

Abstract
Rationale:
Pro-inflammatory cytokines have been identified as potential targets for lowering vascular risk. Experimental evidence and Mendelian randomization suggest a role of monocyte-chemoattractant protein-1 (MCP-1) in atherosclerosis and stroke. However, data from large-scale observational studies are lacking.
Objective:
To determine whether circulating levels of MCP-1 are associated with risk of incident stroke in the general population.
Methods and Results:
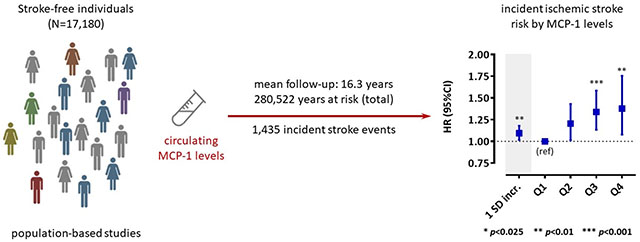
We used previously unpublished data on 17,180 stroke-free individuals (mean age 56.7±8.1 years; 48.8% males) from six population-based prospective cohort studies and explored associations between baseline circulating MCP-1 levels and risk of any stroke, ischemic stroke, and hemorrhagic stroke over a mean follow-up interval of 16.3 years (280,522 person-years at risk; 1,435 incident stroke events). We applied Cox proportional hazard models and pooled hazard ratios (HR) using random-effects meta-analyses. Following adjustments for age, sex, race, and vascular risk factors, higher MCP-1 levels were associated with increased risk of any stroke (HR per 1 SD increment in ln-transformed MCP-1: 1.07, 95%CI: 1.01-1.14). Focusing on stroke subtypes, we found a significant association between baseline MCP-1 levels and higher risk of ischemic stroke (HR: 1.11, [1.02-1.21]), but not hemorrhagic stroke (HR: 1.02, [0.82-1.29]). The results followed a dose-response pattern with a higher risk of ischemic stroke among individuals in the upper quartiles of MCP-1 levels as compared to the 1st quartile (HRs: 2nd quartile: 1.19 [1.00-1.42]; 3rd quartile: 1.35, [1.14-1.59]; 4th quartile: 1.38, [1.07-1.77]). There was no indication for heterogeneity across studies and in a sub-sample of four studies (12,516 individuals) the risk estimates were stable after additional adjustments for circulating levels of interleukin-6 and high-sensitivity C-reactive protein.
Conclusions:
Higher circulating levels of MCP-1 are associated with increased long-term risk of stroke. Our findings along with genetic and experimental evidence suggest that MCP-1-signaling might represent a therapeutic target to lower stroke risk.
Keywords: Epidemiology, Ischemic Stroke, Risk Factors, Monocyte chemoattractant protein-1, CCL2, stroke, cerebrovascular disease/stroke, atherosclerosis, chemokine, inflammation
Graphical Abstract
INTRODUCTION
Stroke is the leading cause of adult disability and the second most common cause of death worldwide.1, 2 Inflammatory mechanisms contribute to the pathogenesis of stroke, most notably to large artery atherosclerotic stroke,3, 4 but the specific pro-inflammatory factors mediating stroke risk are largely elusive. Discordant results from the CANTOS5–8 and CIRT6 randomized controlled trials emphasize the importance of targeting specific mediators and pathways for lowering vascular risk.5–8 Treatment with an anti-interleukin-1β (IL-1β) monoclonal antibody reduced the levels of IL-6 and high-sensitivity C-reactive protein (hsCRP) leading to a reduction in the combined primary endpoint of nonfatal myocardial infarction, nonfatal stroke or cardiovascular death independent of low-density lipoprotein (LDL) cholesterol levels,5 whereas treatment with low-dose methotrexate neither reduced cardiovascular event rates nor the levels of IL-1β, IL-6, and hsCRP.6
In a Mendelian Randomization study on circulating levels of 41 cytokines and growth factors, we recently found genetic predisposition to higher levels of the CC-chemokine monocyte-chemoattractant protein-1 (MCP-1; also known as CC-chemokine ligand 2, CCL2) to be associated with increased risk of stroke, ischemic stroke, coronary artery disease, and myocardial infarction.9 MCP-1 recruits monocytes to the subendothelial space of the atherogenic arterial wall10–12 and studies in experimental models of atherosclerosis suggest that targeting MCP-1 or its receptor CCR2 limits plaque size, plaque progression, and plaque destabilization.13–17 These findings define the MCP-1/CCR2 axis as a potential additional target for reducing residual inflammatory risk in vascular disease. However, data on MCP-1 and vascular risk in humans remain scarce.
Among patients with acute coronary syndromes in the OPUS-TIMI 1618 and A to Z trial,19 high circulating MCP-1 levels were associated with a significantly increased risk of death or myocardial infarction during follow-up, independently of baseline variables including hsCRP levels. In population-based studies higher MCP-1 levels were associated with subclinical atherosclerosis and incident coronary artery disease during follow-up.20, 21 In contrast, the relationship between circulating MCP-1 levels and incident stroke remains unknown as does the relationship between MCP-1, IL-6, and CRP in mediating vascular risk.
Here, leveraging data from six population-based prospective cohort studies encompassing 17,180 stroke-free individuals with long-term follow-up, we set out to: (i) determine the association between circulating MCP-1 levels at baseline and risk of incident stroke, (ii) explore associations of MCP-1 levels with risk of major stroke subtypes (incident ischemic and hemorrhagic stroke), and (iii) assess whether any association with stroke risk is independent of the IL-6 and CRP axis by adjusting for the circulating levels of IL-6 and hsCRP.
METHODS
This study is based on summary statistics produced by the studies included in the systematic review. The main individual-study results are provided as Supplemental material. All summary data that support the findings of this study are further available from the corresponding author upon reasonable request. For accessing individual-level data of the included studies the readers should contact the authors representing the respective studies and follow the required processes.
Systematic review.
