

1. Introduction
Tuberculous meningitis (TBM) is the most lethal form of TB, with a mortality in some circumstances exceeding 30% [1]. Outcomes are particularly severe amongst HIV-infected patients, in whom mortality approaches 60% [2]. The global burden of TBM is difficult to ascertain due to challenges in diagnosis and reporting, but is thought to affect approximately 100,000 people a year [1], representing around 6% of all extrapulmonary TB cases. TBM is a paucibacillary disease, and neuronal and vascular injury are mediated by immunopathological host responses to infection [3]. Clinical complications resulting from tissue-damaging responses (such as stroke and hydrocephalus) are important contributors to poor outcome. Despite the central role of inflammation and its downstream consequences, antimycobacterial treatment remains critical because mortality is nearly universal in its absence. However, even with the currently-used standard antitubercular regimen and widespread use of adjunctive corticosteroids as host-directed therapy (HDT), half of those who survive TBM still suffer significant neurological sequelae. These observations suggest a need for improved therapeutic strategies targeting both pathogen and host. Optimal management of TBM requires a combined approach that incorporates maximally effective antimicrobial therapy, more targeted and nuanced modification of injurious host inflammatory response, and better management of complications (Figure 1).
Figure 1.
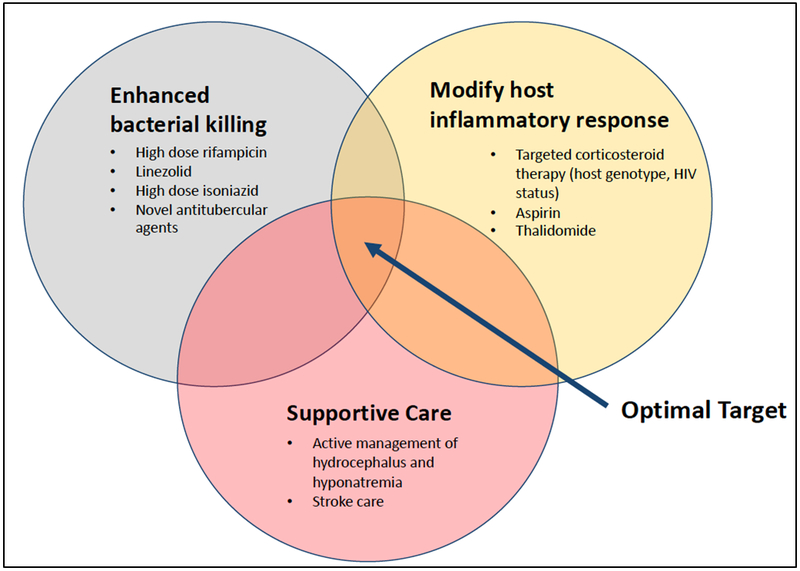
Potential therapeutic strategies for improving outcomes in TBM.
1. Current antimicrobial therapy for TBM is suboptimal
Treatment guidelines for TBM are based on those developed for pulmonary TB; a two-month intensive phase of rifampicin, isoniazid, pyrazinamide, and ethambutol followed by up to ten months of rifampicin and isoniazid. This regimen is based largely on expert opinion and does not take into account the differential ability of antitubercular drugs to penetrate the blood brain barrier (BBB), potentially resulting in suboptimal cerebrospinal fluid (CSF) and disease-site exposures.
Rifampicin is a key agent in TBM therapy; its exclusion from treatment worsens outcomes, and there is high mortality from rifampicin-resistant TBM. Rifampicin is highly protein-bound and the CSF penetration of total (protein-bound plus -unbound) rifampicin is poor. Standard doses (10 mg/kg) achieve total rifampicin CSF concentrations only 10-20% of those in plasma, rarely exceeding the minimum inhibitory concentration (MIC) of Mycobacterium tuberculosis. The effect of standard-dose rifampicin on TBM treatment outcomes, even at sub-optimal CSF concentrations, suggests the possibility of benefit with higher plasma and, by extension, CSF exposures. Isoniazid and pyrazinamide have good CSF penetration with potent early bactericidal activity (EBA) and treatment shortening ability, respectively. Of the currently recommended drugs, the CSF penetration of ethambutol is the poorest, even when the BBB is inflamed, raising questions about its value and need for an alternative agent in TBM treatment. Two favoured strategies have therefore emerged to intensify antitubercular therapy in TBM: use of higher rifampicin doses and adding (or substituting ethambutol with) a more potent fifth drug.
