

Abstract
3-M primordial dwarfism is an inherited disease characterized by severe pre- and postnatal growth retardation and by mutually exclusive mutations in 3 genes, CUL7, OBSL1, and CCDC8. The mechanism underlying 3-M dwarfism is not clear. We showed here that CCDC8, derived from a retrotransposon Gag protein in placental mammals, exclusively localized on the plasma membrane and was phosphorylated by CK2 and GSK3. Phosphorylation of CCDC8 resulted in its binding first with OBSL1, and then CUL7, leading to the membrane assembly of the 3-M E3 ubiquitin ligase complex. We identified LL5β, a plasma membrane protein that regulates cell migration, as a substrate of 3-M ligase. Wnt inhibition of CCDC8 phosphorylation or patient-derived mutations in 3-M genes disrupted membrane localization of the 3-M complex and accumulated LL5β. Deletion of Ccdc8 in mice impaired trophoblast migration and placental development, resulting in intrauterine growth restriction and perinatal lethality. These results identified a mechanism regulating cell migration and placental development that underlies the development of 3-M dwarfism.
Keywords: Development, Genetics
Keywords: Ubiquitin-proteosome system
Introduction
3-M syndrome (OMIM 273750, 612921, 614205) is a rare inherited disorder featured by severe pre- and postnatal growth retardation and was first described by Miller, McKusick, and Malvaux, and is named after their shared initial letter (1). 3-M syndrome is characterized by low body weight and short length at birth, short stature (120–130 cm for adults), widespread skeletal abnormalities, and unusual facial features (2). There is currently no treatment for the disease. The first 3-M syndrome locus was mapped to chromosome 6p21.1, and candidate analysis of the region revealed that loss-of-function mutations in the cullin 7 gene (CUL7) cause 3-M syndrome (3). Subsequent exome sequencing analysis in 3-M patients with wild-type CUL7 identified loss-of-function mutations in 2 additional genes, OBSL1 on 2q35 (4) and CCDC8 on 19q13.32 (5). Notably, these 3 genes are mutated in a mutually exclusive manner, with CUL7 being the most frequently mutated (~65%), followed by OBSL1 (~30%) and CCDC8 (~5%) (5, 6). The mutually exclusive and genetically saturated mutations of these 3 genes in 3-M syndrome patients suggest that these 3 proteins likely function either in a linear pathway or as one functionally minimal and sufficient complex. In addition to 3-M syndrome, CUL7 and OBSL1 mutations were also found in Yakuts syndrome, le Merrer syndrome (also known as gloomy face syndrome), and Silver-Russell syndrome (SRS) (7, 8). These findings expand the scope of 3-M syndrome and link these rare and different primordial growth disorders into a potentially single disease with a common, and yet unknown, underlying molecular and cellular mechanism.
CUL7 is a member of the cullin family of proteins that functions as a scaffold for the assembly of E3 ubiquitin ligases by binding to the small RING finger protein, ROC1 (also known as RBX1), and substrates or substrate receptors. CUL7 (1,698 residues for the human protein) is a large protein that contains multiple functional domains and localizes predominantly in the cytoplasm (9). Deletion of Cul7 in mice leads to intrauterine growth retardation and perinatal death (10), recapitulating some 3-M defects. OBSL1 (obscurin-like 1) is also a large (1,896 residues) protein and was initially identified as a protein related to obscurin, a structural and signaling protein that may interact with the intracellular domains of cell adhesion complexes in myocytes (11). The OBSL1 protein is composed almost entirely of tandemly arranged immunoglobulin-like domains interrupted by a single fibronectin-like adhesive domain and is expressed strongly in the heart and placenta, but expressed at lower levels in many other tissues (11). The CCDC8 gene encodes a coiled-coil domain–containing protein and is epigenetically silenced by DNA methylation in several types of human tumors (12). Subsequent biochemical studies demonstrated the association between CCDC8 and CUL7 (5), between OBSL1 and CUL7 (13), and indeed the formation of the CUL7-OBSL1-CCDC8 ternary complex that we referred to as the 3-M complex (14). However, the biochemical and cellular function of OBSL1 and CCDC8, besides their binding with CUL7, is still not clear.
Of the three 3-M genes, CCDC8 is the youngest evolutionarily and contains only 1 exon. Sequence homology analysis suggested that CCDC8 originated in placental mammals from the domestication of the gag gene of the Ty3/Gypsy LTR retrotransposon during the evolution of Eutheria, but not marsupials or monotremes (15, 16). CUL7 (17) and OBSL1 (11), on the other hand, emerged much earlier in vertebrates, and encode larger, multiple-domain proteins. These findings, together with mutually exclusive mutations in 3-M patients, suggest that OBSL1 and CUL7 may have additional CCDC8-independent functions that are not related to 3-M syndrome, and CCDC8 has an OBSL1- and CUL7-dependent function that is important for placental mammals and, when disrupted, contributes directly to 3-M syndrome development.
Results
Deletion of Ccdc8 caused perinatal lethality, intrauterine growth restriction, and placental defects.
To determine the mechanism underlying the 3-M syndrome, we knocked out Ccdc8 in the C57BL/6J mouse strain by standard homologous recombination methods (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI129107DS1). Deletion of Ccdc8 was validated by genomic Southern blot (Supplemental Figure 1B), RT-qPCR (Supplemental Figure 1C), and RNA in situ hybridization (Supplemental Figure 1D). Heterozygous Ccdc8+/– mice were viable, fertile, and indistinguishable from wild-type littermates. We crossed heterozygous Ccdc8+/– mice and obtained no viable births of Ccdc8–/– mice (Figure 1A). Ccdc8–/– embryos could be identified at late gestational stages up to E18.5, indicating that loss of Ccdc8 caused a perinatal lethality. At E18.5, Ccdc8–/– embryos were significantly smaller than heterozygous or wild-type embryos (Figure 1, B and C), indicating a growth restriction caused by the loss of Ccdc8 as seen in 3-M syndrome patients.
Figure 1. Deletion of Ccdc8 causes perinatal lethality, intrauterine growth restriction, and placental defects.

(A) Genotypes of embryos and live offspring collected from Ccdc8+/– mouse intercrosses. (B) Gross appearance of Ccdc8+/+, Ccdc8+/–, and Ccdc8–/– embryos at E18.5. Scale bar: 1 cm. (C) Body weights of 41 E18.5 embryos collected from 4 Ccdc8+/– mouse intercrosses. Data are represented as mean ± SEM. Significance was determined by 1-way ANOVA and Bonferroni’s multiple-comparisons test. **Adjusted P < 0.01. (D) Quantification of the area of maternal and fetal blood vessels in the labyrinth. Data are represented as mean ± SEM from 5 randomly selected areas. Significance was determined by Student’s t test. ***P < 0.001. (E) H&E staining of E12.5 placentas with Ccdc8+/+ or Ccdc8–/– fetal genotype. Left: Gross staining pattern. Scale bars: 500 μm. Right: Boxed areas in the left panels were enlarged and maternal and fetal blood vessels are marked with red and blue, respectively. Scale bars: 20 μm. (F) Single-molecule RNA in situ hybridization of Ccdc8+/+ and Ccdc8–/– placenta. Yellow dashed line indicates the border between spongiotrophoblast layer (Sp) and labyrinth (Lab). Scale bars: 10 μm. (G) Ccdc8 mRNA expression pattern shown by RNA in situ hybridization (ISH) of a midsagittal section from an E12.5 embryo. H&E staining is from the adjacent section. Scale bars: 1 mm. NS indicates P ≥ 0.05.
Histological examination revealed that the placenta of Ccdc8–/– embryos was thinner than wild-type placenta and exhibited reduced branching of the maternal blood sinus in the labyrinth area (Figure 1, D and E). Ccdc8 is expressed in the labyrinth area (Figure 1F), suggesting that the defects in the labyrinth area were directly caused by the loss of Ccdc8 function. Ccdc8 is also expressed in multiple tissues in the embryo, including the midbrain, hindbrain, pituitary, tongue, heart, cartilage primordium of the vertebral bodies, metanephros, and urogenital sinus (Figure 1G). The growth restriction of Ccdc8-deficient embryos is likely caused by a combination of lack of adequate nutrients resulting from placental defects and additional defects in specific fetal tissues. The placental defects, intrauterine growth restriction (IUGR), and perinatal lethality are similar to those seen in Cul7-knockout mice (10), and mice deficient for Fbxw8 (18, 19), which encodes a CUL7-binding partner (20). However, Ccdc8–/– mouse embryo fibroblasts (MEFs) showed similar proliferation potential as Ccdc8+/+ MEFs (Supplemental Figure 1E). This is distinct from Cul7–/– MEFs, which exhibited a decreased rate of proliferation and premature senescence when compared with wild-type MEFs, as previously reported (10, 18, 19, 21). These results reinforce the notion that CCDC8 function is largely linked to CUL7, but CUL7 has additional, CCDC8-independent functions.
