

SUMMARY
The activation of G proteins by G protein-coupled receptors (GPCRs) underlies the majority of transmembrane signaling by hormones and neurotransmitters. Recent structures of GPCR-G protein complexes obtained by crystallography and cryo-electron microscopy (cryo-EM) reveal similar interactions between GPCRs and the alpha subunit of different G protein isoforms. While some G protein subtype-specific differences are observed, there is no clear structural explanation for G protein subtype-selectivity. All of these complexes are stabilized in the nucleotide-free state, a condition that does not exist in living cells. In an effort to better understand the structural basis of coupling specificity, we used time-resolved structural mass spectrometry techniques to investigate GPCR-G protein complex formation and G-protein activation. Our results suggest that coupling specificity is determined by one or more transient intermediate states that serve as selectivity filters and precede the formation of the stable nucleotide-free GPCR-G protein complexes observed in crystal and cryo-EM structures.
Keywords: G protein-coupled receptor, G protein, hydrogen/deuterium exchange mass spectrometry, hydroxyl radical footprinting mass spectrometry, conformation
Graphical Abstract
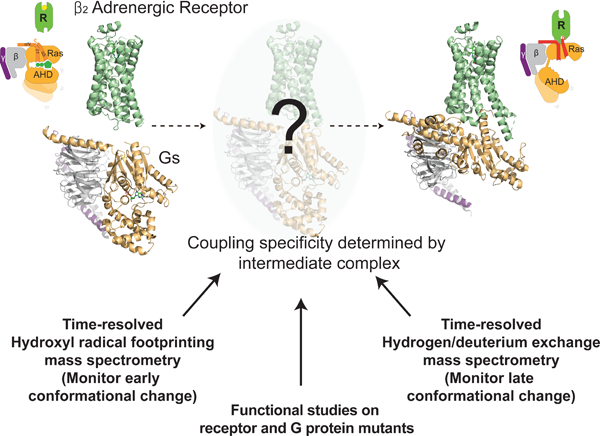
In brief
A time-resolved look at how a GPCR engages a G protein reveals intermediates in the process that dictate both specificity for the interaction and the initial steps kicking off downstream signaling.
INTRODUCTION
G protein-coupled receptors (GPCRs) are the largest family of membrane receptors and modify cellular behavior by activating heterotrimeric G proteins (Lefkowitz, 2007). Heterotrimeric G proteins are composed of α, β, and γ subunits, and the Gα subunit contains a nucleotide-binding pocket between Ras-like and α-helical domains (Figure 1A) (Preininger et al., 2013). In the inactive state, the Gα subunit is bound to GDP and forms a heterotrimer with Gβγ subunits. Upon agonist activation, the GPCR triggers GDP release, and GTP binds. The GTP-bound Gα subunit dissociates from both the receptor and Gβγ subunits to activate downstream signaling pathways. GPCRs generally couple primarily to one of three major G protein subfamilies: Gs, Gi/o and Gq/11.
Figure 1. Structures representing different stages in the G protein cycle.
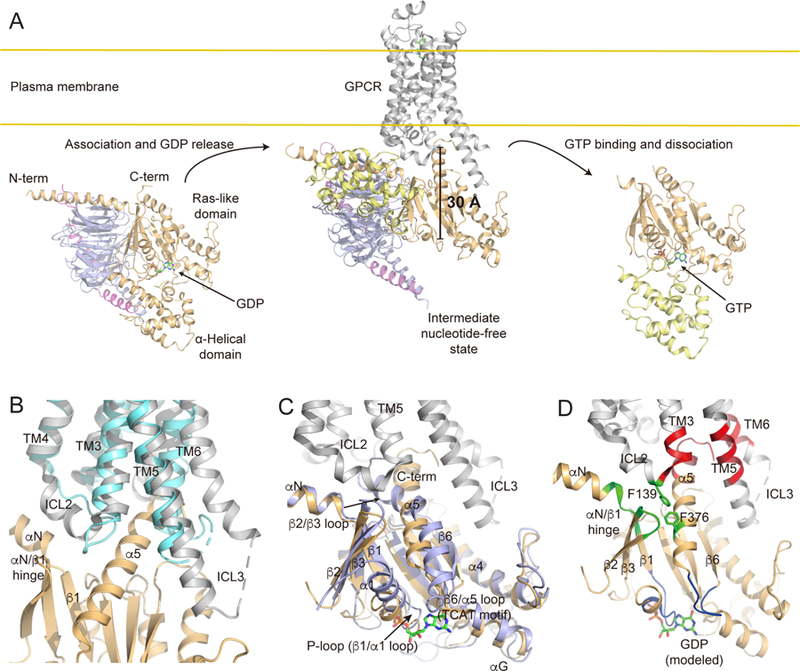
(A) GPCR-mediated G protein activation illustrated with representative crystal structures. Left: The GDP-bound heterotrimeric G protein (Liu et al., companion manuscript) shows the position of nucleotide-binding pocket. GDP is shown as a stick model with carbons colored green. The Ras-like domain of Gα is colored as light orange, the α-helical domain (AHD) of Gα as light yellow, Gβ as light blue, and Gγ as violet. Middle: The X-ray crystal structure of the β2AR-Gs complex (PDB: 3SN6) shows a large movement of AHD and opening of the nucleotide-binding pocket. The β2AR is shown in grey. Nanobody Nb35 and the T4 lysozyme insertion that were essential for crystallization have been omitted for clarity. Right: The GTP-bound Gα subunit structure (PDB: 1AZT) shows the position of GTP (stick model) and closing of the nucleotide binding pocket.
(B) Comparison of the high-resolution X-ray crystal structures of the β2AR in an agonist-bound form (PDB: 3PDS, cyan) and an agonist-bound Gs-coupled form (PDB: 3SN6, grey). The nucleotide-free state of Gα is shown in light orange.
(C) Comparison of the X-ray crystal structures of the Ras-like domain of Gαs in a GDP-bound form (Liu et al., companion manuscript, light blue) and a β2AR-bound nucleotide-free form (PDB: 3SN6, light orange). The active state of the β2AR (PDB: 3SN6) is shown in grey.
(D) Description of the ICL2 and the cytosolic-core links in the nucleotide-free GPCR-G protein complex. Receptor binding signals through the Ras-like domain of Gα to the nucleotide-binding pocket (blue) via the ICL2 (green) and the cytosolic-core (red) links. GDP (stick model) is positioned in the nucleotide-free structure of the β2AR-Gs complex based on structural alignment with the GDP-bound heterotrimeric Gs protein (Liu et al., companion manuscript). The rest of the Ras-like domain of Gα subunit is shown in light orange and the β2AR in grey.
The structural basis of GPCR-mediated G protein activation has been of great interest. The β2 adrenergic receptor-Gs (β2AR-Gs) X-ray crystal structure, the first X-ray crystal structure of a GPCR-G protein complex, revealed the GPCR-Gs interfaces and conformational changes upon complex formation (Rasmussen et al., 2011) (Figure 1A middle). Subsequently, a X-ray crystal structure of the adenosine A2a receptor in complex with an engineered Gs (A2A-miniGs), and single-particle cryo-electron microscopy structures of calcitonin receptor-Gs (CT-Gs), glucagon-like peptide-1 receptor-Gs (GLP1R-Gs), µ opioid receptor-Gi (µOR-Gi), adenosine A1a receptor-Gi (A1A-Gi), serotonin 5HT1B receptor-Gi (5HT1B-Gi), and rhodopsin-Gi complexes have been determined with similar GPCR-G protein interfaces (Carpenter et al., 2016; Draper-Joyce et al., 2018; Garcia-Nafria et al., 2018; Kang et al., 2018; Koehl et al., 2018; Liang et al., 2017; Zhang et al., 2017). These structures provide snap-shots of stable nucleotide-free GPCR-G protein complexes; however, they cannot identify the temporal sequence of coupling events nor do they fully explain the structural basis for coupling specificity, that is, why a particular GPCR prefers a specific G protein isoform.
A comparison of the structures of the inactive-state β2AR and the nucleotide-free β2AR-Gs complex reveal structural changes at the receptor-Gs interfaces (Figure 1B): intracellular loop 2 (ICL2) forms an α-helix; the N-terminus of ICL3 forms an α-helix as an extension of transmembrane domain 5 (TM5); and TM6 moves outward. The ICL2, especially F139, of the β2AR engages the hydrophobic pocket formed by the αN/β1 hinge, β2/β3 loop and F376 at α5 helix of Gαs (Figures 1C and 1D green), while the outward movement of TM6 provides a space in the cytosolic core of the β2AR formed by ICL2 and TMs 3, 5 and 6, in which the C-terminus of Gαs can insert (Figures 1C and 1D red). Understanding the timing and orchestration of these interactions may be key to understand coupling selectivity and the functional activation path.
The distance between the GPCR-G protein interface and the GDP-binding pocket is approximately 30 Å (Figure 1A), and therefore recep tor-mediated GDP release is accomplished by allosteric conformational changes mediated by structural links between the GPCR-G protein interface (Figure 1D green, red) and the nucleotide-binding pocket (Figure 1D blue). The C-terminus of Gα (the α5 helix) links the guanine pocket TCAT motif in the β6/α5 loop with the cytosolic core of the receptor, and undergoes the largest conformational change in the Ras-like domain of Gα upon coupling to the receptor (Figures 1C and 1D). The αN/β1 hinge and β1 strand at the amino terminus of the Ras-like domain links ICL2 with the P-loop that binds the diphosphate of GDP (Figures 1C and 1D). Much has been learned about the detailed structural features and conformational dynamics of these two allosteric pathways using a variety of biophysical, biochemical and computational approaches on different GPCR-G protein pairs (Alexander et al., 2014; Dror et al., 2015; Kaya et al., 2014; Moro et al., 1993; Nanoff et al., 2006; Oldham et al., 2006, 2007; Scheerer et al., 2009; Van Eps et al., 2011; Yano et al., 2017; Zhou et al., 1999). Nevertheless, understanding the temporal sequence of these substantial conformational changes during GPCR-mediated G protein activation are likely to provide additional insights into GPCR-G protein coupling selectivity (Preininger et al., 2013).
In the present study, we employed hydrogen/deuterium exchange mass spectrometry (HDX-MS) and hydroxyl radical mediated protein footprinting with mass spectrometry (HRF-MS) (Figures S1A) in a time-resolved manner to investigate the sequence of events during GPCR-mediated GDP release. HDX-MS and HRF-MS have been proven to be useful complementary techniques to investigate the conformational dynamics of proteins (Bavro et al., 2015; Calabrese and Radford, 2018; Harrison and Engen, 2016; Kang et al., 2015; Kiselar and Chance, 2018; Orban et al., 2012; Rajabi et al., 2015; Wang and Chance, 2017). Both techniques provide information on changes in structure and/or solvent interactions of proteins; HDX-MS probes the exchange between amide hydrogen in the peptide backbone and deuterium in the solvent and is governed by both the stability of the secondary structure elements and the overall tertiary and quaternary conformations. HRF-MS probes the solvent accessibility of side chains of proteins to hydroxyl radicals (generated by radiolysis) derived from bulk or ordered water (Konermann et al., 2008; Zheng et al., 2008). HDX-MS can productively monitor the dynamics of protein secondary structures as these backbone amide hydrogens are involved in the formation of hydrogen bonds in protein secondary structures; the amide hydrogens within structured regions (e.g. α-helix or β-strand) are labeled more slowly than more flexible regions. HRF-MS provides a readout of changes in local solvent structure in a region of interest irrespective of secondary structure per se; residues in close proximity to bulk or ordered water are labeled with a radiolytically derived OH• radical, providing a covalent reporter for both changes in structure and association with/exposure to solvent. These techniques are quite complementary as HDX-MS provides data on structural changes at the second to minute timescale (on the backbone) whereas HRF-MS can provide residue-level solvent accessibility data on timescales from milliseconds to seconds.