2. Higher rifampicin doses achieve more favorable exposures and may improve outcomes
Animal models demonstrate a clear dose-response relationship for rifampicin, including at disease sites, and indicate the standard dose is at the lower end of the dose-response curve. This is replicated in clinical studies of pulmonary TB where there is correlation between rifampicin dose and sputum culture conversion. Reassuringly, no major safety signals have been detected with increased doses 20 - 35 mg/kg in randomized controlled trials (RCTs) for pulmonary TB. A number of studies conducted in South-East Asia have explored the efficacy and PK of higher oral rifampicin doses up to 30 mg/kg compared to the standard 10 mg/kg dose in adult TBM (Table 1). Total CSF concentrations correlated with plasma exposures, and at the 30 mg/kg dose exceeded the MIC of rifampicin,[4] suggesting potential for improved efficacy. A significant mortality benefit was observed with the use of 13 mg/kg given intravenously (equivalent to 20 mg/kg orally) [5] in an Indonesian study [6], and there was a trend towards improved survival in a follow-up trial using oral rifampicin up to 30 mg/kg [4], although neither trial was powered for clinical endpoints. However, the use of oral rifampicin at 15 mg/kg (given with levofloxacin) was not associated with increased survival in a large phase 3 trial in Vietnam [7]. The inconsistent impact of higher rifampicin doses on mortality may be explained by unpredictable dose-response relationships due to PK variability, suggesting that doses higher than 15mg/kg may be required to reduce mortality. Additionally, serial dynamic 11C-labelled rifampicin positron emission tomography (PET) in rabbits suggested that rifampin doses ≥30 mg/kg may be required to achieve adequate intralesional concentrations in a model of pediatric TBM [8]. These observations provide rationale for future clinical trials, which will evaluate the safety, PK and efficacy of 35 mg/kg rifampicin in TBM, in combination with additional new agents.
Table 1.
Summary of completed trials of higher dose rifampicin in TBMa
| Reference number |
Study design | Rifampicin experimental dose |
Population | Key findings |
|---|---|---|---|---|
| 6 | RCT: open label, factorial | 13 mg/kg (600 mg) IV (with or without additional moxifloxacin 400 mg or 800 mg)b |
Adults Indonesia n = 60 HIV 12% |
Higher dose rifampicin associated with reduced mortality (65%) compared to standard dose (35%); p = 0.03 Not powered for clinical endpoint |
| 5 | RCT: open label | 17 mg/kg (750 mg) PO 20 mg/kg (900 mg) PO 13 mg/kg (600 mg) IV |
Adults Indonesia n = 30 HIV 20% |
Similar PK exposures between intravenous (IV) and both oral (PO) doses Clinical efficacy endpoints not assessed |
| 7 | RCT: double blind, placebo-controlled | 15 mg/kg PO (with added levofloxacin)b for 2 months |
Adults Vietnam n = 817 HIV 43% |
No difference in primary mortality endpoint at 9 months |
| 4 | RCT: double-blind, placebo-controlled | 20 mg/kg (900 mg) PO 30 mg/kg (1350 mg) POb |
Adults Indonesia n = 60 HIV 10% |
Trend towards lower mortality with rifampicin 30 mg/kg (15%) vs. standard dose (35%) at 6 months; not significant Not powered for clinical endpoint |
RCT, randomized controlled trial; IV, intravenous; PO, oral.
