [DOI] [PubMed] [Google Scholar]
- 6.Ciudin A, Baena-Fustegueras JA, Fort JM, Encabo G, Mesa J, Lecube A. Successful treatment for the Dunnigan-type familial partial lipodystrophy with Roux-en-Y gastric bypass. Clin Endocrinol (Oxf) 2011;75(3):403–404. doi: 10.1111/j.1365-2265.2011.04057.x. [DOI] [PubMed] [Google Scholar]
- 7.Chang SH, Stoll CR, Song J, Varela JE, Eagon CJ, Colditz GA. The effectiveness and risks of bariatric surgery: an updated systematic review and meta-analysis, 2003-2012. JAMA Surg. 2014;149(3):275–287. doi: 10.1001/jamasurg.2013.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45(9):1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 9.Robbins AL, Savage DB. The genetics of lipid storage and human lipodystrophies. Trends Mol Med. 2015;21(7):433–438. doi: 10.1016/j.molmed.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016;23(5):770–784. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman J. The long road to leptin. J Clin Invest. 2016;126(12):4727–4734. doi: 10.1172/JCI91578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karpe F, Pinnick KE. Biology of upper-body and lower-body adipose tissue — link to whole-body phenotypes. Nat Rev Endocrinol. 2015;11(2):90–100. doi: 10.1038/nrendo.2014.185. [DOI] [PubMed] [Google Scholar]
- 13.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21(6):697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 14.Montague CT, O’Rahilly S. The perils of portliness: causes and consequences of visceral adiposity. Diabetes. 2000;49(6):883–888. doi: 10.2337/diabetes.49.6.883. [DOI] [PubMed] [Google Scholar]
- 15.Jensen MD. Role of body fat distribution and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93(11 suppl 1):S57–S63. doi: 10.1210/jc.2008-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 17.Broekema MF, Savage DB, Monajemi H, Kalkhoven E. Gene-gene and gene-environment interactions in lipodystrophy: lessons learned from natural PPARγ mutants. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(5):715–732. doi: 10.1016/j.bbalip.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications—a review. Nutr J. 2014;13:17. doi: 10.1186/1475-2891-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer S. Ligands for the nuclear peroxisome proliferator-activated receptor gamma. Trends Pharmacol Sci. 2015;36(10):688–704. doi: 10.1016/j.tips.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395(6698):137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 21.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 22.Spiegelman BM. PPAR-γ: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47(4):507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 23.Hu E, Tontonoz P, Spiegelman BM. Transdifferentiation of myoblasts by the adipogenic transcription factors PPARγ and C/EBPα. Proc Natl Acad Sci U S A. 1995;92(21):9856–9860. doi: 10.1073/pnas.92.21.9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 25.Savage DB, Murgatroyd PR, Chatterjee VK, O’Rahilly S. Energy expenditure and adaptive responses to an acute hypercaloric fat load in humans with lipodystrophy. J Clin Endocrinol Metab. 2005;90(3):1446–1452. doi: 10.1210/jc.2004-1494. [DOI] [PubMed] [Google Scholar]
- 26.Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes. 2002;51(12):3586–3590. doi: 10.2337/diabetes.51.12.3586. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab. 2002;87(1):408–411. doi: 10.1210/jcem.87.1.8290. [DOI] [PubMed] [Google Scholar]
- 28.Semple RK, Chatterjee VK, O’Rahilly S. PPARγ and human metabolic disease. J Clin Invest. 2006;116(3):581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lüdtke A, Buettner J, Schmidt HH, Worman HJ. New PPARG mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand. J Med Genet. 2007;44(9):e88. doi: 10.1136/jmg.2007.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lüdtke A, et al. Peroxisome proliferator-activated receptor-γ C190S mutation causes partial lipodystrophy. J Clin Endocrinol Metab. 2007;92(6):2248–2255. doi: 10.1210/jc.2005-2624. [DOI] [PubMed] [Google Scholar]
- 31.Campeau PM, et al. Clinical and molecular characterization of a severe form of partial lipodystrophy expanding the phenotype of PPARγ deficiency. J Lipid Res. 2012;53(9):1968–1978. doi: 10.1194/jlr.P025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francis GA, et al. Peroxisomal proliferator activated receptor-γ deficiency in a Canadian kindred with familial partial lipodystrophy type 3 (FPLD3) BMC Med Genet. 2006;7:3. doi: 10.1186/1471-2350-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monajemi H, et al. Familial partial lipodystrophy phenotype resulting from a single-base mutation in deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-γ. J Clin Endocrinol Metab. 2007;92(5):1606–1612. doi: 10.1210/jc.2006-1807. [DOI] [PubMed] [Google Scholar]
