

Abstract
In a society where physical activity is limited and food supply is abundant, metabolic diseases are becoming a serious epidemic. Metabolic syndrome (MetS) represents a cluster of metabolically related symptoms such as obesity, hypertension, dyslipidemia, and carbohydrate intolerance, and significantly increases type 2 diabetes mellitus risk. Insulin resistance and hyperinsulinemia are consistent characteristics of MetS, but which of these features is the initiating insult is still widely debated. Regardless, both of these conditions trigger adverse responses from the pancreatic β cell, which is responsible for producing, storing, and releasing insulin to maintain glucose homeostasis. The observation that the degree of β cell dysfunction correlates with the severity of MetS highlights the need to better understand β cell dysfunction in the development of MetS. This Review focuses on the current understanding from rodent and human studies of the progression of β cell responses during the development of MetS, as well as recent findings addressing the complexity of β cell identity and heterogeneity within the islet during disease progression. The differential responses observed in β cells together with the heterogeneity in disease phenotypes within the patient population emphasize the need to better understand the mechanisms behind β cell adaptation, identity, and dysfunction in MetS.
Introduction
Nutrient excess and sedentary behaviors of our modern society are forerunners of metabolic syndrome (MetS), a condition that represents a cluster of metabolically related symptoms that can have life-threatening consequences (1). The primary characteristics of MetS are central obesity, hypertension, dyslipidemia, and carbohydrate intolerance, and the presence of at least three of these risk factors constitutes MetS; however, a single precise definition of MetS and the contribution of the underlying components of MetS remains highly debated (2–4). Notably, individuals with MetS have significantly increased risk for type 2 diabetes mellitus (T2DM), independent of many other risk factors (5). Insulin resistance, or the inability of insulin to optimally stimulate glucose uptake into the body’s cells, and hyperinsulinemia both contribute to MetS, but which of these conditions is the primary driver of the syndrome has been debated for decades. The prevailing view is that insulin resistance causes elevation of plasma glucose levels, which promotes increased demand on pancreatic β cells to produce and secrete more insulin (6). This compensatory hyperinsulinemic response by the β cells is multifactorial and initially restores euglycemia in the prediabetic state; however, chronic exposure to excess glucose and lipids eventually leads to β cell dysfunction and/or cell death to cause overt diabetes. During the past several years, however, the idea that insulin resistance precedes β cell dysfunction has been challenged, and there is a growing appreciation that, at least in a subset of patients, the contribution of islet β cell hyperresponsiveness is a primary event in the development of carbohydrate intolerance (7, 8). Furthermore, there is mounting evidence that the hyperinsulinemic state contributes to some of the other disorders associated with MetS, including cardiovascular disease, nonalcoholic fatty liver disease, and polycystic ovarian disease (8). These findings suggest that understanding β cell dysfunction in MetS may inform novel approaches to treating T2DM, cardiovascular disease, and other associated MetS complications. In this Review, we will discuss the well-characterized mechanisms that contribute to disease-mediated β cell dysfunction and death, as well as potential alternative β cell adaptive responses to external stressors associated with MetS based on recent studies in mice and evidence from human cadaveric pancreas tissue.
The pancreatic islet
There are four hormone-producing endocrine cell populations within the adult pancreatic islets of Langerhans: α, β, δ, and PP (pancreatic polypeptide) cells. Islet β cells are defined by their ability to produce, store, and secrete insulin in response to nutrients such as glucose, lipids, and a subset of amino acids. The α and δ cells secrete the hormones glucagon and somatostatin, respectively, to achieve a glucose-homeostatic condition (reviewed in ref. 9). The β cell is exquisitely sensitive to the nutrient environment and can respond to extremely small changes in blood glucose concentrations (between 4.5 and 8 mM), triggering significant changes in insulin secretion within just minutes through the complex process of stimulus-secretion coupling (10). However, because the β cell is fine-tuned for acute fluctuations in nutrient concentrations, chronic exposure to elevated levels of glucose and free fatty acids, as seen in MetS, results in progressive β cell adaptation and failure. As discussed below, β cell response to physiologic and pathophysiologic states of nutrient excess can occur through several mechanisms, including adaptive changes in β cell mass and function. Furthermore, β cell adaptations can occur both prior to and in response to MetS. Both rodent and human studies have contributed to the evolution of our understanding of these processes.
β Cell responses in MetS
Metabolic syndrome presents a set of unique stressors to the β cell, including elevated glucose, increased free fatty acids, and inflammation (11–13). In this adverse environment, β cells initially mount a compensatory response to ramp up β cell functional capacity and insulin secretion to meet the elevated metabolic demand. In some individuals, this initial adaptive phase of insulin hypersecretion triggers insulin resistance that initiates a vicious cycle of enhanced insulin production followed by a progressive loss of β cell mass and/or function to result in impaired β cell metabolic function, depleted cellular defense mechanisms, and disrupted insulin secretory capacity (Figure 1 and refs. 11, 14). On the other hand, in some individuals, excess nutrient conditions first impair insulin responses in peripheral tissues, such as the liver, to initiate the insulin-resistant environment that will subsequently induce a compensatory response by β cells. Ultimately, this again triggers enhanced insulin secretion that can progress to β cell dysfunction in an environment of chronic metabolic demand (Figure 1 and refs. 2, 3). How and why these distinct scenarios are initiated is not well understood, but certainly partially depends on genetic differences between individuals. For example, β cell defects tend to be more important in Asian populations in contrast to the predominance of insulin resistance in White populations (15–17). Ultimately, however, regardless of whether the initial trigger is nutrient excess, insulin resistance, or both, the transition from an adaptive β cell response to a pathological β cell response represents a critical step in the progression to diabetes. For this reason, a greater understanding of the intracellular processes responsible for the β cell’s shift from adaptive to maladaptive responses will facilitate the development of therapeutic interventions.