All participants received standard antitubercular therapy plus corticosteroids
Compared to standard dose rifampicin (10 mg/kg)
3. Additional antimicrobial agents for TBM
Options for antitubercular therapy have been improved by registration of new compounds and introduction of repurposed drugs. Several agents, plus older drugs traditionally used in drug-resistant TB, fulfill the two key requirements for consideration in TBM therapy: enhanced activity against M. tuberculosis plus ability to achieve adequate CSF concentrations. Preliminary data suggest the novel nitroimidazole delamanid has good brain and intralesional penetration in animal models [International Workshop on Clinical Pharmacology of Tuberculosis Drugs; Abstract 18. The Hague, October 2018] and may enter clinical studies for TBM in the future, as may potent new oxazolidinones currently under development. The older second-line agents, cycloserine (or terizidone, a condensation product containing two cycloserine molecules) and ethionamide, have good CSF penetration [9] and have been used as substitutes for ethambutol in TBM; ethionamide-based regimens are associated with particularly good outcomes in pediatric TBM in South Africa [10]. However, use of cycloserine and ethionamide is limited by dose-related neurological and gastrointestinal toxicity, respectively. Fluoroquinolones have undergone some evaluation with disappointing results. Linezolid has emerged as the most promising repurposed antimicrobial for TBM.
4.1. Fluoroquinolones
Moxifloxacin and levofloxacin are highly active against M. tuberculosis and are essential components of treatment regimens for rifampicin-resistant TB. Both have excellent CSF penetration, and one study found a relationship between CSF exposures of levofloxacin and clinical outcomes when added to standard therapy for TBM [11]. Fluoroquinolones were evaluated in combination with high dose rifampicin in two RCTs of TBM in South-East Asia (Table 1). These produced disappointing results, unable to demonstrate an independent effect on TBM outcomes. In a subgroup analysis of the Vietnam trial, administration of the experimental regimen containing levofloxacin (with higher rifampicin doses of 15 mg/kg) before coma onset did result in survival benefit for patients diagnosed with isoniazid mono-resistant TBM [12]. This may suggest that the potent early bactericidal activity (EBA) of fluoroquinolones can substitute benefit provided by isoniazid, but are not additive in this respect. It remains to be determined whether agents with enhanced sterilizing ability could provide additive effect to rifampicin in drug-sensitive TBM.
4.2. Linezolid
Linezolid is well-established in the treatment of drug-resistant pulmonary TB, with two RCTs and multiple observational studies demonstrating improved outcomes when added to treatment. In vitro models have demonstrated potent activity against a non-replicative persister M. tuberculosis phenotype, as well as additive activity when administered with rifampicin, particularly at higher doses, spurring interest in its use as a treatment-shortening agent in drug-sensitive pulmonary TB [13, 14]. Linezolid is attractive for TBM therapy due to moderate EBA and sterilizing ability against M. tuberculosis as well as favorable PK characteristics: linezolid has almost complete oral bioavailability with extensive tissue distribution, including in CSF. This feature has led to successful use in severe central nervous system infections caused by Gram-positive bacteria. Retrospective studies have demonstrated favorable clinical outcomes in children [15] and adults [16] with drug-sensitive TBM and provide further rationale for its inclusion in experimental regimens in upcoming clinical trials.
4. Host-directed therapy
The most widely-researched HDT in TBM are corticosteroids, which improve medium-term survival in HIV-uninfected patients but have no effect on morbidity [17]. The mechanism by which corticosteroids improve mortality is poorly understood. There is interest in a common functional promoter variant in the gene encoding the enzyme leukotriene-A4 hydrolase (LTA4H), which appears to predict response to dexamethasone in HIV-uninfected individuals by altering the balance of pro- and anti-inflammatory eicosanoids. In a post hoc analysis of prospective clinical studies in Vietnam, benefit from dexamethasone was restricted to TBM patients with a LTA4H hyperinflammatory (TT) genotype, with possible harm in those with a hypoinflammatory (CC genotype) [18], suggesting a role for a personalized approach to corticosteroid therapy. An RCT in which participants are stratified by LTA4H genotype is underway in Vietnam to test this ().
Aspirin has dual effects on the pathogenic hallmarks of TBM: at low doses it may prevent ischaemic infarction through inhibition of thromboxane A2 and platelet aggregation; at high doses aspirin inhibits the expression of proinflammatory eicosanoids and tumor necrosis factor (TNF)-α, plus triggers production of molecules that contribute to resolution of inflammation [19]. Three RCTs have investigated aspirin in adult and paediatric TBM, at varying doses. There appears to be clinical benefit, with mortality reduction in one relatively small adult trial [20], but this remains uncertain (Table 2). Although no significant increase in adverse events were observed in these studies, safety concerns exist, particularly with higher doses in the context of concomitant dexamethasone and linezolid. Upcoming phase 2 () and 3 trials are planned to address this equipoise.