- 34.Visser ME, et al. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C) Diabetologia. 2011;54(7):1639–1644. doi: 10.1007/s00125-011-2142-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savage DB, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-γ. Diabetes. 2003;52(4):910–917. doi: 10.2337/diabetes.52.4.910. [DOI] [PubMed] [Google Scholar]
- 36.Agostini M, et al. Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab. 2006;4(4):303–311. doi: 10.1016/j.cmet.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joy TR, Hegele RA. Prevalence of reproductive abnormalities among women with familial partial lipodystrophy. Endocr Pract. 2008;14(9):1126–1132. doi: 10.4158/EP.14.9.1126. [DOI] [PubMed] [Google Scholar]
- 38.Safar Zadeh E, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol. 2013;59(1):131–137. doi: 10.1016/j.jhep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barroso I, et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 40.Broekema MF, et al. Natural helix 9 mutants of PPARγ differently affect its transcriptional activity. Mol Metab. 2019;20:115–127. doi: 10.1016/j.molmet.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majithia AR, et al. Prospective functional classification of all possible missense variants in PPARG. Nat Genet. 2016;48(12):1570–1575. doi: 10.1038/ng.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dyment DA, Gibson WT, Huang L, Bassyouni H, Hegele RA, Innes AM. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur J Med Genet. 2014;57(9):524–526. doi: 10.1016/j.ejmg.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Gouda HN, Sagoo GS, Harding AH, Yates J, Sandhu MS, Higgins JP. The association between the peroxisome proliferator-activated receptor-γ2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am J Epidemiol. 2010;171(6):645–655. doi: 10.1093/aje/kwp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barak Y, et al. PPARγ is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4(4):585–595. doi: 10.1016/S1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 45.Duan SZ, et al. Hypotension, lipodystrophy, and insulin resistance in generalized PPARγ-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117(3):812–822. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koutnikova H, et al. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPARγ hypomorphic mice. Proc Natl Acad Sci U S A. 2003;100(24):14457–14462. doi: 10.1073/pnas.2336090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He W, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100(26):15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai YS, et al. Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARγ. J Clin Invest. 2004;114(2):240–249. doi: 10.1172/JCI20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal AK, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840–4847. doi: 10.1210/jc.2003-030855. [DOI] [PubMed] [Google Scholar]
- 50.Garg A, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84(9):3390–3394. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]
- 51.Eberhardt C, Gray PW, Tjoelker LW. Human Lysophosphatidic Acid. J Biol Chem. 1997;272(32):20299–20305. doi: 10.1074/jbc.272.32.20299. [DOI] [PubMed] [Google Scholar]
- 52.Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003;88(11):5433–5437. doi: 10.1210/jc.2003-030835. [DOI] [PubMed] [Google Scholar]
- 53.Agarwal AK, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31(1):21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- 54.Magré J, et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes. 2003;52(6):1573–1578. doi: 10.2337/diabetes.52.6.1573. [DOI] [PubMed] [Google Scholar]
- 55.Haque W, Garg A, Agarwal AK. Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem Biophys Res Commun. 2005;327(2):446–453. doi: 10.1016/j.bbrc.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 56.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernández-Galilea M, Tapia P, Cautivo K, Morselli E, Cortés VA. AGPAT2 deficiency impairs adipogenic differentiation in primary cultured preadipocytes in a non-autophagy or apoptosis dependent mechanism. Biochem Biophys Res Commun. 2015;467(1):39–45. doi: 10.1016/j.bbrc.2015.09.128. [DOI] [PubMed] [Google Scholar]