We systematically searched PubMed from inception through 15 March 2019 for population-based prospective cohort studies exploring associations between circulating MCP-1 levels and the risk of incident vascular outcomes including coronary artery disease, myocardial infarction, fatal or non-fatal stroke, and peripheral artery disease. The reference lists of the identified studies were further hand searched. The detailed search strategy is available in the Appendix. We subsequently contacted the corresponding authors of the selected studies inquiring about their interest to contribute data for the current meta-analysis examining the association between circulating MCP-1 levels and risk of incident stroke. Investigators of the following six studies agreed to participate and the following studies were thus included in the current meta-analysis: the Atherosclerosis Risk in Communities (ARIC) Study,20 the Dallas Heart Study (DHS),21 the Norfolk arm of the European Prospective Investigation of Cancer (EPIC-Norfolk) study,22 the Offspring Cohort of the Framingham Heart Study (FHS),23 the Monitoring of Trends and Determinants in Cardiovascular Disease (MONICA) subcohort of the Kooperative Gesundheitsforschung in der Region Augsburg (KORA) study,24 and the cardiovascular subcohort of the Malmö Diet and Cancer Study (MDCS).25 With the exception of the FHS Offspring study, which had previously published part of the data included in this analysis (96 vs 172 incident events)23, none of the studies previously published data on the association between circulating MCP-1 levels and risk of incident stroke. The flowchart describing the study selection is depicted in Online Figure I.
Study populations, MCP-1 level measurements and assessment of stroke outcomes.
The study design, population characteristics, methods used for quantifying circulating MCP-1 levels, stroke outcome definitions, and assessments in individual cohorts are detailed in Online Table I. In brief, all studies were population-based prospective cohorts and participants included in the current analyses were selected from these cohorts based on availability of MCP-1 measurements at baseline. Circulating MCP-1 levels were measured in serum or plasma samples drawn during the baseline assessments. As incident stroke was the primary outcome of the current study, all participants with a history of stroke at baseline assessments (prevalent cases) were excluded from subsequent analyses. Stroke occurrence was assessed during follow-up visits over mean intervals of 11 to 23 years based on self-reported information and validation from medical records of the participants. In addition to information on any stroke, all studies further provided information on the major stroke subtypes (ischemic vs hemorrhagic stroke).
Quality assessment.
Study quality was assessed using the cohort subscale of the Newcastle-Ottawa scale.26 The criteria for awarding quality points were the following: a general population sample (representativeness of exposed cohort); selection of patients for inclusion independently of MCP-1 levels (selection of the non-exposed cohort); measurement of MCP-1 levels in the serum or plasma based on a validated assay (ascertainment of exposure); exclusion of patients with prevalent stroke at baseline (outcome not present at start of study); adjustments for age and sex, as well as for conventional vascular risk factors (comparability items); assessment of stroke outcomes blindly to MCP-1 levels with validation based on medical records (assessment of outcome); a follow-up interval longer than 5 years (follow-up duration); and a completion of follow-up rate of >90% (adequacy of follow-up cohorts).
Statistical analysis.
A pre-defined analysis protocol was circulated to investigators of each of the cohort studies requesting summary results for meta-analysis. MCP-1 levels were ln-transformed in all studies for normalization. We did not consider absolute MCP-1 values due to marked differences in mean MCP-1 level values between studies, probably related to different assays used for MCP-1 quantification (Table 1). We first examined descriptive associations between MCP-1 levels and conventional vascular risk factors. We pooled study-specific z-scores reflecting differences of MCP-1 levels from the overall mean of each study with random-effects models across the risk factor categories and statistically examined associations using meta-regression.
Table 1.
Descriptive baseline characteristics of the six included population-based prospective cohort studies.
| Cohort | ARIC | DHS | EPIC-Norfolk | FHS Offspring | MONICA/KORA | MDCS-CV |
|---|---|---|---|---|---|---|
| Geographical setting (baseline assessment) | USA (1986-1989) | USA (2000-2002) | UK (1993-1997) | USA (1998-2001) | Germany (1984-2002) | Sweden (1991-1994) |
| N individuals included in the analysis | 1,234 | 2,931 | 3,182 | 3,069 | 2,055 | 4,709 |
| Follow-up (years) | 23.0 [13.2-27.8] | 11.0 (1.7) | 16.8 (6.4) | 13.8 (3.7) | 15.7 (6.4) | 19.5 (4.9) |
| N incident stroke events | 153 | 64 | 503 | 172 | 116 | 427 |
| N incident ischemic stroke events | 141 | 42 | 458 | 141 | 99 | 352 |
| N incident hemorrhagic stroke events | 12 | 9 | 76 | 22 | 17 | 69 |
| N fatal stroke events | 10 | 6 | 132 | 26 | 22 | 30 |
| Age (years) | 56.9 (5.3) | 44.0 (10.0) | 65.3 (7.8) | 61.6 (9.4) | 52.4 (10.3) | 57.5 (4.9) |
| Male sex (N, %) | 738 (59.8) | 1254 (42.8) | 2009 (63.1) | 1421 (46.3) | 1093 (53.2) | 1873 (39.8) |
| Hypertension (N, %) | 417 (33.9) | 944 (32.7) | 2029 (63.8) | 1378 (44.9) | 877 (42.7) | 2958 (62.8) |
| SBP (mmHg) | 125 (20) | 124 (19) | 141 (18) | 127 (19) | 133 (19) | 141 (19) |
| DBP (mmHg) | 74 (12) | 78 (10) | 85 (11) | 74 (10) | 82 (11) | 87 (9) |
| Diabetes (N, %) | 156 (12.6) | 296 (10.1) | 623 (19.6) | 379 (12.3) | 103 (5.0) | 183 (3.9) |
| Hypercholesterolemia (N, %) | 760 (61.6) | 377 (12.9) | 414 (13.0) | 1615 (52.6) | 1251 (57.4) | 2918 (62.8) |
| LDL cholesterol levels (mg/dL) | 142.8 (39.9) | 107.4 (35.3) | 160.1 (39.4) | 119.9 (32.7) | 148.5 (2.4) | 161.3 (37.9) |
| HDL cholesterol levels (mg/dL) | 49.6 (16.5) | 50.0 (14.6) | 51.8 (15.1) | 53.9 (16.7) | 56.0 (17.0) | 53.8 (14.3) |
| BMI (kg/m2) | 27.4 (5.1) | 29.7 (7.0) | 26.6 (3.6) | 28.1 (5.3) | 27.2 (4.1) | 25.6 (3.9) |
| Smoking status (N, %) | ||||||
| Never smokers | 461 (37.3) | 1639 (55.9) | 1201 (10.3) | 1077 (35.1) | 947 (46.1) | 1916 (40.1) |
| Ex-smokers | 397 (32.2) | 496 (16.9) | 1652 (51.9) | 1604 (52.3) | 591 (28.8) | 1777 (37.8) |
| Current smokers | 376 (30.5) | 796 (27.2) | 329 (37.7) | 388 (12.6) | 517 (25.1) | 1010 (21.5) |
| eGFR (mL/min/1.73 m2) | 100.0 (16.6) | 99.5 (23.7) | 74.5 (24.9) | 83.3 (16.5) | 87.9 (17.4) | 76.9 (15.3) |
| Coronary artery disease (N, %) | 68 (5.5) | 79 (2.7) | 0 (0) | 265 (8.6) | 46 (2.2) | 78 (1.7) |
| Atrial fibrillation (N, %) | 1 (0.1) | 35 (1.2) | n/a | 119 (3.9) | n/a | 34 (0.7) |
| Heart failure (N, %) | 53 (4.3) | 83 (2.8) | 0 (0) | 31 (1.0) | 119 (5.7) | 2 (0.04) |
| hsCRP levels (mg/L) | n/a | 2.8 [1.2-6.8] | 2.0 [1.0-3.8] | 2.2 [1.0-5.1] | 1.4 [0.7-3.3] | 1.3 [0.7-2.7] |
| Sample used for MCP-1 assessment | plasma | plasma | serum | serum | serum | plasma |
| MCP-1 levels (pg/mL) | 398.9 [348.4-467.1] | 166.5 [122.9-224.4] | 51.5 [38.8-68.1] | 313.4 [253.9-382.3] | 298.0 [127.6-323.8] | 2.52 [2.22-2.82]* |
The numbers correspond to N (%) for categorical variables and to mean (SD) or median [25th - 75th percentile] for continuous variables.
