

Abstract
Malignant peripheral nerve sheath tumors contain loss of histone H3K27 trimethylation (H3K27me3) due to driver mutations affecting the polycomb repressive complex 2 (PRC2). Consequently, loss of H3K27me3 staining has served as a diagnostic marker for this tumor type. However, recent reports demonstrate H3K27me3 loss in numerous other tumors, including some in the differential diagnosis of malignant peripheral nerve sheath tumor. Since these tumors lose H3K27me3 through mechanisms distinct from PRC2 loss, we set out to determine whether loss of dimethylation of H3K27, which is also catalyzed by PRC2, might be a more specific marker of PRC2 loss and malignant peripheral nerve sheath tumor. Using mass spectrometry, we identify a near complete loss of H3K27me2 in malignant peripheral nerve sheath tumors and cell lines. Immunohistochemical analysis of 72 malignant peripheral nerve sheath tumors, 7 K27M-mutant gliomas, 43 ependymomas, and 10 Merkel cell carcinomas demonstrates that while H3K27me3 loss is common across these tumor types, H3K27me2 loss is limited to malignant peripheral nerve sheath tumors and is highly concordant with H3K27me3 loss (33/34 cases). Thus, increased specificity does not come at the cost of greatly reduced sensitivity. To further compare H3K27me2 and H3K27me3 immunohistochemistry, we investigated 42 melanomas and 54 synovial sarcomas, histologic mimics of malignant peripheral nerve sheath tumor with varying degrees of H3K27me3 loss in prior reports. While global H3K27me3 loss was not seen in these tumors, weak and limited H3K27me3 staining was common. By contrast, H3K27me2 staining was more clearly retained in all cases, making it a superior binary classifier. This was confirmed by digital image analysis of stained slides. Our findings indicate that H3K27me2 loss is highly specific for PRC2 loss and that PRC2 loss is a rarer phenomenon than H3K27me3 loss. Consequently, H3K27me2 loss is a superior diagnostic marker for malignant peripheral nerve sheath tumor.
Introduction
Malignant peripheral nerve sheath tumors are aggressive sarcomas arising in association with peripheral nerves1,2. They occur both sporadically and in the setting of neurofibromatosis type I (NF1) in roughly equal proportions3. Classically, the diagnosis of malignant peripheral nerve sheath tumor has been difficult in the absence of a clinical history of NF1 due to the lack of reliable immunohistochemical markers4. The relatively recent finding of recurrent loss-of-function alterations in components of the polycomb repressive complex 2 (PRC2) provided hope for a new immunohistochemical marker rooted in the molecular biology of the tumor5–7. These molecular alterations were identified in both sporadic and syndromic cases, and involved the PRC2 components EED and SUZ12. They led to global loss of H3K27me3, which was enthusiastically adopted as a diagnostic marker for malignant peripheral nerve sheath tumor8–11. Despite this initial promise, H3K27me3 immunohistochemistry has been problematic. The reported specificity of H3K27me3 varied across the initial large series, and numerous cases were qualitatively described as exhibiting heterogeneous or ‘mosaic’ staining, or partial loss12,13. This heterogeneity could present challenges in small tissue samples. Our own experience with this marker is consistent with these reports, with some cases showing heterogeneous staining, or poor staining within internal controls (vascular endothelium and infiltrating lymphocytes). Indeed, the variability of H3K27me3 staining and lack of specificity of H3K27me3 loss for malignant peripheral nerve sheath tumor was recently demonstrated in two large-scale analyses which included cases of synovial sarcoma, fibrosarcomatous dermatofibrosarcoma protuberans and melanoma--perhaps the three closest histologic mimics of malignant peripheral nerve sheath tumor12–14. The failure of H3K27me3 to reliably distinguish malignant peripheral nerve sheath tumor from these tumors is particularly problematic.
In addition to the histologic mimics mentioned above, H3K27me3 loss has been described in a number of other tumors. H3K27me3 loss has been shown in histone H3K27M-mutant glioma15–17, a subset of posterior fossa ependymomas18,19, Merkel cell carcinomas20, a subset of meningiomas21, and in rare cases of dedifferentiated chondrosarcoma22. While this does not present a diagnostic challenge, since most of these tumors are not in the differential diagnosis of malignant peripheral nerve sheath tumor, it does suggest that there are multiple discrete pathways by which a tumor might lose H3K27me3, and thus that H3K27me3 loss is not a specific marker of PRC2 loss-of-function.
To identify a more specific marker for PRC2 loss and malignant peripheral nerve sheath tumor, we began with the biologic function of PRC2. PRC2 consists of 4 core subunits: EED, SUZ12, RBBP4/7, and the methyltransferase EZH1/2, which mediates its enzymatic function. It catalyzes the mono-, di-, and trimethylation of histone H3K27. The genomic distribution and the function of each mark is distinct: H3K27me1 is deposited in the gene bodies of actively transcribed genes, H3K27me2 is primarily localized in intergenic regions, and H3K27me3 is generally localized to promoters where it inhibits transcription (reviewed in23). While H3K27me3-mediated gene silencing is the best characterized function of PRC2, H3K27me2 is actually the most abundant form of H3K27 modification, and also plays a role in transcriptional repression24,25. Since, among the tumors with a known mechanism of H3K27me3 loss, malignant peripheral nerve sheath tumor is the only one that has been demonstrated to have recurrent loss-of-function alterations in PRC2 components, we reasoned that malignant peripheral nerve sheath tumors should also display loss of H3K27me2, while other tumors with distinct mechanisms of H3K27me3 loss might not. We therefore set out to determine whether H3K27me2 distinguishes true PRC2 loss from isolated loss of H3K27me3, and, relatedly, whether H3K27me2 might serve as a more specific marker for malignant peripheral nerve sheath tumor.