Figure 1. β Cell compensation and dysfunction in MetS and T2DM.
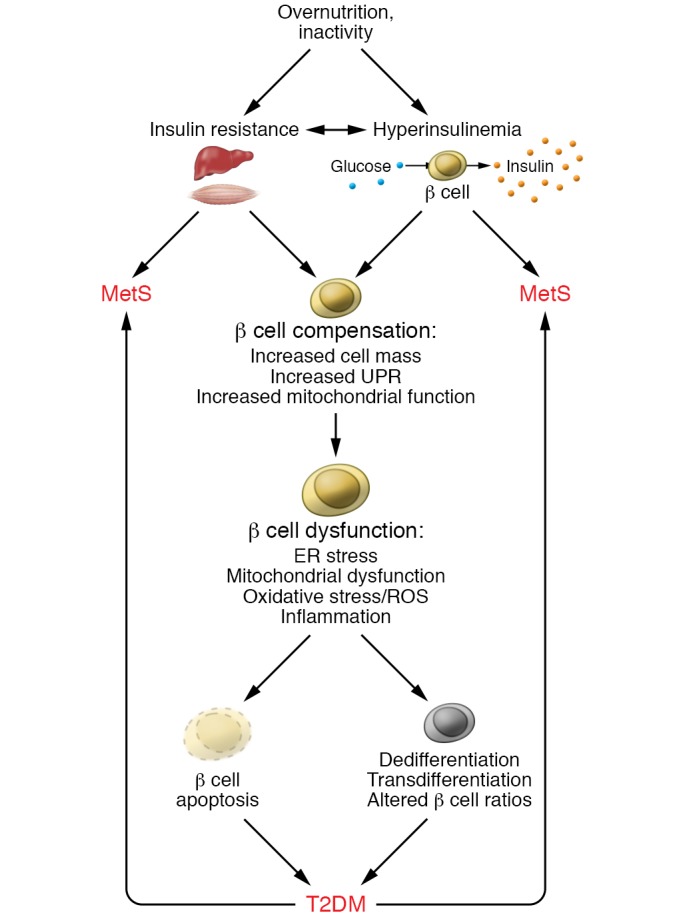
A lifestyle of overnutrition and/or inactivity can give rise to an insulin-resistant condition and/or induce insulin hypersecretion from the pancreatic β cell. In each case, a feedback cycle can be established to exacerbate insulin resistance and increase insulin secretion; both conditions can trigger MetS and its related complications. Initially, β cells are able to functionally compensate for the increased metabolic demand by increasing β cell mass, inducing an unfolded protein response (UPR) and improving mitochondrial function. However, over time, in a subset of individuals, β cell compensation cannot be sustained, and β cells become dysfunctional, presenting with ER stress, mitochondrial dysfunction, oxidative stress, and inflammation. Ultimately, the stressed β cells undergo cell death, dedifferentiation, transdifferentiation, or phenotypic alterations that compromise function. Disrupted β cell function can feed back to exacerbate MetS.
Changes in β cell mass
One of the most well-studied features of β cell adaptation in MetS is β cell proliferation. Studies in rodent models have provided the most compelling data for the role of adaptive β cell mass in environmental conditions mimicking MetS. For example, mice given corticosterone in their drinking water for 5 weeks displayed many features of MetS, including dyslipidemia, insulin resistance, glucose intolerance, and hypertension, and displayed increased islet volume due to β cell proliferation (18). Similarly, in the relatively diabetes-resistant C57BL/6J leptin-deficient (ob/ob) mouse model of obesity, there was a 3.6-fold increase in β cell volume (19). Interestingly, when the leptin mutation is induced in the BTBR (Black and Tan Brachyury) background, the mice are unable to increase β cell proliferation and progress to diabetes (20). The observation that adaptive β cell proliferation plays an essential role in delaying and/or preventing diabetes has been validated by several additional studies of genetic mouse models with impaired β cell replication that progress more rapidly to diabetes in response to high-fat diet (HFD) (21, 22).
Although adaptive increases in β cell proliferation have been well documented in insulin-resistant and HFD-fed rodent models, the extent of these early adaptive changes in β cell mass is more difficult to characterize in humans, in whom there are no available technologies to accurately track longitudinal changes in β cell mass. Currently, measures of human β cell mass are performed at static time points and rely predominantly on the availability of cadaveric tissue samples. One of the earliest studies to examine β cell mass compared autopsy tissues from approximately 20 obese individuals versus lean controls to demonstrate that obese individuals had higher β cell mass than the control group (23). Since that time, several additional cadaveric studies have suggested that the ability to increase β cell mass in the context of obesity and insulin resistance is necessary to prevent the development of diabetes (24, 25). These analyses were reinforced by a recent autopsy study that reported that human obese, nondiabetic patients have a significantly higher β cell mass than individuals with diabetes who are either lean or obese (26). Furthermore, a study that was able to evaluate pancreatic samples from nondiabetic insulin-resistant subjects who had undergone pancreatoduodenectomy also identified a marked increase in β cell numbers (27). However, these human studies remain somewhat controversial. For example, Butler et al. (28) concluded that increased β cell apoptosis was the primary mechanism underlying the development of T2DM, while studies from human cadaveric islet samples have reported substantial variation (0% to 63%) in β cell mass reduction in patients with a T2DM diagnosis (28–31).