CCDC8 localized on and recruits OBSL1 and CUL7 to the plasma membrane.
To elucidate the cellular function of the 3-M complex, we determined the subcellular localization of endogenous CCDC8 and CUL7 proteins. Currently available antibodies for these proteins are either not sufficiently sensitive (for CCDC8, which is expressed at a very low level) or have high immunofluorescence (IF) background (for CUL7). We therefore used the CRISPR/Cas9 method to sequentially knock in 16 tandem copies of the MYC epitope tag (16×MYC), and 21 tandem copies of the FLAG epitope tag into endogenous CUL7 and CCDC8 in U2OS cells, respectively, generating 16×MYC-CUL7 and CCDC8-21×FLAG double-knockin (referred to hereafter as DKI) cells (Figure 2A). We verified the correct homozygous knockin of both CCDC8 and CUL7 by Western blot and intact CUL7-CCDC8 interactions by coimmunoprecipitation (co-IP) assay (Figure 2B). The DKI cells provide a highly sensitive and specific assay to visualize the subcellular localization of these proteins. Using these DKI cells, we found that CCDC8 protein is localized predominantly on the plasma membrane, while CUL7 is localized in both the cytoplasm and plasma membrane (Figure 2C). We further confirmed this finding by subcellular fractionation and Western blotting (Figure 2D). These results provided us the first insight into the potential function of the 3-M complex on the plasma membrane.
Figure 2. CCDC8 localizes on and recruits OBSL1 and CUL7 to the plasma membrane.

(A) Schematic representation of the domain structure and knockin epitope tagging of endogenous CUL7 and CCDC8 to generate 16×MYC-CUL7 and CCDC8-21×FLAG double-knockin (DKI) U2OS cells. (B) Validation of DKI U2OS cells. Cell lysates from DKI and parental U2OS cells were subjected to anti-FLAG immunoprecipitation (IP) and Western blot. (C) Immunostaining of DKI and parental U2OS cells with antibodies specific for MYC, FLAG, or plasma membrane marker Na+/K+-ATPase demonstrates the colocalization of endogenous (endo.) CCDC8 and CUL7 on the plasma membrane. Scale bars: 10 μm. (D) U2OS cells transfected with shRNA targeting CCDC8 were fractionated by centrifugation, and total lysate (Lys), cytoplasm (Cyto), and membrane (Mem) fractions were analyzed by Western blot with antibodies recognizing the indicated proteins. CUL7 and CUL9 were recognized by the same monoclonal antibody. (E) Induced overexpression of CCDC8-mKate localized CUL7-mTagBFP and OBSL1-EGFP onto the plasma membrane. Dox, doxycycline. Scale bars: 10 μm.
To determine how the three 3-M proteins affect the subcellular localization of each other, we ectopically expressed CCDC8 (with mKate), CUL7 (with mTagBFP), and OBSL1 (with EGFP) with different fluorescent tags and put the expression of CCDC8-mKate under the control of the tetracycline-inducible promoter (Tet-On). In the absence of CCDC8 induction (without doxycycline, Dox–), both ectopically expressed CUL7 and OBSL1 accumulated mostly in the cytoplasm (Figure 2E). Induction of CCDC8 by the addition of doxycycline (Dox+) localized both CUL7 and OBSL1 to the plasma membrane. To confirm this, we knocked down CCDC8 in U2OS cells and performed a cell fractionation–Western blot assay. This experiment showed that knocking down CCDC8 completely removed the membrane association of both CUL7 and OBSL1 (Figure 2D). Together, these data suggest that the 3-M complex is assembled on the plasma membrane.
CCDC8 is phosphorylated by CK2 and GSK3.
To determine how 3-M complex formation is regulated, we searched for posttranslational modifications of 3-M proteins and found that CCDC8 is potentially phosphorylated (Figure 3A). Searching the PhosphoSitePlus database (22) revealed that serine 142, 146, 261, and 273 in CCDC8 were identified multiple times by independent mass spectrometric analyses. Close examination of amino acid residues surrounding these sites predicts GSK3, CK2, CDK1, and PKC as potential kinases (Figure 3A). Pharmacological inhibition of GSK3 (by CHIR-98012) or CK2 (by CX4945), but not CDK1 (by RO-3306) or PKC (by sotrastaurin), reduced CCDC8 pan-phosphorylation levels (Figure 3B), indicating that both GSK3 and CK2 phosphorylate CCDC8. Phosphorylation of a substrate by GSK3 often requires a so-called priming phosphorylation at a site located 4 residues carboxyl-terminal to the GSK3 phosphorylation site (23), suggesting the possibility that CK2 phosphorylates Ser146 and primes the phosphorylation of Ser142 by GSK3. We raised 2 antibodies recognizing Ser142- and Ser146-phosphorylated CCDC8 (Supplemental Figure 2). Using these 2 antibodies, we found that inhibition of GSK3 substantially reduced the phosphorylation of Ser142, while inhibition of CK2 reduced both Ser142 and Ser146 phosphorylation (Figure 3C). We then carried out in vitro kinase assays using purified GSK3, CK2, and CCDC8. CK2 can phosphorylate wild-type and S142A mutant CCDC8, but not S146A mutants (Figure 3D). Sequential incubation of immobilized CCDC8 with CK2 and GSK3 showed that GSK3 phosphorylates wild-type CCDC8 only after it was first primed by CK2, but not CCDC8 that was not incubated with CK2 or mutated at either S142, S146, or both sites (Figure 3E). These results demonstrate that CCDC8 is phosphorylated sequentially by CK2 and GSK3.
Figure 3. CCDC8 is phosphorylated by CK2 and GSK3.

(A) Schematic representation of the domain structure and reported serine phosphorylation sites on the CCDC8 protein. The sequences adjacent to the putative phosphorylated serines and their predicted kinases are shown. (B) U2OS and HEK293 cells were treated with kinase inhibitors targeting GSK3 (by CHIR-98012), CK2 (by CX4945), CDK1 (by RO-3306), and PKC (by sotrastaurin), and lysates were separated by either 10% conventional SDS-PAGE or 6% Zn Phos-tag PAGE, followed by Western blotting using an antibody specific for CCDC8. (C) U2OS cells with endogenous CCDC8 3×FLAG-tagged by CRISPR were treated with GSK3 or CK2 inhibitors. Endogenous CCDC8 was precipitated by anti-FLAG antibody, followed by Western blot with anti-CCDC8, anti–phosphorylated serine 142 (α–p-Ser142), or anti–phosphorylated serine 146 (α–p-Ser146) antibodies. (D) Purified recombinant CCDC8 proteins were incubated with or without recombinant CK2 in kinase buffer containing ATP-γ-S, followed by treatment with p-nitrobenzyl mesylate to alkylate thiophosphates and form thiophosphate ester epitopes. In vitro phosphorylation of CCDC8 by CK2 was detected by Western blot with an antibody recognizing the thiophosphate ester. (E) Recombinant CCDC8 proteins were immobilized on magnetic beads, incubated first with CK2 in buffers containing ATP, and then with GSK3 in buffers containing ATP-γ-S. The phosphorylation by GSK3 was detected by alkylation with p-nitrobenzyl mesylate and Western blot with an antibody recognizing the thiophosphate ester.
The Wnt pathway inhibits CCDC8 phosphorylation and 3-M complex assembly.
Ser142 and Ser146 are located in the N-terminal region of CCDC8, which was previously reported to interact with OBSL1 (ref. 24 and Figure 3A), suggesting the possibility that phosphorylation of CCDC8 by CK2 and GSK3 may regulate its binding with OBSL1 and CUL7 and thus the assembly of the 3-M complex. To test this possibility, we performed a co-IP assay which showed that mutation of CK2 and GSK3 phosphorylation sites in CCDC8 (S142A, S146A, or double mutant S142A/S146A [2SA]) disrupt its binding with OBSL1, while phospho-mimicking mutant S142D/S146D (2SD) retained the interaction (Figure 4A). To determine how CK2 and GSK3 affect the interaction of endogenous 3-M proteins, we knocked in 3 tandem copies of FLAG (3×FLAG) epitope tag at the N-terminus of OBSL1 in U2OS cells, which allowed detecting the expression of multiple isoforms of endogenous OBSL1 (Figure 4B). Pharmacological inhibition of either CK2 (Figure 4B) or GSK3 (Figure 4C), or knocking down CK2 or GSK3 expression by siRNA (Supplemental Figure 3), reduced the binding between OBSL1 and CCDC8. S142A, S146A, or S2A CCDC8 lost the ability to recruit OBSL1 and CUL7 to the plasma membrane compared with ectopically expressed wild-type or phospho-mimicking 2SD-mutant CCDC8 (Figure 4D). These results suggest that phosphorylation of CCDC8 at S142 and S146 by CK2 and GSK3 promotes CCDC8 binding to OBSL1 and the recruitment of OBSL1 and CUL7 to colocalize with CCDC8 on the plasma membrane.
