

Abstract
The metabolic syndrome (MetS) is a constellation of risk factors that, if left untreated, will often progress to greater metabolic defects such as type 2 diabetes and nonalcoholic fatty liver disease. While these risk factors have been established for over 40 years, the definition of MetS warrants reconsideration in light of the substantial data that have emerged from studies of the gut microbiome. In this Review we present the existing recent literature that supports the gut microbiome’s potential influence on the various risk factors of MetS. The interplay of the intestinal microbiota with host metabolism has been shown to be mediated by a myriad of factors, including a defective gut barrier, bile acid metabolism, antibiotic use, and the pleiotropic effects of microbially produced metabolites. These data show that events that start in the gut, often in response to external cues such as diet and circadian disruption, have far-reaching effects beyond the gut.
Introduction
The gut microbiome has been implicated in the etiopathogenesis of multiple diseases ranging from the intestinal (inflammatory bowel diseases, colon cancer) to the neurological (Parkinson’s disease, autism). However, definition of the microbiome’s role in metabolic diseases has remained elusive. The primary reason is that the very factors believed to be central drivers of dysmetabolism are also believed to be the primary drivers of our gut microbiome composition: diet and lifestyle. While it is conceptually intuitive that the gut microbiome and host metabolism would be interrelated, disentangling cause and effect remains a challenge. In this Review we will explore the evidence for a link between the gut microbiome and metabolic events that contribute to the metabolic syndrome (MetS). In particular, we will take a gut-centric view of MetS that roots itself in the hypothesis that chronic systemic defects, such as MetS, may begin here, where immune, hormonal, nervous, and microbial signals converge (Figure 1).
Figure 1. A model for the gut microbiome’s interaction with the intestinal epithelial barrier and its contribution to metabolic diseases.
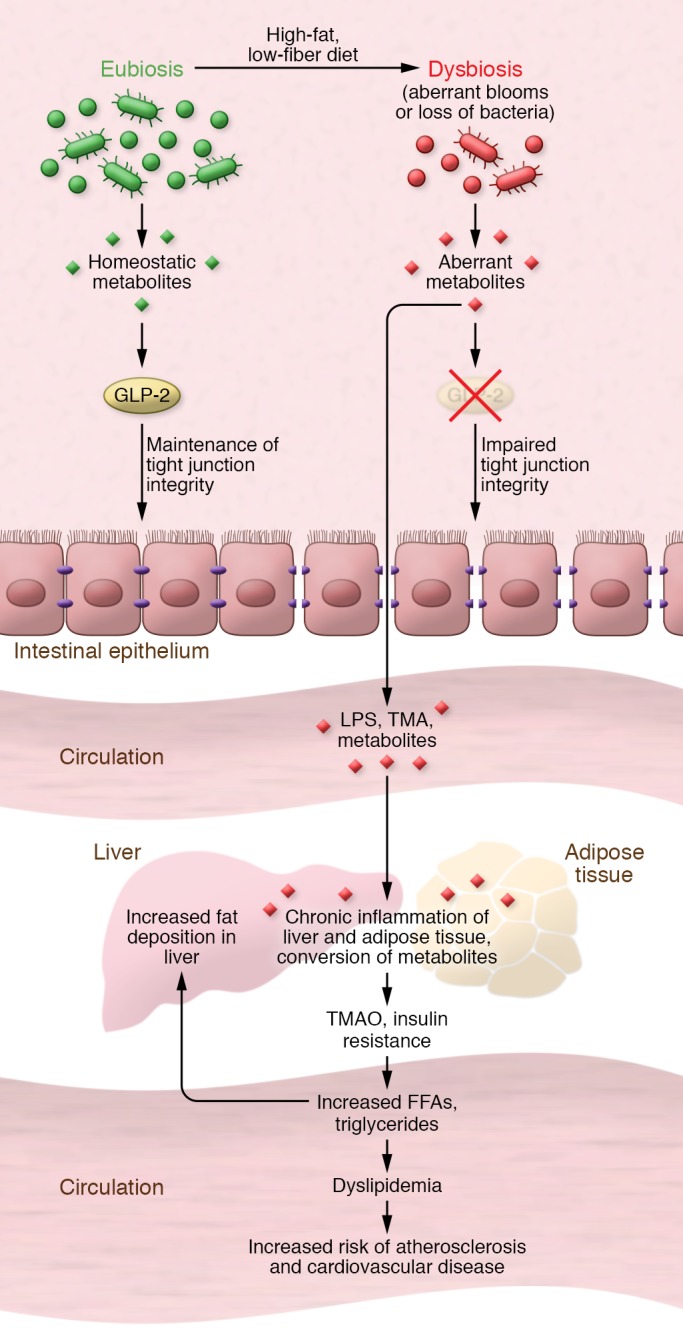
From the top: High-fat, low-fiber diet induces intestinal dysbiosis, resulting in aberrant metabolite concentrations that disrupt GLP-2–mediated tight junction integrity. This loss of integrity makes the gut epithelium more permissive to microbial lipopolysaccharide (LPS), trimethylamine (TMA), and other metabolites entering the circulation and contributing to the chronic inflammation of liver and adipose tissue that is associated with the development of cardiovascular disease, insulin resistance, and other conditions associated with the metabolic syndrome.
Defining and redefining the metabolic syndrome
The term “metabolic syndrome” was first coined in the 1970s by Herman Haller, who was studying the various risks associated with atherosclerosis. However, the association of dysmetabolism with cardiovascular risk factors was documented as early as the 1920s; the link between these cardiovascular risk factors and android adiposity (the accompanying phenotype, characterized by fat distribution in the trunk and upper body) was described shortly thereafter. Then, in 1988, Gerald Reaven offered a new name for MetS, “syndrome X,” which now emphasized insulin resistance among the constellation of risk factors (1).