Compensatory metabolic adaptation of β cells to stress
In addition to the adaptive β cell proliferative response that occurs in a high-nutrient environment, β cells adapt to metabolic challenges by employing intrinsic mechanisms to enhance β cell performance. Increased insulin production/secretion, upregulation of glucose metabolism pathways, and induction of the adaptive unfolded protein response (UPR) have all been shown to transiently increase in conditions of overnutrition and insulin resistance as a defense mechanism to preserve β cell function and survival (32). Studies in Wistar rats demonstrated that nutrient-challenged β cells increased their expression of the glucose transporter GLUT2 and enhanced glucokinase activity to promote insulin hypersecretion (33, 34). Islets of Zucker fatty (ZF) rats also displayed 3- to 10-fold increases in stimulated insulin secretion due to increased glucose utilization and oxidation (35). This study further demonstrated that β cell mitochondrial metabolism was elevated as a result of increased flux through pyruvate carboxylase and the malate-pyruvate and citrate-pyruvate shuttles. In humans, similar compensatory increases in augmented insulin secretion have been reported in obese, nondiabetic patients and individuals with prediabetes (36, 37). In addition to elevated insulin secretion, a study of HFD-fed C57BL/6NTac (B6N) mice demonstrated hyperexpression of genes known to promote β cell function and differentiation, including many key β cell transcriptional factors (11). The same study identified the transient upregulation of UPR genes to protect against decompensating ER stress responses. In an analogous proteomics study, transient increases in molecules involved in protein synthesis and folding and cell survival were also observed (38). Furthermore, an interesting study by Sharma et al. (39) suggested that induction of UPR functioned as a sensor of insulin demand and induced β cell proliferation through the activation of ATF6 in mouse and human islets. These studies all demonstrate the remarkable ability of β cells to adapt to increased metabolic demand and suggest that interventions that promote optimal β cell physiology in the face of increased insulin demand are a potential therapeutic strategy.
While increased insulin secretion allows the β cell to respond to excess metabolic demand, prolonged elevated circulating insulin can impact insulin sensing in peripheral tissues, such as liver and muscle. Although it is clear that insulin resistance is a central component of MetS, there is mounting evidence that prolonged insulin hypersecretion could be an initiating event in the syndrome. Several studies in mice and humans have demonstrated that prolonged elevated insulin secretion in excess nutrient environments precedes and promotes insulin resistance (40, 41). For example, human cross-sectional studies by Ferrannini et al. (42) demonstrated that there were stepwise increases in basal insulin secretion and a decreased proportion of glucose-stimulated insulin secretion as individuals moved from normal glucose tolerance to T2DM (Figure 2). Furthermore, recent data in youth with prediabetes and T2DM from the NIH Restored Insulin Secretion (RISE) study demonstrated that insulin hypersecretion in youth predicted progression to diabetes. Interestingly, this pattern differs in adults who have T2DM or are at risk of developing T2DM (43, 44). These human studies strongly support an initiating role for insulin hypersecretion in the development of MetS. In several rodent models, there is also evidence for transient increases in β cell proliferation and intrinsic β cell functional adaptations in response to increased insulin demand, before substantial changes in insulin resistance can be detected. For example, deletion of the insulin and IGF-1 receptors from β cells or manipulation of IRS1/2 and AKT isoforms in mice triggers insulin resistance and, in some instances, diabetes (29, 45–48). Furthermore, mutations in IRS1 have been linked to β cell dysfunction and association with T2DM in several human populations (49, 50). Regardless of the initiating event, there is considerable evidence that increased functional capacity is an essential component of β cell adaptation in MetS that is triggered to delay and/or prevent progression to diabetes.
Figure 2. Fasting and glucose-stimulated insulin secretion in the progression to T2DM.
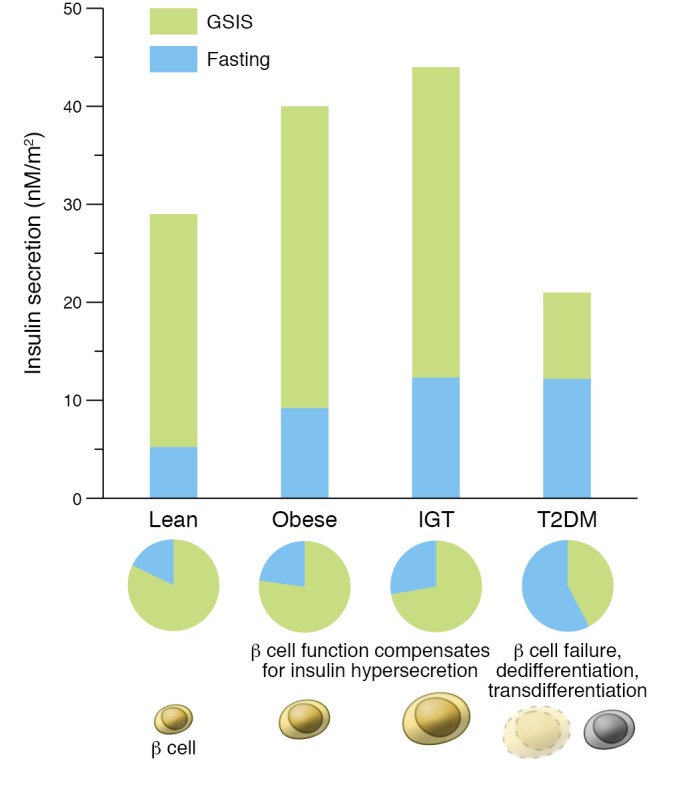
Fasting insulin secretion (blue) increases as people progress from normal glucose tolerance to T2DM; in contrast, glucose-stimulated insulin secretion (GSIS; green) represents a lower percentage of overall insulin secretion in impaired glucose tolerance (IGT) and less than half of insulin secretion in T2DM. Adapted with permission from Frontiers in Endocrinology (41) based on data in ref. 42.