Figure 4. The Wnt pathway inhibits CCDC8 phosphorylation and 3-M complex assembly.

(A) HA-tagged wild-type or mutant CCDC8 was coexpressed with OBSL1-FLAG, followed by IP and Western analyses to examine CCDC8-OBSL1 protein interaction using indicated antibodies. (B and C) U2OS cells with endogenous OBSL1 3×FLAG-tagged by CRISPR were treated with CK2 inhibitor CX-4945 (B) or GSK3 inhibitor CHIR-98014 (C) for the indicated time. The binding between OBSL1 with CUL7 and CCDC8 was examined by IP and Western analyses using indicated antibodies. (D) mKate-fused wild-type or mutant CCDC8 was cotransfected with CUL7-mTagBFP and OBSL1-EGFP into U2OS cells, followed by confocal microscopic examination. Scale bars: 10 μm. (E) U2OS cells were treated with recombinant Wnt3a or IGF1. Cell lysates were subjected to 6% Zn Phos-tag PAGE (top panel) or 10% conventional SDS-PAGE. Indicated proteins were detected by Western blot. (F) U2OS with endogenous CCDC8 3×FLAG-tagged by CRISPR were treated with recombinant Wnt3a. Endogenous CCDC8 was enriched by anti-FLAG immunoprecipitation and CCDC8 phosphorylation was detected by anti–p-Ser142 or anti–p-Ser146 antibody. (G) U2OS cells expressing endogenous 3×FLAG-OBSL1 were treated with recombinant Wnt3a for the indicated time. The binding between OBSL1 with CUL7 and CCDC8 was determined by IP and Western analyses.
The insulin and Wnt signaling pathways are the 2 major pathways that regulate GSK3 (25, 26). Treating U2OS cells with Wnt3a, but not insulin-like growth factor 1 (IGF1), reduced CCDC8 phosphorylation (Figure 4E). Wnt3a treatment also reduced phosphorylation of CCDC8 Ser142 without any effect on the steady-state level of CCDC8 protein (Figure 4F) and the interaction between CCDC8 and OBSL1 (Figure 4G). Together, these results demonstrate that the Wnt signaling pathway, through inhibiting GSK3, negatively regulates CCDC8 phosphorylation and 3-M complex assembly on the plasma membrane.
3-M E3 ligase ubiquitinates and degrades plasma membrane protein LL5β.
To identify potential substrates of the 3-M complex, we carried out an extensive proteomic search for the protein(s) interacting with individual endogenously expressed 3-M proteins. We used CRISPR/Cas9 technology to knock in the 3×FLAG epitope tag to endogenous 3-M genes, CCDC8, OBSL1, and CUL7, and 2 genes, FBXW8 and CUL9, that encode proteins known to interact with CUL7, followed by immunoprecipitation with FLAG antibody and mass spectrometric analyses (Figure 5A). The accuracy and robustness of these analyses were verified by the identification of all known interactions between these 5 proteins, including SKP1 and FBXW8 with all three 3-M proteins, and p53 with both CUL7 and CUL9 (Table 1 and Supplemental Table 1). The specificity of the mass spectrometric analyses was also seen by the mutually exclusive binding of FBXW8 and CUL9 with 3-M proteins, but not with each other, and interaction of p53 with both CUL7 and CUL9, but not with other 3-M proteins, which is consistent with a previous study (27). The plasma membrane–localized protein PHLDB2/LL5β was identified in the FBXW8, CUL7, and OBSL1, but not the CUL9 immunocomplexes (Table 1). LL5β was also identified as a protein of a previously reported 3-M interaction network that includes 131 proteins derived from the mass spectrometric analyses of overexpressed individual 3-M proteins (28). The interaction between endogenous LL5β and 3-M proteins was validated by co-IP of LL5β with CUL7, OBSL1, CCDC8, and FBXW8, but not CUL9 (Figure 5B). These results identify LL5β as a bona fide 3-M complex–interacting protein.
Figure 5. 3-M E3 ligase ubiquitinates and degrades plasma membrane protein LL5β.

(A) Schematic representation of the strategy for identifying proteins interacting with the 3-M complex. Endogenous CCDC8, FBXW8, CUL7, CUL9, and OBSL1 were tagged with 3×FLAG tag by CRISPR in U2OS cells. Individual protein complexes were purified using FLAG antibody and analyzed by mass spectrometry. (B) Validation of LL5β as a 3-M–interacting protein. Endogenous 3×FLAG-tagged CCDC8, FBXW8, CUL7, CUL9, or OBSL1 protein was immunoprecipitated with FLAG antibody, followed by Western blotting (IB) analyses. (C and D) U2OS cells were transfected with plasmid expressing indicated proteins (C) or infected with shRNA lentivirus targeting indicated proteins (D). The ubiquitylation of overexpressed (C) or endogenous (D) LL5β ubiquitylation was determined by IP and Western blotting analyses. (E) U2OS cells were infected with lentivirus expressing shRNA targeting CUL7 or CCDC8, followed by direct Western blotting to detect indicated proteins. The quantitation of relative abundance of LL5β is shown. (F) U2OS cells with endogenous 3×FLAG-OBSL1 tagged by CRISPR were transfected with siRNA targeting OBSL1, followed by direct Western blotting to detect indicated proteins. (G and H) U2OS cells were treated with GSK3 or CK2 inhibitors (G) or Wnt3a (H), followed by direct Western blotting to detect indicated proteins. (I) CUL7 ubiquitinated LL5β in vitro. Indicated proteins were individually immunopurified from 293F cells and eluted using antigenic peptide. Substrate LL5β was incubated with a mixture of purified proteins as indicated in the presence of UBE1, UBE2D3, and HA-ubiquitin (HA-Ub) with or without ATP. After the reaction, LL5β was denatured by boiling in 1% SDS and immunoprecipitated, followed by SDS-PAGE and Western blot.
Table 1. Number of peptides identified by mass spectrometric analysesA.
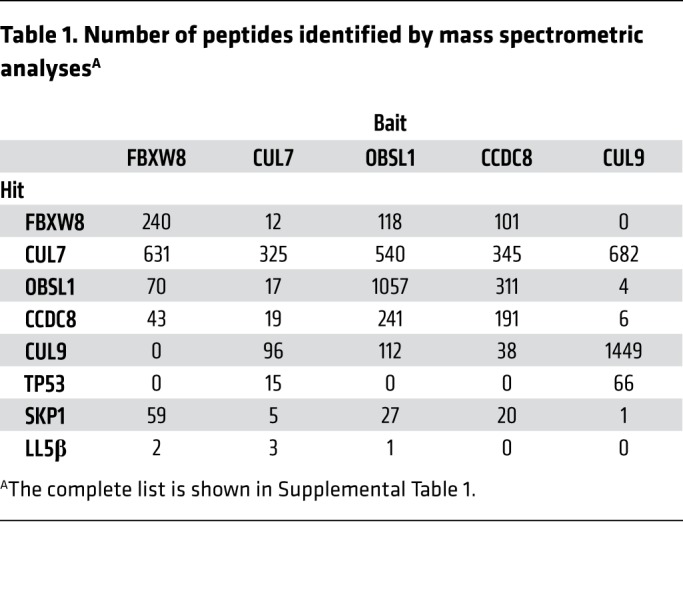
Combined overexpression of FBXW8, CUL7, OBSL1, and CCDC8 promoted the polyubiquitylation of LL5β (Figure 5C), while knockdown of either CUL7 or CCDC8 reduced LL5β polyubiquitylation (Figure 5D). The level of LL5β protein was increased after knocking down CUL7, CCDC8 (Figure 5E), or OBSL1 (Figure 5F), by the treatment of cells with CK2 or GSK3 inhibitor (Figure 5G), or Wnt3a (Figure 5H). In vitro incubation of purified LL5β with a mixture of individually immunopurified 3-M proteins (CCDC8, OBSL1, and CUL7) and FBXW8 resulted in LL5β polyubiquitylation (Figure 5I, lane 2). Omission of ATP (lane 1), substrate LL5β (lane 3), or CUL7 (lane 5) completely abolished the polyubiquitylation smear, demonstrating a CUL7-dependent LL5β polyubiquitylation. Dropout of FBXW8 (lane 4) also significantly reduced LL5β polyubiquitylation, suggesting an important role of FBXW8 in CUL7-dependent LL5β polyubiquitylation. Dropout of either OBSL1 (lane 6) or CCDC8 (lane 7) did not impair LL5β polyubiquitylation in vitro. This finding is consistent with a model in which CCDC8 and OBSL1 promote LL5β polyubiquitylation in vivo by recruiting CUL7 to the plasma membrane, but they are not required for the catalytic activity of CUL7.