Table 2.
Summary of completed trials of adjunctive aspirin in TBM
| Reference number |
Study design | Intervention | Population | Key findings |
|---|---|---|---|---|
| 19 | RCT: double-blind, placebo-controlled | Aspirin 81 mg vs. 1000 mg vs. placebo for 60 daysa | Adults HIV-uninfected Vietnam n = 120 |
No difference in primary efficacy endpoint (new brain infarction or death at 60 days) Reduced infarcts and death with aspirin 81 mg (15%) and 1000 mg (11%) compared to placebo (34%) in subgroup with confirmed TBM; p = 0.06 |
| 20 | RCT: open label, placebo-controlled | Aspirin 150 mg vs. placebo for 3 monthsb | Adults HIV-uninfected India n = 118 |
No difference in primary outcome of stroke (MRI or clinical) at 3 months Significant reduction in mortality with aspirin (22%) compared to placebo (43%); p = 0.02 |
| 24 | RCT: double-blind, placebo-controlled | Aspirin 75 mg vs. aspirin 100 mg/kg vs. placebo for 1 montha | Children HIV-infected (n = 5) and uninfected South Africa n = 146 |
Aspirin not associated with improved neurological or cognitive outcomes, or survival at 6 months Study not powered for clinical endpoint |
RCT, randomized controlled trial
All participants received standard antitubercular therapy plus corticosteroids
Corticosteroids administered only to participants with severe disease
The influence of hyperinflammatory immune phenotype on TBM outcomes supports a strategy to target TNF-α in TBM. Thalidomide, a TNF-α antagonist with anti-angiogenic properties, has emerged as a candidate for HDT, and was safe and well tolerated as an adjunctive therapy to treat children with MRC grade 2 TBM [21]. However, excess adverse events and deaths occurred in the thalidomide arm of a subsequent phase 3 RCT for pediatric TBM, leading to early trial discontinuation [22]. Use of high thalidomide doses (24 mg/kg) and inclusion of more severe disease in the experimental group may have influenced these outcomes. Although no subsequent trials have taken place, observational data suggest thalidomide may have a role in treatment of tuberculous cerebral mass lesions where corticosteroid therapy has failed.
6. Conclusion
Rational strategies proposed for enhanced pharmacotherapy in TBM include: (i) intensified antimicrobial therapy with higher doses of rifampicin plus an additional potent agent with good CSF penetration, and (ii) more targeted and nuanced modification of injurious host inflammatory response with host-directed therapies (HDT). Novel therapeutic approaches in TBM, facilitated in part by availability of new drugs, are being informed by advances in understanding of the biology of TBM and use of PK data to optimize antimicrobial dosing. Several planned clinical trials will provide a better evidence base for TBM treatment over the next 5 years.
7. Expert Opinion
A number of specific uncertainties exist in relation to planned strategies for intensified TBM pharmacotherapy.
There are concerns about linezolid toxicity, which is dose- and exposure-dependent. This may be more pronounced in HIV co-infection due to pre-existing peripheral neuropathy and anemia, potentially limiting use in this population, as well as in combination with HDT with overlapping toxicities. Because of its narrow therapeutic window [23], the optimal dose and duration of linezolid for TBM is unknown, and it is essential to define PK targets that minimize the risk of toxicity while contributing therapeutic benefit. An additional concern is the drug-drug interaction between linezolid and rifampicin, whereby potent induction of CYP3A4 by rifampicin leads to ~30% reduced linezolid exposures when these drugs are co-administered. Furthermore, linezolid is a substrate of the P-glycoprotein drug transporter which is abundant in the BBB and also extensively induced by rifampicin, potentially leading to increased clearance of linezolid from CSF and brain interstitium. The magnitude and clinical impact of rifampicin’s inductive effects are unknown, particularly at higher rifampicin doses where induction may be more pronounced. In view of these uncertainties and lack of clinical data, investigators have proposed an empirical approach to linezolid dosing in TBM: an initial ‘intensive phase’ of higher doses (1200 mg daily) for the first month to maximize efficacy and overcome potential increased metabolism and CNS clearance as a result of rifampicin induction in the initial critical stages of illness, followed by dose reduction in the second month of therapy to reduce risk of toxicity.