- 58.Gale SE, et al. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J Biol Chem. 2006;281(16):11082–11089. doi: 10.1074/jbc.M509612200. [DOI] [PubMed] [Google Scholar]
- 59.Cautivo KM, et al. AGPAT2 is essential for postnatal development and maintenance of white and brown adipose tissue. Mol Metab. 2016;5(7):491–505. doi: 10.1016/j.molmet.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subauste AR, et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes. 2012;61(11):2922–2931. doi: 10.2337/db12-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cortés VA, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 2009;9(2):165–176. doi: 10.1016/j.cmet.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cortés VA, Cautivo KM, Rong S, Garg A, Horton JD, Agarwal AK. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2–/– lipodystrophic mice independent of hepatocyte leptin receptors. J Lipid Res. 2014;55(2):276–288. doi: 10.1194/jlr.M045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266(17):11341–11346. [PubMed] [Google Scholar]
- 64.Jiang HP, Serrero G. Isolation and characterization of a full-length cDNA coding for an adipose differentiation-related protein. Proc Natl Acad Sci U S A. 1992;89(17):7856–7860. doi: 10.1073/pnas.89.17.7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blanchette-Mackie EJ, et al. Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res. 1995;36(6):1211–1226. [PubMed] [Google Scholar]
- 66.Patel S, Yang W, Kozusko K, Saudek V, Savage DB. Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc Natl Acad Sci U S A. 2014;111(25):9163–9168. doi: 10.1073/pnas.1318791111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gandotra S, Lim K, Girousse A, Saudek V, O’Rahilly S, Savage DB. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5) J Biol Chem. 2011;286(40):34998–35006. doi: 10.1074/jbc.M111.278853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kozusko K, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes. 2015;64(1):299–310. doi: 10.2337/db14-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ajjaji D, et al. Dual binding motifs underpin the hierarchical association of perilipins1-3 with lipid droplets. Mol Biol Cell. 2019;30(5):703–716. doi: 10.1091/mbc.E18-08-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gandotra S, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284(50):34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Granneman JG, Moore HP, Mottillo EP, Zhu Z. Functional interactions between Mldp (LSDP5) and Abhd5 in the control of intracellular lipid accumulation. J Biol Chem. 2009;284(5):3049–3057. doi: 10.1074/jbc.M808251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanders MA, et al. Endogenous and synthetic ABHD5 ligands regulate ABHD5-perilipin interactions and lipolysis in fat and muscle. Cell Metab. 2015;22(5):851–860. doi: 10.1016/j.cmet.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Danesch U, Hoeck W, Ringold GM. Cloning and transcriptional regulation of a novel adipocyte-specific gene, FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like proteins interact with sequences required for differentiation-dependent expression. J Biol Chem. 1992;267(10):7185–7193. [PubMed] [Google Scholar]
- 75.Puri V, et al. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem. 2007;282(47):34213–34218. doi: 10.1074/jbc.M707404200. [DOI] [PubMed] [Google Scholar]
- 76.Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279(45):46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 77.Nishino N, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118(8):2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou L, et al. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun. 2015;6:5949. doi: 10.1038/ncomms6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keller P, et al. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem. 2008;283(21):14355–14365. doi: 10.1074/jbc.M708323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, et al. Translocation of CIDEC in hepatocytes depends on fatty acids. Genes Cells. 2014;19(11):793–802. doi: 10.1111/gtc.12180. [DOI] [PubMed] [Google Scholar]