CCDC8 regulates extracellular matrix remodeling and cell migration.
LL5β localizes on the plasma membrane and tethers microtubule plus ends to the cell cortex, thereby regulating microtubule dynamics, extracellular matrix (ECM) remodeling, cell adhesion, and cell migration (29–31). We previously found that depletion of CCDC8, OBSL1, or CUL7 impairs microtubule dynamics (14). We hypothesize that the 3-M E3 ligase, through promoting LL5β ubiquitylation and degradation and controlling microtubule dynamics, regulates ECM modeling and cell migration. To test this, we performed both the Boyden chamber transwell assay and gelatin degradation assays. We found that knocking down CCDC8 inhibited U2OS cell migration and invasion (Figure 6A), and increased degradation of gelatin coated on the cell culture surface (Figure 6, B and C). This observation is very similar to the finding that overexpression of LL5β in U2OS also inhibited cell migration and invasion (Supplemental Figure 4). Ccdc8–/– MEFs showed reduced cell migration and cell invasion compared with the wild-type MEFs (Figure 6D). These results demonstrate a function of the 3-M complex in the regulation of ECM modeling and cell migration.
Figure 6. CCDC8 regulates ECM remodeling and cell migration.

(A) U2OS cells stably expressing shRNA targeting CCDC8 were subjected to transwell cell migration or invasion assay. Data are represented as mean ± SEM from 3 replicates. Significance was determined by 1-way ANOVA and Dunnett’s multiple-comparisons test. **Adjusted P < 0.01, ***adjusted P < 0.001. (B) U2OS cells stably expressing shRNA targeting CCDC8 were cultured on cover glasses precoated with fluorescent gelatin for 8 hours, followed by microscopic examination showing gelatin degradation. Scale bars: 10 μm. (C) Quantification of relative degradation area in B. Fifty cells in each group were chosen randomly. Dots indicate the percentage of area of individual cells being degraded. Lines indicate mean ± SEM. Significance was determined by 1-way ANOVA and Dunnett’s multiple-comparisons test. **Adjusted P < 0.01, ****adjusted P < 0.0001. (D) MEFs of indicated genotypes were subjected to transwell cell migration or invasion assay. Data are represented as mean ± SEM from 3 replicates. Significance was determined by Student’s t test. *P < 0.05, **P < 0.01. (E) RT-qPCR showing Ccdc8 and LL5β mRNA expression in mouse trophoblast stem cells (TSCs) and in vitro–differentiated trophoblast giant cells (TGCs) or syncytiotrophoblast layer II (SynT-II) cells. Cdx2, Prl3d1, and SynB are markers for TSCs, TGCs, and SynT-II, respectively. FGF, fibroblast growth factor; RA, retinoic acid; CHIR, GSK3 inhibitor. (F) TSCs or in vitro–differentiated SynT-II cells were subjected to transwell cell migration or invasion assay. Data are represented as mean ± SEM from 3 replicates. Significance was determined by 1-way ANOVA and Dunnett’s multiple-comparisons test. **Adjusted P < 0.01. (G) LL5β in wild-type or Ccdc8-knockout SynT-II cells was determined by direct Western blotting analysis.
Dysfunction of ECM modeling and cell migration has been shown to lead to placental defects in multiple mouse models (32–34). Intrauterine trophoblast migration is a crucial event in placentation (35). These findings led us to postulate that a defect in ECM modeling and trophoblast cell migration contribute to the placental defects seen in Cul7- (10), Fbxw8- (18, 19), and Ccdc8-mutant (Figure 1) mice and the development of 3-M syndrome. Supporting this notion, CUL7 is expressed in human trophoblasts and, when its expression was induced in vitro in human choriocarcinoma cells, it induced morphological changes characteristic of epithelial-mesenchymal transition and enhanced cell migration and invasion (36).
To determine the function of 3-M genes in trophoblast migration, we utilized a previously established primary trophoblast stem cell (TSC) culture system (37). The TSCs can be maintained in TSC medium in the presence of FGF and heparin and induced to differentiate into trophoblast giant cells or syncytiotrophoblast layer II (SynT-II) cells by removing FGF and supplementing with retinoid acid (RA) or GSK inhibitor (CHIR) (38, 39). Ccdc8 expression was undetectable in TSCs, but they started to express Ccdc8 upon differentiation (Figure 6E). We knocked out Ccdc8 in TSCs by CRIPSR-mediated genome editing (Supplemental Figure 5, A and B). Ccdc8+/+ and Ccdc8–/– TSCs showed similar migration speed in the transwell assay, but after induction to differentiate into SynT-II cells, Ccdc8–/– cells exhibited significantly reduced migration (Figure 6F). Supporting the role of CCDC8 in regulating cell migration by recruiting CUL7 E3 ligase, deletion of Ccdc8 in differentiated SynT-II cells caused LL5β proteins to accumulate (Figure 6G), but had no effect on LL5β gene expression (Supplemental Figure 5C). Together, these results support the model in which CCDC8 recruits the 3-M E3 ligase complex to degrade LL5β and thereby regulates trophoblast cell migration and placental development.
3-M syndrome patient–derived mutations disrupt the assembly of the 3-M complex on the plasma membrane and the regulation of LL5β protein.
Multiple mutations in either CUL7, CCDC8, or OBSL1 genes have been reported in 3-M patients. Many mutations caused a truncation or frameshift, indicating a loss of function in nature. There are also several missense mutations whose mechanistic significance are unclear (Figure 7A). To determine whether development of 3-M syndrome is linked to the defect in the assembly of the 3-M complex on the plasma membrane and CUL7-mediated LL5β degradation, we coexpressed mKate-fused wild-type CCDC8 with wild-type or patient-derived mutant CUL7 or OBSL1 and examined the subcellular localization. This experiment showed that while wild-type OBLS1 and CUL7 were localized to the plasma membrane by the ectopically expressed CCDC8, 3-M patient–derived OBSL1 (R489X) and CUL7 (R1573X) mutant proteins remained localized in the cytoplasm (Figure 7B). Notably, CCDC8 was not able to localize wild-type CUL7 to the plasma membrane when it was coexpressed with mutant OBLS1, supporting the model that OBSL1 recruits CUL7 to membrane-localized CCDC8.
Figure 7. 3-M syndrome patient–derived mutations disrupt the assembly of the 3-M complex on the plasma membrane and the regulation of LL5β.

(A) Schematic representation of domains, binding proteins, and 3-M patient–derived mutations in CUL7. (B) mKate-fused wild-type or mutant CCDC8 was coexpressed with CUL7-mTagBFP and OBSL1-EGFP in U2OS cells, followed by confocal microscopic analyses of protein localization. Scale bars: 10 μm. (C) FLAG-CUL7 and HA-OBSL1 were coexpressed in HEK293 cells. CUL7-OBSL1 association was determined by IP and Western analysis. (D) U2OS cells with CCDC8-21×FLAG and 16×MYC-CUL7 double knockin (DKI, see Figure 2 for details) were subjected to CRISPR-mediated genome editing to introduce a homozygous L1588P mutation. The binding between endogenous CUL7 and CCDC8 was determined by IP and Western. (E) Total cell lysates derived from parental, DKI, and DKI/L1588P U2OS cells were fractionated by centrifugation, and total lysate (Lys), cytoplasm (Cyto), and membrane (Mem) fractions were analyzed by Western blotting. (F) The steady-state level of LL5β protein was determined by direct Western blot in parental, DKI, and DKI/L1588P U2OS cells. (G) Schematic representation of the function and regulation of 3-M E3 ubiquitin ligase. MT, microtubule.
We further characterized 2 additional 3-M patient–derived missense mutations in CUL7, S1536L and L1588P (6), and found that L1588P, but not S1536L, disrupted the binding of CUL7 with OBSL1 (Figure 7C). We then knocked in the L1588P mutation into the CUL7 gene in the U2OS DKI (16×MYC-CUL7 and CCDC8-21×FLAG) cells (Figure 7D and Supplemental Figure 6). We found that the L1588P mutation in CUL7 disrupted its interaction with endogenous CCDC8 (Figure 7D), its association with the membrane (Figure 7E), and the ability to degrade LL5β protein (Figure 7F). Together, these results demonstrate that multiple 3-M patient–derived mutations in either OBLS1 or CUL7 impaired the assembly of the 3-M E3 ligase complex on the plasma membrane and the regulation of LL5β.