β Cell dysfunction in MetS
Despite evidence that β cells can mount a compensatory response to insulin resistance and overnutrition, in individuals who develop T2DM the ability to compensate is transient. Over time, production of large amounts of insulin by the compensating β cells exerts continuous demand on the ER for proper protein synthesis, folding, trafficking, and secretion. Ultimately, β cells are unable to sustain the increased workload, and the initial adaptive responses become progressively maladaptive. β Cell dysfunction has been extensively studied in MetS, insulin-resistant conditions, and T2DM, both in rodent models and in humans, to reveal similar underlying molecular defects (reviewed in refs. 6, 51). In conditions of chronic nutrient exposure, sustained overproduction and secretion of insulin strains the folding capacity of the ER, and misfolded or unfolded proteins accumulate in the ER lumen, resulting in ER stress and activation of β cell apoptosis pathways (reviewed in refs. 52, 53). Consistently, ER stress markers are commonly elevated in pancreatic islets of animals exposed to HFD (32). Studies in islets from human T2DM patients also show a doubling of the ER size compared with controls, indicative of the presence of ER stress responses (54). Increased demand on the β cell also results in increased flux through mitochondria. Eventually, the overworked β cell mitochondria also become dysfunctional, which impairs the coupling of glucose metabolism and insulin secretion. This then triggers oxidative stress and higher levels of reactive oxygen species, which further worsens β cell function and eventually promotes β cell exhaustion and loss (reviewed extensively in refs. 55, 56).
Paradigms of β cell loss
Traditionally, it has been assumed that loss of insulin-producing cells in conditions causing β cell exhaustion was caused by a decrease in β cell numbers through apoptosis and other mechanisms of β cell death. However, our understanding of β cell biology has greatly expanded in recent years, and the simplistic view that β cell death is the primary outcome of β cell exhaustion and dysfunction has evolved considerably. Notably, there is growing evidence that β cells can respond to the environmental stressors by undergoing dedifferentiation and/or transdifferentiation, such that there is minimal alteration in islet cell mass (reviewed in refs. 57). These phenomena have been most extensively explored in rodent models; however, corroborative evidence from analysis of human cadaveric pancreatic tissue is also emerging (Figure 3).
Figure 3. Fates of overexerted β cells.
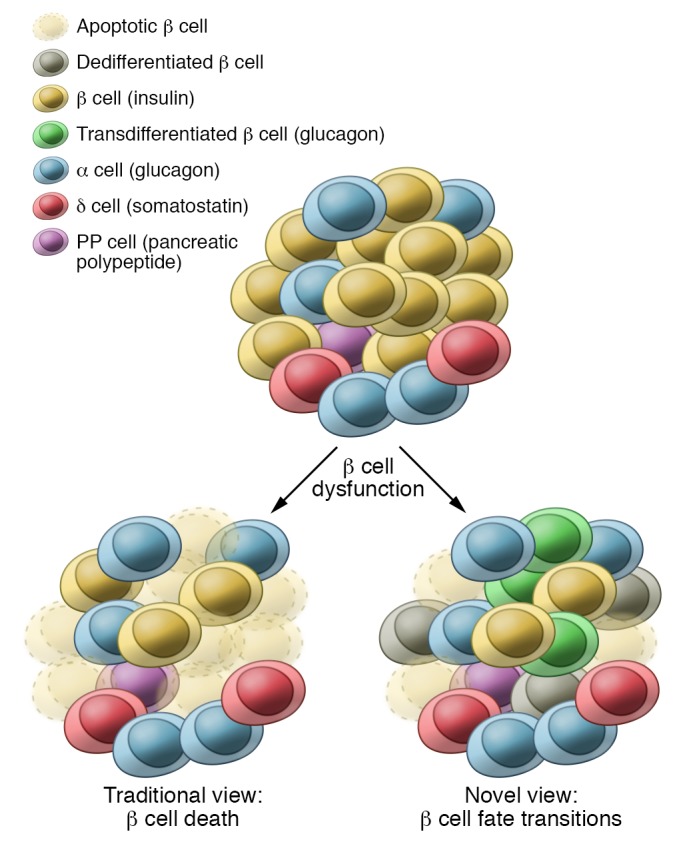
Upon overexertion, in many animal models and humans, β cells initially undergo functional compensation, which can be followed by a pathogenic response. In the past, overexerted β cells were thought to predominantly undergo cell death. More recently, there has been evidence from animal models and human pancreatic tissue that β cells can respond by undergoing dedifferentiation, transdifferentiation, or β cell subtype transitions.