Despite observation of these clinical findings for over 80 years, developing a standard definition for MetS has proved challenging. Multiple organizations, such as the World Health Organization, the American Association for Clinical Endocrinology, and the International Diabetes Federation, have since come out with their own definitions for MetS that, while overlapping, are not identical (2, 3). These definitions all include central obesity, dyslipidemia, insulin resistance, and hypertension, but the definition and cutoffs of these individual risk factors vary. To further complicate, or perhaps shed light on, MetS, additional risk factors may begin to emerge as more studies are conducted and more data generated. For example, the gut has been wholly ignored in the definition of MetS, but data from the last decade, as the microbiome field has come to the fore, have shown that mechanisms leading to MetS risk factors may originate in the gut.
Evidence for a gut-centric theory of MetS emerged in 2007 with a series of studies in rodents and humans showing that chronic consumption of a high-fat diet (HFD) leads to intestinal barrier defects that facilitate the passage of intestinal luminal contents (food antigen, bacterial by-products, bacteria themselves), and bacterial lipopolysaccharide (LPS) in particular, into systemic circulation. The ensuing low-grade inflammation and its perpetuation was coined metabolic endotoxemia for its inhibitory effect on normal glycemic function (4, 5). This was the first report of bacterial LPS activation of Toll-like receptors (TLRs) leading to an innate immune response that impaired insulin sensitivity. As a result, the study of factors affecting intestinal permeability has become one of the most promising areas of microbiome research. Furthermore, areas of research with an established involvement in metabolic diseases, such as the role of bile acids as bioactive molecules, and circadian misalignment, have since been found to be influenced by and have an impact on the gut microbiome and its metabolites. Findings from these compelling areas of research may ultimately lead to additional defining phenotypes of MetS that take into account defects at the level of the gut.
The microbiome: a historical perspective
The emergence of the fields of bacteriology and microbiology in the early 1800s took disease out of the realm of the spiritual, where it was believed to be caused by evil spirits, curses, or punishment for wrongdoings, and into the realm of pathogenesis (6). Early observations by Redi, Virchow, and Pasteur of sterile laboratory conditions disproved the accepted theory that life erupted from nonliving material. This theory of spontaneous generation, based on observations of maggots emerging from old meat, for example, could not be demonstrated in sterile conditions. Pasteur later showed that bacteria exist in “nonliving” materials such as wine (7). What today is called wild fermentation, in which the grape juice is allowed to ferment in the natural yeasts of open air, was commonly practiced at the time. In the 1800s it was believed that the fermentative process was caused by air itself, but Pasteur discovered that the yeasts in the open environment were responsible. Interestingly, however, he found that if bacteria found their way into the wine, the juice would spoil. These were the beginnings of the germ theory of disease, which suggested that environmental microorganisms were the source of most diseases (8).
Two subsequent seminal discoveries bridged the gap between environmental bacteriology and human disease. In 1876 a German physician, Robert Koch, proved that a single bacterium could cause a disease. He discovered rod-shaped structures in the blood of humans and livestock that were mysteriously dying across Europe. Through a series of sophisticated experiments isolating the structures from blood and reintroducing them into healthy animals, he determined they were living bacteria, which he named Bacillus anthracis — what we know today as anthrax (9). The experiments he conducted led to the series of scientific principles, Koch’s postulates, that one uses to determine whether a particular microorganism is responsible for disease. While these postulates have been slightly modified over time, they are still an accepted set of experiments for determining a microbe’s role in disease (10). It wasn’t, however, until Theodor Escherich’s 1885 discovery of what is now called Escherichia coli isolated from the colons of healthy children that evidence for gut-resident bacteria emerged. Escherich determined that some of the discovered E. coli strains were innocuous, while others were responsible for infant gastroenteritis. Here emerged not only the concept of native gut microbiota, but also that of strain variation of a bacterium in different contexts and individuals — a concept heavily investigated in the microbiome field today (11–13).
The nearly 100 years that followed was an era of infectious disease control and vaccine development. These heroic efforts curtailed diseases that once killed by the thousands. This period was followed by a new era, ushered in by Carl Woese and George Fox, that redefined the tree of life on the basis of genetic signature rather than morphology (14). In their seminal paper, arguably one of the most influential microbiology publications to date, Woese and Fox showed that the ribosomal RNA of all living things binned all creatures into three categories, ultimately, archaea, bacteria, and eukarya. The bacterial RNA signature was the 16S rRNA, and subsequent work by Norman Pace, Stanley Falkow, and David Relman showed that molecular tools could be used to reliably identify bacteria in mixed organism communities based on the 16S rRNA (15), and that these tools could be applied to the identification of bacteria in human tissues (16). With this, the genomic revolution of microbiology had begun, converting the study of the trillions of bacteria that live in and on us into an “ome.”
Since the popularization of the term “microbiome” by Joshua Lederberg in 2001, defined as “the ecological community of commensal, symbiotic, and pathogenic microorganisms that literally share our body space” (17), over 15,000 scholarly articles have been published describing the putative roles of the intestinal microbiota in both the maintenance of health and the development of disease. While the microbiome’s reach is now far and wide in terms of its putative involvement in diseases, syndromes, and conditions, pinning down exact mechanisms and patterns in human cohorts has proved challenging because of the microbiome’s exquisite sensitivity to environmental perturbations and the difficulty of accessing certain body sites. Therefore, MetS, being a constellation of risk factors rather than a single condition, has been approached in the microbiome field by first decoupling the risk factors and investigating whether there are microbial links to, for example, cardiovascular disease, or obesity, or dyslipidemia. Therefore, in this Review, we follow the same approach and address some of the most compelling recent findings regarding specific risk factors and their potential relationship with the gut microbiome.