Discussion
The mutually exclusive and genetically saturated mutations in the CUL7 (~65%), OBSL1 (~30%), and CCDC8 (5%) genes in 3-M syndrome patients indicate that the 3-M complex formed by their encoded proteins is a functionally minimal and sufficient complex. CCDC8 is present only in placental mammals, contains a single exon, and encodes a protein predominantly localized on the plasma membrane (Figure 2). Both OBSL1 and CUL7, on the other hand, evolved much earlier (after the appearance of vertebrates), and encode large multiple-domain proteins that localize to the cytoplasm until the signal-induced assembly of the 3-M complex on the plasma membrane. These findings suggest that of the three 3-M genes, OBSL1 and CUL7 have additional, CCDC8-independent functions and the function of CCDC8 is more closely related to 3-M syndrome. We suggest that targeting plasma membrane protein LL5β degradation to promote cell migration represents a major function of the 3-M complex which, when disrupted, causes the development of 3-M syndrome.
We elucidate the biochemical mechanism by which the 3-M E3 ligase regulates cell migration (Figure 7G). CCDC8 localizes predominantly on the plasma membrane and anchors the membrane assembly of the 3-M E3 complex. 3-M complex assembly is triggered by the CK2-primed, GSK3-mediated Ser142 and S146 phosphorylation in CCDC8 (Figure 3). Phosphorylated CCDC8 binds to OBSL1 which, in turn, brings in CUL7 to the plasma membrane (Figure 4). In vitro, CUL7 can catalyze LL5β ubiquitylation in the absence of either OBSL1 or CCDC8 (Figure 5I), suggesting that the function of these 2 proteins is not required for the catalytic activity of CUL7-ROC1 E3 ligase. This mechanism explains the mutually exclusive mutation and thus functional interdependency of the three 3-M genes, as loss of function of any one of them would disrupt the ubiquitylation of membrane-associated LL5β and the regulation of cell migration.
LL5β is a peripheral membrane protein and binds to and recruits the microtubule-stabilizing protein CLASP to the cell cortex (29), thereby negatively regulating focal adhesion turnover (30). Evolutionary, LL5β was present as early as Drosophila, suggesting that CCDC8-promoted LL5β degradation is not an essential regulator of every cell migration machinery. Instead, CCDC8 likely evolved to regulate a specific and recently emerged cellular or developmental process such as placental development that involves active cell migration. During placental development, trophoblast cells migrate and invade through the decidua and maternal uterine spiral arteries to form permissive vessels that facilitate blood flow to and nutrient uptake by the growing fetus (35). Defects in trophoblast migration and invasion, such as that caused by Ccdc8 deficiency (Figure 6), can result in placentation failures and cause pregnancy complications such as preeclampsia, intrauterine growth restriction, placenta accreta, gestational trophoblastic disease, and even maternal or fetal death. Some of these defects are found in the Ccdc8- and Cul7-null mice as well as 3-M patients. We suggest that promoting LL5β degradation and trophoblast cell migration during placental development represents a major, and perhaps the initial, function of CCDC8 and 3-M E3 ligase.
In addition to the placenta, Ccdc8 is expressed in multiple tissues in the embryo (Figure 1G). A recent single-cell transcriptomic analysis demonstrated broad and abundant expression of Ccdc8 in mesenchymal stem cells of adult mice, including those from adipose tissue, muscle, cardiac fibroblast, hematopoietic precursor and progenitor cells, pancreatic A cells, and tracheal mesenchymal cells (40). These findings suggest that CCDC8, and possibly the 3-M E3 ligase, may have additional functions beyond placental development during embryogenesis and later development. For example, ANKRA2, a functionally uncharacterized protein that is abundantly expressed in all tissues examined and associated with the membrane (41), was recently found to bind to a C-terminal region in CCDC8 separate from the N-terminal OBSL1 binding site (24). OBSL1 promotes the formation of the quaternary ANKRA2-CCDC8-OBSL1-CUL7 complex, suggesting the possibility that CUL7 may regulate ANKRA2-associated processes. Additional functions of CCDC8 beyond placental development are also consistent with clinical observations that 3-M patients, unlike fetuses that only had placental defects that caused decreased body weight at birth but usually caught up to their counterparts in the first few years of life after birth, continue to have defects in development after birth. The Ccdc8 mouse model developed here, together with the biochemical and cellular mechanism elucidated, should help to determine the broad function of CCDC8.
Of the three 3-M genes, CUL7 is most frequently mutated and encodes an enzyme. Many efforts made to understand the mechanism of 3-M syndrome have been devoted to identifying the substrates of CUL7. While the function of CUL7 in the regulation of membrane-associated LL5β and cell migration depends on CCDC8 and OBSL1, CUL7 can assemble additional CCDC8-independent complexes which, based on the genetic analyses in human patients, may perform functions that are not directly linked to 3-M syndrome. For example, CUL7 can also form a complex with CUL9 in the cytoplasm (27). Not only do CUL7-CCDC8 and CUL7-CUL9 complexes localize in different compartments, they also interact distinctively with other proteins, such as FBXW8 with CCDC8-CUL7 and p53 with CUL7-CUL9 (Table 1). Deletion of CUL9 results in spontaneous tumor development but has little effect on embryonic development (42), further reinforcing the notion that these 2 CUL7 complexes have distinct functions in vivo. CUL7 E3 ligase was previously reported to ubiquitylate the tumor suppressor p53 (9), insulin receptor substrate IRS-1 to promote its likely degradation in the cytoplasm (21), Golgi protein Grasp65 in neurons (13), hominoid-specific protein TBC1D3 (43), and more recently histone H2B (44). Whether the ubiquitylation of any of these proteins requires CCDC8 and is linked to 3-M syndrome have yet to be determined. It will also be important to determine whether the presence of CUL7 in different E3 ligase complexes coordinates the regulation of different cellular processes.
The canonical Wnt signaling exerts its effect through β-catenin–mediated transcription. In the absence of Wnt signaling, GSK3 phosphorylates β-catenin, generating a phosphor-degron that is recognized by CUL1-dependent β-TrCP E3 ubiquitin ligase (CRL1β-TrCP) and causes β-catenin degradation. Wnt, through an incompletely understood mechanism, inhibits GSK3, leading to the stabilization and nuclear accumulation of β-catenin where it binds members of the T cell factor/lymphocyte enhancer factor family (TCF/LEF) to transcribe Wnt target genes (45). GSK3 is a promiscuous kinase that phosphorylates many additional proteins, some of which are recognized by E3 ligases and stabilized by Wnt signaling, linking Wnt signaling to the regulation of several additional cellular processes, including cell growth, axonal remodeling in postmitotic neurons, and maturation of germ cells (46). The regulation of LL5β represents a potentially novel and β-catenin– and transcription-independent Wnt/GSK3 signaling mechanism. Instead of phosphorylating the substrate directly, GSK3 phosphorylates the scaffold protein CCDC8 to promote the assembly of a functional 3-M E3 complex and the degradation of LL5β.
Wnt signaling has long been implicated in the control of microtubule dynamics by the early findings that GSK3β and 3 major components of the Wnt signaling pathway, adenomatous polyposis coli (APC) protein, disheveled (Dvl), and axin, accumulate at the plus ends of microtubules in migrating cells to regulate cell polarity in a manner that requires Wnt5a (47–49). Mechanistically, how Wnt signaling regulates microtubule dynamics is poorly defined. LL5β localizes on the plasma membrane and tethers microtubule plus ends to the cell cortex, thereby regulating microtubule dynamics, ECM remodeling, cell adhesion, and cell migration (29–31). Depletion of CCDC8, OBSL1, or CUL7 impairs microtubule dynamics (14). The discovery of LL5β regulation by the 3-M E3 ligase complex provides a mechanism linking Wnt signaling to microtubule dynamics and various other cellular processes. The wide expression of the three 3-M genes suggests that Wnt signaling may regulate the function of 3-M E3 ligase in other cellular and developmental processes beyond cell migration during placental development.
While it is well recognized that transposons and their derivatives occupy more than 40% of the human genome and significantly shape our genome architecture and influence gene regulation, only a few human proteins derived from retrotransposons have been functionally and mechanistically studied (50). Two such examples are syncytin, which was derived from the Env protein of an endogenous retrovirus and mediates placental cytotrophoblast fusion and morphogenesis (51, 52), and PEG10 and PEG11/RTL1, which were derived from retrotransposon Gag and Pol proteins, and are essential for placental formation and maintenance, respectively (52, 53). Our study suggests that Gag-derived CCDC8, while retaining membrane-binding activity, has evolved into a scaffold protein that assembles an active E3 ubiquitin ligase in a signal-dependent manner on the plasma membrane to regulate cell migration and placental development. These findings illustrate the importance of recurrent domestications of retrotransposon proteins for placental development (54). Our study also showed that young genes, like Ccdc8 which exist only in placental mammals, could rapidly evolve critical functions, such as the essential role of Ccdc8 in placental and embryonic development.
Methods
Mouse model.