Materials and Methods
Cell culture
Malignant peripheral nerve sheath tumor cell lines were generously provided by Karen Cichowski (90–8TL, S462) and Jeffrey Field (STS26T, ST88–14, T265, HEI193). All cell lines were maintained in DMEM (Corning™10013CM) supplemented with 10% FBS (GE, HyClone, SH30071.03) and the media was changed every 2–3 days. Upon reaching approximately 80% confluence, cells were collected with Trypsin-EDTA 0.25% (Gibco™ 25200056) and split 1:3. Cells were tested for mycoplasma contamination at 6–12 month intervals.
Selection of tissue samples
All human samples were collected and analyzed under protocols approved by the Institutional Review Boards (IRB) of the participating institutions. All tissue experiments were conducted on de-identified human tissue specimens taken as part of a diagnostic or therapeutic procedure independent from the research study and exempt from a separate consent process. Histologic analyses were conducted on a combination of whole-mount slide sections and tissue microarrays. Tumor samples were identified through a database search, and the initial diagnostic histologic sections were reviewed by to confirm the diagnosis. A subset of malignant peripheral nerve sheath tumor samples was designated as arising in the setting of NF1 following chart review to confirm a clinically documented history of NF1. Sporadic cases were confirmed by established morphologic and immunohistochemical criteria, and adequate exclusion of alternative diagnoses. Standard 5 μm tissue sections were used for hematoxylin/eosin staining and immunohistochemical (IHC) analysis. For tissue mass spectrometry analysis, 3 mm tissue cores were taken from representative areas of solid tumor for protein isolation, purification and characterization. All digital images were taken on a Leica DMC 4500 camera and captured and processed using the Leica Digital Application Suite v4.12.
Extraction of histones from formalin-fixed, paraffin-embedded tissues
3 mm diameter cores of formalin-fixed, paraffin-embedded tumor were weighed and cut into small pieces. The pieces were resuspended in a 10x volume of xylene and incubated at 37°C for 10 min with gentle agitation. Following centrifugation and removal of xylene, the process was repeated with xylene and then with 100% ethanol twice, 95% ethanol, 85%, 70%, 50%, 20%, and then water. Samples were then resuspended in 100 mM Tris-HCl pH 8.0 with 100 mM NaCl, 1 mM AEBSF, 5 nM microcystin, and 1 mM sodium butyrate, and homogenized with a mortar and pestle. Samples were then passed through an 18-gauge needle 10x, then a 21-gauge needle 10x. The lysate was sonicated 3 × 30 s at room temperature with a 50% duty ratio. 20% SDS was added to final concentration of 2%, and the sample was placed on a 95°C shaker for 2 hrs total, with another sonication step at 1 hr. The lysate was centrifuged at 17,000–20,000 g for 15 minutes. The concentration of solubilized protein was assessed with Pierce BCA assay kit (Thermo Fisher, 23225). For total protein isolation, 100 μg of total protein was precipitated using methanol and chloroform as described in26. The precipitated protein was resuspended in water and 50 μg was loaded in 2 adjacent wells of a NuPAGE™ 12% Bis-Tris Protein Gel (NP0341BOX). Proteins were separated by SDS-PAGE and Coomassie stained. Histone bands were then excised from the gel and processed as previously described27.
Extraction of histones from cell lines
Histones were extracted as previously described27. Briefly, cells were harvested by trypsinization, washed twice with PBS, and pellets (approx. 1×106 cells) were lysed on ice in nuclear isolation buffer with 0.3% NP-40 alternative. Nuclei were incubated with 0.4 N H2SO4 for 3 hours at 4°C with gentle agitation. 100% trichloroacetic acid (w/v) was added to the acid extract to a final concentration of 20% and samples were incubated on ice at 4°C overnight to precipitate histones. The resulting histone pellets were rinsed with ice cold acetone + 0.1% HCl and then with ice cold acetone before resuspension in water for estimation of protein concentration.
Liquid chromatography-mass spectrometry analysis of histones
Histones were prepared and analyzed by LC-MS/MS as previously described27. Histones were derivatized with propionic anhydride, digested with trypsin, and N-termini were derivatized again. Samples were desalted using C18 stage-tips and resuspended in 0.1% formic acid. Approximately 1 μg of peptides were analyzed by nanoflow liquid chromatography using a Thermo Scientific Easy nLC 1000 equipped with a 75 μm × 20 cm fused silica column packed in-house with Reprosil-Pur C18-AQ (3 μm particle size; Dr. Maisch GmbH, Germany). Buffer A was 0.1% formic acid and buffer B was 0.1% formic acid in 80% acetonitrile. Peptides were resolved using a two-step linear gradient from 5% to 33% B over 45 min, then from 33% to 90% B over 10 min at a flow rate of 300 nL/min. The HPLC was coupled online to a Thermo Q Exactive mass spectrometer operating in the positive mode using a Nanospray Flex Ion Source (Thermo Scientific) at 2.3 kV. The instrument was operated in data-independent acquisition (DIA) mode: the range between 300–1100 m/z was surveyed in windows of 50 m/z, with a full scan after every 8 MS/MS scans. The full scan was acquired at resolution 35,000, AGC target 2e5, and maximum injection time of 200 ms. MS2 scans were acquired with a resolution of 17,500 with AGC target 2e5 and max inject time of 200 ms. Data were analyzed in EpiProfile 2.028.