- 81.Xu Y, et al. Cidec promotes the differentiation of human adipocytes by degradation of AMPKα through ubiquitin-proteasome pathway. Biochim Biophys Acta. 2015;1850(12):2552–2562. doi: 10.1016/j.bbagen.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 82.Walther TC, Chung J, Farese RV. Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubio-Cabezas O, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1(5):280–287. doi: 10.1002/emmm.200900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka N, et al. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J Biol Chem. 2015;290(5):3092–3105. doi: 10.1074/jbc.M114.605980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Van Maldergem L, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen W, et al. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32(6):1099–1111. doi: 10.1128/MCB.06465-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Payne VA, et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008;57(8):2055–2060. doi: 10.2337/db08-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mcilroy GD, et al. Adipose specific disruption of seipin causes early-onset generalised lipodystrophy and altered fuel utilisation without severe metabolic disease. Mol Metab. 2018;10:55–65. doi: 10.1016/j.molmet.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sui X, et al. Cryo-electron microscopy structure of the lipid droplet-formation protein seipin. J Cell Biol. 2018;217(12):4080–4091. doi: 10.1083/jcb.201809067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yan R, et al. Human SEIPIN binds anionic phospholipids. Dev Cell. 2018;47(2):248–256.e4. doi: 10.1016/j.devcel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 91.Han S, Binns DD, Chang YF, Goodman JM. Dissecting seipin function: the localized accumulation of phosphatidic acid at ER/LD junctions in the absence of seipin is suppressed by Sei1p(ΔNterm) only in combination with Ldb16p. BMC Cell Biol. 2015;16:29. doi: 10.1186/s12860-015-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang S, Idrissi FZ, Hermansson M, Grippa A, Ejsing CS, Carvalho P. Seipin and the membrane-shaping protein Pex30 cooperate in organelle budding from the endoplasmic reticulum. Nat Commun. 2018;9(1):2939. doi: 10.1038/s41467-018-05278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang H, et al. Seipin is required for converting nascent to mature lipid droplets. Elife. 2016;5:e16582. doi: 10.7554/eLife.16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 95.Hegele RA, Cao H, Huff MW, Anderson CM. LMNA R482Q mutation in partial lipodystrophy associated with reduced plasma leptin concentration. J Clin Endocrinol Metab. 2000;85(9):3089–3093. doi: 10.1210/jcem.85.9.6768. [DOI] [PubMed] [Google Scholar]
- 96.Shackleton S, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153–156. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]
- 97.Guénantin AC, et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol. 2014;29:148–157. doi: 10.1016/j.semcdb.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 98.Ho R, Hegele RA. Complex effects of laminopathy mutations on nuclear structure and function. Clin Genet. 2019;95(2):199–209. doi: 10.1111/cge.13455. [DOI] [PubMed] [Google Scholar]
- 99.Sawyer SL, et al. Homozygous mutations in MFN2 cause multiple symmetric lipomatosis associated with neuropathy. Hum Mol Genet. 2015;24(18):5109–5114. doi: 10.1093/hmg/ddv229. [DOI] [PubMed] [Google Scholar]
- 100.Rocha N, et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife. 2017;6:e23813. doi: 10.7554/eLife.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Capel E, et al. MFN2-associated lipomatosis: clinical spectrum and impact on adipose tissue. J Clin Lipidol. 2018;12(6):1420–1435. doi: 10.1016/j.jacl.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 102.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Maldergem L, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bi J, et al. Seipin promotes adipose tissue fat storage through the ER Ca2+-ATPase SERCA. Cell Metab. 2014;19(5):861–871. doi: 10.1016/j.cmet.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 105.Chen W, et al. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32(6):1099–1111. doi: 10.1128/MCB.06465-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pagac M, et al. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3-phosphate acyltransferase. Cell Rep. 2016;17(6):1546–1559. doi: 10.1016/j.celrep.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen W, Yechoor VK, Chang BH, Li MV, March KL, Chan L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology. 2009;150(10):4552–4561. doi: 10.1210/en.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Magré J, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–370. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 109.Kim CA, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 110.Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK, University of Washington Center for Mendelian Genomics Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A. 2015;167A(8):1796–1806. doi: 10.1002/ajmg.a.37115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14(2):98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 112.Austin ED, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5(3):336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hayashi YK, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–2633. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A. 2010;152A(9):2245–2253. doi: 10.1002/ajmg.a.33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Köbberling J, Dunnigan MG. Familial partial lipodystrophy: two types of an X linked dominant syndrome, lethal in the hemizygous state. J Med Genet. 1986;23(2):120–127. doi: 10.1136/jmg.23.2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grahn TH, et al. FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem Biophys Res Commun. 2013;432(2):296–301. doi: 10.1016/j.bbrc.2013.01.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zolotov S, Xing C, Mahamid R, Shalata A, Sheikh-Ahmad M, Garg A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am J Med Genet A. 2017;173(1):190–194. doi: 10.1002/ajmg.a.37880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Farhan SM, et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30(12):1649–1654. doi: 10.1016/j.cjca.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 119.George S, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Garg A, Sankella S, Xing C, Agarwal AK. Whole-exome sequencing identifies. JCI Insight. 2016;1(9):e86870. doi: 10.1172/jci.insight.86870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cornell RB, Taneva SG, Dennis MK, Tse R, Dhillon RK, Lee J. Disease-linked mutations in the phosphatidylcholine regulatory enzyme CCTα impair enzymatic activity and fold stability. J Biol Chem. 2019;294(5):1490–1501. doi: 10.1074/jbc.RA118.006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Payne F, et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc Natl Acad Sci U S A. 2014;111(24):8901–8906. doi: 10.1073/pnas.1408523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Testa F, et al. Mutations in the PCYT1A gene are responsible for isolated forms of retinal dystrophy. Eur J Hum Genet. 2017;25(5):651–655. doi: 10.1038/ejhg.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Haider A, et al. PCYT1A regulates phosphatidylcholine homeostasis from the inner nuclear membrane in response to membrane stored curvature elastic stress. Dev Cell. 2018;45(4):481–495.e8. doi: 10.1016/j.devcel.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hoover-Fong J, et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):105–112. doi: 10.1016/j.ajhg.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamamoto GL, et al. Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94(1):113–119. doi: 10.1016/j.ajhg.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Horn D, Robinson PN. Progeroid facial features and lipodystrophy associated with a novel splice site mutation in the final intron of the FBN1 gene. Am J Med Genet A. 2011;155A(4):721–724. doi: 10.1002/ajmg.a.33905. [DOI] [PubMed] [Google Scholar]
- 128.Takenouchi T, et al. Severe congenital lipodystrophy and a progeroid appearance: mutation in the penultimate exon of FBN1 causing a recognizable phenotype. Am J Med Genet A. 2013;161A(12):3057–3062. doi: 10.1002/ajmg.a.36157. [DOI] [PubMed] [Google Scholar]
- 129.Graul-Neumann LM, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3′ terminus of the FBN1-gene. Am J Med Genet A. 2010;152A(11):2749–2755. doi: 10.1002/ajmg.a.33690. [DOI] [PubMed] [Google Scholar]
- 130.Ellis NA, German J. Molecular genetics of Bloom’s syndrome. Hum Mol Genet. 1996;5(Spec No):1457–1463. doi: 10.1093/hmg/5.supplement_1.1457. [DOI] [PubMed] [Google Scholar]
- 131.Goto M, Rubenstein M, Weber J, Woods K, Drayna D. Genetic linkage of Werner’s syndrome to five markers on chromosome 8. Nature. 1992;355(6362):735–738. doi: 10.1038/355735a0. [DOI] [PubMed] [Google Scholar]
- 132.Shen JC, Loeb LA. The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet. 2000;16(5):213–220. doi: 10.1016/S0168-9525(99)01970-8. [DOI] [PubMed] [Google Scholar]