The used assay in MDCS did not provide MCP-1 measurements as absolute values, but as relative expression levels obtained by proximity extension assay (PEA).
Abbreviations: ARIC, Atherosclerosis Risk in Communities Study; DHS, Dallas Heart Study; EPIC-Norfolk, European Prospective Investigation of Cancer, Norfolk; FHS Offspring, Framingham Heart Study- Offspring Cohort; MONICA/KORA, Monitoring of Trends and Determinants in Cardiovascular Disease - Kooperative Gesundheitsforschung in der Region Augsburg; MDCS-CV, Malmö Diet and Cancer Study – Cardiovascular sub-cohort; BMI, body mass index; hsCRP, high-sensitivity C-reactive protein; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; HDL, high-density lipoprotein; LDL, low-density liporprotein; MCP-1, monocyte chemoattractant protein- 1; SBP, systolic blood pressure.
To examine associations between baseline MCP-1 levels and incident stroke, Cox proportional hazard models were fit in each study. MCP-1 levels were included in the models as either a continuous variable (1 SD increment in ln-transformed MCP-1 levels) or categorized in 4 quartiles (1st quartile as reference category) to also assess for potential non-linear associations. We applied three models with different levels of adjustments: model 1 was adjusted for age, sex, and race; model 2 was additionally adjusted for conventional vascular risk factors (hypertension, diabetes mellitus, hypercholesterolemia, body mass index [BMI], smoking [current vs. non-current], estimated glomerular filtration rate [eGFR], coronary artery disease, atrial fibrillation, and heart failure); and model 3 was further adjusted for circulating hsCRP levels on top of these variables. Model 2 was pre-defined as our main model for analyses. In these models, we defined hypertension as a history of physician-diagnosed hypertension, systolic blood pressure (SBP) ≥140 mmHg, diastolic blood pressure (DBP) ≥90 mmHg, or use of one or more antihypertensive medications.27 We defined diabetes mellitus as a history of physician-diagnosed diabetes mellitus, glycosylated hemoglobin type A1C (HbA1c) ≥6.5%, fasting glucose ≥126 mg/dL, random glucose levels ≥200 mg/dL, or use of glucose-lowering medications.28 Hypercholesterolemia was defined as LDL cholesterol levels ≥130 mg/dL, total cholesterol levels ≥200 mg/dL (if LDL cholesterol not available) or use of lipid-lowering drugs,29 and chronic kidney disease as eGFR <60 ml/min/1.73 m2.30 In an alternative model (alternative model 2), we directly adjusted for the components of these definitions instead of the binary variables: thus, instead of hypertension, diabetes mellitus, hypercholesterolemia, and chronic kidney disease, we included SBP (as continuous variable), use of antihypertensive medications, fasting glucose levels (as continuous), use of glucose-lowering medications, LDL cholesterol levels (as continuous), administration of lipid-lowering medications, and eGFR (as continuous).
The purpose of the main models was to explore MCP-1 as a potentially causal risk factor for stroke and not to evaluate the predictive values of its levels. In subsequent models, we aimed to explore whether the association between MCP-1 levels and risk of stroke is independent of the IL-6/CRP pathway that was recently shown to provide an efficient drug target for reducing vascular risk.31 To indirectly examine this, we applied additional adjustments for circulating IL-6 and hsCRP levels. In one model, we included IL-6 on top of age, sex, race, and vascular risk factors, and in a subsequent model we included both IL-6 and hsCRP levels. We did this because CRP is a downstream effector of IL-6, but also comprises a more general marker of inflammation, and thus the alternative adjustments provide different levels of information regarding the involved inflammatory pathways. Data for IL-6 circulating levels were not available in ARIC and the EPIC-Norfolk. Thus, these cohorts were not included in these analyses.
Analyses were conducted separately for any stroke, ischemic stroke, and hemorrhagic stroke. DHS was excluded from the analysis for hemorrhagic stroke, where MCP-1 was examined in quartiles, due to the low numbers of incident events across the quartile categories of MCP-1 levels. The hazard ratios (HR) and the 95% confidence intervals (95%CIs) derived from each study were pooled with random-effects (DerSimonian-Laird) meta-analyses to allow for heterogeneity across studies related to the different baseline characteristics and the different methods of MCP-1 assessment. Heterogeneity across studies was assessed with the I2 and the Cochran’s Q statistic (I2 >50% and p<0.10 were considered statistically significant).
To examine whether the pooled risk estimates were driven by any individual study, we also applied sensitivity analyses by pooling the risk estimates across studies after excluding one study at a time. To explore potential interactions between MCP-1 levels and known cardiovascular risk factors, we performed meta-regression analyses examining how the prevalence of cardiovascular risk factors or the mean or median values of biomarkers, were associated with the risk estimates for stroke in each study. We further performed subgroup analyses by sex, presence of hypertension, presence of diabetes mellitus, and BMI levels (<30 vs. ≥30 kg/m2). Differences in the effect sizes across the subgroup categories were examined by assessing heterogeneity (I2 >50% and p<0.10 were considered statistically significant). Finally, we performed separate analyses for fatal and non-fatal stroke (fatal stroke defined as death occurring within 30 days after the stroke event).