The microbiome and obesity
The prevalence of obesity in Western nations and, increasingly, in non-Western nations is a driving force behind the heightened medical interest in and recognition of MetS (18). Before 1980, roughly 15% of the US adult population was overweight (19). By 2015 to 2016, the prevalence of obesity among adults in the US reached a documented high of 39.8% — a nearly 25% increase over a span of 35 years (19). This rapid rise in obesity rates, coupled with increasing evidence that central obesity is a major risk factor for type 2 diabetes and cardiovascular disease, has warranted the use of the term “epidemic” and made obesity and MetS one of our nation’s largest public health issues. While the development of obesity has historically been thought of as an imbalance between energy expenditure and intake, a substantial body of literature suggests that the regulation of body weight is more complex (20). In the context of MetS, additional consideration should be made of an individual’s unique metabolic processing of foods, genetics, lifestyle choices, and gut microbiome.
While the gut microbiome is responsive to large swings in caloric intake (21, 22), multiple studies show it is most sensitive to diet composition. This was nicely demonstrated by David et al. in a human study in which volunteers were placed on either a plant-based diet (grains, legumes, fruits, and vegetables) or an animal-based diet (meats, eggs, and cheeses) for 5 consecutive days. After these 5 days, there were significant temporal alterations in their microbial communities. Participants placed on animal-based diets experienced a bloom of bile-tolerant microbes (Alistipes, Bilophila, and Bacteroides) and a decrease in the abundance of fiber-fermenting bacteria (22). Additional studies have demonstrated microbial sensitivity to nuances in dietary fat composition (23, 24), fiber types (25, 26), and food additives (27, 28). Dietary fats are particularly interesting, as some fatty acids possess antimicrobial activity, but this property depends on the number of carbons and the number, position, and orientation of the C=C double bonds (29). Furthermore, Caesar et al. showed that rats fed a lard-based diet experienced a bloom of the proinflammatory Bilophila wadsworthia, while those fed a fish oil–based diet experienced blooms of Lactobacillus and Akkermansia muciniphila (30). Blooms of A. muciniphila have been negatively correlated with obesity, treatment-naive type 2 diabetes, and hypertension (31–33). Recently, the first human trial involving oral A. muciniphila supplementation was conducted in overweight/obese insulin-resistant individuals. This double-blind, placebo-controlled, randomized crossover study involved daily supplementation for 3 months, which resulted in significantly improved insulin sensitivity and reduced insulinemia and plasma total cholesterol (34). Therefore, while the relationship between diet and the microbiome may be involved in developing obesity, it may also be a part of the solution.
Further evidence of this was seen in a study in which rodents fed a diet with a high level of fermentable dietary fiber were found to be protected against diet-induced obesity and related metabolic defects (5). In this study, rats pretreated for 35 days with oligofructose, a dietary fiber, did not exhibit hyperphagia, weight gain, or increased serum triglycerides when exposed to an HFD. The authors determined that metabolites produced by microbial fermentation of fibers induced the production of the endogenous peptides glucagon-like peptide-1 (GLP-1) and GLP-2. GLP-1 is known to have beneficial effects on glucose metabolism, and GLP-2 has been shown to improve intestinal epithelial tight junction integrity (5). Other studies supporting the role of the gut microbiome in developing obesity demonstrated that mice prone to obesity and hepatosteatosis when vendor-bred lost these phenotypes after three generations of breeding in a research vivarium (35, 36). Given evidence that the microbiome is heavily influenced by diet and the environment, these data suggest a unique susceptibility of certain microbial signatures to metabolic disease.
While diet is the major influencer of our gut microbiome’s composition and behavior, the microbiome is also sensitive to other external cues from our environment that can exacerbate the purported detrimental effects of high-fat, high-sugar Western diets. Technology has allowed humans to manipulate their light/dark cycle, rather than the other way around, as illustrated by overnight shift work and international travel. However, the body and its central and peripheral circadian clocks evolved in and still operate on a natural light/dark cycle that follows the 24-hour rotation of the earth (37). We now know that many of the body’s natural biological processes, including hormone release and blood glucose levels, exhibit cyclical patterns during a 24-hour period (38, 39). Interference with this cycle has been shown to promote obesity (40, 41), and affect insulin sensitivity (42) and lipid metabolism (43).
At the level of the gut, intestinal hormones and immune cells exhibit natural diurnal fluxes under homeostatic conditions (44, 45). Recent findings have revealed that disturbances in the gut microbiota, either through antibiotic depletion or chronic consumption of an HFD, can lead to local circadian disruption that promotes weight gain or abnormal glucose fluxes (46–49). For example, Leone et al. found that typical diurnal clock gene expression in the brain and liver was absent in germ-free mice, regardless of low-fat or high-fat diet (49). However, conventionally raised mice with an intact microbiota retained normal homeostatic clock gene expression, but only when fed a low-fat diet (49). If these mice were fed an HFD, circadian disruption was observed. Interestingly, it was previously reported that the abundance of several genera of the gut microbiota naturally experiences diurnal shifts in abundance (49). In this study, and in others, it was found that chronic ad libitum feeding, especially of an HFD, creates an arrhythmic microbiome, which in turn disrupts the liver clock genes that direct free fatty acid uptake and release (47, 48). Furthermore, Thaiss et al. showed in mice that the mucus barrier of the gut also exhibits cyclical shifts in thickness that correspond to these shifts in microbial abundance (45). These data suggest that disrupting the gut microbiome’s circadian rhythms can also potentially lead to barrier defects.