There are two different labeling approaches when employing either HDX-MS or HRF-MS. Continuous labeling studies (exposure to deuterated buffer or X-rays for a certain period of time) simply compares two or more different states of proteins (Figure S1B) (Chung et al., 2011; Orban et al., 2012), while pulse-labeling (i.e. time-resolved) studies monitor the sequential events of conformational changes (Figure S1C) (Konermann and Simmons, 2003). In the pulse-labeling study, the conformational changes of a protein are triggered (i.e. the addition of an agonist-activated GPCR to GDP-bound G protein), and the sequential events are detected by performing a brief deuterium or x-ray pulse with samples recovered at various time points post initiation (and quenched) to capture conformational changes as a function of time. While HDX-MS is challenging for monitoring conformational changes at the millisecond timescales available to HRF-MS, it is complementary, monitoring the dynamics of secondary structural elements at later time points (i.e. seconds to minutes).
In this study, we used the Gαsβ1γ2 heterotrimer (Gs) as a model G protein and the β2AR and the A2A as model GPCRs and analyzed them via HDX-MS and HRF-MS. The β2AR-Gs complex is well-characterized by X-ray crystallography (Rasmussen et al., 2011) and continuous labeling HDX-MS (Chung et al., 2011), allowing easier interpretation of the pulse-labeling HDX-MS data. The A2A is another well-known Gs-coupled receptor, and the X-ray crystal structure with engineered Gαs has been recently reported (Carpenter et al., 2016). The β2AR and the A2A belong to the class A GPCRs, and therefore, studying the β2AR and the A2A can be expected to elucidate the general Gs-coupling mechanism of the class A GPCRs.
We observe conformational changes by HRF-MS at the C-terminal end of the a5 helix, followed by coordinated changes in the proximal a5 helix and the b3 strand that would likely alter the GDP-binding pocket, leading to GDP release. Of interest, based upon the HDX studies presented here, the conformational changes at the C-terminal end of the a5 helix, that are expected based on crystal and cryo-EM structures, do not occur until long after GDP release. These results suggest that the first complex formed by Gs and the β2AR or by Gs and the A2A differ from their respective crystal structures, yet may be responsible for GDP release and coupling specificity. In a companion manuscript we present the crystal structure revealing an alternate interaction between the β2AR and the a5 helix of Gs, along with functional evidence that this interaction is important for GDP release (Liu et al., companion manuscript).
RESULTS AND DISCUSSION
Continuous labeling HDX profile of the stable GPCR-Gs complex
Prior to investigating the time-resolved conformational changes upon GPCR-Gs coupling, we examined the stably-formed, nucleotide-free β2AR-Gs and A2A-Gs complexes by continuous labeling HDX-MS (Figure S1B) to determine the shortest D2O pulse duration that could detect a difference between the GPCR-Gs complex and the individual proteins. Moreover, we wanted to determine if the A2A-Gs complex formation leads to changes in Gαs that are similar to those observed in the β2AR-Gs complex (Chung et al., 2011). A stable nucleotide-free A2A-Gs complex was formed under conditions previously used to study the β2AR-Gs complex (Figure S2) (Chung et al., 2011; Rasmussen et al., 2011). This involved incubation of GPCR and Gs for 90 min followed by the addition of apyrase to hydrolyze released GDP and incubation for an additional 90 min. The GDP-bound Gs or the A2A-Gs complex were incubated in the D2O buffer for 10, 100, 1000, or 10,000 sec at room temperature, and the HDX levels were analyzed. The HDX changes in Gαs upon forming a complex with the A2A (Figure S3A) are similar to those we previously observed with the β2AR-Gs complex (Figure S3B) (Chung et al., 2011); the differences between Figure S3A and S3B are likely due to different peptic peptides generated by different pepsin columns and the use of different LC-MS systems to analyze the peptides (Figure S3 legend).
To complement HDX-MS data on Gs, we sought to monitor HDX changes in the β2AR and the A2A. In the previous continuous labeling HDX-MS study with β2AR-Gs, the sequence coverage of the β2AR was only approximately 12%, and we could not obtain significant conformational information (Chung et al., 2011). In the present study, we were able to observe HDX data for ~65% of the β2AR with majority of cytoplasmic regions covered except for the C-terminal part of ICL3 and the TM7/H8 hinge (Figure S3C). With continuous labeling HDX-MS, ICL2 and ICL3 of the β2AR showed decreased HDX upon interaction with Gs (Figure S3C) reflecting α-helix formation after interaction with Gs (Figure 1B). TM4 underwent a slight increase in HDX, while helix 8 (H8) showed a slight decrease in HDX upon interaction with Gs (Figure S3C). Sequence coverage of the A2A was inadequate to assess conformational changes in this receptor by HDX.
Time-resolved structural changes in the β2AR during GPCR-Gs coupling
To study the process of GPCR-Gs complex formation, we rapidly mixed the detergent-solubilized GPCR (A2A or β2AR) with purified Gs at a 1.15:1 stoichiometry, then monitored complex formation using pulse-labeling HDX-MS or HRF-MS techniques (Figure S1C). Our ability to control the time and stoichiometry of mixing necessitated using detergent-solubilized GPCRs and Gs as was employed for their X-ray crystal structure determinations and prior GPCR HDX and HRF studies (Angel et al., 2009; Orban et al., 2012). Nevertheless, complex formation is efficient under these conditions, and we observed complete [3H]GDP release within 10 seconds, the earliest time point that can be reliably measured (Figure 2A). Assuming GDP release is an exponential process, the t1/2 for GDP release would be less than 2 seconds.
Figure 2. Time-resolved analysis of GPCR-Gs complex formation by HDX-MS.
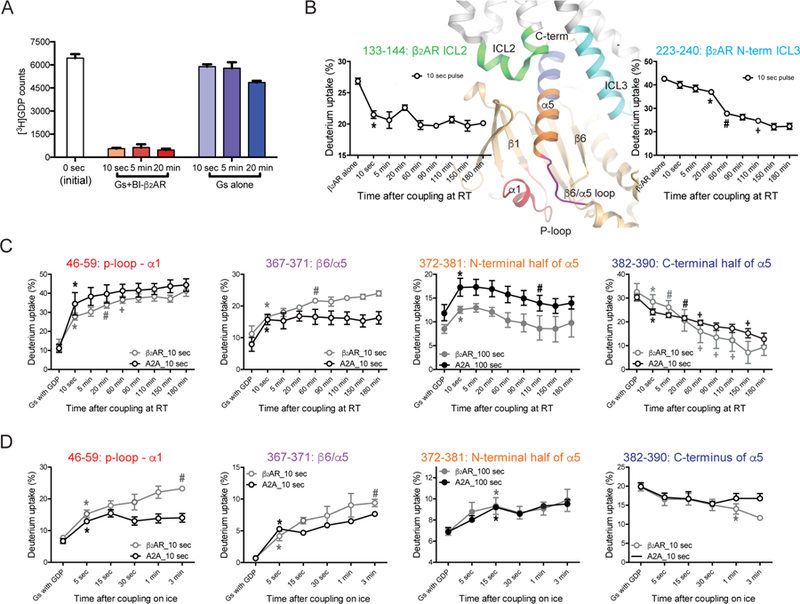
(A) GDP release was complete within 10 sec of incubation with BI-167107-bound β2AR.
(B–D) HDX profiles of selected peptides from the β2AR (B) and Gαs (C and D). The analyzed peptides are indicated as colored regions on the X-ray crystal structure of the β2AR-Gs complex (PDB: 3SN6). The HDX profile changes of the β2AR upon incubation with Gs at the room temperature are analyzed by a 10 sec D2O pulse (B). HDX profile changes of Gαs upon co-incubation with the β2AR or A2A at the room temperature (C) or on ice (D) were analyzed with 10 sec or 100 sec D2O pulses. Statistical significance of the incubation time dependent HDX changes were analyzed by repeated measures ANOVA (rANOVA), and the results from rANOVA analysis are presented in Table S1. To compare time points, a paired t-test was used and p<0.05 was considered to be statistically significant. *, the first incubation time point that shows statistical difference from the β2AR alone or the GDP-bound state of Gs. #, the first incubation time point that shows statistical difference from the * time point. +, the first incubation time point that shows statistical difference from the # or previous + time point. Error bars represent the s.e.m. Please note that the data is plotted using a non-linear/non-logarithmic scale.
We determined the shortest possible D2O pulse to minimize conformational changes during the pulse. Based upon the continuous labeling HDX-MS data (Figure S3), 10 sec or 100 sec of D2O incubation at room temperature allowed us to observe clear HDX differences between the GDP-bound Gs and the receptor-Gs complex. Thus, we chose 10 and 100 sec deuterium pulses for the pulse-labeling HDX-MS. We examined the HDX behavior of the receptor and Gs before incubation and at 10 sec, 5 min, 20 min, 60 min, 90 min, 110 min, 150 min, and 180 min after co-incubation of the receptor and Gs at room temperature (Figure 2B and 2C) or at 5 sec, 10 sec, 30 sec, 1 min, and 3 min after co-incubation on ice (Figure 2D). For incubations at room temperature, apyrase was added at 90 min to hydrolyze released GDP. It should be noted that a few coupling time points (i.e. 10 sec at room temperature and 5, 15, 30, and 60 sec on ice experiments) are shorter than the 100 sec D2O pulse duration; therefore, we used the 10 sec pulse data except for the N-terminal half of the α5 helix (peptide 372–381) because the 10 sec deuterium pulse was not enough to detect deuterium uptake differences between GDP-bound Gs and receptor-bound Gs (Figure S3A).
To monitor the conformational changes at very early time points, we employed HRF-MS and investigated the radiolytic oxidation profiles of protein side chains at 20, 40, 80, 150, 400, 800, 1,500, 5,000, and 10,000, and 30,000 ms after mixing of the receptor and Gs (Figure 3A). For each of these time points, the duration of exposure to X-rays was approximately 50 µs; this dose was optimized to permit sufficient labeling while assuring minimal overall perturbation of the protein. Due to the inherent delay volume of the stopped flow mixing device employed for HRF studies, we were unable to reliably observe time points prior to 20 ms as full mixing must occur prior to X-ray exposure.
Figure 3. Time-resolved analysis of GPCR-Gs complex formation by HRF-MS.
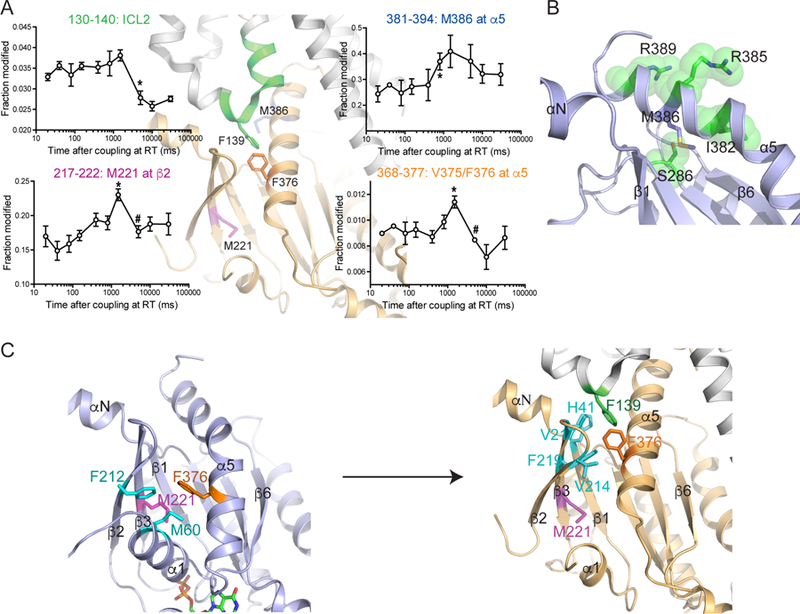
(A) X-ray generated radiolytic oxidative modification profiles of selected peptides or residues from the β2AR or Gαs. Oxidative modification changes of Gαs upon incubation with the β2AR were analyzed. The modified peptides or residues are indicated as colored regions or sticks on the X-ray crystal structure of the β2AR-Gs complex (PDB: 3SN6). Statistical significance of the incubation time dependent changes were analyzed by rANOVA, and the results from rANOVA analysis are presented in Table S1. To compare time points, a t-test was used and p<0.05 was considered to be statistically significant. *, the first incubation time point that shows statistical difference from the β2AR alone or the GDP-bound state. #, the first incubation time point that shows statistical difference from the * time point. Error bars represent the s.e.m. The time series of M386 (381–394) is not significant by rANOVA (p=0.110), but t-test showed that 800 ms is significantly different from initial 20 ms (p=0.03). This is not to say that the t-test is incorrect, only that the entire series is too variant in spread at each time point to support a statement that the entire series is significant to a reasonable α value.