Statistical significance was set at a two-sided p-value <0.05 for the main analysis for any stroke. For the subsequent analysis for stroke subtypes, we corrected for multiple comparisons based on the Bonferroni method (p <0.05/2 stroke subtypes=0.025). Finally, we corrected for multiple comparisons in the descriptive analyses exploring the correlations between MCP-1 levels and baseline variables (threshold for statistical significance at p <0.05/12 variables=0.004). All analyses were conducted with SAS (v9.4) and Stata (v13.0).
RESULTS
Following a systematic review and contact with the lead investigators, six population-based prospective cohort studies contributed previously unpublished data for this meta-analysis. All studies scored high in quality as they fulfilled the full set of Newcastle-Ottawa scale criteria (Online Table II). The baseline characteristics of each study are presented in Table 1. In total, 17,180 individuals (mean age 56.7 ± 8.1 years; 48.8% males), who were stroke-free at baseline, were followed for a mean interval of 16.3 years (range of mean follow-up: 11 to 23 years) with 280,522 person-years at risk. A total of 1,435 incident stroke cases were diagnosed during follow-up, which were classified as ischemic in 1,233 cases and as hemorrhagic in 205 cases. Two hundred twenty-six (15.7%) of the incident stroke events were fatal. Median MCP-1 levels differed between studies possibly reflecting differences in the methods used for MCP-1 quantification (Online Table I). Figure 1 displays associations of standardized MCP-1 levels with conventional vascular risk factors in the pooled sample. We found the following baseline factors to be associated with higher circulating MCP-1 levels: older age, male sex, higher systolic blood pressure, presence of diabetes mellitus, higher LDL cholesterol levels, higher HDL cholesterol levels, higher BMI, current smoking, lower estimated glomerular filtration rate (eGFR), history of coronary artery disease (CAD), higher hsCRP levels, and higher IL-6 levels.
Figure 1.
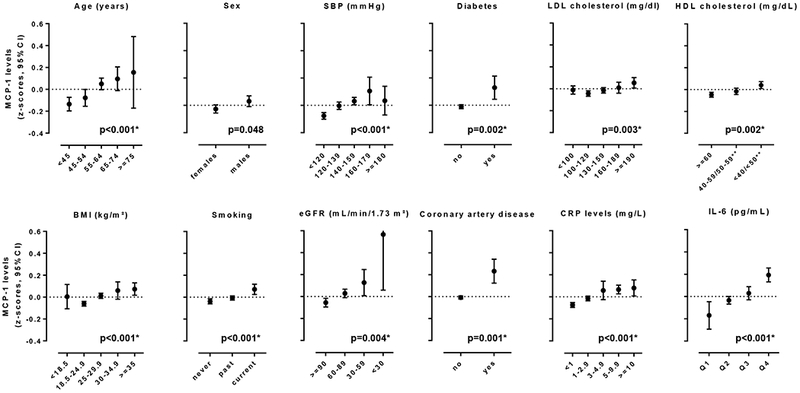
Cross-sectional associations between baseline circulating MCP-1 levels, demographic factors, conventional vascular risk factors, and inflammatory biomarkers. Shown are the results from the pooled sample consisting of six population-based studies.
* statistically significant results (after correction for multiple comparisons statistical significance was set at p <0.05/12=0.004).
** <40 and 40-59 mg/dL for men, <50 and 50-59 mg/dL for women.
Z-score for circulating MCP-1 levels correspond to differences from the mean value of each study. P-values are derived from meta-regression.
Abbreviations: BMI, body mass index; HDL, high-density lipoprotein; hsCRP, high-sensitivity C-reactive protein; eGFR, estimated glomerular filtration rate; LDL, low-density lipoprotein; MCP-1, monocyte chemoattractant protein- 1; SBP, systolic blood pressure.
In the pooled analysis, we found higher MCP-1 levels at baseline to be associated with an increased risk of any stroke both in a model adjusted for age, sex, and race (model 1: HR per 1 SD increment in ln-transformed MCP-1: 1.10, 95%CI: 1.01-1.19, p=0.02) and in the main model further adjusted for vascular risk factors (model 2, HR: 1.07, 95%CI: 1.01-1.14, p=0.03) (Figure 2 and Online Table III). In analyses comparing MCP-1 quartiles, we found the association between MCP-1 levels and risk of stroke to follow a dose-response pattern with a higher risk among individuals in the upper quartiles of circulating MCP-1 levels as compared to the 1st quartile (HRs from model 2: 2nd quartile, 1.16, 95%CI: 0.99-1.36, p=0.07; 3rd quartile 1.31, 95%CI: 1.12-1.53; p=0.001; 4th quartile, 1.33, 95%CI: 1.05-1.68; p=0.008). The results were further stable in a model additionally adjusting for circulating hsCRP levels (model 3 in Figure 2 and Online Table III).
Figure 2.
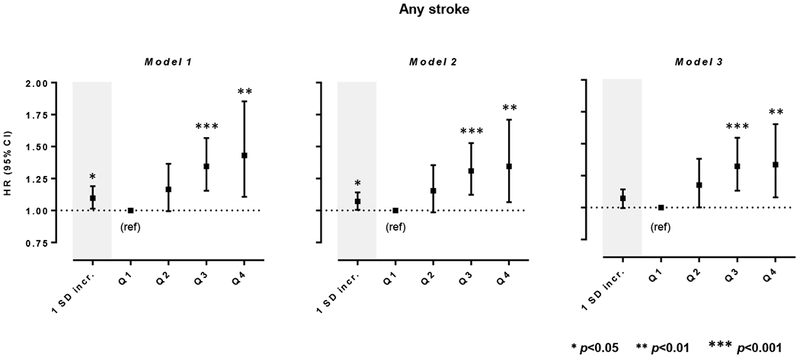
Associations between baseline circulating MCP-1 levels and risk of any stroke. Shown are the results from random-effects meta-analyses of the pooled sample consisting of six population-based studies.Model 1 is adjusted for age, sex, and race. Model 2 is adjusted for age, sex, race, and vascular risk factors including body mass index (1 kg/m2 increment), smoking (current vs. non-current), estimated glomerular filtration rate (1 mL/min/1.73 m2 increment), history of coronary artery disease, diabetes mellitus, hypercholesterolemia, hypertension, atrial fibrillation, and heart failure at baseline. Model 3 is additionally adjusted for circulating high-sensitivity C-reactive protein (hsCRP) levels.