- 133.Shastry S, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192–E197. doi: 10.1210/jc.2010-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Weedon MN, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–950. doi: 10.1038/ng.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thauvin-Robinet C, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141–149. doi: 10.1016/j.ajhg.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dyment DA, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158–166. doi: 10.1016/j.ajhg.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu Y, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Torrelo A, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. 2010;62(3):489–495. doi: 10.1016/j.jaad.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 139.Cavalcante MP, et al. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature—a rare case with a novel mutation. Eur J Pediatr. 2016;175(5):735–740. doi: 10.1007/s00431-015-2668-4. [DOI] [PubMed] [Google Scholar]
- 140.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83(1):18–34. doi: 10.1097/01.md.0000111061.69212.59. [DOI] [PubMed] [Google Scholar]
- 141.Carr A, et al. An objective case definition of lipodystrophy in HIV-infected adults: a case-control study. Lancet. 2003;361(9359):726–735. doi: 10.1016/S0140-6736(03)12656-6. [DOI] [PubMed] [Google Scholar]
- 142.Sekhar RV, et al. Metabolic basis of HIV-lipodystrophy syndrome. Am J Physiol Endocrinol Metab. 2002;283(2):E332–E337. doi: 10.1152/ajpendo.00058.2002. [DOI] [PubMed] [Google Scholar]
- 143.Brinkman K, Smeitink JA, Romijn JA, Reiss P. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet. 1999;354(9184):1112–1115. doi: 10.1016/S0140-6736(99)06102-4. [DOI] [PubMed] [Google Scholar]
- 144.Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106(4):453–458. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bingham A, et al. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore) 2008;87(2):70–86. doi: 10.1097/MD.0b013e31816bc604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ley CJ, Lees B, Stevenson JC. Sex- and menopause-associated changes in body-fat distribution. Am J Clin Nutr. 1992;55(5):950–954. doi: 10.1093/ajcn/55.5.950. [DOI] [PubMed] [Google Scholar]
- 147.Schlögl H, et al. Leptin substitution in patients with lipodystrophy: neural correlates for long-term success in the normalization of eating behavior. Diabetes. 2016;65(8):2179–2186. doi: 10.2337/db15-1550. [DOI] [PubMed] [Google Scholar]
- 148.Aotani D, et al. Functional magnetic resonance imaging analysis of food-related brain activity in patients with lipodystrophy undergoing leptin replacement therapy. J Clin Endocrinol Metab. 2012;97(10):3663–3671. doi: 10.1210/jc.2012-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lüdtke A, et al. Hepatic steatosis in Dunnigan-type familial partial lipodystrophy. Am J Gastroenterol. 2005;100(10):2218–2224. doi: 10.1111/j.1572-0241.2005.00234.x. [DOI] [PubMed] [Google Scholar]
- 150.Javor ED, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41(4):753–760. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- 151.Alidjan F, Langendonk J. Familial partial lipodystrophy (type dunnigan) results in extreme premature cardiovascular disease. phenotypic variation in a kindred study. Atherosclerosis. 2015;241(1):e38 [Google Scholar]
- 152.Bidault G, et al. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33(9):2162–2171. doi: 10.1161/ATVBAHA.113.301933. [DOI] [PubMed] [Google Scholar]
- 153.Zhang X, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57(5):1246–1253. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 154.Mullican SE, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med. 2017;23(10):1150–1157. doi: 10.1038/nm.4392. [DOI] [PubMed] [Google Scholar]
- 155.Miehle K, et al. Serum concentrations of fibroblast growth factor 21 are elevated in patients with congenital or acquired lipodystrophy. Cytokine. 2016;83:239–244. doi: 10.1016/j.cyto.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 156.Patel S, et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 2019;29(3):707–718.e8. doi: 10.1016/j.cmet.2018.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Matsubara M, Maruoka S, Katayose S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol. 2002;147(2):173–180. doi: 10.1530/eje.0.1470173. [DOI] [PubMed] [Google Scholar]
- 158.Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1(12):1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 159.Pan WW, Myers MG. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci. 2018;19(2):95–105. doi: 10.1038/nrn.2017.168. [DOI] [PubMed] [Google Scholar]
- 160.Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395. doi: 10.1210/jcem.87.5.8624. [DOI] [PubMed] [Google Scholar]