Collectively, these data suggest that the gut microbiome may mediate or even orchestrate events locally in the intestines, through the influence of diet, that alter their metabolite signaling to the rest of the body. This signaling interacts with host metabolism in ways we are just beginning to understand. Further research will be needed to understand how to leverage the microbiome in the prevention and treatment of obesity.
Bacterial metabolites and dyslipidemia
Dyslipidemia is an umbrella term used to describe an abnormal amount of lipids in the blood. Diagnostic criteria for MetS define dyslipidemia as an elevation in circulating triglycerides or a reduction in circulating high-density lipoprotein (HDL) (50). Both the National Cholesterol Education Program ATPIII guidelines and the International Diabetes Federation state that HDL cholesterol less than 1.0 mmol/L (40 mg/dL) in men and less than 1.3 mmol/L (50 mg/dL) in women, or blood triglycerides greater than 1.7 mmol/L (150 mg/dL), should be flagged as abnormal values. Dyslipidemia and the resulting atherosclerotic plaques remain major risk factors for cardiovascular disease and are often intricately linked with impaired glucose metabolism and obesity (51).
In an effort to disentangle cause and effect in dyslipidemia, scientists have turned to the microbiome for clues and possible solutions. Short-chain fatty acids (SCFAs) are the metabolic end products of microbial fermentation of dietary fibers and present a potentially promising therapeutic role in mitigating the consequences of long-standing dyslipidemia (52). The human gut does not have the enzymatic capacity to break down certain foods, namely, complex carbohydrates in the form of dietary fiber. However, unique taxa of anaerobic bacteria that reside in the cecum and the large intestine can ferment these fibers into a wide variety of by-products. The most heavily studied of these by-products are the SCFAs.
The three most abundant SCFAs in the human colon are acetate, propionate, and butyrate, which are found at an approximate molar ratio of 60:20:20, respectively (52). While acetate is produced in the largest proportion, a majority of it bypasses the splanchnic circulation to be oxidized by muscle or used by adipocytes for lipogenesis, while the remaining acetate is converted to butyrate by luminal bacteria (53–55). Butyrate has been of particular interest for its known benefits in providing energy to intestinal epithelial cells, promoting colonocyte health, and maintaining intestinal epithelial integrity (56). Butyrate levels in patients with inflammatory diseases are also reported to be significantly lower than those in healthy controls (56, 57). Butyrate supplementation has also been shown to directly induce intestinal gluconeogenesis (IGN) genes in human Caco-2 cells via a cAMP-mediated mechanism (58). This study also showed that intestinal glucose production promoted beneficial effects on food intake and glucose metabolism via the peripheral nervous system. Propionate, on the other hand, was able to indirectly induce IGN via a gut-brain neural connection. Interestingly, propionate has also been shown to stimulate intestinal release of the satiety hormone peptide YY (PYY) and GLP-1, leading to reduced energy intake in humans. After 24 weeks of propionate supplementation, subjects experienced reductions in weight, abdominal adipose tissue, and hepatic fat, and maintained insulin sensitivity (59).
A separate, double-blind, randomized crossover study was conducted to determine the preventative effects of lupin kernel fiber, a fiber derived from legumes, or citrus fiber on cardiovascular disease (60). The authors found that after three interventional feeding periods of 4 weeks in duration, study volunteers placed on the high-fiber diets (i.e., lupin or citrus fiber) experienced a reduction in C-reactive protein, systolic blood pressure, and circulating blood lipids, in addition to experiencing higher satiety, weight loss, and a reduction in waist circumference. The authors hypothesized that the lipid-lowering effects of a high-fiber diet were the result of SCFA production. This claim is supported by participants’ fecal SCFA content, particularly acetate and propionate, which were increased significantly in both of the high-fiber diets in comparison with control and individual baselines (60).
However, Perry et al. found that acetate, when produced in elevated quantities by the gut microbiota, could activate the parasympathetic nervous system, leading to increased glucose-stimulated insulin secretion, ghrelin secretion, hyperphagia, and obesity (61). When acetate was infused directly into rats, they exhibited impaired glucose disposal and impaired insulin suppression of hepatic gluconeogenesis during a hyperglycemic-euglycemic clamp. These rats also exhibited increases in plasma, liver, and skeletal muscle triglyceride content (61). These data highlight that microbe-derived SCFAs are bioactive by-products that interact with host metabolism in complex ways. Whether the end effect is positive or negative is likely highly context-dependent.
Microbial components can also present risk for MetS. While SCFAs can produce large tropic effects on the body and health, components of the bacterial cell wall like LPS and peptidoglycan can be recognized by the host immune system and contribute to cardiovascular disease risk (62). For example, mice injected with LPS exhibited a reduction in plasma HDL cholesterol and elevations in plasma triglycerides (63). This association was not limited to rodents, as a retrospective human study conducted on 587 individuals from the Finnish Diabetic Neuropathy cohort revealed that those with the highest levels of serum LPS also presented with significantly higher levels of serum triglycerides and blood pressure (64).