β Cell dedifferentiation
One of the first studies to challenge the paradigm of β cell death was the Weir group’s demonstration that chronic mild to severe hyperglycemia in pancreatectomized rats triggered loss of β cell differentiation (58). This observation was reinforced in a genetic model when Talchai et al. (59) generated a β cell–specific deletion of the transcription factor FoxO1 (FoxO1 βCKO mice), a major target of insulin signaling and regulator of metabolic homeostasis in many tissues. Similar to the previous studies, FoxO1 βCKO mice that were subjected to pathophysiologic models of β cell stress, such as aging and multiple pregnancies, had impaired glucose tolerance, reduced insulin secretion, and a 30% decrease in β cell mass. However, lineage tracing revealed that the mutant β cells were not lost through apoptosis, but became dedifferentiated to revert to a more progenitor-like state. This observation has been bolstered by a number of studies that have demonstrated that adoption of a dedifferentiated phenotype was a protective mechanism to support β cell survival under conditions of stress (60). There have since been many additional reports of β cell dedifferentiation in response to genetic and environmental perturbations — in both mice and humans. Inducible ablation of the transcription factor LDB1 in mature β cells also resulted in impaired insulin secretion and glucose homeostasis due to a reduction in β cell identity genes and induction of the endocrine progenitor marker Neurogenin3 (NEUROG3), again suggesting that β cells have the ability to become dedifferentiated (61). Using the well-established T2D mouse model db/db, which has a mutation in the leptin receptor, Neelankal et al. used transcriptome analysis of 12-week-old islets from WT, db heterozygotes, and db/db mutants to demonstrate that dedifferentiation was also occurring in a model of insulin resistance (62). Evidence of dedifferentiated β cells, defined by the presence of hormone-negative endocrine cells, was also reported in a study of human T2D patients with adequate glucose control (63). However, without lineage analyses and more extensive progenitor marker analyses, dedifferentiation is more difficult to prove in humans. To overcome this challenge, Diedisheim et al. (64) used the functional human β cell line EndoC-βH1 to show that treatment with the growth factors FGF1 and FGF2 induces β cell dedifferentiation and that this process was reversible under specific conditions. They were also able to identify novel human β cell dedifferentiation markers SOX9, HES1, MYC, PYY, GAST, and NEUROG3 in addition to the previously reported ALDH1A3 marker described by Cinti et al. (65). In summary, there is considerable evidence for the occurrence of dedifferentiation in stressed rodent and human β cells as a potential adaptive mechanism. Although in many instances, dedifferentiation is induced in conditions of prolonged insulin demand, the precise molecular signals that trigger a dedifferentiation pathway are still not well characterized (60).
β Cell transdifferentiation
In addition to evidence of dedifferentiation, several groups have demonstrated that β cells can lose their identity by being converted to different hormone-expressing cell types. Gutiérrez et al. (66) demonstrated that β cell–specific deletion of Nkx2.2, a transcription factor essential for β cell development and function, led to both partial and complete transdifferentiation of β cells; a subset of β cells coexpressed insulin and the other islet endocrine hormones, whereas genetic lineage tracing revealed that a subset of β cells had lost their β cell characteristics and were reprogrammed to α or δ cells. Interestingly, unlike FoxO1 mutations, there was no evidence that these cells first underwent a dedifferentiation event. Similarly, β cell–specific deletion of Pax6, a transcription factor critical for the maintenance of β cell identity, resulted in direct β cell transdifferentiation to markedly upregulate expression of ghrelin (67), a hormone that is not normally expressed in the adult pancreas. Moreover, deletion of Pdx1 from adult β cells resulted in loss of β cell identity markers that was accompanied by expression of glucagon and acquisition of α cell characteristics (68). Although it is not possible to perform similar genetic lineage tracing analysis in vivo in humans, long-term culture of human islets suggested there was spontaneous β cell transdifferentiation into the ductal lineage, suggesting that human β cells might also have the ability to undergo reprogramming (69). Additionally, islet samples from T1D and T2D patients have provided evidence of the existence of insulin-producing β cells expressing multiple hormones, indicating their ability to at least partially lose their β cell identity (63). However, without lineage analyses, it is difficult to prove that the polyhormonal cells arose from monohormonal β cells.
β Cell plasticity
Evidence of β cell dedifferentiation and transdifferentiation in T2DM highlights the previously underappreciated plasticity of endocrine cells and suggests that β cells undergo a variety of changes in response to stress that are not limited to cell death. This represents a critical discovery that may allow for the development of alternative approaches to restore β cell function in MetS. A major question that remains is why β cells variably respond to environmental insults by dedifferentiating, transdifferentiating, or dying. In mouse models, the response may depend on the particular genetic disruption; however, this has yet to be resolved. Alternatively, the response may depend on the type of nutritive or environmental stress, or may be associated with particular islet dysfunctions, such as ER stress versus mitochondrial dysfunction. In humans, it is likely that the differential β cell responses have a genetic basis that perhaps has adapted and evolved in response to changing environmental conditions.
β Cell heterogeneity
An emerging concept to explain the differential responses of β cells is the existence of β cell functional heterogeneity within a single islet. Although this is a relatively old concept (70–72), technological advances have provided appreciable insight into the existence of different β cell subpopulations that are distinguished by function, morphology, location, expression profiles, and connectivity (reviewed in refs. 66, 73). A particularly seminal study identified the presence of four antigenically diverse β cell subpopulations, β1 through β4, within human islets (74). Interestingly, each subpopulation had distinct gene expression profiles and insulin secretion capabilities; the most abundant β1 population had the lowest basal insulin secretion, whereas the β4 population displayed the highest insulin secretion capabilities. Furthermore, in samples from T2DM patients versus matched controls, there were substantial increases in the high-secreting β4 population, without significant changes in the other subpopulations. β Cell heterogeneity has also been observed at the level of islet electrical excitability and connectivity. β Cell heterogeneity was shown to be required to accurately reproduce Ca++ wave propagation dynamics; loss of stimulus-induced insulin secretion coupling has been detected early in disease progression and is thought to be an important mechanism contributing to β cell dysfunction (75). In addition, a recent study identified a population of β cells termed “extreme” β cells located in the center of the islet. These cells were characterized by a distinct polarization pattern and higher proinsulin and ribosomal RNA content (76). Additional subpopulations of β cells, including “hub” β cells that demonstrate pacemaker activities to coordinate insulin response to glucose and “virgin” β cells, which are postulated to represent a population of immature β cells that form a neogenic niche at the periphery of the islet, have also been identified (77, 78); however, the extent to which these populations represent distinct β cell subtypes versus a phenotypic continuum of a homogeneous cell population remains to be seen. Furthermore, the functional importance of β cell heterogeneity remains to be addressed; however, it is possible that each β cell subtype’s differential response to increased insulin demand would determine whether a β cell dedifferentiates, becomes reprogrammed, or undergoes apoptosis in conditions of metabolic stress.