(B) The surrounding environment of M386. In the GDP-bound Gs structure, M386 is located within a pocket formed by four amino acids (green spheres) with limited solvent exposure
(C) Rearrangement of interactions with M221 and F376 of Gas following formation of the nucleotide-free β2AR-Gs complex. In the GDP-bound Gs structure, M221 and F376 form interactions with residues within β2-β3 strands and α1 helix (left), which are lost in the β2AR-bound nucleotide-free structure (PDB: 3SN6) (right). In the β2AR-bound nucleotide-free structure (PDB: 3SN6), F376 forms new interactions with F139 of the β2AR and amino acids in the αN/β1 hinge and β2/β3 loop (right).
When the β2AR is incubated with Gs at room temperature, the HDX level of ICL2 decreases within 10 sec of incubation (Figure 2B, peptide 133–144); consistent with this hydroxyl radical modifications of ICL2 decrease between 1,500 and 5,000 ms of incubation (Figure 3A, peptide 130–140). In contrast, the HDX level of the N-terminus of ICL3 decreases gradually over 110 min (Figure 2B, peptide 223–240). This data suggests that ICL2 undergoes conformational changes much earlier than the N-terminus of ICL3 when β2AR interacts with Gs. Unfortunately, we were unable to get an HRF-MS kinetic profile of ICL3. The decrease in HDX in ICL2 most likely reflects the transition from an unstructured loop (observed in the inactive state crystal structure) to an α-helix and interaction with Gs as observed in the β2AR-Gs crystal structure (Figure 1B). The decline in HDX at the N-terminal end of ICL3 is consistent with the extension of the α-helix from TM5 observed in the β2AR-Gs structure (Figure 1B).
Time-resolved structural changes in Gas during GPCR-Gs coupling
The pulse-labeling HDX profiles of Gαs are monitored after stimulation by β2AR or A2A (Figures 2C and 2D). The regions near the nucleotide-binding pocket (P-loop and β6/α5 loop: peptides 46–59 and 367–371) of Gαs showed rapid and almost complete HDX changes within 10 sec of co-incubation of Gs with the receptors at room temperature (Figure 2C), which is on a timescale similar to ICL2 of the β2AR (Figure 2B). Of note, the conformational changes of Gαs in these regions show slightly different HDX profile change trends between β2AR and A2A stimulation; A2A induced complete conformational change within 10 sec while β2AR induced a relatively large conformational change within 10 sec followed by small changes until 60 min (Figure 2C). The rapid HDX level increased within 10 sec in these peptides, which reflects the empty nucleotide-binding pocket in the GPCR-Gs complex, consistent with rapid [3H]GDP release from Gs following addition of the purified agonist-bound β2AR under conditions similar to those used for the HDX-MS experiments (Figure 2A). Similarly, other regions near the nucleotide-binding pocket and the interfaces between the Ras-like and the helical domains (i.e., Switch III and αG through the αG/α4 loop) also underwent rapid HDX profile changes (Figure S4B), consistent with the hypothesis of a rapid release of GDP. The differences between β2AR and A2A-induced Gαs conformational changes in peptides 46–59 and 367–371 at later time points may be due to differences in the receptor amino acids that interact with Gs or to differences in the extent to which agonists stabilize the active conformation.
In contrast to the rapid changes observed in the nucleotide-binding pocket, the maximal changes in deuterium uptake occurred on a much slower timescale in the C-terminal half of the α5 helix (peptide 382–390; note that the very last 4 residues at the C-terminus were not detected). We observed a gradual decline in HDX until 110–150 min after co-incubation of Gs with the receptors at room temperature (Figure 2C). Interestingly, this HDX profile of the C-terminal half of the α5 helix is similar to that of the N-terminus of ICL3 of the β2AR (Figure 2B), suggesting that the N-terminus of ICL3 of the β2AR and the C-terminal half of the α5 helix of Gαs undergo conformational changes on a similar timescale. The slow and continuous decrease of HDX levels of these regions may reflect slow and continuous stabilization of the peptide backbone by forming α-helices as observed in the high-resolution structure of the receptor-Gs complex (Figure 1B).
We incubated the receptor and Gs on ice to slow the complex formation in an effort to monitor earlier events. Size exclusion chromatography and bimane assays confirmed that GPCR-Gs complexes can be formed at 0°C (Figure S2). While the peptides from the nucleotide-binding pocket showed an increase in HDX levels within 5–15 sec incubation with receptor on ice, the C-terminal half of the α5 helix showed no HDX profile changes up to 1–3 min incubation with receptors (Figure 2D). This data confirms that HDX profile changes occur faster in the nucleotide-binding pocket than the C-terminal half of the α5 helix.
In contrast to the slow HDX changes at the C-terminal half of the α5 helix, HDX level change at the N-terminal half of the α5 helix (peptide 372–381) occurred more rapidly (i.e. increase in HDX level within 10 sec at room temperature and 15 sec on ice) (Figures 2C and 2D). Of note, we observed significant changes only with the 100 sec pulse, as the 10 sec pulse is too short to detect the differences between GDP-bound Gs and receptor-bound Gs (Figure S3A). This region is particularly interesting as it contains F376 that forms part of the hydrophobic pocket that interacts with F139 in ICL2 of the β2AR (Figure 1D), and therefore the altered HDX may reflect the allosteric conformational changes that establish interactions with F139 in ICL2. Unfortunately, we could not obtain peptide mass spectra from the αN/β1 hinge through β1 strand (Figure S3A), the most direct allosteric link between ICL2 and the P-loop. Nevertheless, the N-terminal part of the αN helix showed rapid increase of HDX levels upon co-incubation of the receptor and Gs on a timescale similar to that observed for changes in ICL2 of the β2AR (Figures S5 and 2B).
To characterize conformational changes at timescales inaccessible to HDX-MS, we employed time-resolved HRF-MS. With HRF-MS, we were able to detect time-dependent changes in oxidation of residues from multiple sites on Gαs (i.e. M221, V375/F376, and M386) (Figure 3A). The earliest HRF changes were observed in M386, a residue located at the C-terminal half of the α5 helix (Figure 3B). As discussed in more detail below, in GDP-bound Gs, this end of the α5 helix folds over the aN-b1 junction (Liu et al., companion manuscript), stabilizing the GDP-binding pocket. M386 is located in a pocket with limited solvent exposure in GDP-bound Gs based on the crystal structure (Figure 3B). In HRF studies, M386 shows a significant increase in oxidative modification from 20–800 ms. This increase in reactivity observed within 800 ms suggests that interactions of the C-terminus of Gαs with the β2AR dislodges the M386 side chain from this pocket over this time period. While these changes appear to be inconsistent with the pulse-labeling HDX-MS result of the peptide 382–390, where this peptide showed a slow and continuous decrease in HDX (Figure 2C), it should be noted that because HDX-MS measures average HDX at the backbone amides of the whole peptide while HRF-MS is reporting oxidative labeling at the side chain of a specific residue. Thus, this discrepancy suggests that C-terminal half of the α5 helix undergoes a two-phase conformational change; from initial encounter with β2AR up to 800 ms, the α5 helix undergoes conformational changes which result in the exposure of the M386 side chain to solvent, while stable helix formation of the C-terminus of α5 helix occurs over a longer timescale.
M221 and V375/F376 showed similar HRF-MS profiles; the oxidative labeling increased to a maximum within 1,500 ms followed by a sharp decrease (Figure 3A). In the GDP-bound inactive state (Liu et al., companion manuscript), M221 and F376 form part of a hydrophobic pocket linking the N-terminus of the α5 helix with the β2-β3 strands and the α1 helix (Figure 3C left). Upon binding to the receptor, this hydrophobic pocket undergoes a major structural change due to the movement of the α5 helix. After this conformational change, M221 establishes new hydrophobic interactions with residues in β1, β2 and β3 strands and the α1 helix, while F376 moves to form part of a hydrophobic pocket with F139 of ICL2 and V214 and V217 in the β2/β3 loop and F219 at the N-terminal end of the β3 strand (Figure 3C right). This data suggests that the transient increase in oxidative modification of M221 and F376 at 1,500 ms (Figure 3A) may be due to a transient increase in exposure to solvent as the G protein transitions from inactive to active states. Consistent with this hypothesis, the decrease in oxidative modification of these residues between 1,500 and 5,000 ms correlates with the decrease in oxidative labeling of the β2AR ICL2 over the same time period (Figure 3A), which may reflect the formation of α-helix and engagement of F139 of the β2AR ICL2 with the hydrophobic pocket containing F376 on the α5 helix of Gαs.
Interactions with the C-terminus of Gas are necessary but not sufficient for activation
The studies presented above suggest that the C-terminal half of the α5 helix (containing M386) initially interacts with the receptor; however, the C-terminus of the α5 helix remains dynamic (as reflected in high HDX) long after GDP release is complete. Thus, the interaction between the β2AR and the α5 helix observed in the β2AR-Gs crystal structure may not reflect the initial interactions that trigger GDP release. To confirm the importance of the C-terminus of the α5 helix in our biochemical system, we truncated the last 5 residues from Gs (Gs_∆5). As expected from previous studies (DeMars et al., 2011), we observe no receptor-mediated GDP release with the truncated G protein (Figure 4A). Moreover, we cannot detect any interaction between the β2AR and the truncated Gs in a spectroscopic assay that monitors conformational changes in TM6 (Figures 4B, discussed below). However, we do observe a small, but significant decrease in agonist binding affinity (Figure 4C), suggesting that Gs may interact with the β2AR through interactions that do not involved the C-terminal α5 helix.
Figure 4. Functional analysis of the roles of Gαs C-terminus and β2AR ICL2.
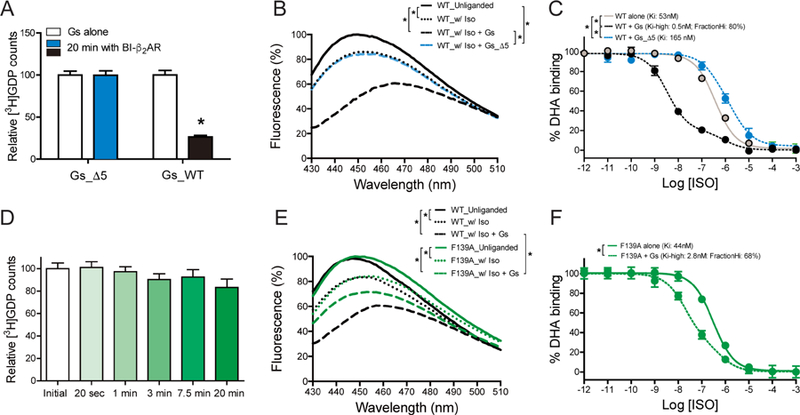
(A–C) Effect of Gαs C-terminal five-residue truncation (Gs_∆5) on β2AR-induced GDP release activity (A), Gs-stabilized movement of TM6 of the β2AR (B), and agonist affinity change (C). The C-terminal five-residue truncation mutant failed to release GDP upon co-incubation with the β2AR (A), failed to induce a bimane fluorescence change (B), and failed to stabilize the high-affinity agonist-binding state in the β2AR (C).
(D–F) Effect of β2AR F139A mutation on β2AR-induced GDP release from Gs (D), Gs-induced movement of TM6 of the β2AR (E), and agonist affinity change (F). β2AR F139A cannot catalyze the release of GDP from Gs (D). Changes in bimane fluorescence (B) and agonist affinity (E) suggest that β2AR F139A forms a ternary complex with Gs, but the conformation is different from that observed with the wild-type β2AR.