The peptide that is generated upon trypsin cleavage of derivatized histone H3 spans amino acids 27–40. This peptide contains two lysine residues that can be post-translationally modified (lysine 27 and lysine 36). K27 can be unmodified (K27unmod), monomethylated (K27me1), dimethylated (K27me2), trimethylated (K27me3), or acetylated (K27ac). K36 can be unmodified (K36unmod), monomethylated (K36me1), dimethylated (K36me2), or trimethylated (K36me3). All possible combinations of modifications were included in the search. The area under the curve of each identified form of the histone H3 27–40 peptide was summed, and the values were normalized such that the sum of all modified forms was equal to 100%. Combinatorial modifications containing K27 methylation were combined so that the total abundance of each K27 methylation state could be compared. For example, the total abundance of the single modification K27me1 is equal to the sum of the abundances of K27me1K36unmod, K27me1K36me1, K27me1K36me2, and K27me1K36me3. The total abundance of K27me2 is equal to the sum of K27me2K36unmod, K27me2K36me1, K27me2K36me2, and K27me2K36me3. The total abundance of K27me3 is equal to the sum of K27me3K36unmod, K27me3K36me1, K27me3K36me2, and K27me3K36me3.
Reanalysis of publicly available histone modification data
Histone modification data was downloaded from two previously published manuscripts29,30. The first dataset came from LeRoy et al. 2013, “Additional File 1,” titled “13072_2013_292_MOESM1_ESM.xlsx” and was downloaded from https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/1756-8935-6-20. This file contains post-translational modification (PTM) abundances for the H3 aa 27–40 peptide. These combinatorial modification abundances were converted to single PTM abundances using the procedure described above. The second dataset came from Stafford et al. 2018, “Data file S2,” downloaded from http://advances.sciencemag.org/content/4/10/eaau5935/tab-figures-data. The histone PTM values, which were already in single PTM format, were copied from sheet 4 of this file, titled “H31_K27-K36.” To generate the scatterplot, the data from these two resources were combined with the new data presented herein and the resulting table was z-score normalized. Both the raw table and the z-score normalized table are included in Supplementary Table 1.
Immunohistochemistry
Immunohistochemical staining was conducted on 5 μm sections of formalin-fixed, paraffin-embedded tissue using a Leica Bond-IIITM instrument employing the Bond Polymer Refine Detection System (Leica Microsystems DS9800). Heat-induced epitope retrieval was conducted with ER1 or ER2 solution for the times listed with the antibodies (Leica Microsystems). Incubation with Primary antibody (H3K27me3, Cell-Signaling Technologies, AB#9733, 1:200, ER2/30 minutes; H3K27me2, abcam ab24684, 1:500, ER1/20 minutes) was 15 min. This was followed by polymer detection according to the manufacturer’s recommended protocol (Leica Microsystems RE7230-CE). All experiments were conducted at room temperature. Slides are washed three times between each step with bond wash buffer or water.
For the binary classification of positive/negative shown in Table 1, tumors showing loss of nuclear reactivity in >90% of tumor cells with retained internal control staining in stromal cells were categorized as negative. For semi-quantitative classification shown in Table 2, cells were scored as in14, on the basis of fraction of cells staining, regardless of intensity, within tissue sections with adequate internal control staining. Point scores were as follows: 3+ (>95%), 2+ (50–95%), 1+ (5–50%), 0+ (<5%). Quantitative immunohistochemical analysis for H-score calculation was carried out on selected cases of malignant peripheral nerve sheath tumor, melanoma and synovial sarcoma using QuPath software31. Mean optical density for nuclear H3K27me2 and H3K27me3 immunohistochemical staining was measured on digital images using a nuclear size threshold of 100–400 μm2. H-scores were calculated on the basis of mean OD values from 500 randomly selected tumor nuclei. Mean OD values were binned into four categories: 0+(<0.15); 1+(0.15–0.30); 2+(0.30–0.45); 3+(>0.45). The final numerical value was calculated as the sum of: (1* (#cells with 1+ staining)) + (2* (#cells with 2+ staining)) + (3* (#cells with 3+ staining)). Plots were generated using GraphPad Prism v7.0. Boxplot error-bar parameters are according to the standard Tukey method.
Table 1:
H3K27me3 and H3K27me2 IHC in selected tumors
| Tumor type | H3K27me3 loss | H3K27me2 loss |
|---|---|---|
| Malignant peripheral nerve sheath tumor, total (N=72) | 34 (47%) | 33 (46%) |
| H3.3K27M-mutant glioma (N=7) | 7 (100%) | 0 (0%) |
| Ependymoma (N=43) | 21 (48.8%) | 0 (0%) |
| Merkel cell carcinoma (N=10) | 9 (90%) | 0 (0%) |
Table 2:
H3K27me3 and H3K27me2 IHC in Malignant peripheral nerve sheath tumor, Melanoma and Synovial sarcoma
| Tumor type | H3K27me3 | H3K27me2 | ||||||
|---|---|---|---|---|---|---|---|---|
| 0 (<5%) | 1+ (5–50%) | 2+ (50–95%) | 3+ (>95%) | 0 (<5%) | 1+ (5–50%) | 2+ (50–95%) | 3+ (>95%) | |
| Malignant peripheral nerve sheath tumor (N=72) | 34 (47%) | 11 (15%) | 23 (32%) | 4 (6%) | 33 (46%) | 2 (3%) | 18 (25%) | 19 (26%) |
| Melanoma | ||||||||
| BRAF mutant (N=20) | 0 (0%) | 4 (20%) | 9 (45%) | 7 (35%) | 0 (0%) | 0 (0%) | 5 (25%) | 15 (75%) |
| NRAS mutant (N=22) | 0 (0%) | 4 (18%) | 13 (59%) | 5 (23%) | 0 (0%) | 0 (0%) | 5 (23%) | 17 (77%) |
| Synovial Sarcoma (N=54) | 0 (0%) | 24 (44%) | 21 (39%) | 9 (17%) | 0 (0%) | 0 (0%) | 20 (37%) | 34 (63%) |
Results
PRC2 loss in malignant peripheral nerve sheath tumor is associated with global loss of H3K27me3 and H3K27me2