Analyses for 1 SD increment correspond to ln-transformed MCP-1 levels.
We next examined the associations of circulating MCP-1 levels at baseline with stroke subtypes (Figure 3 and Online Table III) and found significant associations of higher MCP-1 levels at baseline with the risk of ischemic stroke (HR per 1 SD increment in ln-MCP-1 from model 2: 1.11, 95%CI: 1.02-1.21, p=0.009), but not with hemorrhagic stroke (model: HR: 1.02, 95%CI: 0.82-1.29, p=0.83). MCP-1 levels in the 2nd, 3rd, and 4th quartiles, as compared to the 1st, were associated with a higher risk for ischemic stroke after adjusting for age, sex, race, and vascular risk factors (model 2, HRs: 2nd quartile, 1.19, 95%CI: 1.00-1.42, p=0.05; 3rd quartile 1.35, 95%CI: 1.14-1.59; p<0.001; 4th quartile, 1.38, 95%CI: 1.07-1.77; p=0.008). The results were highly consistent in the model additionally adjusting for circulating hsCRP levels on top of the vascular risk factors (model 3 in Figure 3 and Online Table IV).
Figure 3.
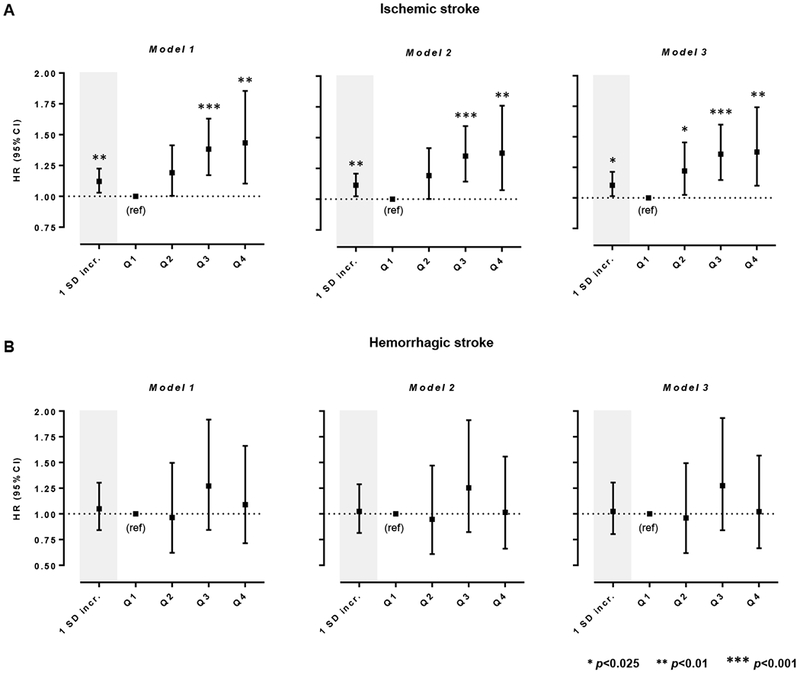
Associations between baseline circulating MCP-1 levels and risk of (A) ischemic stroke and (B) hemorrhagic stroke. Shown are the results from random-effects meta-analyses of the pooled sample consisting of six population-based studies.
* Statistical significance threshold was set at p <0.05/2=0.025 after correction for multiple comparisons (two stroke subtypes).
Model 1 is adjusted for age, sex, and race. Model 2 is adjusted for age, sex, race, and vascular risk factors including body mass index (1 kg/m2 increment), smoking (current vs. non-current), estimated glomerular filtration rate (1 mL/min/1.73 m2 increment), history of coronary artery disease, diabetes mellitus, hypercholesterolemia, hypertension, atrial fibrillation, and heart failure at baseline. Model 3 is additionally adjusted for circulating high-sensitivity C-reactive protein (hsCRP) levels.
Analyses for 1 SD increment correspond to ln-transformed MCP-1 levels.
Study-specific risk estimates are depicted in Online Figures II–IV. There was no evidence of heterogeneity in any of the analyses (I2 <50% and Cochran Q-derived p>0.10), except for moderate heterogeneity in the analysis of the upper 4th MCP-1 quartile for any stroke and ischemic stroke (I2=49.8%; p=0.08 and I2=46.1%; p=0.10, respectively). The results were similar for both fatal and non-fatal stroke (I2=0% for between-subgroup comparisons), although the confidence intervals for fatal stroke were wider probably because of lower statistical power (Online Figure V). The association estimates remained consistent in alternative models directly adjusting for the crude components of vascular risk factors (SBP, fasting glucose levels, LDL cholesterol, eGFR) and use of antihypertensive, glucose-lowering, or lipid-lowering medications (alternative model 2; Online Tables III–V). Furthermore, the results remained stable in sensitivity analyses omitting one study per time (leave-one-out analysis) showing that the results were not driven by any individual study (Online Figures VI–VIII). Meta-regression analyses showed that none of the examined study population characteristics nor the sample source (serum vs. plasma) modified the associations of MCP-1 with the risk of any stroke, ischemic stroke, or hemorrhagic stroke (Online Table VI). Finally, in subgroup analyses stratifying for sex, hypertension, diabetes mellitus, and BMI (≥30 vs. <30 kg/m2) there was no indication for heterogeneity in the risk estimates for any stroke, ischemic stroke, and hemorrhagic stroke between subgroups (I2=0%) (Online Figure IX).
As a last step, we performed analyses with additional adjustments for IL-6 and hsCRP levels in four studies (12,516 individuals; 758 incident stroke events) with available data. Adjustment for IL-6 levels showed that the risk estimates between MCP-1 levels and risk of stroke and stroke subtypes remained stable, although with wider confidence intervals than the main analysis, as would be expected given the smaller sample sizes (Online Table VII). Similarly, simultaneous adjustments for both IL-6 and hsCRP did not alter the risk estimates between MCP-1 and risk of stroke or stroke subtypes, even though both variables were associated with the risk of any stroke and ischemic stroke (Online Table VII).
DISCUSSION
Pooling data from six population-based cohort studies involving 17,180 stroke-free individuals, we found higher circulating levels of MCP-1 at baseline to be associated with a higher long-term risk of stroke after accounting for age, sex, race, and vascular risk factors. In analyses for stroke subtypes, MCP-1 levels were specifically associated with the risk of ischemic stroke, but not with hemorrhagic stroke. These associations followed a dose-response pattern and risk estimates were stable after additional adjustments for serum levels of IL-6 or hsCRP.