- 161.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32(4):498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 162.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119(2):315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Semple RK, et al. Paradoxical elevation of high-molecular weight adiponectin in acquired extreme insulin resistance due to insulin receptor antibodies. Diabetes. 2007;56(6):1712–1717. doi: 10.2337/db06-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. 2017;127(1):74–82. doi: 10.1172/JCI88883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.du Plessis J, et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology. 2015;149(3):635–48.e14. doi: 10.1053/j.gastro.2015.05.044. [DOI] [PubMed] [Google Scholar]
- 168.Kim JI, et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol. 2015;35(10):1686–1699. doi: 10.1128/MCB.01321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Clément K, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18(14):1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 170.Araújo-Vilar D, et al. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clin Endocrinol (Oxf) 2012;76(6):816–824. doi: 10.1111/j.1365-2265.2011.04208.x. [DOI] [PubMed] [Google Scholar]
- 171.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106(2):171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Virtue S, Vidal-Puig A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008;6(9):e237. doi: 10.1371/journal.pbio.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Danforth E. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26(1):13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 174.Scott RA, et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes. 2014;63(12):4378–4387. doi: 10.2337/db14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lotta LA, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. doi: 10.1038/ng.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shungin D, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187–196. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Emdin CA, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA. 2017;317(6):626–634. doi: 10.1001/jama.2016.21042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Banerji MA, Faridi N, Atluri R, Chaiken RL, Lebovitz HE. Body composition, visceral fat, leptin, and insulin resistance in Asian Indian men. J Clin Endocrinol Metab. 1999;84(1):137–144. doi: 10.1210/jcem.84.1.5371. [DOI] [PubMed] [Google Scholar]
- 179.Barzilai N, et al. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes. 1999;48(1):94–98. doi: 10.2337/diabetes.48.1.94. [DOI] [PubMed] [Google Scholar]
- 180.Gastaldelli A, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496–506. doi: 10.1053/j.gastro.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 181.Amato MC, et al. Visceral Adiposity Index: a reliable indicator of visceral fat function associated with cardiometabolic risk. Diabetes Care. 2010;33(4):920–922. doi: 10.2337/dc09-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wahrenberg H, Hertel K, Leijonhufvud BM, Persson LG, Toft E, Arner P. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363–1364. doi: 10.1136/bmj.38429.473310.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lotta LA, et al. Association of genetic variants related to gluteofemoral vs abdominal fat distribution with type 2 diabetes, coronary disease, and cardiovascular risk factors. JAMA. 2018;320(24):2553–2563. doi: 10.1001/jama.2018.19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gavrilova O, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105(3):271–278. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wang H, et al. Adipose tissue transplantation ameliorates lipodystrophy-associated metabolic disorders in seipin-deficient mice. Am J Physiol Endocrinol Metab. 2019;316(1):E54–E62. doi: 10.1152/ajpendo.00180.2018. [DOI] [PubMed] [Google Scholar]
- 186.Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351(11):1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 187.Steven S, et al. Very low-calorie diet and 6 months of weight stability in type 2 diabetes: pathophysiological changes in responders and nonresponders. Diabetes Care. 2016;39(5):808–815. doi: 10.2337/dc15-1942. [DOI] [PubMed] [Google Scholar]
- 188.Sjöström L, et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med. 2007;357(8):741–752. doi: 10.1056/NEJMoa066254. [DOI] [PubMed] [Google Scholar]
- 189.Oral EA, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 190.Rosenbaum M, Leibel RL. 20 years of leptin: role of leptin in energy homeostasis in humans. J Endocrinol. 2014;223(1):T83–T96. doi: 10.1530/JOE-14-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]