Mass spectrometry permits the simultaneous evaluation of multiple histone post-translational modifications (PTMs) on the same histone tail in a semi-quantitative fashion. In order to better understand the extent to which PRC2 loss affects all levels of H3K27 methylation in malignant peripheral nerve sheath tumor, we performed mass spectrometry on histones isolated from 6 nerve sheath tumor cell lines, 2 of which had retained PRC2 activity (STS26T—malignant peripheral nerve sheath tumor, HEI93—schwannoma) and 4 of which were derived from SUZ12-mutant malignant peripheral nerve sheath tumors (ST88–14, 90–8TL, T265, S462). Quantification of histone H3K27 methylation revealed a striking loss not only of H3K27me3, but also of H3K27me2 and H3K27me1 in the PRC2 loss malignant peripheral nerve sheath tumor lines (Figure 1a). In order to determine how these histone methylation levels compare to levels reported for other cell lines, we next analyzed two publicly available histone MS datasets29,30 (Figure 1b; Supplementary Table 1). Among the 39 cell lines analyzed, the PRC2 loss malignant peripheral nerve sheath tumor cells displayed the lowest levels of all three methylation marks. Notably, the next-lowest levels of H3K27 methylation were observed in histone H3K27M-mutant glioma cells, consistent with inhibition of PRC2 by the mutant histone15,30. Interestingly, the H3K27me3 levels were similar across both tumor types, but H3K27me1 and H3K27me2 were markedly lower in malignant peripheral nerve sheath tumor lines, suggesting that H3K27me2 loss may better separate malignant peripheral nerve sheath tumor from other tumors with polycomb dysfunction than H3K27me3 loss.
Figure 1.

K27 methylation is severely impaired in malignant peripheral nerve sheath tumor cell lines and tumors. (a) Quantification of histone PTMs from two nerve sheath tumors with normal polycomb function (STS26T - malignant peripheral nerve sheath tumor, HEI93 - Schwannoma; light gray) and from four malignant peripheral nerve sheath tumors with polycomb loss (ST88–14, 90–8TL, T265, and S462; dark gray). (b) Scatterplot based on z-scores from H3K27 methylation levels across 39 cell lines. (c) Quantification of histone PTMs from cores of formalin-fixed, paraffin-embedded tissue derived from 3 neurofibromas and 8 malignant peripheral nerve sheath tumors (3 Neurofibromas; white, 3 PRC2-retained; light gray and 5 PRC2-loss; dark gray).
Next, in order to determine whether the epigenetic changes observed in malignant peripheral nerve sheath tumor cell lines were conserved in human tumors, we performed mass spectrometry on histones isolated from 8 archival, formalin-fixed paraffin-embedded malignant peripheral nerve sheath tumor tumors (3 PRC2 retained, 5 PRC2 loss) and 3 neurofibromas (Figure 1c). Consistent with the cell line data, the levels of H3K27me2 and H3K27me3 were both markedly reduced in malignant peripheral nerve sheath tumor with PRC2 loss relative to malignant peripheral nerve sheath tumor with PRC2 retained and benign neurofibromas. Both of these modifications were slightly higher in the tumor samples than in the cell lines, perhaps due to the presence of stromal cells and infiltrating immune cells in the tissue cores. Alternatively, or perhaps in addition, this observation could be explained by a recent study that directly compared the levels of histone modifications in cancer cells grown in culture versus grown in vivo as murine xenografts. The study reported that the practice of cell culture alone causes a modest decrease in the abundance of the levels of H3K27me2/332. The observation that H3K27me1 levels were similar across both PRC2 retained and PRC2 loss tumors, however, was unexpected. While this may have been due in part to artefactual monomethylation related to the formalin fixation and paraffin embedding process33, retention of H3K27me1 and loss of H3K27me2/3 has been observed previously in both SUZ12-null24,25 and in EED-null mouse embryonic stem cells25,34,35. This has led to the hypothesis that there may be a PRC2-independent monomethyltransferase for H3K27, but whether such an enzyme exists is unknown23,36.
The mass spectrometry data suggested that PRC2 loss is characterized by loss of both H3K27me2 and H3K27me3. To determine whether this translates to the setting of immunohistochemistry, and to confirm concordance between H3K27me3 loss and H3K27me2 loss in a larger cohort of malignant peripheral nerve sheath tumors, we conducted H3K27me3 and H3K27me2 immunohistochemical staining in parallel on a set of 72 malignant peripheral nerve sheath tumor tumors including sporadic and syndromic cases (Table 1; Figure 2). H3K27me3 and H3K27me2 staining were highly concordant in malignant peripheral nerve sheath tumor. Thirty-two cases showed retained nuclear staining for both H3K27me3 and H3K27me2. H3K27me2 staining was generally more intense and uniform, likely reflecting the greater abundance of this modification. Of the thirty-four cases with loss of H3K27me3, all but one also exhibited complete loss of H3K27me2. The lone exception showed only weak staining in a small subset of cells, markedly reduced relative to internal control staining (Figure 2b, lower right, high-power inset).
Figure 2.

H3K27me3 and H3K27me2 immunohistochemical staining of malignant peripheral nerve sheath tumors. (a) Three representative malignant peripheral nerve sheath tumors showing retained H3K27me3 nuclear staining also show strong staining for H3K27me2. (b) Similarly, malignant peripheral nerve sheath tumors with loss of H3K27me3 staining within tumor cells, show loss of H3K27me2. The sole case scored as H3K27me3-/H3K27me2+ showed only weak nuclear staining, less intense than infiltrating inflammatory cells (arrows, high-power inset, lower right).