The data represent the mean ± SD of three independent measurements. Statistical significance of data in Figures 4A, 4B, 4C, and 4E were analyzed by one-way ANOVA followed by Tukey’s post-test, Figure 4D by rANOVA followed by Tukey’s post-test, and Figure 4F by unpaired t-test. *p value of <0.01. Figures 4B and 4E are representative traces from three independent experiments.
We next sought to determine the importance of interactions between Gs and ICL2 in GDP release. It has previously been shown that mutation of F139A impaired cAMP accumulation in cultured cells (Yano et al., 2017). As shown in Figure 4D, the F139A mutant was unable to trigger GDP release. To determine if β2AR-F139A could still couple to the C-terminus of Gs, we examined the effect of Gs on the fluorescence intensity and λmax of the β2AR labeled at C265 with monobromobimane (denoted hereafter as bimane), an environmentally sensitive fluorophore (Yao et al., 2009). We have previously shown that bimane covalently bound to C265 at the cytoplasmic end of TM6 (bimane-labeled β2AR) is very sensitive to both agonist and G protein stabilized conformational changes. As shown in Figure 4E, the addition of isoproterenol to bimane-labeled β2AR and bimane-labeled β2AR-F139A resulted in a decrease in intensity and a red-shift in λmax. The addition of Gs to wild-type β2AR led to a further decrease in intensity and further shift in λmax (Figure 4E). The addition of Gs to the bimane-labeled β2AR-F139A led to a decrease in intensity, but no change in λmax (Figure 4E). This observation suggests that Gs (probably the C-terminus of α5 helix) binds the β2AR-F139A mutant, but stabilizes a different conformation at the end of TM6. To further examine the effect of the F139A mutant on coupling to Gs, we examined the ability of Gs to stabilize a high-affinity state in wild-type β2AR and β2AR-F139A. Figures 4C and F show that Gs stabilized a high-affinity state in both receptors, but the effect was greater in wild type β2AR (Kihigh = 0.5 nM, 80% high-state) than in β2AR-F139A (Kihigh = 2.8 nM, 68% high-state). Therefore, the binding results are in agreement with the bimane studies; β2AR-F139A can bind Gs through interactions between the α5 helix of Gαs and the TM core of the receptor, but cannot trigger GDP release. These results are also in agreement with recent studies that monitored conformational changes in TM6 of β2AR using single-molecule fluoresce resonance energy transfer (FRET) and revealed an intermediate conformation of TM6 in the presence of GDP-bound Gs (Gregorio et al., 2017).
Sequence of Gs activation
The studies presented suggest a sequence of events leading to nucleotide release (Figure 5A). Agonist-bound β2AR initially engages the C-terminal half of the α5 helix including the relatively flexible last 5 residues of the C-terminal tail of GDP-bound Gαs (Figure 5A i-ii). We do not have any coverage for the C-terminal tail by either HDX-MS or HRF-MS, but it is likely to be required for the initial interaction with the β2AR transmembrane core since, as discussed above, without these amino acids (Gs_∆5), we cannot detect any receptor mediated GDP release or an interaction between the β2AR and Gs_∆5 in the bimane assay (Figures 4A–4C). The first event detected by HRF-MS is an increase in HRF modification of M386 first detected by 800 ms (Figure 3A). This increase in reactivity suggests that interactions of the C-terminal half of the α5 helix with the β2AR dislodges the M386 side chain from a pocket thus exposing it to solvent (Figure 3B). Interaction with the β2AR may also destabilize other interactions between the α5 helix and the αN/β1 hinge and β sheet (Figure 5A ii). In the crystal structure of GDP-bound Gs (Liu et al., companion manuscript), interactions between amino acids in the α5 helix with amino acids in the αN/β1 hinge and β sheet stabilize the GDP-binding pocket (Figure 5B). Disruption of these interactions may be sufficient to destabilize the nucleotide binding pocket. Consistent with this hypothesis, mutation of H387 and Q390 to Ala, which should disrupt two of these stabilizing interactions, led to a 4-fold increase in basal GDP release in Gαs (Liu et al., companion manuscript).
Figure 5. Proposed model for Gs activation by the β2AR.
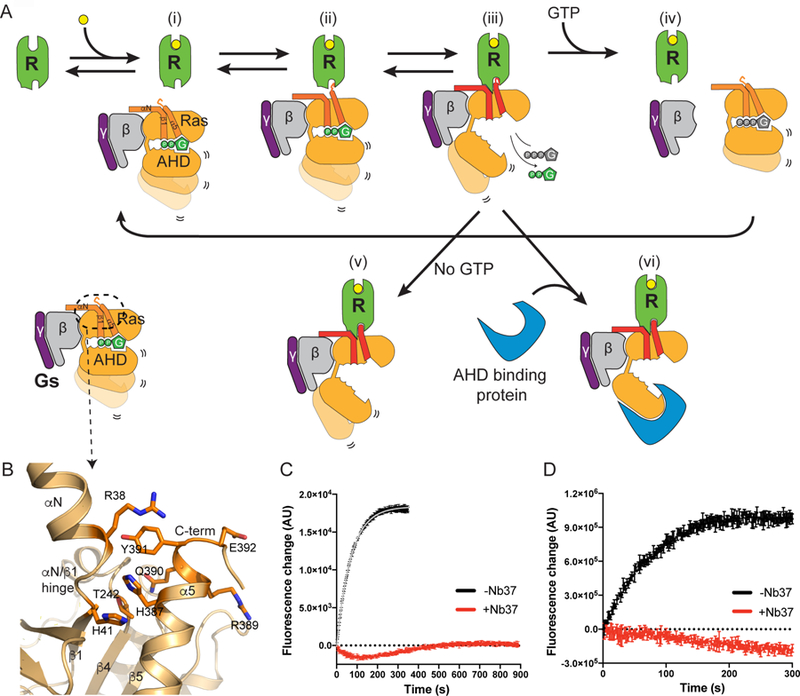
(A) Summary cartoon illustrating proposed sequence of events during GPCR-Gs complex formation and GDP release from Gα subunit. Ras indicates Ras-like domain and AHD indicates α-helical domain.
(B) Intra molecular interactions between αN/β1 hinge and α5 in the GDP-bound Gs. R389 in and E392 are solvent exposed and available for interactions with agonist-bound β2AR.
(C) Effect of Nb37 on the kinetics of GTPγS-induced complex dissociation assayed by bimane fluorescence. The data shows a representative of three independent experiments.
(D) Effect of Nb37 on the kinetics of BODIPY-FL GTPγS binding of the β2AR-Gs complex. The data shows a representative of three independent experiments.
HRF shows a transient increase in oxidative labeling of M221 and F376 that peaks at 1,500 ms (Figure 3A), suggesting a transient increase in solvent exposure for M221 and F376. This may occur during the upward movement of the α5 helix into the core of the receptor (Figures 3C and 5A iii). This is followed by changes in ICL2 consistent with formation of α-helical structure that would favor insertion of F139 in ICL2 into the hydrophobic pocket formed by the α5 helix and the β sheet. These changes would further destabilize the interactions of GDP with the P-loop and TCAT motif leading to GDP release, which we observe to be complete within 10 sec (Figure 2A), followed by GTP binding and complex dissociation (Figure 5A iii-iv).
Our results suggest that the initial engagement between the β2AR and GDP-bound Gs may involve different amino acid interactions than observed in the X-ray crystal structure of the nucleotide-free β2AR-Gs complex. We recently obtained a structure of the β2AR in complex with the last 14 amino acids of the α5 helix (Liu et al., companion manuscript). In this structure, R389 and E392 of the α5 helix engage D1303.49 and R1313.50 of the β2AR ionic lock, while R389 and E392 are solvent exposed in the β2AR-Gs structure. Of interest, in the β2AR-GsGDP structure, R389 and E392 face the solvent (Figure 5B, Liu et al., companion manuscript) and are therefore more readily accessible to engage agonist-bound β2AR.
The most unexpected finding in our study is the very slow rate at which the distal α5 helix and the cytoplasmic end of TM5 undergo conformational changes consistent with the nucleotide-free β2AR-Gs structure (Figure 2B and C, and 5A v). In this structure, residues 382–390 are an α-helix and buried in the core of the receptor, consistent with the low rate of deuterium exchange observed with the continuous labeling HDX-MS and pulse-labeling HDX-MS experiments after 150 min incubation with the receptor (Figures 2C and S3A). A similar helical structure is observed in the A2A-miniGs structure and the structures of the CT-Gs and the GLP1R-Gs complexes (Carpenter et al., 2016; Liang et al., 2017; Zhang et al., 2017). It should be noted that all of these complexes were obtained after prolonged incubation in the presence of apyrase that hydrolyzes free GDP, resulting in a nucleotide-free G protein subunit. Based on our studies, we would not expect to observe a complex represented by the crystal structure of the β2AR-Gs complex at physiological concentrations of GDP and GTP (36 mM and 305 mM in humans, respectively) (Traut, 1994).
The α-helical domain (AHD) plays a role in GDP binding and GTP hydrolysis. When the AHD and Ras-like domains are expressed as separate proteins, GTPγS binding to the Ras-like domain is dependent on co-incubation with the AHD (Markby et al., 1993). It is possible that a cytosolic protein could bind to the AHD and prevent it from closing (Figure 5A vi). This could alter the affinity for GTP and thereby slow GTP-mediated dissociation. To test the hypothesis that preventing closure of the AHD could prolong the stability of the β2AR-Gs complex in the presence of GTP, we monitored GTP-induced complex dissociation in the presence or absence of nanobody 37 (Nb37). Nb37 has previously been shown in negative-stain electron microscopy studies to bind to the tip of the AHD preventing closure (Westfield et al., 2011). To monitor complex dissociation, bimane-labeled β2AR on C265 was used. The binding of Gs to agonist-occupied bimane-labeled β2AR led to a decrease in intensity and a red-shift in λmax (Figure 4B). The addition of GTP led to dissociation of the β2AR-Gs complex resulting in a rapid increase in bimane fluorescence indicating a dissociation half-life of 48 sec (Figure 5C). In the presence of Nb37, we observed no dissociation over 900 sec. Moreover, we could not detect binding to the fluorescent GTP analog BODIPY-GTPγS (Figure 5D). Thus, it is possible that a cytosolic protein binding to the AHD may prevent the closed conformation and therefore prolong the nucleotide-free conformation. There are currently no proteins known to bind to the AHD of Gs in the nucleotide-free state, so the significance of this observation is not known.
In conclusion, we used a combination of two complementary time-resolved MS techniques (HDX-MS and HRF-MS) to monitor the structural dynamics in the G protein Gs upon coupling with the β2AR to understand the functional path of signaling. The initial engagement with the carboxyl-terminal end of the α5 helix is necessary, but not sufficient to promote GDP release. Interaction with F139 in ICL2 is a crucial step for GDP release and probably necessary for the stabilization of the nucleotide-free β2AR-Gs complex. As a result of these findings, we suggest that the conformation of the initial β2AR-Gs structure is likely to differ from that observed in the high-resolution structure of the stable nucleotide-free β2AR-Gs complex, and this initial transient interaction may be critical for determining coupling selectivity. Due to the limited sequence coverage of the β2AR, we could not determine the initial Gs-contacting site on the receptor and further studies are needed to precisely map the interfaces during the initial transient interaction.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Brian K. Kobilka (kobilka@stanford.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human β2AR and A2A receptors were expressed in Sf9 insect cells infected with BestBac recombinant baculovirus (Expression Systems). Human Gs heterotrimer was expressed in Trichoplusia ni insect cells (Expression Systems). Human mutant Gαs was expressed in BL21 (DE3) bacterial strain.