Our results, which were obtained in studies with long-term follow-up, confirm and extend our recent Mendelian randomization finding of a higher stroke risk among individuals with genetic predisposition to higher lifetime MCP-1 levels.9 The results were remarkably consistent between the two approaches: with Mendelian randomization the odds ratio for stroke was 1.06 per SD increment in genetically determined MCP-1 levels, which is almost identical to the hazard ratio for incident stroke observed in the current meta-analysis of observational studies. In accord with the Mendelian randomization results, higher MCP-1 levels were further associated with a higher risk of incident ischemic stroke, but not hemorrhagic stroke, which is consistent with the established role of MCP-1 in experimental atherosclerosis. The magnitude of association of MCP-1 with incident ischemic stroke was modest suggesting that MCP-1 measurement is not likely to be of value as a risk marker for stroke although this would need to be formally examined. Of note however, risk estimates compare well with those for lipoprotein (a),32, 33 which is established as a causal risk factor for atherosclerosis currently under investigation in clinical trials.34, 35 When viewed together with the genetic9 and experimental data13–17 our findings provide triangulation of evidence regarding a role of MCP-1 as a causal risk factor for stroke.
Only limited human data exist supporting vascular benefits by reducing inflammation. Secondary analyses from the CANTOS trial showed that the reductions in vascular event rates after IL-1β inhibition were restricted to individuals with a substantial decrease in IL-6 or hsCRP levels.31, 36 Importantly, the risk estimates for stroke by MCP-1 levels in our study remained stable after additional adjustments for the baseline levels of IL-6, hsCRP, and both IL-6 and hsCRP. This observation provides indirect evidence suggesting that elevated levels of MCP-1 might influence risk of stroke independently of the IL-1β/IL-6/CRP axis. Thus, targeting the MCP-1/CCR2 pathway might serve as an alternative anti-inflammatory strategy with independent and complementary effects in reducing vascular event rates on top of current approaches.
Deficiency of either MCP-115, 17 or its receptor CCR216 decreases plaque burden and limits lipid deposition and macrophage infiltration in experimental models of atherosclerosis. Similar effects are observed with pharmacological treatment using MCP-1 competitors13 or CCR2 antagonists.14, 37–39 In contrast, overexpression of MCP-1 promotes oxidized lipid accumulation, macrophage infiltration, and smooth muscle cell proliferation, thus accelerating atheroscleoris.40 To our knowledge, there has been only one small phase II randomized controlled trial in the context of atherosclerosis in humans that targeted the MCP-1/CCR2 axis. Among 108 patients with cardiovascular risk factors and hsCRP levels >3 mg/L, those treated with a single intravenous infusion of MLN1202, a humanized monoclonal antibody against CCR2, exhibited significant reductions in hsCRP levels after 4 weeks and continuing through 12 weeks after dosing.41 However, this study did not assess clinical outcomes, which would need to be examined in a larger trial.41
Our study has several strengths. The pooled analysis was based on a large sample size of >17,000 individuals from six previously unpublished population-based prospective studies with long follow-up intervals and a large number of incident events, thus providing sufficient statistical power to identify robust associations. The included studies fulfilled all of the criteria of quality assessment, which minimized the risk of several sources of bias. We further applied extensive adjustments for demographic and vascular risk factors thus accounting for confounding and enabling the identification of independent associations between MCP-1 levels and risk of stroke. Finally, in four of the cohorts we had available data on IL-6 and hsCRP measurements, which allowed examining the associations between MCP-1 and stroke after adjusting for these biomarkers.
Our study also has limitations. First, the different assays used by individual studies to quantify circulating MCP-1 levels and the different sample sources (plasma vs. serum) resulted in substantial variations in MCP-1 levels between studies. Although our analyses standardized MCP-1 levels across studies, it was not possible to explore associations between absolute MCP-1 values and risk of stroke. Second, studies differed in terms of demographic characteristics and prevalence of vascular risk factors. While we found no evidence of substantial heterogeneity between studies, there was moderate heterogeneity in the analyses for the highest quartiles of MCP-1, which could possibly be explained by the differences in baseline MCP-1 levels and in vascular risk profiles between studies. Third, we could not explore associations between MCP-1 levels and risk of ischemic stroke subtypes (large artery, cardioembolic, small vessel stroke) as information on deeper phenotyping was not available for the majority of studies. Fourth, our analyses were based on predominantly European ancestry individuals, and do thus not necessarily apply to other ethnic groups. Fifth, we cannot exclude residual confounding. Finally, based on our a priori determined approach and power calculations, we corrected for multiple comparisons within each level of analysis but not across all analyses. Although this would not be expected to have any impact on the findings, future studies with even larger sample sizes would be useful in replicating our results
In conclusion, this meta-analysis demonstrates that higher circulating levels of MCP-1 among stroke-free individuals are associated with increased long-term risk of ischemic stroke. The results extend and corroborate experimental and genetic evidence suggesting a key role of MCP-1 in atherosclerosis and stroke. Additional work is needed to examine whether interventions aimed at interfering with MCP-1 signaling would lower stroke risk.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Inflammatory mechanisms contribute to the pathogenesis of vascular disease and inflammatory cytokines have been identified as potential therapeutic targets for lowering vascular risk.
Using genetic data, we recently showed in Mendelian randomization that lifetime higher monocyte-chemoattractant protein 1 (MCP-1) levels are associated with a higher risk of ischemic stroke.
Preclinical studies in animal models of experimental atherosclerosis further suggest a critical role of MCP-1 in the initiation and propagation of atherosclerosis
What New Information Does This Article Contribute?
We performed a meta-analysis of six population-based cohort studies involving 17,000 stroke-free individuals that were followed up for 16 years.
After adjustment for traditional vascular risk factors, higher baseline MCP-1 levels were associated with a higher risk of any stroke and ischemic stroke, but not hemorrhagic stroke over follow-up.