Loss of H3K27me2 is more specific for PRC2 loss than loss of H3K27me3
To determine whether loss of H3K27me2 is seen in tumors without known loss-of-function in PRC2, we investigated H3K27me2 and H3K27me3 immunohistochemistry in parallel on tumors characterized by H3K27me3 loss through mechanisms distinct from PRC2 loss (H3K27M gliomas; ependymomas) or through unknown mechanisms (Merkel cell carcinoma) (Table 1, Figure 3). All seven H3K27M glioma cases and all ten Merkel cell carcinoma cases showed markedly reduced/complete loss of H3K27me3 in tumor cells, but exhibited strong, uniform nuclear reactivity for H3K27me2. Of forty-three total ependymomas, twenty-one showed loss of H3K27me3 staining (21/43). All twenty-one had retained staining for H3K27me2. Together with the mass spectrometry and immunohistochemical data from malignant peripheral nerve sheath tumor, these results demonstrate that PRC2 loss results in global loss of both H3K27me2 and H3K27me3, and that loss of H3K27me2 distinguishes true PRC2 loss from loss of H3K27me3, which may occur through other mechanisms. The results also suggest that the mechanism for H3K27me3 loss in Merkel cell carcinoma, heretofore uncharacterized, is unlikely to involve biallelic loss of core PRC2 components.
Figure 3.
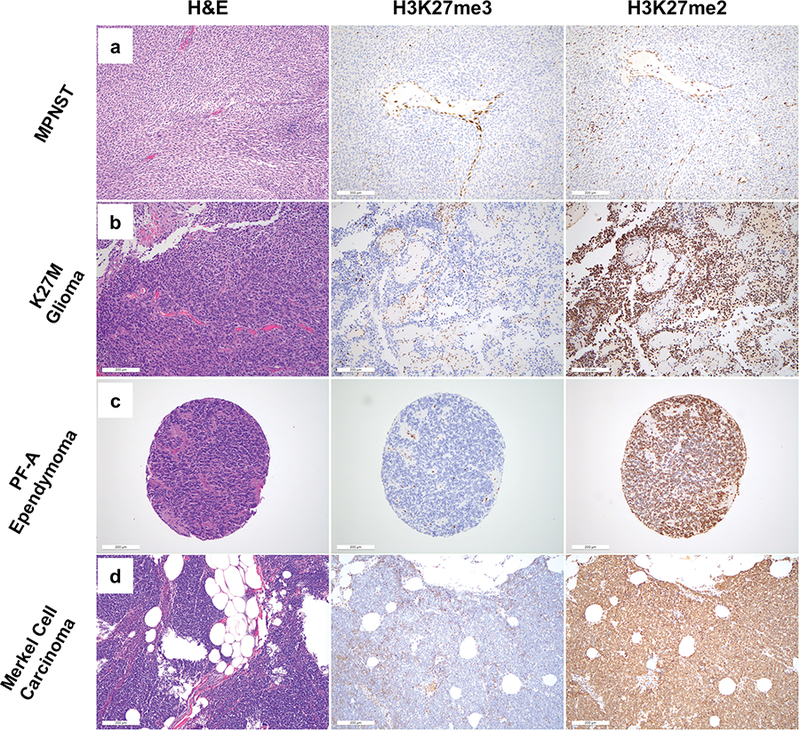
H&E (left) and H3K27me3 (middle) and H3K27me2 (right) immunohistochemical staining for representative cases of tumors characterized by loss of H3K27me3. (a) Malignant peripheral nerve sheath tumor (b) K27M glioma (c) PF-A Ependymoma (d) Merkel cell carcinoma. Of these tumors, only malignant peripheral nerve sheath tumor exhibits loss of nuclear H3K27me2 staining within tumor cells.
Loss of H3K27me2 outperforms loss of H3K27me3 in distinguishing malignant peripheral nerve sheath tumor from histologic mimics
While initial reports suggested that H3K27me3 loss was highly specific for malignant peripheral nerve sheath tumor, subsequent studies have challenged this notion. Most concerningly from a diagnostic standpoint, several of these reports have shown H3K27me3 loss in subsets of tumors in the morphologic differential diagnosis of malignant peripheral nerve sheath tumor--most notably synovial sarcoma and melanoma. Having demonstrated that PRC2 loss in malignant peripheral nerve sheath tumor is characterized by loss of both H3K27me3 and H3K27me2, we set out to determine whether H3K27me2 loss might serve as a more reliable marker of malignant peripheral nerve sheath tumor by comparing staining patterns of H3K27me2 and H3K27me3 malignant peripheral nerve sheath tumor with cases of synovial sarcoma and melanoma. Since prior reports had also posited a link between NRAS mutation and H3K27me3 loss in melanoma37, we ensured that our cohort contained roughly equal numbers of BRAF-mutant and NRAS-mutant melanomas.
With respect to H3K27me3 staining, we did not identify cases with a total absence of staining within our cohort of melanomas and synovial sarcomas (Table 2). We also failed to observe any difference in staining between melanomas with driver BRAF mutations and NRAS mutations. However, we did note significant variability in the fraction of cells of each tumor-type exhibiting nuclear reactivity for H3K27me3, and in the strength of the signal in positive cases (Table 2, Figures 4 and 5). In several cases, staining was limited to 5–10% of tumor cells, and weak enough to create ambiguity with respect to interpretation (Figures 4c and 5c, for example). By contrast, H3K27me2 showed staining in >50% of cells (2+ or greater) in all melanoma and synovial sarcoma cases. Moreover, at each level of H3K27me3 staining, the intensity of staining was generally stronger for H3K27me2 (Figures 4 and 5).
Figure 4.