METHODS DETAILS
Expression and purification of Gs
Bovine Gαs short, His6-rat Gβ1 and bovine Gγ2 were co-expressed in Trichoplusia ni insect cells grown in ESF 921 media (Expression Systems). One-liter cell cultures were grown to a density of 3 million per ml and then infected with two separate Autographa californica nuclear polyhedrosis viruses containing the Gαs and Gβγ genes. The optimal mixing ratio between the viruses was determined by SDS-PAGE and Coomassie Blue staining following small scale expression and purification. After 48 hrs of incubation the infected cells were harvested by centrifugation and frozen at −80°C until use. Cells were resuspended in 400 ml ice-cold lysis buffer (10 mM Tris pH 7.4, 100 µM MgCl2, 5 mM β-ME, 10 µM GDP, 2.5 µg/ml leupeptin and 160 µg/ml benzamidine) for 30 min at RT or until pellets thawed. The lysate was spun for 15 min at 18,000 g, and then pellet was homogenized in 300 ml solubilization buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 1% sodium cholate, 0.05% dodecylmaltoside (DDM), 5 mM MgCl2, 2 µl calf intestinal alkaline phosphatase (CIP), 5 mM β-ME, 10 µM GDP, 5 mM imidazole, 2.5 µg/ml leupeptin, and 160 µg/ml benzamidine) using a 100-ml Dounce-homogenizer and the tight pestle. The sample was stirred for 40 min at 4°C and then spun 20 min at 18,000 g to remove insoluble debris. Five ml pre-equilibrated nickel bound Chelating Sepharose Fast Flow was added to solubilized supernatant, the mixture was incubated on rotation for another 2 hrs at 4°C. The Gs-bound resin was collected by fast spin for 10 min at 4,000 g and washed with solubilization buffer 3 times in a 50 ml conical tube. Next, two kinds of exchange buffers were prepared: E1 buffer (solubilization buffer plus 15 mM imidazole); E2 buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 0.1% DDM, 1 mM MgCl2, 5 mM β-ME, 10 µM GDP, 20 mM imidazole and 2.5 µg/ml leupeptin and 160 µg/ml benzamidine). Washing buffer was made by mixing ice-cold E1 and E2 buffer in the following ratios: 10ml:10ml, 5ml:15ml, 2ml:18ml, 1ml:19ml, respectively. The detergent-exchange buffers were flowed over the nickel resin at 60 ml/hr (20 min per exchange). The protein was eluted in E2 buffer supplemented with 250 mM imidazole, and 5 µl λPPase, 1 µl CIP, and 1 µl Antarctic Phosphatase (NEB) was added and treated for 1 hr at 4°C. The eluate was diluted two-fold to decrease imidazole concentration, passed through a 0.22-µm filter, and loaded onto a pre-equilibrated 2 ml Q Sepharose resin. The resin was washed in 15 ml buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.1% DDM, 1 mM MgCl2, 100 µM TCEP, 10 µM GDP) at 30 ml/hr at 4°C. The protein was eluted in buffer with reduced DDM concentration (20 mM HEPES pH 7.5, 350 mM NaCl, 0.036% DDM, 1 mM MgCl2, 100 µM TCEP, 10 µM GDP). The fractions containing Gs was pooled and diluted three-fold in (20 mM HEPES pH 7.5, 1.15 mM MgCl2, 1.5 mM EDTA, 100 µM TCEP, 1 µM GDP) in a drop-wise manner to adjust final buffer concentration to 20 mM HEPES pH 7.5, 115 mM NaCl, 0.012% DDM, 1.1 mM MgCl2, 1 mM EDTA, 100 µM TCEP, 4 µM GDP. The protein was passed through a 0.22-µm filter and spin-concentrated in a 10-kDa MWCO concentrator (Millipore) to approximately 20 mg/ml. Glycerol was added to 20 % and the protein was flash frozen in liquid nitrogen and stored at −80°C. To further isolate α subunit from G β1γ2 subunits expressed in insect cells for GDP release assay, the purified G protein heterotrimers are loaded onto Nickel-NTA resin, incubated with 10 mM NaF, 30 µM AlCl3, and 10 mM MgCl2 for 2 hr at 4°C, and the flow-through collected containing untagged α subunit. The expression and purification of lipidated Gβ1γ2 is very similar to Gs heterotrimer, except GDP and MgCl2 are removed from the buffer formulation.
Expression and purification of mutant Gαs
Recombinant Gαs containing N-terminal His-tag and HRV 3C protease cleavage site were constructed in pET28a. Gαs_∆5 was made by site-directed point mutation using PCR. The wild-type and mutant construct were transformed into Escherichia coli BL21 (DE3) for protein expression. The cells were grown in Terrific Broth in the presence of antibiotic at 37°C until OD600 reached 0.6 – 0.8. Protein expression was induced by adding 0.5 mM IPTG, and the cells were harvested after incubation at 25°C for 18 hrs. For protein purification, the bacterial pellets were suspended in the lysis buffer (20mM HEPES pH 7.4, 300 mM NaCl, 2 mM MgCl2, 30 µM GDP, 3 mg/ml lysozyme, 1:1000 protease inhibitor cocktail, 2.5 µg/ml leupeptin, 10 µg/ml benzamidine, 100 µM TCEP, 15% glycerol) and incubated for 30 min at room temperature. The lysates were spun down at 13,000 g for 30 min at 4°C, and the supernatant were collected and loaded into a Ni-NTA column pre-equilibrated with the lysis buffer supplemented with 20mM imidazole. The resin was washed by wash buffer (20mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 20 µM GDP, 100 µM TCEP, 20mM Imidazole). The bound proteins were eluted with elution buffer (20mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 20 µM GDP, 100 µM TCEP, 250 mM Imidazole). The Ni-NTA purified protein fractions were cleaved to remove the His-tag by incubation with 3C protease at 4°C overnight. The cleaved products were loaded onto a Superdex-200 (10/300) column equipped in an ÄKTA FPLC system (GE Healthcare Life Sciences, Sweden), and the purified proteins were eluted with second elution buffer (20mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 20 µM GDP, 100 µM TCEP). Protein fractions were collected by monitoring the absorbance at 280 nm.
Expression and purification of the β2AR and F139A mutant
Recombinant baculovirus was prepared using Bestbac expression system (Expression Systems) by using pVL1392 as transfer vector. The full-length β2AR was expressed by infecting Sf9 cells at a density of 4.5 million per ml with second-passage baculovirus stock using 20 ml of virus stock per 1 liter of cell culture. 2 µM alprenolol (a β2AR antagonist) was added to stabilize the receptor during expression. The infected cells were harvested after 48 hrs of incubation at 27°C. Cell pellets were lysed by stirring in lysis buffer (20 mM HEPES pH 7.5, 5 mM EDTA, 1 µM alprenolol, 2.5 µg/ml leupeptin and 160 µg/ml benzamidine) at 10 ml of buffer per gram of cell pellet for 15 min. The receptor was extracted from the cell membrane by using a douncing homogenization in solubilization buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 1% DDM, 1 µM alprenolol, 2.5 µg/ml leupeptin and 160 µg/ml benzamidine) for 1 hr at room temperature. Ten ml of solubilization buffer was added per gram of cell pellet. After further addition of 2 mM CaCl2, the solubilized receptor was clarified by high-speed centrifugation at 18,000 g for 30 min. The receptor bearing the N-terminal FLAG tag was then captured by M1 antibody affinity chromatography (Sigma). The column was extensively washed with HMS-CHS buffer (20 mM HEPES pH 7.5, 350 mM NaCl, 0.1% DDM, 0.01% cholesterol hemisuccinate) plus 2 mM CaCl2 to remove impurities and alprenolol. The receptor was then eluted with HMS-CHS buffer supplemented with 5 mM EDTA and 200 µg/ml free FLAG peptide. The protein sample will be frozen for later use. FLAG-pure receptor was thawed in water bath, and the sample was further purified by affinity chromatography using alprenolol-Sepharose as previously described (Rosenbaum et al., 2007) to select functional receptors. HMS-CHS buffer supplemented with 300 µM alprenolol was used to elute the protein. The eluted receptor with alprenolol around was tandem linked to M1 FLAG column, and washed with HMS-CHS buffer for removal of alprenolol to achieve unliganded receptor. The receptor was then eluted from M1 resin with HMS-CHS buffer supplemented with 5 mM EDTA, 200 µg/ml free FLAG peptide, 10 µM BI-167107. Size-exclusion chromatography (SEC) with Superdex-200 column (GE healthcare) equilibrated in HLS-CHS buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% DDM, 0.01% cholesterol hemisuccinate, 2 µM BI-167107) was finally used to polish the receptor. BI-167107 is a high-affinity β2AR agonist which was used to define the high-resolution crystal structure of β2AR (Rasmussen et al., 2011). The receptor was concentrated to 150 µM for preparing samples for HDX-MS studies. The purity of sample was higher than 95%, as assessed by SDS-PAGE.
Expression and purification of the A2A
The A2A was cloned into the pVL1393 vector containing an N-terminal signal sequence and FLAG epitope and a C-terminal 9His tag. The BestBac expression system was used to generate recombinant baculovirus of the construct through co-infection of Sf9 insect cells grown in ESF 921 media (Expression Systems). One-liter Sf9 cells grown to densities of 3.5 milion per ml in 2800 ml Erlenmeyer flasks shaking at 130 rpm at 27°C were infected with 20 ml of a P2 virus stock prepared according to Expression Systems virus amplification protocol. The infected cultures were harvested after 48 hrs by centrifugation for 15 min at 4,000 g. Yields are typically around 20 ml pelleted cells per liter culture. The pelleted cells were frozen at −80°C until use. Cells were thawed out in 400 ml ice-cold lysis buffer (10 mM HEPES pH 7.5, 20 mM KCl, 10 mM MgCl2, 10 µg/ml leupeptin and 160 µg/ml benzamidine) and homogenized with 25 strokes using a 100-ml Dounce homogenizer and the tight pestle. The homogenized material was pelleted by centrifugation at 18,000 rpm for 20 min at 4°C in a Sorvall RC6+ centrifuge. The pelleted membranes were resuspended in 200 ml lysis buffer, dounce homogenized, and pelleted by centrifugation again. The membrane washing procedure was repeated two more times in 200 ml high salt buffer (1M NaCl, 20 mM KCl, 10 mM HEPES pH7.5, 10 mM MgCl2, 10 µg/ml leupeptin and 160 µg/ml benzamidine). The membranes were resuspended in a cryo buffer (10 mM HEPES pH 7.5, 20mM KCl, 10 MgCl2, 40% glycerol) containing 4 mM theophylline, then flash frozen in liquid nitrogen and stored at −80°C until use. Thawed out membranes were stirred for 1 hr at 4°C in 300 ml ice-cold solubilization buffer (1 % DDM, 0.1% CHS, 20 mM HEPES pH 7.5, 800 mM NaCl, 10% glycerol, 4 mM theophylline, 10 µg/ml leupeptin and 160 µg/ml benzamidine, 2 mg/ml iodoacetamide, 100 µM TCEP) followed by centrifugation at 18,000 rpm for 30 min. The recovered solubilized protein incubated under rotation at 4°C for 2 hrs in the presence of 10 ml Ni-NTA resin and 20 mM imidazole. The Ni-NTA resin was washed in 50 ml high salt (HS) wash buffer (0.1% DDM, 0.01% CHS, 20 mM HEPES pH 7.5, 800 mM NaCl, 10% glycerol, 10 µg/ml leupeptin, and 160 µg/ml benzamidine) containing 4 mM theophylline and 25 mM imidazole followed by washing in 50 ml HS containing 4 mM theophylline and 50 mM imidazole. The protein was eluted in HS buffer with 200 mM imidazole and 4 mM theophylline. 2 mM CaCl2 was added and the protein was loaded onto a 5 ml anti-FLAG M1 antibody affinity resin (Sigma) at room temperature. The M1 resin was washed at room temperature in 30 ml HS buffer containing 2 mM CaCl2 and 15 mM UK432097 agonist to replace A2A bound theophylline. UK432097 is a potent and selective A2A agonist which was used to define the high-resolution crystal structure of A2A (Xu et al., 2011). The agonist bound receptor was eluted in HS buffer containing 5 mM EDTA, 0.2 mg/ml FLAG peptide, and 1 µM UK432097. EDTA and FLAG peptide was removed by dialysis and the A2A was spin-concentrated ~3-fold to 7.5 mg/ml and flash frozen in HS buffer containing 15% glycerol.