Ccdc8-knockout mice were generated by deleting the only exon of the mouse Ccdc8 gene by homologous recombination (Supplemental Figure 1A). A 21.4-kb mouse genomic DNA fragment spanning the Ccdc8 locus was isolated from mouse embryonic (ES) stem cell genomic DNA and subcloned into the target vector. LoxP sites were introduced flanking the only exon of CCDC8, and a neomycin resistance gene flanked by frt sites was inserted upstream of the loxP site. The linearized targeting vector was electroporated into E14 ES cells, followed with selection by G418 and ganciclovir. Double-resistant ES clones carrying the correct recombination was validated by PCR and the neomycin resistance gene was removed by Flp recombinase. ES clones were injected into C57BL/6J blastocysts and chimeric mice were crossed with C57BL/6J to generate Ccdc8fl/+ heterozygotes. Ccdc8 was knocked out by crossing Ccdc8fl/+ heterozygotes with B6.FVB-Tg (EIIa-cre) (Jackson Laboratory). All mice used in this study had been backcrossed for at least 8 generations with C57BL/6J mice.
Cell lines and primary cells.
U2OS, HEK293, and HEK293T cell lines were purchased from ATCC. 293F cells were purchased from Invitrogen. U2OS cells were cultured in McCoy’s 5A supplemented with 10% fetal bovine serum (FBS). HEK293 and HEK293T were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS. 293F were cultured as floating cells in Freestyle 293F Expression medium (Gibco) following the manufacturer’s instructions. All cells were maintained in a humidified incubator at 37°C with 5% CO2. U2OS and HEK293 used for mechanistic studies were validated by short tandem repeat profiling.
Primary mouse TSCs isolated from blastocysts of C57BL/6J × CAST/EiJ were a gift from Mauro Calabrese at the University of North Carolina at Chapel Hill and maintained as previously described (55). TSCs were cocultured with mitomycin C–treated MEF feeder cells in 30% fresh TS medium (RPMI1640 with 20% ES-quality FBS) plus 70% MEF-conditioned TS medium, supplemented with 25 ng/mL FGF4 and 1 μg/mL heparin.
Antibodies.
Rabbit polyclonal antibodies recognizing CCDC8 p-Ser142 and p-Ser146 were generated by GeneScript. Briefly, peptide antigen containing phosphorylated serine was conjugated to KLH and used to immunize New Zealand rabbits. Antibodies were affinity purified by a column conjugated with antigenic peptide, and antibodies recognizing nonphosphorylated CCDC8 were removed by adsorbing to a column conjugated with nonphosphorylated peptide. Purified antibodies were validated by dot blot with peptide antigen and corresponding nonphosphorylated peptide (Supplemental Figure 2). Peptide sequences are listed in Supplemental Table 2.
Primary antibodies against FLAG tag (Sigma-Aldrich, clone M2), MYC tag (Cell Signaling Technology, clone 9B11), HA tag (Roche, clone 3F10), Na,K-ATPase α1 (Cell Signaling Technology, D4Y7E), CCDC8 (Bethyl, polyclonal), tubulin (Santa Cruz Biotechnology, clone B-5-1-2), thiophosphate ester (Abcam, clone 51-8), phospho-LRP6 Ser1490 (Cell Signaling Technology, polyclonal), phospho-IGF-I receptor β Tyr1135/1136 (Cell Signaling Technology, clone 19H7), LL5β (Bethyl, polyclonal, for human protein detection), LL5β (Sigma-Aldrich, polyclonal, for mouse protein detection), and EGFP (Santa Cruz Biotechnology, clone B-2) were purchased commercially. Monoclonal antibody AB38 against both CUL7 and CUL9 was a gift from Loren Field (University of Indiana at Indianapolis) (56).
CRISPR-mediated genome editing.
CRISPR-mediated genome editing was performed as previously described (57). Briefly, sgRNA was subcloned into px458 vectors, and transfected into U2OS cells or TSCs. For the knockin experiment, a 200-mer oligo or pUC19-based donor plasmid was cotransfected as donor template, and DNA ligase IV inhibitor Scr7 was included in culture medium to increase homologous recombination efficiency (58, 59). 21×FLAG tag or 18×MYC tag was generated by repetitive ligation of 3×FLAG or 2×MYC oligos. Single cells were sorted into 96-well plates by FACSAria II cell sorter (BD). Clones were validated by Western blot or genomic PCR and Sanger sequencing. sgRNA and donor template sequences are listed in Supplemental Table 2.
Transfection and lentiviral transduction.
For plasmid transfection, U2OS and HEK293 cells were transfected by FuGene 6 (Roche) following the manufacturer’s instructions. 293F cells were transfected in suspension with homemade PEI (Polyscience) transfection reagent. TSCs were transfected using Lipofectamine 2000 (Invitrogen) by a floating transfection method as previously reported (55). siRNA was transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions.
Lentivirus was produced by cotransfecting pLIX402, pLKO.1, or pLentiCMV-puro along with packaging plasmids psPAX2 and pMD2G (all plasmids from Addgene) into HEK293T cells. Medium containing lentivirus was used to infect target cells. Polybrene (4 μg/mL) was included in culture medium to increase infection efficiency. Puromycin (2.5 μg/mL) was used for selecting infected cells.
PAGE, Western blot, and peptide dot blot.
Protein lysate was mixed with one-third volume of 4× Laemmli Sample Buffer (Bio-Rad) and boiled for 5 minutes. Samples were separated by electrophoresis using Mini-PROTEAN TGX precast gels (Bio-Rad), and then transferred to a polyvinylidene difluoride (PVDF) membrane using a Trans-Blot Turbo Transfer System (Bio-Rad) following the manufacturer’s instructions.
Zinc Phos-tag gels were prepared with Phos-tag acrylamide (Wako) following the manufacturer’s instructions. Zinc ion was removed by soaking the gels in Tris-glycine buffer containing 1 mM EDTA before transferring to the PVDF membrane.
The membranes were blocked in 5% fat-free milk in PBST, and then sequentially incubated with primary antibody and secondary antibody conjugated with horseradish peroxidase (HRP). Chemiluminescence was developed with LumiGLO Reserve Chemiluminescence Substrate Kit (KPL) and pictures were obtained by ChemiDoc gel imaging system (Bio-Rad).
For dot blot of peptide, the indicated amount of peptide was dotted on a nitrocellulose membrane. Blocking, antibody incubation, chemiluminescence, and imaging used the same method as Western blot.
Immunoprecipitation.
Cells were lysed in NP40 lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.2% NP40 supplemented with Pierce Halt Protease Inhibitor Cocktail) at 4°C for 30 minutes. Cell lysates were centrifuged at 15,000 g for 10 minutes, and supernatants were incubated with antibody-conjugated agarose beads for 3 hours with rotation at 4°C. Beads were washed 4 times with NP40 buffer and were boiled in 1× Laemmli Sample Buffer (Bio-Rad) for Western blot analysis.
Immunofluorescence and confocal microscopy.
For immunofluorescence, cells were cultured on glass-bottom dishes (MatTek). Cells were fixed in 4% paraformaldehyde (PFA, diluted from 16% solution from Electron Microscopy Sciences) at room temperature for 10 minutes and permeabilized in 0.1% Triton X-100 in PBS for 5 minutes. Cells were blocked in 1% bovine serum albumin (BSA) in PBS for 1 hour, and sequentially incubated with primary antibody and Alexa Fluor (Molecular Probes) dye–conjugated secondary antibody in blocking buffer. After staining, samples were mounted in ProLong Gold Antifade Mountant with DAPI (Invitrogen), and pictures were obtained with an FV1000 confocal microscope (Olympus).
Reverse transcription and quantitative PCR.
RNA was isolated by TRIzol reagent (Invitrogen). Reverse transcription was carried out with SuperScript III First-Strand Synthesis System (Invitrogen). Quantitative PCR was carried out with PowerUp SYBR Green Master Mix (Invitrogen) reagents on a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) following the manufacturer’s instructions. PCR primer sequences are listed in Supplemental Table 2.
Southern blot.
Genomic DNA was digested with enzyme (Fermentas) at 37°C overnight and subjected to agarose electrophoresis. DNA was transferred to positively charged nylon membranes (Roche), and Southern blotting was carried out with a DIG-High Prime DNA Labeling and Detection Starter Kit II (Roche) following the manufacturer’s instructions. Primer sequences used for synthesizing probes are listed in Supplemental Table 2.
Paraffin section, hematoxylin and eosin staining, and RNA in situ hybridization.
Mouse E12.5 embryos and placenta were harvested and fixed in 10% formalin. Sections (5 μm) of tissue were stained with routine hematoxylin and eosin (H&E) or subjected to RNA in situ hybridization with RNAscope 2.5 LS probes targeting mouse Ccdc8 (ACD Bio) following the manufacturer’s instructions. Slides were scanned with an Aperio Whole Slide Scanner (Leica).
Mass spectrometric analysis and database search.