Compelling lines of evidence further implicate the microbiome in promoting cardiovascular disease risk. Studies in antibiotic-treated and germ-free mice suggest an obligate role for the gut microbiome in converting dietary phosphatidylcholine to the pro-atherosclerotic molecule trimethylamine N-oxide (TMAO) (65, 66). In this context, microbes cleave dietary choline from animal products to trimethylamine (TMA), which is in turn oxidized by the liver, via the hepatic flavin monooxygenase 3 (FMO3), to form TMAO. Patients with atherosclerosis had significantly higher levels of circulating TMAO compared with healthy controls (66). Furthermore, in a recent human study, vegans and omnivores challenged with dietary choline supplementation demonstrated dose-dependent increases of TMAO in the circulation (67). Interestingly, the prothrombotic phenotype is completely abolished upon treatment with broad-spectrum antibiotics, strongly suggesting a role for the microbiome in this phenomenon.
Furthermore, it has been identified that the nuclear farnesoid X receptor (FXR), which is involved in bile acid metabolism in the liver and small intestines, can regulate FMO3 (68). Bennet et al. nicely demonstrated that FXR ligands administered to wild-type mice could induce FMO3 expression and increase TMAO levels, but this effect was abrogated in Fxr–/– mice (68). Treatment with dietary carnitine, a substrate for bacterial production of TMA, or TMAO directly, resulted in a reduction of the bile acid pool in mice (69). This would have likely effects on FXR activation. This potential interplay in the liver between TMAO and FXR is intriguing given the attention that has been paid to microbial activation of FXR, which is expressed in several metabolically active tissues, such as the liver and small intestines. At the same time, FXR agonists, such as obeticholic acid (OBA), are currently being tested as a treatment in patients with nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Mudaliar et al. in 2013 found that administering OBA for 6 weeks to patients with NAFLD and type 2 diabetes mellitus resulted in improved insulin sensitivity, compared with placebo (70). Furthermore, in NASH, Neuschwander-Tetri et al. found that patients with NASH exhibited significant improvement in the NASH activity score after 72 weeks of OBA treatment (71). However, it is unclear how the gut microbiome may be mediating these effects.
Altogether, this collection of data suggests that microbial metabolites, namely SCFAs derived from the fermentation of dietary fiber, could prove protective in the development of dyslipidemia and MetS, while other bacterial metabolites, such as TMA, secondary bile acids, or components of the bacterial cell wall such as LPS, may be drivers or strong contributors.
The microbiome, low-grade inflammation, and insulin resistance
While chronic low-grade inflammation is not an established factor defining MetS, it is an established factor in the etiopathogenesis of obesity and insulin resistance, and therefore is intimately related to the metabolic defects observed with MetS. The central role of intestinal permeability in chronic low-grade inflammation therefore makes the microbiome a central player in the inflammation-induced onslaught of metabolic defects.
Inflammation is described as a series of responses by vascularized tissue to injury or infection (72). These responses are a necessary protective response for survival, but prolonged exposure to stimuli and mobilization of immune cells can be detrimental. The first study linking chronic low-grade inflammation to obesity and insulin resistance was reported by Hotamisligl et al. in 1993; TNF-α overexpression in the adipose tissue of rodents led to insulin resistance (73). This study was later extended to obese humans, in whom the authors discovered increased expression of TNF-α mRNA in obese adipose tissue in comparison with controls (74). The authors stated that metabolic cells such as adipocytes and hepatocytes are in close contact with immune cells and blood vessels. This proximity establishes constant communication between metabolism and immune responses (75).
Indeed, several studies have shown increased proinflammatory markers in adipose tissue of obese individuals (76) with enlarged adipocytes in mice and humans exhibiting macrophage infiltration (77) and dying adipocytes surrounded by crown-like structures consisting of phagocytosing macrophages. These adipocytes themselves, along with the macrophages, produce proinflammatory cytokines and chemokines (78, 79). Cai et al. further discovered that HFD-induced obesity leads to chronic activation of NF-κB in the liver. This subacute inflammation led to hyperglycemia and profound hepatic insulin resistance (80). These seminal studies established chronic low-grade inflammation as a central factor contributing to the development of diabetes.
HFDs are often used experimentally to trigger inflammatory events leading to altered metabolism; however, substantial efforts are under way to understand what factors, in addition to HFDs, are inflammatory triggers. Here, the gut microbiome, specifically intestinal pathobionts (commensal bacteria that can become pathogenic), is proving to be a promising avenue. A study by Vaziri et al. in chronic kidney disease showed that the microbiome might contribute to oxidative stress conditions by releasing uremic toxins such as indoxyl sulfate and p-cresyl sulfate, adding to oxidative stress and inflammation in the kidneys (81). Thus, it can be speculated that a specific subpopulation of the microbiome might thrive in these oxidative conditions, in turn contributing to the disease state. This concept was explored more deeply by Byndloss et al. in a study whereby the natural anaerobic homeostasis of the intestine was intentionally disrupted, resulting in the discovery that an oxygenated intestine promotes expansion of pathobionts and leads to inflammation (82). The elegant mechanism leverages the fact that intestinal PPARγ limits bioavailability of oxygen in the lumen via β-oxidation in colonocytes. The stimulant for this natural process is bacterially produced butyrate. When butyrate-producing bacteria were eradicated from the gut by antibiotics, luminal oxygen levels increased and pathogenic forms of E. coli and Salmonella bloomed (82). Another intriguing gut-centric mechanism that may lead to dysmetabolism induced by low-grade inflammation is the role of the intracellular endocannabinoid (ECB) system. Obese individuals have increased levels of ECB in adipose tissue and plasma, and bacterial LPS is known to be a potent stimulator of ECB synthesis (83). Pharmacological or genetic inhibition of the cannabinoid receptor CB1 protects against obesity, hepatic steatosis, and low-grade inflammation. In subsequent rodent studies, the investigators concluded that the gut microbiome controls ECB activity in the colon and adipose tissue by comparing germ-free with conventional mice, as well as by testing dietary factors and genetically modified mice that are known to have altered gut microbiome composition (84). The putative role of the ECB system in healthy humans is still not well understood, but it is clear that it can be stimulated by bacterial LPS and leads to metabolic dysfunction in model systems.