H&E (left) and H3K27me3 (middle) and H3K27me2 (right) immunohistochemical staining for representative melanoma cases with strong (a) moderate (b) and weak/absent (c) H3K27me3 staining within tumor nuclei. All cases show strong retained nuclear H3K27me2 staining within tumor cells.
Figure 5.

H&E (left) and H3K27me3 (middle) and H3K27me2 (right) immunohistochemical staining for representative synovial sarcoma cases with strong (a) moderate (b) and weak/absent (c) H3K27me3 staining within tumor nuclei. All cases show strong retained nuclear H3K27me2 staining within tumor cells.
To get a quantitative sense of the increased discriminatory power of H3K27me2 as compared H3K27me3 immunohistochemistry, we evaluated mean staining intensity of tumor nuclei using QuPath31. From these intensities, we calculated H-scores for H3K27me2 and H3K27me3 staining for each case. Each H-score is a weighted measure of both strength of staining and uniformity, with higher scores indicating more tumor nuclei staining more intensely. H3K27me3 H-scores showed a broad distribution, with numerous tumors of each histotype having scores close to zero. In contrast, H3K27me2 H-scores more effectively segregated tumors into binary ‘positive’ and ‘negative’ categories (Figure 6). BRAF-mutant and NRAS-mutant melanomas showed no difference in mean H-score for either H3K27me2 or H3K27me3 staining (p=0.25 and 0.54 for K27me2 and K27me3, respectively).
Figure 6.

Numerical H-scores calculated from digital image analysis of nuclear immunohistochemical staining for (a) H3K27me3 and (b) H3K27me2. Selected malignant peripheral nerve sheath tumor cases, divided into H3K27me3 loss (MPNSTloss) and H3K27me3-retained (MPNSTret), selected melanoma cases, divided into BRAF-mutant (Melanoma BRAF) and NRAS-mutant (Melanoma NRAS), and selected synovial sarcoma cases.
Our immunohistochemical results suggest that, while complete loss of H3K27me3 is uncommon in melanoma and synovial sarcoma, markedly reduced, weak staining occurs at relatively high rates, which may cause interpretive difficulties when trying to distinguish these entities from malignant peripheral nerve sheath tumor. This suggests that this marker is not a robust discriminator, and subtly different antibody performance or conditions might lead to complete loss of staining within tumor cells. This lack of robustness may be responsible for the high variability in reported rates of PRC2 loss in malignant peripheral nerve sheath tumor in the literature (Table 3). Collectively, our results suggest that H3K27me2 loss is a robust and specific indicator of PRC2 loss. Immunohistochemical loss of H3K27me2 does not occur in melanoma or synovial sarcoma, and all tested tumors exhibited 2+ staining or greater. From a biological standpoint, this data also suggests that the reduced H3K27me3 staining seen in these tumors is unlikely to be related to loss-of-function alterations in PRC2. Finally, in our hands, the levels of H3K27me2 and H3K27me3 staining were consistent across both BRAF and NRAS-mutant melanomas, which does not support a driver-specific role for loss of histone H3K27 methylation in melanoma.
Table 3:
Collective H3K27me3 IHC results from prior malignant peripheral nerve sheath tumor studies
| Study | Sporadic | Syndromic | Radiation | Reference |
|---|---|---|---|---|
| Asano et al | 14/20 (70%) | 16/34 (47%) | - | 13 |
| Cleven et al | 17/54 (31%) | 33/80 (41%) | - | 10 |
| Le Guellec et al | 38/53 (72%) | 46/65 (71%) | 4/4 (100%) | 12 |
| Otsuka et al | 18/50 (36%) | 19/43 (44%) | - | 11 |
| Pekmezci et al | 4/13 (31%) | 13/26 (50%) | - | 14 |
| Prieto-Granada et al | 16/18 (89%) | 15/33 (45%) | 11/12 (92%) | 8 |
| Schaefer et al | 34/70 (49%) | 7/10 (70%) | 10/10 (100%) | 9 |
| This study | 12/36 (33%) | 17/30 (57%) | 5/6 (83%) | |
| total | 153/314 (49%) | 166/321 (52%) | 30/32 (94%) |
Discussion
The results of both H3K27me3 and H3K27me2 presented in this work are in agreement with other large-scale studies of H3K27me3 immunohistochemistry in malignant peripheral nerve sheath tumor (Table 3), indicating that H3K27me3 loss reflects true PRC2 loss in these tumors, and suggesting that the concordance between H3K27me2 and H3K27me3 is likely to hold in larger series. In our series, as with others, though the number of cases is smaller, rates of PRC2 loss as reflected in H3K27me2/3 immunohistochemistry seem to be highest in the subset of tumors arising following radiation therapy. In addition, despite variation in the individual series, the total fraction of both sporadic and syndromic malignant peripheral nerve sheath tumor harboring PRC2 loss appears to be roughly half of all cases. Thus, while H3K27me2 is highly specific and appears to be the best marker to date for malignant peripheral nerve sheath tumor, novel markers with greater sensitivity are still needed to aid in arriving at this notoriously challenging diagnosis.
While it was initially thought to be a specific marker for malignant peripheral nerve sheath tumor, there is an increasing awareness that some degree of loss of H3K27me3 is a relatively common event across diverse tumor types. By contrast, H3K27me2 loss is specific for malignant peripheral nerve sheath tumor, because it appears to require PRC2 loss-of-function. This requirement is likely attributable to the greater baseline abundance of H3K27me2 in cells, such that only complete loss of PRC2 activity is sufficient to reduce H3K27me2 below the level of IHC detection. The higher baseline levels of H3K27me2 were evident across virtually all of our samples, and likely contribute to the robustness of the stain and ease of interpretation as compared to H3K27me3 immunohistochemistry. Moreover, while the focus of this paper was on tumor staining, the stronger signal also enhanced the internal control staining, promoting greater ease of interpretation.