On top of experimental and genetic data, our findings provide additional evidence supporting MCP-1 signaling as a promising target for lowering stroke risk
In view of recent findings suggesting the efficacy of anti-inflammatory approaches in lowering vascular risk, there is a need for identification of specific inflammatory mediators that show promise as potential therapeutic targets. Experimental and genetic evidence suggests MCP-1, a chemokine involved in monocyte recruitment, to play a critical role in atherosclerosis and stroke. Here, we aimed to amplify this concept by exploring in a meta-analysis of 6 previously unpublished cohort studies whether MCP-1 levels are associated with risk of stroke. Following up 17,000 stroke-free individuals for a mean of 16 years, we found baseline MCP-1 levels to be associated with a higher risk of any stroke, independently of traditional vascular risk factors. Across stroke subtypes, there was a significant association of MCP-1 levels with the risk of ischemic stroke, but not hemorrhagic stroke. Adjustments for interleukin-6 (IL-6) and C-reactive protein (CRP) levels did not attenuate these associations, thus indicating that MCP-1 signalling might contribute to stroke risk independently of the well-established IL-6-CRP axis. Along with genetic and experimental data, our findings provide triangulation of evidence suggesting MCP-1-as a causal risk factor for stroke and MCP-1 signaling as a potential therapeutic target.
ACKNOWLEDGEMENTS
The authors thank the staff and participants of all the included studies for their important contributions.
SOURCES OF FUNDING
M. Georgakis is funded by scholarships from the German Academic Exchange Service (DAAD) and Onassis Foundation. The ARIC study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract nos. (HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700005I, HHSN268201700004I). The DHS study was funded by a grant from the Donald W. Reynolds Foundation. The EPIC-Norfolk study is funded by grants from the Medical Research Council UK (G9502233, G0401527) and Cancer Research UK (C864/A8257, C864/A2883). FHS is supported by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195 and No. HHSN268201500001I and 75N92019D00031), received funding by grants from the National Institute of Aging (R01s AG054076, AG049607, AG059421, U01-AG049505, AG058589 and AG052409) and the National Institute of Neurological Disorders and Stroke (R01 NS017950, UH2 NS100605), as well as grants for the MCP-1 measurements by NIH (1RO1 HL64753, R01 HL076784, 1 R01 AG028321). The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. The MDCS-CV study has been supported with funding from the Swedish Research Council, Swedish Heart and Lung Foundations, and the Swedish Foundation for Strategic Research. This project has received funding from the European Union’s Horizon 2020 research and innovation programme (No 666881), SVDs@target (to M. Dichgans) and No 667375, CoSTREAM (to M. Dichgans); the DFG as part of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) and the CRC 1123 (B3) (to M. Dichgans); the Corona Foundation (to M. Dichgans); the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain)(to M. Dichgans); the e:Med program (e:AtheroSysMed) (to M. Dichgans) and the FP7/2007-2103 European Union project CVgenes@target (grant agreement number Health-F2-2013-601456) (to M. Dichgans).
The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
DISCLOSURES
Dr. de Lemos reports research grants from Abbott Diagnostics and Roche Diagnostics, consulting from Ortho Clinical Diagnostics and Jannsen, honoraria for steering committee membership from Amgen, and DSMB membership from Novo Nordisc and Regeneron. Dr. Koenig reports personal fees for consulting from AstraZeneca, Novartis, DalCor, Kowa, Amgen, and Sanofi, grants and non-financial support from Roche Diagnostics, Beckmann, Singulex, and Abbott. Dr. Ayers reports statistical consulting fees from the National Institutes of Health. Dr. Hoogeveen reports research grants and personal fees for consulting from Denka Seiken. The other authors have nothing to disclose.
Nonstandard Abbreviations and Acronyms:
- ARIC
Atherosclerosis Risk in Communities
- BMI
body mass index
- CCL2
CC-chemokine ligand 2
- DHS
Dallas Heart Study
- eGFR
estimated glomerular filtration rate
- EPIC
European Prospective Investigation of Cancer
- FHS
Framingham Heart Study
- HR
hazard ratio
- hsCRP
high-sensitivity C-reactive protein
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- LDL-C
low-density lipoprotein cholesterol
- KORA
Kooperative Gesundheitsforschung in der Region Augsburg
- MONICA
Monitoring of Trends and Determinants in Cardiovascular Disease
- MCP-1
monocyte-chemoattractant protein-1
- MDCS
Malmö Diet and Cancer Study
- SBP
systolic blood pressure
REFERENCES
- 1.DALYs GBD, Collaborators H. Global, regional, and national disability-adjusted life-years (dalys) for 315 diseases and injuries and healthy life expectancy (hale), 1990-2015: A systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1603–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortality GBD, Causes of Death C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: A systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1459–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esenwa CC, Elkind MS. Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol. 2016;12:594–604 [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on A. Inflammation in atherosclerosis: From pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Cantos Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, Hasan A, Rosenberg Y, Iturriaga E, Gupta M, Tsigoulis M, Verma S, Clearfield M, Libby P, Goldhaber SZ, Seagle R, Ofori C, Saklayen M, Butman S, Singh N, Le May M, Bertrand O, Johnston J, Paynter NP, Glynn RJ, Investigators C. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. 2019;380:752–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ridker PM. Anticytokine agents: Targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ Res. 2019;124:437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aday AW, Ridker PM. Targeting residual inflammatory risk: A shifting paradigm for atherosclerotic disease. Front Cardiovasc Med. 2019;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgakis MK, Gill D, Rannikmae K, Traylor M, Anderson CD, Lee JM, Kamatani Y, Hopewell JC, Worrall BB, Bernhagen J, Sudlow CLM, Malik R, Dichgans M. Genetically determined levels of circulating cytokines and risk of stroke. Circulation. 2019;139:256–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin J, Kakkar V, Lu X. Impact of mcp-1 in atherosclerosis. Curr Pharm Des. 2014;20:4580–4588 [DOI] [PubMed] [Google Scholar]