The LC-MS/MS setup for protein identification was as previously described (60). In short, the protein samples were first reduced with DTT and derivatized with iodoacetamide, and then were separated by SDS-PAGE. Proteins from Coomassie blue–stained gel bands were in-gel digested with trypsin following protocols published previously (61). The generated peptides were desalted with StageTip and then analyzed by LC-MS/MS using a hybrid quadrupole-Orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific) as described previously (60). MS/MS spectra were assigned by searching against the UniProt Human database (20,195 sequences; reviewed sequences only; version 2015_06) using the SEQUEST algorithm embedded in Proteome Discoverer software (version 1.4, Thermo Fisher Scientific).
Cell fractionation.
U2OS cells were crosslinked with 1 mM dithiobis(succinimidyl propionate) (DSP) in PBS at room temperature for 30 minutes. After crosslinking, cells were collected by a scraper and were homogenized by passing through a 25-gauge needle 15 times in fractionation buffer (20 mM HEPES pH 7.5, 10 mM KCl, 250 mM sucrose, 1 mM EGTA, and 2 mM MgCl2 supplemented with Pierce Halt Protease Inhibitor Cocktail). Nuclei and undisrupted cells were removed by centrifugation at 10,000 g for 10 minutes at 4°C. Membrane and cytoplasm fractions were separated by centrifugation at 100,000 g for 1 hour at 4°C. Fractions were boiled in 1× Laemmli Sample Buffer (Bio-Rad) for Western blot analysis.
In vitro kinase assay.
CCDC8 protein was expressed and purified from 293F cells. A radioactive-free in vitro kinase assay was carried out by incubating CCDC8 with CK2 (NEB) or GSK3 (Abcam) in kinase buffer (20 mM Tris, pH 7.5, 50 mM KCl, 10 mM MgCl2) with 2 mM ATP (Sigma-Aldrich) or ATP-γ-S (Abcam) for 1 hour at 30°C. After the reaction, the thiophosphorylation sites on the substrates were alkylated by incubating with 2.5 mM p-nitrobenzyl mesylate for 1 hour at room temperature, followed by detection of the alkylated product by a thiophosphate ester–specific rabbit monoclonal antibody (Abcam).
Protein purification and in vitro ubiquitination assay.
SBP-FLAG–tagged CUL7, FBXW8, OBSL1, CCDC8, and LL5β proteins were overexpressed in 293F cells by transient transfection. Indicated proteins were affinity purified by incubating 293F cell lysate with streptavidin-Sepharose high-performance beads (GE Healthcare), elution with biotin, and concentrating with Amicon ultracentrifugal filters (Millipore). UBE1, UBE2D3, and HA-ubiquitin were purchased from Boston Biochem. The in vitro ubiquitination assay was carried out by incubating indicated 3-M components and LL5β with 1 μM UBE1, 5 μM UBE2D3, and 20 μM HA-ubiquitin in ubiquitination buffer (20 mM HEPES, pH 7.0, 20 mM NaCl, 2 mM MgCl2, 2 mM ATP) for 1 hour at 37°C. After the reaction, the reaction mixture was denatured by adding 1% SDS and 5 mM DTT and boiling for 10 minutes, followed with the enrichment of LL5β by diluting the reaction mixture 10 times with PBS and immunoprecipitation with anti-LL5β antibody. The ubiquitination was detected by Western blot with anti-HA antibody.
Gelatin degradation assay.
Cover glasses were coated with Oregon Green 488–conjugated gelatin as previously described (62). U2OS cells were cultured for 18 hours on gelatin-coated cover glasses. Cover glasses were fixed in 4% PFA for 10 minutes and stained with phalloidin–Alexa Fluor 594 (Invitrogen) for 30 minutes in PBS containing 0.1% saponin, mounted with ProLong Gold Antifade Mountant with DAPI (Invitrogen), and pictures were obtained with an FV1000 confocal microscope (Olympus).
Transwell assay.
Boyden chamber transwell assays were carried out with Costar Transwell cell culture inserts (Corning) with 8-μm pore size. Inserts uncoated or coated with 0.5 mg/mL Matrigel (Corning) were used from migration or invasion assay, respectively. A total of 100,000 U2OS cells, MEFs, or SynT-II cells differentiated from TSCs were cultured in the upper chamber in serum-free medium for 24, 3, or 8 hours, respectively. Culture medium with serum was added to the lower chamber as attractant. After culture for the indicated time, cells retained in the upper chamber were removed by cotton swab. The inserts were fixed in 4% PFA and stained with 0.1% crystal violet for 30 minutes. After rinsing with water, the transwell inserts were dried at room temperature. To quantify the relative numbers of cells that migrated through the membrane, crystal violet retained in migrated cells was dissolved by soaking Transwell inserts in 10% acetic acid and quantified by reading at absorbance of 595 nm in a plate reader (BioTek).
Statistics.
Quantification of maternal and fetal blood vessel area (Figure 1, D and E) was done by ImageJ software (NIH). Maternal and fetal blood vessels were distinguished by whether the red blood cells had a nucleus. The border of blood vessels was drawn manually, and Figure 1G shows the mean ± SEM of the percentage of area occupied by maternal or fetal blood vessels from 5 randomly selected areas.
Fluorescence microscopy images were processed and analyzed by ImageJ software. For gelatin degradation assay in Figure 6, B and C, the quantification method was previously reported (63).
Two-tailed Student’s t test was carried out to analyze Figure 1D, Figure 6D, and Supplemental Figure 4B. One-way ANOVA and Dunnett’s multiple-comparisons test were carried out to analyze Figure 6A, Figure 6C, and Figure 6F. One-way ANOVA and Bonferroni’s multiple-comparisons test were carried out to analyze Figure 1C. All analyses were done in Prism 8 software (GraphPad). Significance was defined as P < 0.05. Sample numbers, definition of center, and dispersion and precision measures are shown in figure legends.
Study approval.
The Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill (UNC-Chapel Hill) approved all procedures involving experimental animals.
Author contributions
YX and PW designed the experiments. PW, FY, and ZL established the mouse model. YY performed mass spectrometric analysis. SEP analyzed the embryo phenotype. PW performed the rest of the experiments and wrote the manuscript with YX.
Supplementary Material
Acknowledgments
We thank Loren Field (University of Indiana at Indianapolis) for providing anti-CUL7/CUL9 Ab38 monoclonal antibody; Mauro Calabrese (UNC-Chapel Hill) for providing TSCs; Bin Gu (The Hospital for Sick Children, Toronto) for advising on TSC culturing; the UNC Animal Models Core for generating the Ccdc8-mutant mouse; UNC Translational Pathology Lab for histological analyses; and Jim Bears, Keith Burridge, Mark Peifer, Kun-Liang Guan, and members of the Xiong Lab for discussion throughout this study. This study was supported by NIH grants GM067113 and CA068377 (to YX).
Version 1. 07/25/2019
In-Press Preview
Version 2. 09/09/2019
Electronic publication
Version 3. 10/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4393–4407.https://doi.org/10.1172/JCI129107.
Contributor Information
Pu Wang, Email: PuWang@unc.edu.
Feng Yan, Email: feng-yan@metonpharma.com.
Zhijun Li, Email: zhijunl@email.unc.edu.
Yanbao Yu, Email: yayu@jcvi.org.
Scott E. Parnell, Email: scott_parnell@med.unc.edu.
Yue Xiong, Email: yxiong@email.unc.edu.