GPCR-Gs complex formation and HDX-MS
To investigate the complex formation at room temperature (25°C), 65 µM of Gs was mixed with BI-167107 bound β2AR or UK-432097 bound A2A in 1.15-fold molar excess of receptors at room temperature. The final DDM concentration in the complex formation reaction was approximately 0.4%. Apyrase (200 mU/ml) was added after 90 mins of incubation to hydrolyze GDP released from Gαs. For continuous labeling deuterium exchange, 4 µl of GPCRs-Gs complex, agonist bound receptors, or GDP-bound Gs was mixed with 26 µl of D2O buffer and incubated for 10 sec, 100 sec, 1,000 sec, and 10,000 sec at room temperature. For pulse-labeling deuterium exchange, Gs and GPCRs were mixed at room temperature or on ice and 4 µl aliquots were taken at indicated time points (before mixing, 10 sec, 5 min, 20 min, 60 min, 90 min, 110 min, 150 min, and 180 min after incubation for room temperature experiment and before mixing, 5 sec, 15 sec, 30 sec, 1 min, and 3 min after incubation for on ice experiment), mixed with 26 µl of D2O buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 2 µM agonist, 100 µM TCEP, 0.1% DDM in D2O), and incubated for 10 sec or 100 sec at room temperature or on ice. All deuterium exchanged samples described above were quenched by 30 µl of ice-cold quench buffer (0.1 M NaH2PO4, 20 mM TCEP, pH 2.01), immediately frozen on dry ice, and stored at −80°C. For the samples incubated on ice, the experiments were performed in a room maintained at 4°C to minimize temperature fluctuation during sample mixing. For non-deuterated (ND) samples, 4 µl of protein samples (65 µM) were mixed with 26 µl of H2O buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% DDM in H2O), to which 30 µl of ice-cold quench buffer was added, and snap-frozen on dry ice.
The quenched samples were digested and analyzed by HDX-UPLC-ESI-MS system (Waters) as previously described (Duc et al., 2015). Briefly, quenched samples were thawed and immediately injected to immobilized pepsin column (2.1 × 30 mm) (Life Technologies) in 100 µl/min of flow rate with 0.05% formic acid in H2O at 10°C. Peptide fragments were subsequently collected on a C18 VanGuard trap column (1.7 µm x 30 mm) (Waters) for desalting with 0.05% formic acid in H2O and then isolated by ultra-pressure liquid chromatography using an Acquity UPLC C18 column (1.7 µm, 1.0 × 100 mm) (Waters) at a flow rate of 40 µl/min with an acetonitrile gradient starting with 8% and increasing to 85% over 8.5 minutes. To minimize the back-exchange of deuterium to hydrogen, the system including trapping column and UPLC column were maintained at 0.5°C during the analysis, and all buffers was adjusted to pH 2.5.
Mass spectral analyses were performed with a Xevo G2 quadruple-time of flight (Q-TOF) equipped with a standard electrospray ionization (ESI) source in MSE mode (Waters) in positive ion mode. All settings/conditions for the system were as previously reported (Kim et al., 2015). Peptic peptides were identified in non-deuterated samples with ProteinLynx Global Server 2.4 (Waters). Searches were run with variable methionine oxidation modification, and the peptides were filtered on a peptide score of 6. To process HDX-MS data, the amount of deuterium in each peptide was determined by measuring the centroid of the isotopic distribution using the DynamX 2.0 software package (Waters). The average back-exchange level in our system was ~30%, but we did not correct for back-exchange because the analyses compared different states of proteins. All of the data was derived from at least three independent experiments.
Hydroxyl radical footprinting of samples
The β2AR and Gs heterotrimer were purified as described above, and dialyzed into 100 mM sodium phosphate buffer at pH 7.5. We implemented a syringe-pump driven stopped-flow mixing device at the NSLS-II XFP beamline to reduce sample consumption while enabling ms scale delays prior to exposure (Asuru et al., in press). This setup pushes two samples together into a turbulent mixer and then into a 200 nm internal diameter quartz capillary where X-ray exposure occurs. Controlling X-ray dosage (and thus hydroxyl radical dosage) at sample is controlled by flow rate of mixed sample through the zone of X-ray exposure defined by X-ray slits (which in this experiment were set to 220 µm) as well as attenuation of the X-ray beam (time of exposure for all sample was ~50 µs). Because the half-life of the OH radical in solution is on the order of 10−9 s (Pryor, 1986), the modification of sample in these experiments happens during the 50 µs exposure to X-rays, and does not significantly persist in solution beyond this time period. Pilot studies carried out with 10 nM Alexa 488 dye were employed to determine the thickness of aluminum necessary to attenuate the beam; this dye extinction enables visualization of hydroxyl radical labeling efficiency which can be affected by buffer components, X-ray dosage and protein concentration (Asuru et al., in press; Gupta et al., 2007). We measured several different levels of attenuation using experimental buffer containing 10 nM alexa 488. After exposure to X-rays, the samples were measured in a fluorimeter and based upon the residual fluorescence in the sample, we selected an attenuation level which resulted in ~60% loss of alexa fluorescence. This level of attenuation provides a dosage that has been shown in previous studies of GPCRs and other membrane proteins to result in adequate probe coverage of footprinted proteins and to minimize multiple labeling events (Gupta et al., 2014). The experiment was conducted as follows: The two sides of the sample apparatus were loaded with 20 µl of 15 µM β2AR bound to high affinity agonist, BI167107 or 13 µM Gs heterotrimer, respectively; 100 µl of protein buffer (50 mM sodium phosphate pH 8.0, 30 mM NaCl, 0.1 % DDM, 0.001% cholesterol hemi-succinate, 1µM BI167107) was used to push the two samples through the turbulent mixer and X-ray exposure capillary, and collected into a 1.5 ml tube containing 13 µl of 10 mM methionine amide. The methionine amide serves to reduce secondary modification by longer lived peroxides which are generated by collapse of unreacted OH radicals and other reactive species which are generated during protein modification (Xu et al., 2005). These species are less reactive, but could potentially modify protein during downstream sample processing if not removed from the solution. Immediately after X-ray exposure, samples were frozen in liquid nitrogen until further analysis. Time points chosen for analysis were 0, 20, 40, 80, 150, 400, 800, and 1,500 ms using a delay loop between mixer and exposure cell of varying length at a constant flow rate of 8.5 ml/minute. For the three longer time points that were also measured (5, 10, and 30 sec), a push/mix-pause-expose regime was employed as the length of delay loops required were too long, and lead to high backpressure when performed at constant flow conditions.
TCA/Acetone sample cleanup and protein proteolysis
Sample (10 µl) was denatured by addition of 20 µl of 8 M urea containing 100 mM DTT and incubated at 37°C for 60 min. This solution was precipitated by addition of 800 µl of acetone solution containing 10% TCA (−20°C) and kept at −20°C overnight. Sample was centrifuged at 17,000 x g for 30 min at 4°C. Supernatant was removed without disturbing the pellet, and washed 3 times with ice cold acetone. Samples were air dried at room temperature for 30 min to remove residual acetone. Dried sample pellets were resuspended in 10 µl of buffer containing 10 mM Tris-Cl, pH 8.5, 10 mM DTT and 3 M urea by a 15 min sonication. Samples were treated with Iodoacetate (IA) by addition 1ul of 250 mM freshly prepared IA and incubated in the dark at 37°C for 60 mins. Samples were then digested at 37°C overnight with LysC at a protein to enzyme ratio of 10:1 followed by a 6 hr AspN (200 ng per sample) digestion. Samples were stored at −80°C until analysis.
Mass spectrometry of HRF samples and data analysis
For separation and detection of protein digests a UPLC (Waters, Milford, MA) coupled to an Orbitrap Elite hybrid mass spectrometer (Thermo Finnigan, San Jose, CA) was utilized. Peptide digests were loaded onto a nano-ACUITY UPLC 2G-V/MC18 desalting trap column and separated on a Nano-ACQUITY UPLC BEH300 C18 column at a flow rate of 0.3 µl/min. A linear gradient consisting of mobile phase A (0.1% formic acid in water) and B (100% acetonitrile), starting with 1% mobile phase B and gradually increasing to 35% at 62 min to separate peptides, then increased to 90% over 1 min and held at 90% for 10 min to clean the column. All MS data were acquired in positive ion mode. A full MS scan (m/z 350–1800) at a resolution of 120,000 was conducted, twenty MS2 scans (m/z 350–1800) were selected from the twenty most intense peptide peaks of full MS scans. CID cleavage mode was performed at a normalized collision energy of 35%.
LC/MS data were searched against a database composed of sequences of 1D4-tagged β2Adrenergic receptor, Gsα, Gβ and Gγ proteins using the MassMatrix bioinformatics suite (Xu and Freitas, 2007; Xu et al., 2008). Footprinting oxidative labeling of the residues was selected as variable modifications during search. LysC and AspN were selected as in-silico enzymes to cleave proteins after Lys and AspN. Search criteria were 10 ppm mass accuracy for precursor ion and 0.8 Da for product ions.
Based on the search results, LC peak area of each peptide (modified and unmodified) was manually extracted and fraction of a modified peptide was calculated by dividing chromatographic peak area of the modified peptide with sum of chromatographic peak areas of the modified and unmodified species. To account for differences in X-ray exposure/hydroxyl radical dosage, the fraction of modified peptides was normalized by the equation:
Where x is fraction of a modified peptide before normalization, A(x)i is average fraction of the modified peptide through all the mixing time points in the replicate i, average of all four A(x)is were calculated as four replicates were applied each time point of the experiment individually (n=4). Kinetic curves of β2AR-Gs association were plotted by fraction of modified peptides F(x) versus mixing time ranging from 10 ms to 10 sec mixing times (10, 20, 40, 80, 150, 400, 800, 1,500, 5,000, 10,000 and 30,000 ms). Kinetic analysis was constrained to peptides with a single oxidation event to mitigate any conformational changes that might be due to prior oxidative modification.
Size exclusion chromatography
A2A-Gs complexes were formed at room temperature or on ice under similar conditions as the samples prepared for HDX in terms of protein concentration and buffer composition, with the exception of using more material (900 µg Gs with excess amount of A2A, 580 µg) to ensure a detectable signal. The A2A-Gs complex formed at room temperature and on ice incubated for 3 hrs with addition of apyrase after 90 min before analysis by SEC. In addition, the complex was formed on ice for 10 min without addition of apyrase prior to SEC analysis. The SEC was performed on an ÄKTA purifier using a Superdex 200 10/300 GL column in buffer containing 0.1% DDM, 20 mM HEPES pH 7.5, 100 mM NaCl, 100 µM TCEP, 100 nM UK432097. The eluting A2A-Gs complexes were collected, spin concentrated and analyzed again by SEC in the absence or presence of 200 µM GTP to confirm dissociation of the complex.
GDP release assay
[3H]GDP was purchased from Perkin Elmer (NET966250UC) with specific activity of 40 Ci/mmol. To prepare [3H]GDP-bound Gs heterotrimer, 200 nM purified Gs subunit of each G proteins was firstly mixed with 50 nM [3H]GDP for 1 hr at room temperature in the buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% DDM, 100 mM TCEP and 2 mM GDP, and then 2 mM purified Gβγ was further added for incubation of 10 min. 50 mM (final) BI-167107-bound β2AR or the corresponding DDM buffer of same volume was further added to initiate GDP release in the presence of 1 µM GDP. The reaction mixture was aliquoted at indicated time points, and immediately loaded onto calibrated G-50 columns. The follow-through was collected with 1 ml buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% DDM), and Gs-bound [3H]GDP was measured with scintillation counter (Beckman) after adding 15 ml scintillation fluid. The initial sample represents [3H]GDP binding capacity of Gs before initiation of GDP release.
Competition binding
The β2AR-rHDL particles were reconstituted in vitro by mixing POPC, POPG, human ApoA-1 and the β2AR as described (Whorton et al., 2007); [3H]DHA-binding was performed as previously described (Swaminath et al., 2002). For competition binding, the β2AR-rHDL was incubated with [3H]DHA (1.1nM final) and increasing concentration of isoproterenol for 1 hr before harvesting onto GF/B filters. Competition data were fitted to a two-site binding model and isoproterenol high and low Ki values and fractions calculated using GraphPad prism 7.0 software.