- 11.Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutgens E, Faber B, Schapira K, Evelo CT, van Haaften R, Heeneman S, Cleutjens KB, Bijnens AP, Beckers L, Porter JG, Mackay CR, Rennert P, Bailly V, Jarpe M, Dolinski B, Koteliansky V, de Fougerolles T, Daemen MJ. Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: Validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation. 2005;111:3443–3452 [DOI] [PubMed] [Google Scholar]
- 13.Liehn EA, Piccinini AM, Koenen RR, Soehnlein O, Adage T, Fatu R, Curaj A, Popescu A, Zernecke A, Kungl AJ, Weber C. A new monocyte chemotactic protein-1/chemokine cc motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. 2010;56:1847–1857 [DOI] [PubMed] [Google Scholar]
- 14.Bot I, Ortiz Zacarias NV, de Witte WE, de Vries H, van Santbrink PJ, van der Velden D, Kroner MJ, van der Berg DJ, Stamos D, de Lange EC, Kuiper J, AP IJ, Heitman LH. A novel ccr2 antagonist inhibits atherogenesis in apoe deficient mice by achieving high receptor occupancy. Sci Rep. 2017;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281 [DOI] [PubMed] [Google Scholar]
- 16.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in ccr2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897 [DOI] [PubMed] [Google Scholar]
- 17.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of ccl2, cx3cr1, and ccr5 abrogates ly6c(hi) and ly6c(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657 [DOI] [PubMed] [Google Scholar]
- 18.de Lemos JA, Morrow DA, Sabatine MS, Murphy SA, Gibson CM, Antman EM, McCabe CH, Cannon CP, Braunwald E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation. 2003;107:690–695 [DOI] [PubMed] [Google Scholar]
- 19.de Lemos JA, Morrow DA, Blazing MA, Jarolim P, Wiviott SD, Sabatine MS, Califf RM, Braunwald E. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: Results from the a to z trial. J Am Coll Cardiol. 2007;50:2117–2124 [DOI] [PubMed] [Google Scholar]
- 20.Hoogeveen RC, Morrison A, Boerwinkle E, Miles JS, Rhodes CE, Sharrett AR, Ballantyne CM. Plasma mcp-1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis risk in communities study. Atherosclerosis. 2005;183:301–307 [DOI] [PubMed] [Google Scholar]
- 21.Deo R, Khera A, McGuire DK, Murphy SA, Meo Neto Jde P, Morrow DA, de Lemos JA. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44:1812–1818 [DOI] [PubMed] [Google Scholar]
- 22.van Wijk DF, van Leuven SI, Sandhu MS, Tanck MW, Hutten BA, Wareham NJ, Kastelein JJ, Stroes ES, Khaw KT, Boekholdt SM. Chemokine ligand 2 genetic variants, serum monocyte chemoattractant protein-1 levels, and the risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010;30:1460–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoamanesh A, Preis SR, Beiser AS, Kase CS, Wolf PA, Vasan RS, Benjamin EJ, Seshadri S, Romero JR. Circulating biomarkers and incident ischemic stroke in the framingham offspring study. Neurology. 2016;87:1206–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herder C, Baumert J, Thorand B, Martin S, Lowel H, Kolb H, Koenig W. Chemokines and incident coronary heart disease: Results from the monica/kora augsburg case-cohort study, 1984-2002. Arterioscler Thromb Vasc Biol. 2006;26:2147–2152 [DOI] [PubMed] [Google Scholar]
- 25.Schiopu A, Bengtsson E, Goncalves I, Nilsson J, Fredrikson GN, Bjorkbacka H. Associations between macrophage colony-stimulating factor and monocyte chemotactic protein 1 in plasma and first-time coronary events: A nested case-control study. J Am Heart Assoc. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wells GA, Shea B, O’Connell D, Peterson J, Welch V, Losos M. The newcastle-ottawa scale (nos) for assessing the quality if nonrandomized studies in meta-analyses. . 2014;2018
- 27.Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, Kahan T, Mahfoud F, Redon J, Ruilope L, Zanchetti A, Kerins M, Kjeldsen SE, Kreutz R, Laurent S, Lip GYH, McManus R, Narkiewicz K, Ruschitzka F, Schmieder RE, Shlyakhto E, Tsioufis C, Aboyans V, Desormais I, Group ESCSD. 2018 esc/esh guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–3104 [DOI] [PubMed] [Google Scholar]
- 28.American Diabetes A 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2019. Diabetes Care. 2019;42:S13–S28 [DOI] [PubMed] [Google Scholar]
- 29.Jellinger PS, Handelsman Y, Rosenblit PD, Bloomgarden ZT, Fonseca VA, Garber AJ, Grunberger G, Guerin CK, Bell DSH, Mechanick JI, Pessah-Pollack R, Wyne K, Smith D, Brinton EA, Fazio S, Davidson M. American association of clinical endocrinologists and american college of endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract. 2017;23:1–87 [DOI] [PubMed] [Google Scholar]
- 30.Stevens PE, Levin A, Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group M. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158:825–830 [DOI] [PubMed] [Google Scholar]
- 31.Ridker PM, Libby P, MacFadyen JG, Thuren T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett BM, Glynn RJ. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the canakinumab anti-inflammatory thrombosis outcomes study (cantos). Eur Heart J. 2018;39:3499–3507 [DOI] [PubMed] [Google Scholar]
- 32.Emerging Risk Factors C, Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR, Schwartz GG, Olsson AG, Colhoun HM, Kronenberg F, Drechsler C, Wanner C, Mora S, Lesogor A, Tsimikas S. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392:1311–1320 [DOI] [PubMed] [Google Scholar]
- 34.Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57:1953–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khera AV, Kathiresan S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Cantos Trial Group. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet. 2018;391:319–328 [DOI] [PubMed] [Google Scholar]
- 37.Winter C, Silvestre-Roig C, Ortega-Gomez A, Lemnitzer P, Poelman H, Schumski A, Winter J, Drechsler M, de Jong R, Immler R, Sperandio M, Hristov M, Zeller T, Nicolaes GAF, Weber C, Viola JR, Hidalgo A, Scheiermann C, Soehnlein O. Chrono-pharmacological targeting of the ccl2-ccr2 axis ameliorates atherosclerosis. Cell Metab. 2018;28:175–182 e175 [DOI] [PubMed] [Google Scholar]
- 38.Okamoto M, Fuchigami M, Suzuki T, Watanabe N. A novel c-c chemokine receptor 2 antagonist prevents progression of albuminuria and atherosclerosis in mouse models. Biol Pharm Bull. 2012;35:2069–2074 [DOI] [PubMed] [Google Scholar]
- 39.Yamashita T, Kawashima S, Ozaki M, Namiki M, Inoue N, Hirata K, Yokoyama M. Propagermanium reduces atherosclerosis in apolipoprotein e knockout mice via inhibition of macrophage infiltration. Arterioscler Thromb Vasc Biol. 2002;22:969–974 [DOI] [PubMed] [Google Scholar]
- 40.Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, Milos PM. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–1525 [DOI] [PubMed] [Google Scholar]
- 41.Gilbert J, Lekstrom-Himes J, Donaldson D, Lee Y, Hu M, Xu J, Wyant T, Davidson M, Group MLNS. Effect of cc chemokine receptor 2 ccr2 blockade on serum c-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol. 2011;107:906–911 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.