The greater frequency of H3K27me3 loss as compared to PRC2 loss suggests that there are multiple molecular pathways to polycomb dysfunction, not all of which require the global loss-of-function that is seen in malignant peripheral nerve sheath tumor5–7. In H3K27M-mutant glioma, H3K27me3 loss occurs because the mutant histone potently inhibits PRC2 activity via an interaction with the EZH2 subunit15,30. In posterior fossa ependymoma, loss of H3K27me3 appears to be related to mutations in or overexpression of CXorf67, an uncharacterized gene whose protein product physically interacts with PRC238. In both of these tumor types, some degree of PRC2 activity remains, as evinced by the data presented here and by prior reports noting high levels of H3K27me3 at tumor suppressor genes as detected by chromatin immunoprecipitation coupled to high-throughput sequencing (ChIP-seq)17,18,39.
In synovial sarcoma and Merkel cell carcinoma, the existing molecular data provide some room to speculate on the cause of reduced levels of H3K27me3. In synovial sarcoma the fusion oncoprotein hijacks the SWI/SNF chromatin remodeling apparatus to more effectively antagonize polycomb function and reduce H3K27me3 levels at particular sites40. It is unclear, however, whether this is sufficient to globally reduce H3K27me3 levels. In Merkel cell carcinomas, H3K27me3 loss is unique to tumors harboring Merkel polyomavirus (MPV) infection which may suggest that the virus somehow drives H3K27me3 loss20. Alternatively, lower levels of H3K27me3 may be a reflection of the cell of origin, rather than tumor promoting processes, since suppression of H3K27me3 drives the formation of ectopic Merkel cells in murine models of skin development41. These and other possible explanations require further investigation.
The mechanism for H3K27me3 loss in melanomas is unclear. In a survey of 265 melanomas, 37% had complete H3K27me3 loss and 51% had a partial loss12. In a separate study, loss of H3K27me3 was observed in 4/5 nodular melanomas arising from congenital melanocytic nevi, but was not observed in any of 20 benign proliferative nodules, suggesting that loss may contribute to cell transformation. Of the 4 melanomas that lost H3K27me3, 3 were NRAS-mutant, leading some to suggest that NRAS mutation might create selective pressure for H3K27me3 loss37. In our series, while a subset of melanomas showed somewhat reduced levels of H3K27me3 staining, global loss was rare/absent and H3K27me2 was completely intact. This was true in both NRAS-mutant and BRAF-mutant melanomas. It is unclear whether the discrepant results between our work and this prior series points to different rates of polycomb dysfunction in the cases, to a lack of robustness in H3K27me3 as a marker, or to some combination of these factors. Whatever the cause of H3K27me3 reduction or loss in melanoma, the uniformly strong staining for H3K27me2 suggests that melanomas do not exhibit true PRC2 loss and that H3K27me2 may prove a more useful marker in the distinction between malignant peripheral nerve sheath tumor and melanoma in challenging cases.
In addition to its diagnostic applications, immunohistochemistry for H3K27me2, perhaps in conjunction with H3K27me3 immunohistochemistry, may aid in predicting therapeutic vulnerabilities. Tumor suppressor silencing by residual H3K27me3 appears to be a key mechanism supporting tumor growth in K27M gliomas, which, despite low-levels of H3K27me3 globally, are highly sensitive to EZH2 inhibition39,42. By contrast, malignant peripheral nerve sheath tumor cell lines with PRC2 loss-of-function do not respond to EZH2 inhibition, since they have no residual functional PRC243,44. Collectively, these observations suggest that isolated H3K27me3 loss may be an indicator of sensitivity to EZH2 inhibitors. On the other hand, isolated H3K27me3 loss and global PRC2 loss may both indicate sensitivity to other classes of drugs targeting epigenetic regulators. In both K27M gliomas and malignant peripheral nerve sheath tumors, sensitivity to HDAC inhibitors and related compounds that dysregulate transcription are linked to polycomb dysfunction45–47.
Finally, in rare instances, tumors other than malignant peripheral nerve sheath tumor have exhibited loss of H3K27me3, and this has been linked to a homozygous defect in PRC2 components. To date, these reports are rare, and include a single case each of extraskeletal osteosarcoma and dedifferentiated liposarcoma, both of which had homozygous deletions in EED48,49. We think it likely that other such cases will also be identified. If they do exhibit bona fide PRC2 loss, they are likely to lose H3K27me2 as well. Loss of H3K27me2 by immunohistochemistry, therefore, is unlikely to be perfectly specific for malignant peripheral nerve sheath tumor. It is, however, a better surrogate for global PRC2 loss, and as such, a superior marker for malignant peripheral nerve sheath tumor.
Supplementary Material
Acknowledgements
This research was supported by US National Institutes of Health (NIH) grants (GM110174, AI118891 and CA196539 to B.A.G. and P01CA196539 to B.A.G.; 5K12CA076931 to J.B.W.; T32GM008275 and TL1TR001880 to D.M.M). J.B.W. is also supported by the University of Pennsylvania Department of Pathology and Laboratory Medicine Institutional lab startup funds. B.A.G. is also supported by a Robert Arceci Scholar award from the Leukemia and Lymphoma Society. The authors would also like to acknowledge Dr. Kumarasen Cooper in the Department of Pathology at the Hospital of the University of Pennsylvania for his support and helpful discussions about the content of the manuscript.