Bimane fluorescence assay
50 nM bimane (Invitrogen) labeled β2AR reconstituted in rHDL was incubated with either 3 µM isoproterenol (ISO) for 10 min at room temperature. After further addition of 6 µM Gs, the protein samples were incubated for another 45 min. The bimane fluorescence was measured by excitation at 370 nm, and emission spectra was recorded from 430 to 510 nm at 1-nm increments with 0.5 nm s−1 integration on a Spex FluoroMax-3 spectrofluorometer (Jobin Yvon Inc.) in photon counting mode set at a 4-nm emission bandwidth pass. The bimane assay for complex dissociation studies is described below.
Nucleotide binding studies
For the nucleotide binding experiment, purified β2AR was diluted to 20 µM in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.1% DDM, 0.01% CHS, 100 µM TCEP, 1 mM magnesium chloride, and 100 µM isoproterenol and incubated for 1 hr at RT. Gs heterotrimer was then added to 10 µM and incubated with the receptor for 1 hr at RT. After complex formation, the sample was divided into two and Nb37 was added to 1 mM to one sample while the other one was treated with Nb37 storage buffer (20 mM HEPES pH 7.5 and 100 mM sodium chloride). Both samples were incubated for 1 hr at RT. Nucleotide binding to the β2AR/Gs complex with or without Nb37 was followed by a change in fluorescence intensity of BODIPY-FL-GTPγS (Thermo Fisher Scientific). Fluorescence was recorded with a Horiba Fluorolog spectrofluorometer. The fluorophore was exited at 495 nm and emission was detected at 508 nm at 22°C. Slit widths were set to 0.5 nm (excitat ion) and 10 nm (emission). All experiments were performed in imaging buffer comprised of 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 0.1% DDM, 0.01% CHS, 10 mM magnesium chloride, and 100 µM TCEP. Kinetics data were collected with 1 µM BODIPY-FL-GTPγS in imaging buffer in the absence of complex for 100 sec to establish the baseline fluorescence intensity. β2AR-Gs complex was added with a 1:10 dilution (1 µM final G protein concentration) and rapidly mixed in the fluorescence cuvette without halting data collection (t=0 s). Data points were acquired every second for 300 s. The resulting kinetics spectra were plotted using GraphPad Prism 7.0 software.
Complex dissociation studies
For complex dissociation experiments, purified β2AR labeled at position C265 with bimane was incubated with 100 µM isoproterenol for 1h in 20 mM HEPES pH 7.5, 100 mM sodium chloride, 0.1% DDM, 0.01% CHS, 100 µM TCEP, 1 mM magnesium chloride. Gs heterotrimer was then added with 1.2 molar excess (2 µM final concentration of β2AR and 2.4 µM Gs) and incubated for 1 hr at RT. After complex formation, Nb37 was added to 1 mM to one half of the sample, while the other half was treated with Nb37 storage buffer (20 mM HEPES pH 7.5 and 100 mM NaCl). Both samples were incubated for 1 hr at RT. Bimane fluorescence was measured with a Horiba Fluorolog spectrofluorometer with an excitation wavelength of 370 nm and emission wavelength of 440 nm at 22°C. Slit widths were set to 4 nm. For collection of kinetics data, β2AR/Gs complex with and without Nb37 was diluted 1:10 into buffer comprised of 20 mM HEPES, pH 7.5, 100 mM sodium chloride, 0.1% DDM, 0.01% CHS, 10 mM magnesium chloride, and 100 µM TCEP at 25°C and bimane fluorescence was recorded for 100 sec to establish the baseline fluorescence intensity. GTPγS (Abcam Biochemicals) was added to 500 µM and rapidly mixed in the fluorescence cuvette without halting data collection (t=0 sec). Data points were acquired every second and the resulting kinetics spectra were plotted and fit to a one-phase association function using GraphPad Prism 7.0 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
For analysis of the time series data, repeated measures ANOVA (rANOVA) was employed at an α level =.01 and the F statistic calculated; time series as a whole were considered to be significant if the F statistic was greater than 1 at the significance level tested. An equivalent p value was also reported in Table S1 when appropriate. T-test was used to determine the significance between individual time points of the series. When time series data did not meet the threshold of significance by rANOVA, an unpaired or paired (Student’s) t-test was employed to assess the significance between time points. GraphPad Prism was used for the statistical analysis.
Supplementary Material
Figure S1. Description of the continuous-labeling and the pulse-labeling, Related to Figures 2 and 3
(A) Description of surface labeling and mass spectrometry analysis to monitor protein conformation. Both HDX and HRF employ similar post modification analytics. In short, the sample is labeled with compound (either through amide hydrogen exchange with D2O in the case of HDX or through covalent oxidative modification of protein side chain by hydroxyl radical in the case of HRF); sample is then digested with protease, separated on column and LC-MS/MS employed to obtain mass spectra; analysis of spectra reveals state or time dependent differential labeling.
(B) Description of continuous-labeling studies. To compare the conformations of two different states of a protein (for example, GDP-bound Gs vs. GPCR-bound nucleotide-free Gs), the labeling kinetics of each state is analyzed by exposing proteins in each state to D2O or X-ray for various durations.
(C) Description of pulse-labeling studies. To analyze the sequence of conformational changes of a protein transitioning from one state to another, the protein is triggered to undergo conformational change (for example, addition of agonist-bound GPCRs to GDP-bound Gs) followed by D2O or X-ray purse for a short period at specific time delays.
Figure S2. Analysis of GPCR-Gs complex formation and the effect of temperature, Related to Figure 2
(A and B) Analytical size exclusion chromatography shows the A2A-Gs complex formation (A) and GTP-induced complex dissociation (B) at different temperatures.
(C) The formation of the ternary complex on ice was confirmed by the bimane assay. Bimane-labeled β2AR was used to measure changes of fluorescence spectra induced by local conformational changes around the cytoplasmic end of TM6 upon ligand and G protein binding. The outward movement of TM6 is reflected in a decrease in the intensity and a red-shift in the maximal emission of bimane. The formation of the β2AR-Gs complex was slower at 0°C (on ice) than at room temperature (data not shown), but the same level of complex formation was observed after 1 hr of incubation. Emission spectra were obtained for 50 nM bimane-labeled receptor in 500 µl buffer (0.1% DDM, 20 mM HEPES pH 7.5, 10 mM NaCl) under 3 conditions: in the absence of ligand, after 15 min incubation with 2 µM isoproterenol (Iso), and after 1h incubation with 2 µM Iso and 250 nM Gs at the indicated temperatures. The bimane fluorescence was measured by excitation at 370 nm, and emission spectra was recorded from 430 to 510 nm at 1-nm increments with 0.5 nm s−1 integration on a Spex FluoroMax-3 spectrofluorometer (Jobin Yvon Inc.) in photon counting mode set at a 4-nm emission bandwidth pass. The data shows a representative in three independent experiments.
Figure S3. Continuous labeling HDX-MS analysis of Gαs before and after interaction with the A2A and the β2AR before and after interaction with Gs, Related to Figure 2
(A) Deuterium uptake profile changes of Gαs upon interaction with the A2A were color-coded onto the Ras-like domain of the X-ray crystal structure of Gαs (PDB: 3SN6): white indicates no MS data was obtained; light orange indicates no HDX change upon complex formation; blue indicates decreased HDX upon complex formation; and red indicates increased HDX upon complex formation. Detailed deuterium uptake levels of selected peptides are shown as uptake plots, which were generated by three independent experiments. Error bars represent the s.e.m. *p<0.05.
(B) Deuterium uptake profile changes of Gαs upon interaction with the β2AR were color-coded onto the Ras-like domain of the X-ray crystal structure of Gαs (PDB: 3SN6): color codes are same as (A). Data is reproduced from (Chung et al., 2011). It should be noted that there are differences in a few regions between Figure S3A and S3B, probably due to different peptic peptides analyzed because of the different pepsin column and LC-MS system used. For example, a peptide from αN was detected in the present work but no peptide from αN was identified in the previous study. No peptide from β1 was identified in the current work while a peptide from β1 was detected in the previous study. β6 shows no change in the current study while it showed higher HDX in the complex in the previous study, which is probably due to different peptides analyzed (i.e. the peptide from the current study does not contain β6/α5 loop while the peptide from the previous study contain β6/α5 loop). The α4 region showed higher HDX in the complex in the current study while it showed no change in the previous study, which might be due to the fact that different receptors interact with G proteins slightly differently, possibly due to differences in the length of ICL3. (C) Deuterium uptake profiles of the β2AR before or after interaction with Gs were color-coded onto the snake map of the β2AR: no HDX-MS with white, no HDX change upon complex formation with grey, decreased HDX upon complex formation with blue, and increased HDX upon complex formation with red. Detailed deuterium uptake levels of selected peptides are shown as uptake plots, which were generated by three independent experiments. Error bars represent the s.e.m. *p<0.05.
Figure S4. Pulse-labeling HDX-MS analysis of Gαs upon incubation with β2AR or A2A, Related to Figure 2
(A) Pulse-labeling HDX-MS analysis (100 sec D2O pulse) of Gαs upon β2AR or A2A binding at the room temperature.
(B) Pulse-labeling HDX-MS analysis of other regions of Gαs Ras-like domain during GPCR-Gs complex formation. Uptake plots of selected peptides from Gαs during incubation with the A2A or the β2AR at room temperature or on ice with 10 sec or 100 sec pulse are presented.
Statistical significance of the incubation time dependent HDX changes were analyzed as described in Figure 2. *, the first incubation time point that shows statistical difference from the GDP-bound state. #, the first incubation time point that shows statistical difference from the * time point. +, the first incubation time point that shows statistical difference from the # or previous + time point. Black symbols are for A2A-induced changes and grey symbols are for β2AR-induced changes. Error bars represent the s.e.m. Please note that the data is plotted using a non-linear/non-logarithmic scale.
Highlights.
Temporal assembly of a GPCR-Gs complex revealed by time-resolved mass spectrometry.
The sequence of GPCR-mediated G protein activation was elucidated.
Key structural elements were found to dictate nucleotide release.
A stable Gαs C-terminal helix is not required for GDP release.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea funded by the Korean government (NRF-2018R1A2B6001554 and NRF-2012R1A5A2A28671860) (K.Y.C.), the Lundbeck Foundation (S.G.F.R.), the Danish Medical Research Council (S.G.F.R.), the Carlsberg Foundation (S.G.F.R.), and the UNIK Center for Synthetic Biology (S.G.F.R.), the German Academic Exchange Service (DAAD) (D.H.), the National Institutes of Health grants R00EY09718 (D.T.L), P30EB09998 (M.R.C.) and R01GM083118 (B.K.K.) and an National Science Foundation grant DBI-1228549 (M.R.C.). B.K.K is a Chan Zuckerberg Biohub investigator. This research used Beamline BM-17 (XFP) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We thank Betsy White for technical assistance with β2AR and Nb37 expression and purification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes four figures, one table and can be found with this article online.
DECLARATION OF INTERESTS
Brian Kobilka is a co-founder of and a consultant for ConformeRx, Inc.