The reemergence of the concept of β cell heterogeneity has allowed us to move away from a simplistic view of β cells simultaneously responding in an identical way to metabolic insults. This has also impacted how we think about the ways in which β cell subpopulations mediate glucose homeostasis in health and disease. For example, immature proliferative β cells are more resistant to cell death, while increased metabolic and electrical activity can increase susceptibility to cell death (reviewed in ref. 75). Additionally, functionally mature β cells are more sensitive to the inflammatory environment and the effects of the cytokines IL-1β and IL-6 and therefore would be expected to fail earlier in disease as a result of ER stress. Ultimately, understanding the mechanisms behind the different functions and responses of β cells to disease will be crucial to our ability to prevent β cell loss and/or dysfunction in MetS.
Conclusions and perspectives
Based on the wealth of emerging information about the different β cell responses to increased nutrient stress in MetS, it is clear that understanding and targeting both the adaptive and pathogenic changes in β cell function is a rational approach for preventing the progression of MetS to T2DM. Earlier clinical reports and recent clinical deep phenotyping in the RISE study in adults and youth in prediabetes or early T2DM indicate a large spectrum of insulin secretory capacity, basal insulin secretion, and glucose-stimulated insulin secretion responses, as well as heterogeneous responses to interventions with insulin replacement, weight loss, and exercise (43, 44). Two recent studies provide clinical proof of concept that intervening at the behavioral level of MetS/prediabetes using short-term diet or exercise augments glucose-stimulated insulin secretion in human subjects (79, 80). These studies support the idea that early intervention is critical to our ability to prevent or delay the progression to T2DM, but they also highlight the heterogeneity of these responses within different patients and the need to better understand particular islet compositions, β cell population compositions, and the respective responses of these heterogeneous cell populations. Furthermore, there needs to be an increased emphasis on understanding the mechanisms of the early responses of β cells to the different environmental insults in the different ethnic populations. Being able to address these factors during the β cell compensation phase before dysfunction occurs (Figure 1) should be a primary focus of the field.
In addition to a greater appreciation for heterogeneity of β cell responses between individuals, as discussed in this Review, there is growing evidence of β cell plasticity and differential β cell responses to the different metabolic insults, suggesting that potential treatments should include protocols that protect and/or reestablish β cell identity, rather than focusing solely on β cell replacement therapies. The idea that endocrine cells, and specifically β cells, are an extremely heterogeneous and plastic population of cells should also affect our therapeutic approaches. For example, if we are able to determine why a certain subset of β cells responds to a particular insult while others do not, we can focus on enhancing or protecting the optimal β cell subset. Alternatively, we will need to better understand why there is evidence for dedifferentiation versus transdifferentiation and/or reprogramming, and whether these outcomes are linked to different β cell subpopulations or a specific metabolic insult. Increased knowledge about the specific β cell defect that needs to be corrected in an individual patient will influence the treatment options. In addition, it would be useful to identify biomarker combinations that would longitudinally predict the health and status of the entire β cell population as well as the different subsets of β cells as a potential indicator of what specific interventions should occur. While many of these studies have been pioneered in mice, there is a need for additional human studies to ensure a seamless transition from findings in mice to a treatment of human disease. Technological advances and greater access to human tissues, as well as improved in vitro cell reprogramming, are getting us closer to those goals. In conclusion, addressing and understanding the adaptations and subtle changes that β cells undergo in response to metabolic changes is an important area of focus that will give us a more complete set of customizable tools to use in response to MetS.
Acknowledgments
We acknowledge support from the P30DK048520 Colorado Nutrition Obesity Research Center (NORC) and a Children’s Diabetes Foundation ACORN grant (to LH); VA Merit (CX001532, BX002046), NIH UL1 TR002535, Center for Woman’s Health Research (to JEBR); and NIH R01 DK082590, U01 DK072504, R01 DK63016219, and R01 DK118155 (to LS).
Version 1. 08/19/2019
Electronic publication
Version 2. 10/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4001–4008.https://doi.org/10.1172/JCI129188.