A recent example that further extends the relationship between inflammation, altered glycemia, and intestinal permeability can be found in the elegant study by Thaiss et al. Here the authors posit that dysmetabolism can be both a driver and the result of impaired barrier function. They carefully dissect the roles of leptin, the gut microbiome, and obesity in barrier function through systematic screening in multiple animal models and humans and ultimately determine that intracellular hyperglycemia in the intestinal epithelium, among the many factors that define dysmetabolism, is the driver (85). Furthermore, they address an often overlooked side effect of intestinal permeability: increased susceptibility to enteric infections. In all of the models in which they induced intestinal hyperglycemia, the additional introduction of the Citrobacter rodentium pathogen resulted in increased systemic dissemination of the bacterium in comparison with controls. In humans, they measured 27 serum markers and correlated them with serum pathogen recognition receptor (PRR) ligands (i.e., surrogate markers of intestinal permeability) and found that the only marker that was positively associated with an increase in circulating PRR ligands was hemoglobin A1c (85). Collectively, these findings suggest that the relationship between chronic hyperglycemia, intestinal permeability, and increased risk for systemic infections may need to be considered part of the evolving MetS definition.
Fecal transplants and metabolic correction
Perhaps one of the most direct ways microbiome-induced phenotypes have been tested is through fecal microbiota transplants (FMTs). In this system, feces from a donor are transferred to a recipient via nasogastric tube, colonoscope, enema, capsule, or a combination of these. Therapeutically, this method has demonstrated extraordinary success in curing colitis due to recurrent Clostridium difficile infection (86). The exact mechanism of its success is still not fully understood, but in this case FMT is essentially restoring diversity of the recipient’s microbiome that was eradicated by long-term use of broad-spectrum antibiotics, and outcompeting C. difficile for vital niches in the intestine. This raises the question of whether FMT can be used for other indications — for example, to transfer a metabolically healthy microbiome into a recipient with MetS in the hope that the healthy phenotype will be transferred. This has been demonstrated in many rodent studies. Among the first was a study from Bäckhed et al., in which microbiota transferred from conventional mice into germ-free mice resulted in a 60% increase in body fat content and insulin resistance within 14 days despite reduced food intake (87). In humans, allogenic (but not autologous) transfer of microbiota from lean donors to obese individuals with MetS led to an increase in insulin sensitivity within 6 weeks, as well as an increase in butyrate-producing bacteria (88, 89). However, in a follow-up study that extended the observation period to 18 weeks after FMT, the gut microbiota composition and insulin sensitivity returned to baseline levels (89).
These data suggest that a metabolically healthy microbiome may exist, and that the associated phenotype can be transferred, via the microbiome, to individuals with MetS. Finally, some studies have emerged looking at whether the beneficial effects of pharmacological treatment for dysmetabolism could be transferred to individuals with MetS. Metformin, for example, can promote blooms of Akkermansia muciniphila, typically regarded as a beneficial bacterium, through an unknown mechanism (90, 91). When the metformin-treated human microbiome was transplanted into germ-free mice with glucose intolerance, the observed glucose defects were corrected (92). As compelling as these data may be, more studies with larger sample sizes and longer duration are required to determine the long-term stability of donor engraftment and associated phenotypes.
Conclusions
The factors contributing to MetS are the result of complex host intrinsic factors such as genetics and the gut microbiome, and extrinsic factors such as diet and lifestyle. While genetics are essentially fixed in time, the gut microbiome affords an opportunity to manipulate how the body processes external cues and potentially mitigate risk for MetS. Similarly, the data presented here also show that interactions of certain members of the gut microbiota community can also process external cues in a way that contributes to chronic low-grade inflammation, obesity, hyperglycemia, and dyslipidemia. Interestingly, dietary factors have been most widely implicated in causing metabolic defects, and also are accepted as the primary driver of our gut microbiota composition. The exquisite sensitivity of the gut microbiome not only to amount of food, but to dietary composition, acting as dietary biosensors for the host, makes it nearly impossible to divorce the role of the gut microbiome from considerations of how food choices affect host metabolism. However, additional environmental cues such as shifts in light/dark cycles leading to circadian disruption can now be seen to disrupt the natural diurnal variation in the gut microbiota, which also has an impact on host metabolism. However, all the data to date still show that these effects are worsened if combined with a high-fat or Western-type diet.
The microbiome field is still in its infancy compared with the long-standing study of endocrinology and metabolic diseases. Therefore, this discordance in the amount of data available on the microbiome’s role in metabolic diseases should elicit hope rather than frustration, as it only suggests that many more avenues of discovery still remain.
Address correspondence to: Suzanne Devkota, 110 George Burns Road, Davis Building Room 4011, Los Angeles, California 90048, USA. Phone: 310.423.4325; Email: Suzanne.Devkota@cshs.org.
Version 1. 10/01/2019
Print issue publication
Footnotes
Conflict of interest: SD is a consultant for the Janssen Human Microbiome Institute and a named inventor on a patent application related to intestinal microbiota.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4050–4057.https://doi.org/10.1172/JCI129194.
Contributor Information
Kruttika Dabke, Email: Kruttika.Dabke@cshs.org.
Gustaf Hendrick, Email: Gustaf.Hendrick@cshs.org.