With regard to high dose rifampicin, questions exist about whether oral dosing will achieve similar exposures to intravenous use, which has been associated with mortality benefit in TBM [6]. This has important implications for the deployment of intensified antimicrobial therapy for TBM in resource-limited settings as intravenous rifampicin would be associated with increased cost, prolonged hospitalization, and complications relating to peripheral intravenous catherization. Based on existing population PK models of rifampicin and data from a clinical trial showing equivalent AUC (which drives rifampicin effect) between 13 mg/kg given IV and 20 mg/kg given orally [5], it is likely that exposures will be similar between oral 35 mg/kg and IV 20mg/kg: this will be tested in two upcoming RCTs.
There is recognition that treatment strategies may need to be personalized in order to maximize benefit. For example, adjunctive corticosteroids have not yet been shown to have an effect on death or disability in HIV co-infected TBM patients and among HIV-uninfected patients appear to only offer survival benefit in those with the homozygous TT LTA4H genotype; the prevalence of both these factors have large regional variations. In addition to interventions described above, other novel strategies being evaluated include high dose isoniazid stratified by N-acetylcysteine (NAT2) acetylator status () and use of efflux pump inhibitors to boost antitubercular drug concentrations in CSF. This new wave of interest in improving pharmacotherapy for TBM is welcome and will provide a better evidence base for treatment of this devastating condition.
Acknowledgments
Funding:
S Wasserman is supported by the European & Developing Countries Clinical Trials Partnership (Grant number CDF1018) and by the Wellcome Trust (Grant number 203135/Z/16/Z). A Davis is supported by a Wellcome Clinicians Ph.D. grant provided to the University College London. RJ Wilkinson is supported by the Francis Crick Institute which receives its core funding from Cancer Research UK, the UK Medical Research Council and Wellcome Trust (FC00110218). He also receives support from the Wellcome Trust (104803, 203135) and the National Institutes of Health (AJ115940). Meanwhile, G Meintjes was supported by the Wellcome Trust (Grant numbers 098316 and 203135/Z/16/Z), the South African Research Chairs Initiative of the Department of Science and Technology and the National Research Foundation (NRF) of South Africa (Grant numbers 64787), through National Research Foundation incentive funding (UID: 85858) and the South African Medical Research Council through its TB and HIV Collaborating Centres Programme with funds received from the National Department of Health/South African Medical Research Council (RFA# SAMRC-RFA-CC: TB/HIV/AIDS-01-2014).
Footnotes
Declaration of Interest:
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
REFERENCES
- 1.Wilkinson RJ, Rohlwink U, Misra UK, et al. Tuberculous meningitis. Nature Rev Neurol 2017;13:581–98.** Comprehensive and state of the art review of tuberculous meningitis
- 2.Marais S, Pepper DJ, Marais BJ, Torok ME. HIV-associated tuberculous meningitis--diagnostic and therapeutic challenges. Tuberculosis (Edinb). 2010;90(6):367–74. [DOI] [PubMed] [Google Scholar]
- 3.Davis A, Meintjes G, Wilkinson RJ. Treatment of tuberculous meningitis and its complications in adults. Curr Treat Options Neurol 2018;20:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dian S, Yunivita V, Ganiem AR, et al. Double-blind, randomized, placebo-controlled phase II dose-finding study to evaluate high-dose rifampin for tuberculous meningitis. Antimicrob Agents Chemother 2018;62:pii: e01014-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yunivita V, Dian S, Ganiem AR, et al. Pharmacokinetics and safety/tolerability of higher oral and intravenous doses of rifampicin in adult tuberculous meningitis patients. Int J Antimicrob Agents 2016;48:415–21. [DOI] [PubMed] [Google Scholar]