References
- 1.Smith GI, Mittendorfer B, Klein S. Metabolically healthy obesity: facts and fantasies. J Clin Invest. 2019;129(10):3978–3989. doi: 10.1172/JCI129186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 3.Reaven GM. Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu Rev Med. 1993;44:121–131. doi: 10.1146/annurev.me.44.020193.001005. [DOI] [PubMed] [Google Scholar]
- 4.O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1–12. doi: 10.1111/obr.12229. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzo C, Okoloise M, Williams K, Stern MP, Haffner SM, San Antonio Heart Study The metabolic syndrome as predictor of type 2 diabetes: the San Antonio heart study. Diabetes Care. 2003;26(11):3153–3159. doi: 10.2337/diacare.26.11.3153. [DOI] [PubMed] [Google Scholar]
- 6.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corkey BE. Diabetes: have we got it all wrong? Insulin hypersecretion and food additives: cause of obesity and diabetes? Diabetes Care. 2012;35(12):2432–2437. doi: 10.2337/dc12-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res. 2019;16(2):118–127. doi: 10.1177/1479164119827611. [DOI] [PubMed] [Google Scholar]
- 9.Mastracci TL, Sussel L. The endocrine pancreas: insights into development, differentiation, and diabetes. Wiley Interdiscip Rev Dev Biol. 2012;1(5):609–628. doi: 10.1002/wdev.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heimberg H, De Vos A, Vandercammen A, Van Schaftingen E, Pipeleers D, Schuit F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993;12(7):2873–2879. doi: 10.1002/j.1460-2075.1993.tb05949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta D, et al. Temporal characterization of β cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J Biol Chem. 2017;292(30):12449–12459. doi: 10.1074/jbc.M117.781047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontés G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2010;1801(3):289–298. doi: 10.1016/j.bbalip.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 14.Halban PA, et al. β-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37(6):1751–1758. doi: 10.2337/dc14-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porte D. Mechanisms for hyperglycemia in the metabolic syndrome. The key role of beta-cell dysfunction. Ann N Y Acad Sci. 1999;892:73–83. doi: 10.1111/j.1749-6632.1999.tb07786.x. [DOI] [PubMed] [Google Scholar]
- 16.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46(1):3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 17.Yabe D, Seino Y, Fukushima M, Seino S. β cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Curr Diab Rep. 2015;15(6):602. doi: 10.1007/s11892-015-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fransson L, Franzén S, Rosengren V, Wolbert P, Sjöholm Å, Ortsäter H. β-Cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J Endocrinol. 2013;219(3):231–241. doi: 10.1530/JOE-13-0189. [DOI] [PubMed] [Google Scholar]
- 19.Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes. 2003;52(7):1716–1722. doi: 10.2337/diabetes.52.7.1716. [DOI] [PubMed] [Google Scholar]
- 20.Keller MP, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18(5):706–716. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Ouaamari A, et al. SerpinB1 promotes pancreatic β cell proliferation. Cell Metab. 2016;23(1):194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirakawa J, et al. Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation. Cell Metab. 2017;25(4):868–882.e5. doi: 10.1016/j.cmet.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogilvie RF. The islands of Langerhans in 19 cases of obesity. J Pathol Bacteriol. 1933;37(3):473–481. doi: 10.1002/path.1700370314. [DOI] [Google Scholar]
- 24.Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Survey and synthesis of pathology research. 1985;4(2):110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- 25.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 26.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mezza T, et al. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63(3):994–1007. doi: 10.2337/db13-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 29.Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia. 1983;24(5):366–371. doi: 10.1007/BF00251826. [DOI] [PubMed] [Google Scholar]
- 30.Maclean N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes. 1955;4(5):367–376. doi: 10.2337/diab.4.5.367. [DOI] [PubMed] [Google Scholar]
- 31.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45(1):85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 32.Oh YS, Bae GD, Baek DJ, Park EY, Jun HS. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front Endocrinol (Lausanne) 2018;9:384. doi: 10.3389/fendo.2018.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, Hosokawa H, Bumbalo LM, Leahy JL. Regulatory effects of glucose on the catalytic activity and cellular content of glucokinase in the pancreatic beta cell. Study using cultured rat islets. J Clin Invest. 1994;94(4):1616–1620. doi: 10.1172/JCI117503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larqué C, et al. Early endocrine and molecular changes in metabolic syndrome models. IUBMB Life. 2011;63(10):831–839. doi: 10.1002/iub.544. [DOI] [PubMed] [Google Scholar]
- 35.Liu YQ, Jetton TL, Leahy JL. β-Cell adaptation to insulin resistance. Increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic Zucker fatty rats. J Biol Chem. 2002;277(42):39163–39168. doi: 10.1074/jbc.M207157200. [DOI] [PubMed] [Google Scholar]
- 36.Chandrashekar L, Kumarit GR, Rajappa M, Revathy G, Munisamy M, Thappa DM. 25-hydroxy vitamin D and ischaemia-modified albumin levels in psoriasis and their association with disease severity. Br J Biomed Sci. 2015;72(2):56–60. doi: 10.1080/09674845.2015.11666797. [DOI] [PubMed] [Google Scholar]
- 37.Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81(2):442–448. doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Ouaamari A, et al. Compensatory islet response to insulin resistance revealed by quantitative proteomics. J Proteome Res. 2015;14(8):3111–3122. doi: 10.1021/acs.jproteome.5b00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma RB, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831–3846. doi: 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res. 2019;16(2):118–127. doi: 10.1177/1479164119827611. [DOI] [PubMed] [Google Scholar]
- 41.Erion K, Corkey BE. β-Cell failure or β-cell abuse? Front Endocrinol (Lausanne) 2018;9:532. doi: 10.3389/fendo.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. β-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90(1):493–500. doi: 10.1210/jc.2004-1133. [DOI] [PubMed] [Google Scholar]