References
- 1.Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012;123:295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hornick JL. Limited biopsies of soft tissue tumors: the contemporary role of immunohistochemistry and molecular diagnostics. Mod Pathol 2019. doi: 10.1038/s41379-018-0139-y. [DOI] [PubMed] [Google Scholar]
- 3.Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 2009;249:1014–1022. [DOI] [PubMed] [Google Scholar]
- 4.Le Guellec S, Decouvelaere A-V, Filleron T, et al. Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am J Surg Pathol 2016;40:896–908. [DOI] [PubMed] [Google Scholar]
- 5.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014;514:247–251. [DOI] [PubMed] [Google Scholar]
- 7.Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1170–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol 2016;40:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer I-M, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol 2016;29:4–13. [DOI] [PubMed] [Google Scholar]
- 10.Cleven AHG, Sannaa GAA, Briaire-de Bruijn I, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 2016;29:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otsuka H, Kohashi K, Yoshimoto M, et al. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol Res Pract 2018;214:417–425. [DOI] [PubMed] [Google Scholar]
- 12.Le Guellec S, Macagno N, Velasco V, et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol 2017;30:1677–1687. [DOI] [PubMed] [Google Scholar]
- 13.Asano N, Yoshida A, Ichikawa H, et al. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017;70:385–393. [DOI] [PubMed] [Google Scholar]
- 14.Pekmezci M, Cuevas-Ocampo AK, Perry A, et al. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod Pathol 2017;30:1710–1719. [DOI] [PubMed] [Google Scholar]
- 15.Lewis PW, Müller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013;340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013;24:660–672. [DOI] [PubMed] [Google Scholar]
- 17.Chan K-M, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013;27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayliss J, Mukherjee P, Lu C, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 2016;8:366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panwalkar P, Clark J, Ramaswamy V, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 2017;134:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busam KJ, Pulitzer MP, Coit DC, et al. Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Mod Pathol 2017;30:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz LM, Hielscher T, Liechty B, et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol 2018;135:955–963. [DOI] [PubMed] [Google Scholar]
- 22.Makise N, Sekimizu M, Konishi E, et al. H3K27me3 deficiency defines a subset of dedifferentiated chondrosarcomas with characteristic clinicopathological features. Mod Pathol 2018. doi: 10.1038/s41379-018-0140-5. [DOI] [PubMed] [Google Scholar]
- 23.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016;352:aad9780. [DOI] [PubMed] [Google Scholar]
- 24.Pasini D, Bracken AP, Jensen MR, et al. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 2004;23:4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari KJ, Scelfo A, Jammula S, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 2014;53:49–62. [DOI] [PubMed] [Google Scholar]
- 26.Wessel D, Flügge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 1984;138:141–143. [DOI] [PubMed] [Google Scholar]
- 27.Sidoli S, Garcia BA. Characterization of Individual Histone Posttranslational Modifications and Their Combinatorial Patterns by Mass Spectrometry-Based Proteomics Strategies. Methods Mol Biol 2017;1528:121–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan Z-F, Sidoli S, Marchione DM, et al. EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J Proteome Res 2018;17:2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy G, Dimaggio PA, Chan EY, et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stafford JM, Lee C-H, Voigt P, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 2018;4:eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noberini R, Osti D, Miccolo C, et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res 2018;46:3817–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Muller M, Xu B, et al. Unrestricted modification search reveals lysine methylation as major modification induced by tissue formalin fixation and paraffin embedding. Proteomics 2015;15:2568–2579. [DOI] [PubMed] [Google Scholar]
- 34.Schoeftner S, Sengupta AK, Kubicek S, et al. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 2006;25:3110–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen X, Liu Y, Hsu Y-J, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008;32:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters AHFM, Kubicek S, Mechtler K, et al. Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin. Mol Cell 2003;12:1577–1589. [DOI] [PubMed] [Google Scholar]
- 37.Busam KJ, Shah KN, Gerami P, et al. Reduced H3K27me3 Expression Is Common in Nodular Melanomas of Childhood Associated With Congenital Melanocytic Nevi But Not in Proliferative Nodules. Am J Surg Pathol 2017;41:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pajtler KW, Wen J, Sill M, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 2018;136:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 2017;23:483–492. [DOI] [PubMed] [Google Scholar]
- 40.Kadoch C, Williams RT, Calarco JP, et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet 2017;49:213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perdigoto CN, Dauber KL, Bar C, et al. Polycomb-Mediated Repression and Sonic Hedgehog Signaling Interact to Regulate Merkel Cell Specification during Skin Development. PLoS Genet 2016;12:e1006151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piunti A, Hashizume R, Morgan MA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 2017;23:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amirnasr A, Verdijk RM, van Kuijk PF, et al. Expression and inhibition of BRD4, EZH2 and TOP2A in neurofibromas and malignant peripheral nerve sheath tumors. PLoS One 2017;12:e0183155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wassef M, Luscan A, Aflaki S, et al. EZH1/2 function mostly within canonical PRC2 and exhibit proliferation-dependent redundancy that shapes mutational signatures in cancer. Proc Natl Acad Sci U S A 2019. doi: 10.1073/pnas.1814634116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 2015;21:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagaraja S, Vitanza NA, Woo PJ, et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017;31:635–652.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wojcik JB, Marchione DM, Sidoli S, et al. Epigenomic reordering induced by Polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res 2019;:canres.3704.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makise N, Sekimizu M, Kubo T, et al. Extraskeletal osteosarcoma: MDM2 and H3K27me3 analysis of 19 cases suggest disease heterogeneity. Histopathology 2018;73:147–156. [DOI] [PubMed] [Google Scholar]
- 49.Makise N, Sekimizu M, Kubo T, et al. Clarifying the Distinction Between Malignant Peripheral Nerve Sheath Tumor and Dedifferentiated Liposarcoma: A Critical Reappraisal of the Diagnostic Utility of MDM2 and H3K27me3 Status. Am J Surg Pathol 2018;42:656–664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.