REFERENCES
- Alexander NS, Preininger AM, Kaya AI, Stein RA, Hamm HE, and Meiler J (2014). Energetic analysis of the rhodopsin-G-protein complex links the alpha5 helix to GDP release. Nat Struct Mol Biol 21, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel TE, Gupta S, Jastrzebska B, Palczewski K, and Chance MR (2009). Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A 106, 14367–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuru A, Farquhar E, Sullivan M, Abel D, Toomey J, Chance MR, and Bohon J (in press). The XFP (17-BM) beamline for X-ray footprinting at NSLS-II. J Synch Rad [DOI] [PMC free article] [PubMed]
- Bavro VN, Gupta S, and Ralston C (2015). Oxidative footprinting in the study of structure and function of membrane proteins: current state and perspectives. Biochem Soc Trans 43, 983–994. [DOI] [PubMed] [Google Scholar]
- Calabrese AN, and Radford SE (2018). Mass spectrometry-enabled structural biology of membrane proteins. Methods 147, 187–205. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Nehme R, Warne T, Leslie AG, and Tate CG (2016). Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL Jr., and Sunahara RK (2011). Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477, 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars G, Fanelli F, and Puett D (2011). The extreme C-terminal region of Galphas differentially couples to the luteinizing hormone and beta2-adrenergic receptors. Mol Endocrinol 25, 1416–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce CJ, Khoshouei M, Thal DM, Liang YL, Nguyen ATN, Furness SGB, Venugopal H, Baltos JA, Plitzko JM, Danev R, et al. (2018). Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558, 559–563. [DOI] [PubMed] [Google Scholar]
- Dror RO, Mildorf TJ, Hilger D, Manglik A, Borhani DW, Arlow DH, Philippsen A, Villanueva N, Yang Z, Lerch MT, et al. (2015). SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duc NM, Du Y, Zhang C, Lee SY, Thorsen TS, Kobilka BK, and Chung KY (2015). Effective application of bicelles for conformational analysis of G protein-coupled receptors by hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom 26, 808–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nafria J, Nehme R, Edwards PC, and Tate CG (2018). Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Chai J, Cheng J, D’Mello R, Chance MR, and Fu D (2014). Visualizing the kinetic power stroke that drives proton-coupled zinc(II) transport. Nature 512, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Sullivan M, Toomey J, Kiselar J, and Chance MR (2007). The Beamline X28C of the Center for Synchrotron Biosciences: a national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J Synchrotron Radiat 14, 233–243. [DOI] [PubMed] [Google Scholar]
- Harrison RA, and Engen JR (2016). Conformational insight into multi-protein signaling assemblies by hydrogen-deuterium exchange mass spectrometry. Curr Opin Struct Biol 41, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, et al. (2018). Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya AI, Lokits AD, Gilbert JA, Iverson TM, Meiler J, and Hamm HE (2014). A conserved phenylalanine as a relay between the alpha5 helix and the GDP binding region of heterotrimeric Gi protein alpha subunit. J Biol Chem 289, 24475–24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Yun Y, Kim HR, Seo MD, and Chung KY (2015). Different conformational dynamics of various active states of beta-arrestin1 analyzed by hydrogen/deuterium exchange mass spectrometry. J Struct Biol 190, 250–259. [DOI] [PubMed] [Google Scholar]
- Kiselar J, and Chance MR (2018). High-Resolution Hydroxyl Radical Protein Footprinting: Biophysics Tool for Drug Discovery. Annu Rev Biophys [DOI] [PubMed]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. (2018). Structure of the micro-opioid receptor-Gi protein complex. Nature 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L, and Simmons DA (2003). Protein-folding kinetics and mechanisms studied by pulse-labeling and mass spectrometry. Mass Spectrom Rev 22, 1–26. [DOI] [PubMed] [Google Scholar]
- Konermann L, Tong X, and Pan Y (2008). Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J Mass Spectrom 43, 1021–1036. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ (2007). Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 190, 9–19. [DOI] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, et al. (2017). Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markby DW, Onrust R, and Bourne HR (1993). Separate GTP binding and GTPase activating domains of a G alpha subunit. Science 262, 1895–1901. [DOI] [PubMed] [Google Scholar]
- Moro O, Lameh J, Hogger P, and Sadee W (1993). Hydrophobic amino acid in the i2 loop plays a key role in receptor-G protein coupling. J Biol Chem 268, 22273–22276. [PubMed] [Google Scholar]
- Nanoff C, Koppensteiner R, Yang Q, Fuerst E, Ahorn H, and Freissmuth M (2006). The carboxyl terminus of the Galpha-subunit is the latch for triggered activation of heterotrimeric G proteins. Mol Pharmacol 69, 397–405. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Van Eps N, Preininger AM, Hubbell WL, and Hamm HE (2006). Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol 13, 772–777. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Van Eps N, Preininger AM, Hubbell WL, and Hamm HE (2007). Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc Natl Acad Sci U S A 104, 7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban T, Jastrzebska B, Gupta S, Wang B, Miyagi M, Chance MR, and Palczewski K (2012). Conformational dynamics of activation for the pentameric complex of dimeric G protein-coupled receptor and heterotrimeric G protein. Structure 20, 826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preininger AM, Meiler J, and Hamm HE (2013). Conformational flexibility and structural dynamics in GPCR-mediated G protein activation: a perspective. J Mol Biol 425, 2288–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor WA (1986). Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol 48, 657–667. [DOI] [PubMed] [Google Scholar]
- Rajabi K, Ashcroft AE, and Radford SE (2015). Mass spectrometric methods to analyze the structural organization of macromolecular complexes. Methods 89, 13–21. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. (2007). GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Scheerer P, Heck M, Goede A, Park JH, Choe HW, Ernst OP, Hofmann KP, and Hildebrand PW (2009). Structural and kinetic modeling of an activating helix switch in the rhodopsin-transducin interface. Proc Natl Acad Sci U S A 106, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminath G, Steenhuis J, Kobilka B, and Lee TW (2002). Allosteric modulation of beta2-adrenergic receptor by Zn(2+). Mol Pharmacol 61, 65–72. [DOI] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Van Eps N, Preininger AM, Alexander N, Kaya AI, Meier S, Meiler J, Hamm HE, and Hubbell WL (2011). Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci U S A 108, 9420–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, and Chance MR (2017). Protein Footprinting Comes of Age: Mass Spectrometry for Biophysical Structure Assessment. Mol Cell Proteomics 16, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfield GH, Rasmussen SG, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, et al. (2011). Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci U S A 108, 16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, and Sunahara RK (2007). A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A 104, 7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, and Stevens RC (2011). Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Kiselar J, He Q, and Chance MR (2005). Secondary reactions and strategies to improve quantitative protein footprinting. Anal Chem 77, 3029–3037. [DOI] [PubMed] [Google Scholar]
- Xu H, and Freitas MA (2007). A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics 8, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhang L, and Freitas MA (2008). Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J Proteome Res 7, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano A, Takahashi Y, Moriguchi H, Inazumi T, Koga T, Otaka A, and Sugimoto Y (2017). An aromatic amino acid within intracellular loop 2 of the prostaglandin EP2 receptor is a prerequisite for selective association and activation of Galphas. Biochim Biophys Acta 1862, 615–622. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Velez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, and Kobilka B (2009). The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci U S A 106, 9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, et al. (2017). Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. N ature 546, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Wintrode PL, and Chance MR (2008). Complementary structural mass spectrometry techniques reveal local dynamics in functionally important regions of a metastable serpin. Structure 16, 38–51. [DOI] [PubMed] [Google Scholar]
- Zhou H, Yan F, Yamamoto S, and Tai HH (1999). Phenylalanine 138 in the second intracellular loop of human thromboxane receptor is critical for receptor-G-protein coupling. Biochem Biophys Res Commun 264, 171–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Description of the continuous-labeling and the pulse-labeling, Related to Figures 2 and 3
(A) Description of surface labeling and mass spectrometry analysis to monitor protein conformation. Both HDX and HRF employ similar post modification analytics. In short, the sample is labeled with compound (either through amide hydrogen exchange with D2O in the case of HDX or through covalent oxidative modification of protein side chain by hydroxyl radical in the case of HRF); sample is then digested with protease, separated on column and LC-MS/MS employed to obtain mass spectra; analysis of spectra reveals state or time dependent differential labeling.
(B) Description of continuous-labeling studies. To compare the conformations of two different states of a protein (for example, GDP-bound Gs vs. GPCR-bound nucleotide-free Gs), the labeling kinetics of each state is analyzed by exposing proteins in each state to D2O or X-ray for various durations.
(C) Description of pulse-labeling studies. To analyze the sequence of conformational changes of a protein transitioning from one state to another, the protein is triggered to undergo conformational change (for example, addition of agonist-bound GPCRs to GDP-bound Gs) followed by D2O or X-ray purse for a short period at specific time delays.
Figure S2. Analysis of GPCR-Gs complex formation and the effect of temperature, Related to Figure 2
(A and B) Analytical size exclusion chromatography shows the A2A-Gs complex formation (A) and GTP-induced complex dissociation (B) at different temperatures.
(C) The formation of the ternary complex on ice was confirmed by the bimane assay. Bimane-labeled β2AR was used to measure changes of fluorescence spectra induced by local conformational changes around the cytoplasmic end of TM6 upon ligand and G protein binding. The outward movement of TM6 is reflected in a decrease in the intensity and a red-shift in the maximal emission of bimane. The formation of the β2AR-Gs complex was slower at 0°C (on ice) than at room temperature (data not shown), but the same level of complex formation was observed after 1 hr of incubation. Emission spectra were obtained for 50 nM bimane-labeled receptor in 500 µl buffer (0.1% DDM, 20 mM HEPES pH 7.5, 10 mM NaCl) under 3 conditions: in the absence of ligand, after 15 min incubation with 2 µM isoproterenol (Iso), and after 1h incubation with 2 µM Iso and 250 nM Gs at the indicated temperatures. The bimane fluorescence was measured by excitation at 370 nm, and emission spectra was recorded from 430 to 510 nm at 1-nm increments with 0.5 nm s−1 integration on a Spex FluoroMax-3 spectrofluorometer (Jobin Yvon Inc.) in photon counting mode set at a 4-nm emission bandwidth pass. The data shows a representative in three independent experiments.
Figure S3. Continuous labeling HDX-MS analysis of Gαs before and after interaction with the A2A and the β2AR before and after interaction with Gs, Related to Figure 2
(A) Deuterium uptake profile changes of Gαs upon interaction with the A2A were color-coded onto the Ras-like domain of the X-ray crystal structure of Gαs (PDB: 3SN6): white indicates no MS data was obtained; light orange indicates no HDX change upon complex formation; blue indicates decreased HDX upon complex formation; and red indicates increased HDX upon complex formation. Detailed deuterium uptake levels of selected peptides are shown as uptake plots, which were generated by three independent experiments. Error bars represent the s.e.m. *p<0.05.
(B) Deuterium uptake profile changes of Gαs upon interaction with the β2AR were color-coded onto the Ras-like domain of the X-ray crystal structure of Gαs (PDB: 3SN6): color codes are same as (A). Data is reproduced from (Chung et al., 2011). It should be noted that there are differences in a few regions between Figure S3A and S3B, probably due to different peptic peptides analyzed because of the different pepsin column and LC-MS system used. For example, a peptide from αN was detected in the present work but no peptide from αN was identified in the previous study. No peptide from β1 was identified in the current work while a peptide from β1 was detected in the previous study. β6 shows no change in the current study while it showed higher HDX in the complex in the previous study, which is probably due to different peptides analyzed (i.e. the peptide from the current study does not contain β6/α5 loop while the peptide from the previous study contain β6/α5 loop). The α4 region showed higher HDX in the complex in the current study while it showed no change in the previous study, which might be due to the fact that different receptors interact with G proteins slightly differently, possibly due to differences in the length of ICL3. (C) Deuterium uptake profiles of the β2AR before or after interaction with Gs were color-coded onto the snake map of the β2AR: no HDX-MS with white, no HDX change upon complex formation with grey, decreased HDX upon complex formation with blue, and increased HDX upon complex formation with red. Detailed deuterium uptake levels of selected peptides are shown as uptake plots, which were generated by three independent experiments. Error bars represent the s.e.m. *p<0.05.
Figure S4. Pulse-labeling HDX-MS analysis of Gαs upon incubation with β2AR or A2A, Related to Figure 2
(A) Pulse-labeling HDX-MS analysis (100 sec D2O pulse) of Gαs upon β2AR or A2A binding at the room temperature.
(B) Pulse-labeling HDX-MS analysis of other regions of Gαs Ras-like domain during GPCR-Gs complex formation. Uptake plots of selected peptides from Gαs during incubation with the A2A or the β2AR at room temperature or on ice with 10 sec or 100 sec pulse are presented.
Statistical significance of the incubation time dependent HDX changes were analyzed as described in Figure 2. *, the first incubation time point that shows statistical difference from the GDP-bound state. #, the first incubation time point that shows statistical difference from the * time point. +, the first incubation time point that shows statistical difference from the # or previous + time point. Black symbols are for A2A-induced changes and grey symbols are for β2AR-induced changes. Error bars represent the s.e.m. Please note that the data is plotted using a non-linear/non-logarithmic scale.