- 43.RISE Consortium. Metabolic contrasts between youth and adults with impaired glucose tolerance or recently diagnosed type 2 diabetes: I. Observations using the hyperglycemic clamp. Diabetes Care. 2018;41(8):1696–1706. doi: 10.2337/dc18-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.RISE Consortium. Restoring Insulin Secretion (RISE): design of studies of β-cell preservation in prediabetes and early type 2 diabetes across the life span. Diabetes Care. 2014;37(3):780–788. doi: 10.2337/dc13-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet. 1999;23(1):32–40. doi: 10.1038/12631. [DOI] [PubMed] [Google Scholar]
- 46.Ueki K, et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet. 2006;38(5):583–588. doi: 10.1038/ng1787. [DOI] [PubMed] [Google Scholar]
- 47.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329–339. doi: 10.1016/S0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 48.Kitamura T, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110(12):1839–1847. doi: 10.1172/JCI16857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keshavarzi F, Golsheh S. IRS1- rs10498210 G/A and CCR5-59029 A/G polymorphisms in patients with type 2 diabetes in Kurdistan. Mol Genet Genomic Med. 2019;7(5):e631. doi: 10.1002/mgg3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodhini D, Radha V, Mohan V. Association study of IRS1 gene polymorphisms with type 2 diabetes in south Indians. Diabetes Technol Ther. 2011;13(7):767–772. doi: 10.1089/dia.2011.0017. [DOI] [PubMed] [Google Scholar]
- 51.Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human beta cell mass and function in diabetes: recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab. 2017;6(9):943–957. doi: 10.1016/j.molmet.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabhi N, Salas E, Froguel P, Annicotte JS. Role of the unfolded protein response in beta cell compensation and failure during diabetes. J Diabetes Res. 2014;2014:795171. doi: 10.1155/2014/795171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 54.Marchetti P, Bugliani M, De Tata V, Suleiman M, Marselli L. Pancreatic beta cell identity in humans and the role of type 2 diabetes. Front Cell Dev Biol. 2017;5:55. doi: 10.3389/fcell.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol Metab. 2012;23(9):477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501–518. doi: 10.1089/ars.2016.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Remedi MS, Emfinger C. Pancreatic β-cell identity in diabetes. Diabetes Obes Metab. 2016;18(suppl 1):110–116. doi: 10.1111/dom.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonas JC, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem. 1999;274(20):14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- 59.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol. 2018;236(2):R109–R143. doi: 10.1530/JOE-17-0516. [DOI] [PubMed] [Google Scholar]
- 61.Ediger BN, et al. LIM domain-binding 1 maintains the terminally differentiated state of pancreatic β cells. J Clin Invest. 2017;127(1):215–229. doi: 10.1172/JCI88016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neelankal John A, Ram R, Jiang FX. RNA-Seq analysis of islets to characterise the dedifferentiation in type 2 diabetes model mice db/db. Endocr Pathol. 2018;29(3):207–221. doi: 10.1007/s12022-018-9523-x. [DOI] [PubMed] [Google Scholar]
- 63.Sun J, et al. β-Cell dedifferentiation in patients with T2D with adequate glucose control and nondiabetic chronic pancreatitis. J Clin Endocrinol Metab. 2019;104(1):83–94. doi: 10.1210/jc.2018-00968. [DOI] [PubMed] [Google Scholar]
- 64.Diedisheim M, et al. Modeling human pancreatic beta cell dedifferentiation. Mol Metab. 2018;10:74–86. doi: 10.1016/j.molmet.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cinti F, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab. 2016;101(3):1044–1054. doi: 10.1210/jc.2015-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gutierrez GD, Gromada J, Sussel L. Heterogeneity of the pancreatic beta cell. Front Genet. 2017;8:22. doi: 10.3389/fgene.2017.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swisa A, et al. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J Clin Invest. 2017;127(1):230–243. doi: 10.1172/JCI88015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao T, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014;19(2):259–271. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmied BM, et al. Transdifferentiation of human islet cells in a long-term culture. Pancreas. 2001;23(2):157–171. doi: 10.1097/00006676-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 70.Hellerstrom C, Petersson B, Hellman B. Some properties of the B cells in the islet of Langerhans studied with regard to the position of the cells. Acta Endocrinol. 1960;34:449–456. doi: 10.1530/acta.0.xxxiv0449. [DOI] [PubMed] [Google Scholar]
- 71.Meda P, Denef JF, Perrelet A, Orci L. Nonrandom distribution of gap junctions between pancreatic beta-cells. Am J Physiol. 1980;238(3):C114–C119. doi: 10.1152/ajpcell.1980.238.3.C114. [DOI] [PubMed] [Google Scholar]
- 72.Meda P, Michaels RL, Halban PA, Orci L, Sheridan JD. In vivo modulation of gap junctions and dye coupling between B-cells of the intact pancreatic islet. Diabetes. 1983;32(9):858–868. doi: 10.2337/diab.32.9.858. [DOI] [PubMed] [Google Scholar]
- 73.Benninger RKP, Dorrell C, Hodson DJ, Rutter GA. The impact of pancreatic beta cell heterogeneity on type 1 diabetes pathogenesis. Curr Diab Rep. 2018;18(11):112. doi: 10.1007/s11892-018-1085-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorrell C, et al. Human islets contain four distinct subtypes of β cells. Nat Commun. 2016;7:11756. doi: 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benninger RK, et al. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca²+ wave dynamics. Biophys J. 2014;107(11):2723–2733. doi: 10.1016/j.bpj.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Farack L, et al. Transcriptional heterogeneity of beta cells in the intact pancreas. Dev Cell. 2019;48(1):115–125.e4. doi: 10.1016/j.devcel.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 77.van der Meulen T, et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metab. 2017;25(4):911–926.e6. doi: 10.1016/j.cmet.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnston NR, et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 2016;24(3):389–401. doi: 10.1016/j.cmet.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malin SK, Kullman EL, Scelsi AR, Godin JP, Ross AB, Kirwan JP. A whole-grain diet increases glucose-stimulated insulin secretion independent of gut hormones in adults at risk for type 2 diabetes. Mol Nutr Food Res. 2019;63(7):e1800967. doi: 10.1002/mnfr.201800967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Malin SK, et al. Impact of short-term exercise training intensity on β-cell function in older obese adults with prediabetes. J Appl Physiol. 2018;125(6):1979–1986. doi: 10.1152/japplphysiol.00680.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]