References
- 1.Miller JD, McKusick VA, Malvaux P, Temtamy S, Salinas C. The 3-M syndrome: a heritable low birthweight dwarfism. Birth Defects Orig Artic Ser. 1975;11(5):39–47. [PubMed] [Google Scholar]
- 2.Clayton PE, et al. Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol (Oxf) 2012;77(3):335–342. doi: 10.1111/j.1365-2265.2012.04428.x. [DOI] [PubMed] [Google Scholar]
- 3.Huber C, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37(10):1119–1124. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 4.Hanson D, et al. The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am J Hum Genet. 2009;84(6):801–806. doi: 10.1016/j.ajhg.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson D, et al. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011;89(1):148–153. doi: 10.1016/j.ajhg.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber C, et al. A large-scale mutation search reveals genetic heterogeneity in 3M syndrome. Eur J Hum Genet. 2009;17(3):395–400. doi: 10.1038/ejhg.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akawi NA, Ali BR, Hamamy H, Al-Hadidy A, Al-Gazali L. Is autosomal recessive Silver-Russel syndrome a separate entity or is it part of the 3-M syndrome spectrum? Am J Med Genet A. 2011;155A(6):1236–1245. doi: 10.1002/ajmg.a.34009. [DOI] [PubMed] [Google Scholar]
- 8.Maksimova N, et al. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J Med Genet. 2007;44(12):772–778. doi: 10.1136/jmg.2007.051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene. 2006;25(33):4534–4548. doi: 10.1038/sj.onc.1209490. [DOI] [PubMed] [Google Scholar]
- 10.Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci U S A. 2003;100(17):9855–9860. doi: 10.1073/pnas.1733908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geisler SB, et al. Obscurin-like 1, OBSL1, is a novel cytoskeletal protein related to obscurin. Genomics. 2007;89(4):521–531. doi: 10.1016/j.ygeno.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris MR, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30(12):1390–1401. doi: 10.1038/onc.2010.525. [DOI] [PubMed] [Google Scholar]
- 13.Litterman N, et al. An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011;9(5):e1001060. doi: 10.1371/journal.pbio.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan J, et al. The 3M complex maintains microtubule and genome integrity. Mol Cell. 2014;54(5):791–804. doi: 10.1016/j.molcel.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaneko-Ishino T, Ishino F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol. 2012;3:262. doi: 10.3389/fmicb.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang SW, Lahiri C, Poh CL, Tan KO. PNMA family: Protein interaction network and cell signalling pathways implicated in cancer and apoptosis. Cell Signal. 2018;45:54–62. doi: 10.1016/j.cellsig.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Marín I. Diversification of the cullin family. BMC Evol Biol. 2009;9:267. doi: 10.1186/1471-2148-9-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsunematsu R, Nishiyama M, Kotoshiba S, Saiga T, Kamura T, Nakayama KI. Fbxw8 is essential for Cul1-Cul7 complex formation and for placental development. Mol Cell Biol. 2006;26(16):6157–6169. doi: 10.1128/MCB.00595-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsutsumi T, Kuwabara H, Arai T, Xiao Y, Decaprio JA. Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol Cell Biol. 2008;28(2):743–751. doi: 10.1128/MCB.01665-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci U S A. 2002;99(26):16601–16606. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30(4):403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(Database issue):D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie J, et al. Ankyrin repeats of ANKRA2 recognize a PxLPxL motif on the 3M syndrome protein CCDC8. Structure. 2015;23(4):700–712. doi: 10.1016/j.str.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2(10):769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 26.Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. GSK-3 - at the crossroads of cell death and survival. J Cell Sci. 2014;127(Pt 7):1369–1378. doi: 10.1242/jcs.138057. [DOI] [PubMed] [Google Scholar]
- 27.Skaar JR, et al. PARC and CUL7 form atypical cullin RING ligase complexes. Cancer Res. 2007;67(5):2006–2014. doi: 10.1158/0008-5472.CAN-06-3241. [DOI] [PubMed] [Google Scholar]
- 28.Hanson D, Stevens A, Murray PG, Black GC, Clayton PE. Identifying biological pathways that underlie primordial short stature using network analysis. J Mol Endocrinol. 2014;52(3):333–344. doi: 10.1530/JME-14-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lansbergen G, et al. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev Cell. 2006;11(1):21–32. doi: 10.1016/j.devcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 30.Stehbens SJ, Paszek M, Pemble H, Ettinger A, Gierke S, Wittmann T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat Cell Biol. 2014;16(6):561–573. doi: 10.1038/ncb2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Astro V, Chiaretti S, Magistrati E, Fivaz M, de Curtis I. Liprin-α1, ERC1 and LL5 define polarized and dynamic structures that are implicated in cell migration. J Cell Sci. 2014;127(Pt 17):3862–3876. doi: 10.1242/jcs.155663. [DOI] [PubMed] [Google Scholar]
- 32.Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet. 2001;2(7):538–548. doi: 10.1038/35080570. [DOI] [PubMed] [Google Scholar]
- 33.Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology (Bethesda) 2005;20:180–193. doi: 10.1152/physiol.00001.2005. [DOI] [PubMed] [Google Scholar]
- 34.Walentin K, Hinze C, Schmidt-Ott KM. The basal chorionic trophoblast cell layer: An emerging coordinator of placenta development. Bioessays. 2016;38(3):254–265. doi: 10.1002/bies.201500087. [DOI] [PubMed] [Google Scholar]
- 35.Silva JF, Serakides R. Intrauterine trophoblast migration: A comparative view of humans and rodents. Cell Adh Migr. 2016;10(1–2):88–110. doi: 10.1080/19336918.2015.1120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu J, et al. Ubiquitin ligase cullin 7 induces epithelial-mesenchymal transition in human choriocarcinoma cells. J Biol Chem. 2010;285(14):10870–10879. doi: 10.1074/jbc.M109.004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282(5396):2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- 38.Yan J, Tanaka S, Oda M, Makino T, Ohgane J, Shiota K. Retinoic acid promotes differentiation of trophoblast stem cells to a giant cell fate. Dev Biol. 2001;235(2):422–432. doi: 10.1006/dbio.2001.0300. [DOI] [PubMed] [Google Scholar]
- 39.Zhu D, Gong X, Miao L, Fang J, Zhang J. Efficient induction of syncytiotrophoblast layer II cells from trophoblast stem cells by canonical Wnt signaling activation. Stem Cell Reports. 2017;9(6):2034–2049. doi: 10.1016/j.stemcr.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabula Muris Consortium. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562(7727):367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rader K, Orlando RA, Lou X, Farquhar MG. Characterization of ANKRA, a novel ankyrin repeat protein that interacts with the cytoplasmic domain of megalin. J Am Soc Nephrol. 2000;11(12):2167–2178. doi: 10.1681/ASN.V11122167. [DOI] [PubMed] [Google Scholar]
- 42.Pei XH, et al. Cytoplasmic CUL9/PARC ubiquitin ligase is a tumor suppressor and promotes p53-dependent apoptosis. Cancer Res. 2011;71(8):2969–2977. doi: 10.1158/0008-5472.CAN-10-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kong C, et al. Ubiquitination and degradation of the hominoid-specific oncoprotein TBC1D3 is mediated by CUL7 E3 ligase. PLoS ONE. 2012;7(9):e46485. doi: 10.1371/journal.pone.0046485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah VJ, Maddika S. CRL7SMU1 E3 ligase complex-driven H2B ubiquitylation functions in sister chromatid cohesion by regulating SMC1 expression. J Cell Sci. 2018;131(8):jcs213868. doi: 10.1242/jcs.213868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 46.Acebron SP, Niehrs C. β-Catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol. 2016;26(12):956–967. doi: 10.1016/j.tcb.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421(6924):753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 48.Ciani L, Krylova O, Smalley MJ, Dale TC, Salinas PC. A divergent canonical WNT-signaling pathway regulates microtubule dynamics: dishevelled signals locally to stabilize microtubules. J Cell Biol. 2004;164(2):243–253. doi: 10.1083/jcb.200309096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlessinger K, McManus EJ, Hall A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J Cell Biol. 2007;178(3):355–361. doi: 10.1083/jcb.200701083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jangam D, Feschotte C, Betrán E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017;33(11):817–831. doi: 10.1016/j.tig.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mi S, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403(6771):785–789. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- 52.Sekita Y, et al. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet. 2008;40(2):243–248. doi: 10.1038/ng.2007.51. [DOI] [PubMed] [Google Scholar]
- 53.Ono R, et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat Genet. 2006;38(1):101–106. doi: 10.1038/ng1699. [DOI] [PubMed] [Google Scholar]
- 54.Imakawa K, Nakagawa S. The phylogeny of placental evolution through dynamic integrations of retrotransposons. Prog Mol Biol Transl Sci. 2017;145:89–109. doi: 10.1016/bs.pmbts.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 55.Himeno E, Tanaka S, Kunath T. Isolation and manipulation of mouse trophoblast stem cells. Curr Protoc Stem Cell Biol. 2008;Chapter 1:Unit 1E.4. doi: 10.1002/9780470151808.sc01e04s7. [DOI] [PubMed] [Google Scholar]
- 56.Dowell JD, et al. Expression of a mutant p193/CUL7 molecule confers resistance to MG132- and etoposide-induced apoptosis independent of p53 or Parc binding. Biochim Biophys Acta. 2007;1773(3):358–366. doi: 10.1016/j.bbamcr.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu VT, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33(5):543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 60.Yu Y, et al. Comprehensive metaproteomic analyses of urine in the presence and absence of neutrophil-associated inflammation in the urinary tract. Theranostics. 2017;7(2):238–252. doi: 10.7150/thno.16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 62.Vázquez-Vela E, Vázquez-Vela G. Acetabular reaction to the Bateman bipolar prosthesis. Clin Orthop Relat Res. 1990;(251):87–91. [PubMed] [Google Scholar]
- 63.Martin KH, Hayes KE, Walk EL, Ammer AG, Markwell SM, Weed SA. Quantitative measurement of invadopodia-mediated extracellular matrix proteolysis in single and multicellular contexts. J Vis Exp. 2012;(66):e4119. doi: 10.3791/4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.