- 6.Ruslami R, Ganiem AR, Dian S, et al. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open-label, randomised controlled phase 2 trial. Lancet Infect Dis 2013;13:27–35. [DOI] [PubMed] [Google Scholar]
- 7.Heemskerk AD, Bang ND, Mai NT, et al. Intensified antituberculosis therapy in adults with tuberculous meningitis. N Engl J Med 2016;374:124–34. [DOI] [PubMed] [Google Scholar]
- 8.Tucker EW, Guglieri-Lopez B, Ordonez AA, et al. Noninvasive 11C-rifampin positron emission tomography reveals drug biodistribution in tuberculous meningitis. Sci Transl Med 2018;10:eaau0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donald PR. Cerebrospinal fluid concentrations of antituberculosis agents in adults and children. Tuberculosis (Edinb). 2010;90(5):279–92. [DOI] [PubMed] [Google Scholar]
- 10.van Well GT, Paes BF, Terwee CB, Springer P, Roord JJ, Donald PR, et al. Twenty years of pediatric tuberculous meningitis: a retrospective cohort study in the western cape of South Africa. Pediatrics. 2009;123(1):e1–8. [DOI] [PubMed] [Google Scholar]
- 11.Thwaites GE, Bhavnani SM, Chau TT, et al. Randomized pharmacokinetic and pharmacodynamic comparison of fluoroquinolones for tuberculous meningitis. Antimicrob Agents Chemother. 2011;55:3244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heemskerk AD, Nguyen MTH, Dang HTM, et al. Clinical outcomes of patients with drug-resistant tuberculous meningitis treated with an intensified antituberculosis regimen. Clin Infect Dis 2017;65:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drusano GL, Myrick J, Maynard M, et al. Linezolid kills acid phase and non-replicative persister phase mycobacterium tuberculosis in a hollow fiber infection model. Antimicrob Agents Chemother 2018;62:pii: e00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drusano GL, Neely M, Van Guilder M, et al. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PloS One 2014;9:e101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Lu J, Liu J, Zhao Y, et al. Linezolid is associated with improved early outcomes of childhood tuberculous meningitis. Pediatr Infect Dis J 2016;35:607–10. [DOI] [PubMed] [Google Scholar]
- 16.Sun F, Ruan Q, Wang J, et al. Linezolid manifests a rapid and dramatic therapeutic effect for patients with life-threatening tuberculous meningitis. Antimicrob Agents Chemother 2014;58:6297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasad K, Singh MB, Ryan H. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev 2016;4:Cd002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobin DM, Roca FJ, Oh SF, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 2012;148:434–46.*Study demonstrating effect of host genotype on corticosteroid response in an animal model and in clinical trial participants
- 19.Mai NT, Dobbs N, Phu NH, et al. A randomised double blind placebo controlled phase 2 trial of adjunctive aspirin for tuberculous meningitis in HIV-uninfected adults. eLife 2018; 7:pii: e33478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misra UK, Kalita J, Nair PP. Role of aspirin in tuberculous meningitis: a randomized open label placebo controlled trial. J Neurol Sci 2010;293:12–7. [DOI] [PubMed] [Google Scholar]
- 21.Schoeman JF, Springer P, Ravenscroft A, et al. Adjunctive thalidomide therapy of childhood tuberculous meningitis: possible anti-inflammatory role. J Child Neurol 2000;15:497–503. [DOI] [PubMed] [Google Scholar]
- 22.Schoeman JF, Springer P, van Rensburg AJ, et al. Adjunctive thalidomide therapy for childhood tuberculous meningitis: results of a randomized study. J Child Neurol 2004;19:250–7. [DOI] [PubMed] [Google Scholar]
- 23.Wasserman S, Meintjes G, Maartens G. Linezolid in the treatment of drug-resistant tuberculosis: the challenge of its narrow therapeutic index. Expert Rev Anti-infect Ther 2016;14:901–15. [DOI] [PubMed] [Google Scholar]
- 24.Schoeman JF, Janse van Rensburg A, Laubscher JA, Springer P. The role of aspirin in childhood tuberculous meningitis.J Child Neurol 2011;26:956–62. [DOI] [PubMed] [Google Scholar]