Suzanne Devkota, Email: Suzanne.Devkota@cshs.org.
References
- 1.Sarafidis PA, Nilsson PM. The metabolic syndrome: a glance at its history. J Hypertens. 2006;24(4):621–626. doi: 10.1097/01.hjh.0000217840.26971.b6. [DOI] [PubMed] [Google Scholar]
- 2.Miranda PJ, DeFronzo RA, Califf RM, Guyton JR. Metabolic syndrome: definition, pathophysiology, and mechanisms. Am Heart J. 2005;149(1):33–45. doi: 10.1016/j.ahj.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. doi: 10.1007/s11906-018-0812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 5.Cani PD, Neyrinck AM, Maton N, Delzenne NM. Oligofructose promotes satiety in rats fed a high-fat diet: involvement of glucagon-like Peptide-1. Obes Res. 2005;13(6):1000–1007. doi: 10.1038/oby.2005.117. [DOI] [PubMed] [Google Scholar]
- 6. Murphy D. Concepts of disease and health. In: Zalta EN, ed. Stanford Encyclopedia of Philosophy. Stanford, California, USA: Metaphysics Research Lab, Center for the Study of Language and Information, Stanford University; 2015. [Google Scholar]
- 7.Schwartz M. The life and works of Louis Pasteur. J Appl Microbiol. 2001;91(4):597–601. doi: 10.1046/j.1365-2672.2001.01495.x. [DOI] [PubMed] [Google Scholar]
- 8.Bastian HC. The germ-theory of disease: being a discussion of the relation of bacteria and allied organisms to virulent inflammations and specific contagious fevers. Br Med J. 1875;1(745):469–476. doi: 10.1136/bmj.1.745.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blevins SM, Bronze MS. Robert Koch and the ‘golden age’ of bacteriology. Int J Infect Dis. 2010;14(9):e744–e751. doi: 10.1016/j.ijid.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(suppl 2):S274–S276. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- 11.Greenblum S, Carr R, Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160(4):583–594. doi: 10.1016/j.cell.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vatanen T, et al. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat Microbiol. 2019;4(3):470–479. doi: 10.1038/s41564-018-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd-Price J, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550(7674):61–66. doi: 10.1038/nature23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977;74(11):5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A. 1985;82(20):6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med. 1992;327(5):293–301. doi: 10.1056/NEJM199207303270501. [DOI] [PubMed] [Google Scholar]
- 17.Lederberg J, McCray AT. ’Ome sweet ’omics — a genealogical treasury of words. Scientist. 2001;15(7):8 [Google Scholar]
- 18.Hill JO. Understanding and addressing the epidemic of obesity: an energy balance perspective. Endocr Rev. 2006;27(7):750–761. doi: 10.1210/er.2006-0032. [DOI] [PubMed] [Google Scholar]
- 19.Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief. 2017;(288):1–8. [PubMed] [Google Scholar]
- 20.Beli E, et al. Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in db/db mice. Diabetes. 2018;67(9):1867–1879. doi: 10.2337/db18-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 22.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10–/– mice. Nature. 2012;487(7405):104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan Y, et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut. 2019;68(8):1417–1429. doi: 10.1136/gutjnl-2018-317609. [DOI] [PubMed] [Google Scholar]
- 25.Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529(7585):212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai MS, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167(5):1339–1353.e21. doi: 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chassaing B, Van de Wiele T, De Bodt J, Marzorati M, Gewirtz AT. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut. 2017;66(8):1414–1427. doi: 10.1136/gutjnl-2016-313099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suez J, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514(7521):181–186. doi: 10.1038/nature13793. [DOI] [PubMed] [Google Scholar]
- 29.Desbois AP, Smith VJ. Antibacterial free fatty acids: activities, mechanisms of action and biotechnological potential. Appl Microbiol Biotechnol. 2010;85(6):1629–1642. doi: 10.1007/s00253-009-2355-3. [DOI] [PubMed] [Google Scholar]
- 30.Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Bäckhed F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015;22(4):658–668. doi: 10.1016/j.cmet.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One. 2013;8(8):e71108. doi: 10.1371/journal.pone.0071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yassour M, et al. Sub-clinical detection of gut microbial biomarkers of obesity and type 2 diabetes. Genome Med. 2016;8(1):17. doi: 10.1186/s13073-016-0271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Depommier C, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25(7):1096–1103. doi: 10.1038/s41591-019-0495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ussar S, et al. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015;22(3):516–530. doi: 10.1016/j.cmet.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujisaka S, et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J Clin Invest. 2016;126(12):4430–4443. doi: 10.1172/JCI86674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright KP, McHill AW, Birks BR, Griffin BR, Rusterholz T, Chinoy ED. Entrainment of the human circadian clock to the natural light-dark cycle. Curr Biol. 2013;23(16):1554–1558. doi: 10.1016/j.cub.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badia P, Myers B, Boecker M, Culpepper J, Harsh JR. Bright light effects on body temperature, alertness, EEG and behavior. Physiol Behav. 1991;50(3):583–588. doi: 10.1016/0031-9384(91)90549-4. [DOI] [PubMed] [Google Scholar]
- 39.Vandewalle G, Maquet P, Dijk DJ. Light as a modulator of cognitive brain function. Trends Cogn Sci (Regul Ed) 2009;13(10):429–438. doi: 10.1016/j.tics.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Broussard JL, Van Cauter E. Disturbances of sleep and circadian rhythms: novel risk factors for obesity. Curr Opin Endocrinol Diabetes Obes. 2016;23(5):353–359. doi: 10.1097/MED.0000000000000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann Intern Med. 2012;157(8):549–557. doi: 10.7326/0003-4819-157-8-201210160-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pamidi S, et al. Obstructive sleep apnea in young lean men: impact on insulin sensitivity and secretion. Diabetes Care. 2012;35(11):2384–2389. doi: 10.2337/dc12-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broussard JL, et al. Sleep restriction increases free fatty acids in healthy men. Diabetologia. 2015;58(4):791–798. doi: 10.1007/s00125-015-3500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013;153(4):812–827. doi: 10.1016/j.cell.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 45.Thaiss CA, et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell. 2016;167(6):1495–1510.e12. doi: 10.1016/j.cell.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science. 2017;357(6354):912–916. doi: 10.1126/science.aan0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thaiss CA, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell. 2014;159(3):514–529. doi: 10.1016/j.cell.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 48.Zarrinpar A, Chaix A, Yooseph S, Panda S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014;20(6):1006–1017. doi: 10.1016/j.cmet.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leone V, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17(5):681–689. doi: 10.1016/j.chom.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jellinger PS, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Dyslipidemia and Prevention of Cardiovascular Disease - Executive Summary. Endocr Pract. 2017;23(4):479–497. doi: 10.4158/EP171764.GL. [DOI] [PubMed] [Google Scholar]
- 51.Matey-Hernandez ML, Williams FMK, Potter T, Valdes AM, Spector TD, Menni C. Genetic and microbiome influence on lipid metabolism and dyslipidemia. Physiol Genomics. 2018;50(2):117–126. doi: 10.1152/physiolgenomics.00053.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. 2015;11(10):577–591. doi: 10.1038/nrendo.2015.128. [DOI] [PubMed] [Google Scholar]
- 53.Chambers ES, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. 2015;64(11):1744–1754. doi: 10.1136/gutjnl-2014-307913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zambell KL, Fitch MD, Fleming SE. Acetate and butyrate are the major substrates for de novo lipogenesis in rat colonic epithelial cells. J Nutr. 2003;133(11):3509–3515. doi: 10.1093/jn/133.11.3509. [DOI] [PubMed] [Google Scholar]
- 55.Collins JM, et al. De novo lipogenesis in the differentiating human adipocyte can provide all fatty acids necessary for maturation. J Lipid Res. 2011;52(9):1683–1692. doi: 10.1194/jlr.M012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathewson ND, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. 2016;17(5):505–513. doi: 10.1038/ni.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dillon SM, et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS. 2017;31(4):511–521. doi: 10.1097/QAD.0000000000001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segain JP, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47(3):397–403. doi: 10.1136/gut.47.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Vadder F, et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014;156(1–2):84–96. doi: 10.1016/j.cell.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Fechner A, Kiehntopf M, Jahreis G. The formation of short-chain fatty acids is positively associated with the blood lipid-lowering effect of lupin kernel fiber in moderately hypercholesterolemic adults. J Nutr. 2014;144(5):599–607. doi: 10.3945/jn.113.186858. [DOI] [PubMed] [Google Scholar]
- 61.Perry RJ, et al. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature. 2016;534(7606):213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown JM, Hazen SL. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu Rev Med. 2015;66:343–359. doi: 10.1146/annurev-med-060513-093205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao HB, Sun ZL, Zhang HB, Zhang DS. Berberine inhibits dyslipidemia in C57BL/6 mice with lipopolysaccharide induced inflammation. Pharmacol Rep. 2012;64(4):889–895. doi: 10.1016/S1734-1140(12)70883-6. [DOI] [PubMed] [Google Scholar]
- 64.Lassenius MI, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34(8):1809–1815. doi: 10.2337/dc10-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang WH, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu W, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bennett BJ, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mudaliar S, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574–582.e1. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 71.Neuschwander-Tetri BA, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Larsen GL, Henson PM. Mediators of inflammation. Annu Rev Immunol. 1983;1:335–359. doi: 10.1146/annurev.iy.01.040183.002003. [DOI] [PubMed] [Google Scholar]
- 73.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 74.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J Clin Invest. 1995;95(5):2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi: 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- 76.O’Rourke RW, et al. Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells. Diabetologia. 2011;54(6):1480–1490. doi: 10.1007/s00125-011-2103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92(3):1023–1033. doi: 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- 79.Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105(2):141–150. doi: 10.1016/j.diabres.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 80.Cai D, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vaziri ND, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83(2):308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 82.Byndloss MX, et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357(6351):570–575. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muccioli GG, et al. The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol. 2010;6:392. doi: 10.1038/msb.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geurts L, et al. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front Microbiol. 2011;2:149. doi: 10.3389/fmicb.2011.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thaiss CA, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359(6382):1376–1383. doi: 10.1126/science.aar3318. [DOI] [PubMed] [Google Scholar]
- 86.Cammarota G, et al. Randomised clinical trial: faecal microbiota transplantation by colonoscopy vs. vancomycin for the treatment of recurrent Clostridium difficile infection. Aliment Pharmacol Ther. 2015;41(9):835–843. doi: 10.1111/apt.13144. [DOI] [PubMed] [Google Scholar]
- 87.Bäckhed F, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vrieze A, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(4):913–916.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 89.Kootte RS, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 2017;26(4):611–619.e6. doi: 10.1016/j.cmet.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 90.Shin NR, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63(5):727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- 91.de la Cuesta-Zuluaga J, et al. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care. 2017;40(1):54–62. doi: 10.2337/dc16-1324. [DOI] [PubMed] [Google Scholar]
- 92.Wu H, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23(7):850–858. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]